

Abstract
In the yeast Saccharomyces cerevisiae, the ScGDH1 and ScGDH3 encoded glutamate dehydrogenases (NADP‐GDHs) catalyze the synthesis of glutamate from ammonium and α‐ketoglutarate (α‐KG). Previous kinetic characterization showed that these enzymes displayed different allosteric properties and respectively high or low rate of α‐KG utilization. Accordingly, the coordinated action of ScGdh1 and ScGdh3, regulated balanced α‐KG utilization for glutamate biosynthesis under either fermentative or respiratory conditions, safeguarding energy provision. Here, we have addressed the question of whether there is a correlation between the regulation and kinetic properties of the NADP‐GDH isozymes present in S. cerevisiae (ScGdh1 and ScGdh3), Kluyveromyces lactis (KlGdh1), and Lachancea kluyveri (LkGdh1) and their evolutionary history. Our results show that the kinetic properties of K. lactis and L. kluyveri single NADP‐GDHs are respectively similar to either ScGDH3 or ScGDH1, which arose from the whole genome duplication event of the S. cerevisiae lineage, although, KlGDH1 and LkGDH1 originated from a GDH clade, through an ancient interspecies hybridization event that preceded the divergence between the Saccharomyces clade and the one containing the genera Kluyveromyces, Lachancea, and Eremothecium. Thus, the kinetic properties which determine the NADP‐GDHs capacity to utilize α‐KG and synthesize glutamate do not correlate with their evolutionary origin.
Keywords: functional diversification, glutamate dehydrogenase, kinetics, paralogous enzymes, phylogeny, yeast gene duplication
1. Introduction
Two pathways determine glutamate biosynthesis in fungi: the NADP‐dependent glutamate dehydrogenase (NADP‐GDH) and the concerted action of glutamine synthetase (GS) and glutamate synthase (GOGAT) (Magasanik, 2003). These enzymes assimilate ammonium into glutamate and glutamine, whose amino groups are subsequently distributed to other compounds. The five‐carbon skeleton of these amino acids derives from α‐ketoglutarate (α‐KG), an intermediate of the tricarboxylic acid cycle. Thus, glutamate biosynthesis represents a crucial intersection of carbon and nitrogen metabolism and, as such, its regulation must balance biosynthetic needs and energy production (DeLuna, Avendano, Riego, & Gonzalez, 2001). Redox homeostasis and defense against oxidative stress are also influenced by glutamate biosynthesis since this amino acid is a glutathione precursor (Guillamon, van Riel, Giuseppin, & Verrips, 2001; Lee, Kim, Kang, Kim, & Maeng, 2012).
Although the relative contribution of the two glutamate‐producing pathways to the biosynthesis of this amino acid varies among species and growth conditions, it has been reported that NADP‐GDH (1.4.1.4) plays a leading role in Schizosaccharomyces pombe, Aspergillus nidulans, Neurospora crassa, and Saccharomyces cerevisiae grown on ammonium as sole nitrogen source, in which inactivation of the NADP‐GDH encoding genes, dramatically reduces growth rate (Fincham, 1951; Macheda, Hynes, & Davis, 1999; Magasanik, 2003; Perysinakis, Kinghorn, & Drainas, 1994). Modulation of NADP‐GDH activity in vivo coordinates metabolic fluxes according to modifications in the availability of nitrogen and carbon sources and contributes to the maintenance of an equilibrated redox state. In Candida albicans, S. pombe, and several Aspergillus species, this modulation involves rate of α‐ ketoglutarate (KG) utilization through allosteric regulation and cooperative kinetics (Holmes, Collings, Farnden, & Shepherd, 1989; Noor & Punekar, 2005; Perysinakis et al., 1994), whereas in Candida tropicalis, Candida pseudotropicalis, Candida parapsilosis, Debaryomyces hansenii, and Aspergillus terreus, NADP‐GDH hyperbolic kinetics determines intermediate utilization (Alba‐Lois et al., 2004; Choudhury & Punekar, 2009; Holmes et al., 1989).
S. cerevisiae has two NADP‐GDH isoforms namely ScGdh1 and ScGdh3 (Avendano, Deluna, Olivera, Valenzuela, & Gonzalez, 1997). ScGdh1 shows hyperbolic kinetics for α‐KG saturation and is the predominant isoform under exponential growth on glucose and when acetate plus raffinose are used as carbon sources (DeLuna et al., 2001; Tang, Sieg, & Trotter, 2011). While ScGdh3 is a cooperative enzyme displaying sigmoidal kinetics for α‐KG utilization, this isoform contributes significantly to NADP‐GDH activity during growth on ethanol as sole carbon source (Avendano et al., 2005; DeLuna et al., 2001) and becomes the predominant isoform during stationary phase (Lee et al., 2012). Accordingly, transcription of the ScGDH3 gene is strongly induced during growth on ethanol and is nearly absent on glucose. This carbon‐mediated regulation is overimposed to the transcriptional activation by low nitrogen availability (Avendano et al., 2005). Although transcription of the ScGDH1 gene is not repressed on ethanol, the relative contribution of the ScGdh1 enzyme to the overall NADP‐GDH activity is much lower than that of ScGdh3 under this condition (DeLuna et al., 2001; Riego, Avendano, DeLuna, Rodríguez, & González, 2002). It is worth mentioning that the NADP‐GDHs are not involved in glutamate catabolism, instead, the NAD‐dependent glutamate dehydrogenase (1.4.1.2) catalyzes the deamination of glutamate to ammonium and α‐KG in yeast (Miller & Magasanik, 1990).
It has been proposed that ScGdh1 and ScGdh3 kinetic differences control α‐KG utilization for biosynthetic purposes without compromising flux trough the tricarboxylic acid cycle for energy production during growth on ethanol as sole carbon source (DeLuna et al., 2001). The non‐redundant roles of ScGdh1 and ScGdh3 may be the result of an evolutionary process in which duplication of an ancestral gene and divergence of the resulting paralogous led to specialization in glutamate production under different conditions associated with the peculiar facultative metabolism of S. cerevisiae (Avendano et al., 2005).
It has been proposed that in the S. cerevisiae lineage, a whole genome duplication (WGD) event took place (Wolfe & Shields, 1997) and that a selected group of the resulting duplicated genes have been retained in two copies among which are the paralogous ScGDH1 and ScGDH3 genes (Seoighe & Wolfe, 1999). However, the evolutionary studies of the fungal NADP‐GDHs have not addressed the characteristics of the pre‐WGD ancestral‐type genes which did not originate through WGD, and those present in the Saccharomycetes, which arose through WGD. The Saccharomycetales (or Hemyascomycetes) group includes species closely related to S. cerevisiae for which the genome sequence and genetic manipulation resources are available, representing a valuable tool for functional evolutionary studies. The yeasts Kluyveromyces lactis and Lachancea kluyveri descend from the pre‐WGD ancestor, and have a single NADP‐GDH‐encoding gene, suggesting that no sporadic duplications have occurred in this gene. With regard to the carbon metabolism operating in these yeasts, it is evident that each one shows different levels of adaptation to the fermentative lifestyle: K. lactis metabolism is constitutively respiratory, for this reason, it cannot grow anaerobically and does not produce respiratory‐deficient mutants (Breunig et al., 2000). L. kluyveri displays an intermediate fermentative capacity between K. lactis and S. cerevisiae, it can grow anaerobically and produce respiratory‐deficient mutants on sugar‐rich media, but it only ferments in the absence of oxygen (Moller, Olsson, & Piskur, 2001; Moller et al., 2002), whereas in S. cerevisiae fermentative metabolism predominates whenever high sugar concentration is available regardless of oxygen disponibility. It even represses respiratory metabolism in the presence of high glucose or fructose concentration, through carbon catabolite repression (Gancedo, 1998). This yeast can grow anaerobically and produce respiratory‐deficient mutants (Gancedo, 1998). One of the most prominent features of baker′s yeast is the rapid conversion of sugars to ethanol and carbon dioxide under both anaerobic an aerobic conditions; this phenomenon is called Crabtree effect (Hagman, Säll, & Piskur, 2014) and is present in yeast species well adapted to the fermentative life style (Pfeiffer & Morley,2014). According to this classification, S. cerevisiae and L. kluyveri are Crabtree positive, whereas K. lactis is Crabtree negative.
This work addresses the question of whether the evolutionary origin of S. cerevisiae ScGdh1 and ScGdh3 NADP‐GDH and their corresponding orthologs in K. lactis and L. kluyveri has influenced their kinetic and transcriptional regulation. Our results show that such regulation does not correlate with the evolutionary origin of the corresponding genes, confirming that gene duplication and further functional diversification play a key role in metabolic remodeling and evolution, regardless of the origin of paralogous gene pair.
2. Experimental Procedures
2.1. Strains
Table 1 describes the characteristics of the strains used in the present work. All strains constructed for this study were derivatives of CLA1 (ura3 leu2), Lk156‐1 (ura3) or KlWM37‐1 (his3 ura3) for S. cerevisiae, L. kluyveri and K. lactis, respectively. Mutants in Scgdh1Δ::kanMx4 (CLA2), Scgdh3Δ::LEU2 (CLA3), Scglt1Δ::URA3 (CLA4), Scgdh1Δ::kanMx4 Scgdh3Δ::LEU2 (CLA5), Scgdh1Δ::kanMx4 Scgdh3Δ::LEU2 Scglt1Δ::URA3 (CLA6) and Klglt1Δ::kanMx4 (KlWM37‐3) have been previously described (Avendano et al., 1997; DeLuna et al., 2001; Valenzuela et al., 1995).
Table 1.
S. cerevisiae, L. kluyveri, and K. lactis strains used in this work
| Strain | Relevant genotype | Source |
|---|---|---|
| CLA1 | MATa ScGDH1 ScGDH3 ScGLT1 ura3 leu2 | Avendano et al., 1997 |
| CLA2 | MATa Scgdh1Δ::kanMx4 ScGDH3 ScGLT1 ura3 leu2 | DeLuna et al., 2001 |
| CLA3 | MATa ScGDH1 Scgdh3Δ::LEU2 ScGLT1 ura3 | Avendano et al., 1997 |
| CLA4 | MATa ScGDH1 ScGDH3 Scglt1Δ::URA3 leu2 | Avendano et al., 1997 |
| CLA5 | MATa Scgdh1Δ::kanMx4 Scgdh3Δ::LEU2 ScGLT1 ura3 | DeLuna et al., 2001 |
| CLA6 | MATa Scgdh1Δ::kanMx4 Scgdh3Δ::LEU2 Scglt1Δ::URA3 | DeLuna et al., 2001 |
| CLA1‐1 | CLA1/pRS416 leu2 | This study |
| CLA2‐1 | CLA2/pRS416 leu2 | This study |
| CLA2‐2 | CLA2/pRS416‐ScGDH1 leu2 | This study |
| CLA2‐3 | CLA2/pRS416‐ScGDH3 leu2 | This study |
| CLA2‐4 | CLA2/pRS416‐LkGDH1 leu2 | This study |
| CLA2‐5 | CLA2/pRS416‐KlGDH1 leu2 | This study |
| CLA5‐1 | CLA5/pRS416 | This study |
| CLA5‐2 | CLA5/pRS416‐ScGDH1 | This study |
| CLA5‐3 | CLA5/pRS416‐ScGDH3 | This study |
| CLA5‐4 | CLA5/pRS416‐LkGDH1 | This study |
| CLA5‐5 | CLA5/pRS416‐KlGDH1 | This study |
| Lk156‐1 | Mata LkGDH1 LkGLT1 ura3 | Montalvo, J. et al. 2015 |
| Lk156‐2 | Mata Lkgdh1Δ::kanMx4 LkGLT1 ura3 | This study |
| Lk156‐3 | Mata LkGDH1 Lkglt1Δ::natMx4 ura3 | This study |
| Lk156‐4 | Mata Lkgdh1Δ::kanMx4 Lkglt1Δ::natMx4 ura3 | This study |
| Lk156‐1‐1 | Lk156‐1/pLk‐EE | This study |
| Lk156‐2‐1 | Lk156‐2/pLk‐EE | This study |
| Lk156‐2‐2 | Lk156‐2/pLk‐EE ‐ScGDH1 | This study |
| Lk156‐2‐3 | Lk156‐2/pLk‐EE ‐ScGDH3 | This study |
| Lk156‐2‐4 | Lk156‐2/pLk‐EE ‐LkGDH1 | This study |
| Lk156‐2‐5 | Lk156‐2/pLk‐EE ‐KlGDH1 | This study |
| KlWM37‐1 | Mata KlGDH1 KlGLT1 his3 ura3 | Valenzuela et al., 1995; |
| KlWM37‐2 | Mata Klgdh1Δ::natMx4 KlGLT1 his3 ura3 | This study |
| KlWM37‐3 | Mata KlGDH1 Klglt1Δ::kanMx4 his3 ura3 | Valenzuela et al., 1995 |
| KlWM37‐4 | Mata Klgdh1Δ::natMx4, Klglt1Δ::kanMx4 his3 ura3 | This study |
| KlWM37‐1‐1 | KlWM37‐1/ YEpKD352 his3 | This study |
| KlWM37‐2‐1 | KlWM37‐2/ YEpKD352 his3 | This study |
| KlWM37‐2‐2 | KlWM37‐2/ YEpKD352‐ScGDH1 his3 | This study |
| KlWM37‐2‐3 | KlWM37‐2/ YEpKD352‐ScGDH3 his3 | This study |
| KlWM37‐2‐4 | KlWM37‐2/ YEpKD352‐LkGDH1 his3 | This study |
| KlWM37‐2‐5 | KlWM37‐2/ YEpKD352‐KlGDH1 his3 | This study |
The L. kluyveri Lkgdh1Δ (Lk156‐2) mutant strain was obtained by replacing the ORF of LkGDH1 with the selectable marker kanMx4. The LkGDH1 gene was replaced by homologous recombination using a module containing the kanMX4 cassette (1469 bp) flanked by 1067 bp of 5′UTR (−1074 to −7) and 1146 bp of 3′UTR (+1368 to +2514) sequences of LkGDH1. This module (3320 bp) was amplified by overlapped extension PCR with deoxyoligonucleotides 107 and 108 (−979 to +2341) using a template built up by three independent modules: (1) the LkGDH1 5′UTR amplified using the 101 and 102 deoxyoligonucleotides and genomic DNA from strain Lk156‐1 as a template, (2) the kanMX4 module which was amplified from the pFA6a plasmid using deoxyoligonucleotides 105 and 106, and (3) the LkGDH1 3′UTR amplified using deoxyoligonucleotides 103 and 104 and genomic DNA from strain Lk156‐1 as a template. The PCR product was transformed into the Lk156‐1 strain. Transformants were selected for G418 resistance (200 μg ml−1). Deoxyoligonucleotides 108‐1 and 108‐2 were used to verify the construction Lkgdh1Δ::kanMx4, these primers generated a module of 1517 bp (+216 of kanMx4 to +2648 of 3′UTR of LkGDH1) using genomic DNA of the G418‐resistant transformants as a template. The deoxyoligonucleotides sequences are indicated in Table S1.
The L. kluyveri Lkglt1Δ (Lk156‐3) mutant strain was obtained by replacing the ORF of LkGLT1 with the selectable marker natMx4. The LkGLT1 gene was replaced by homologous recombination using a module containing the natMX4 cassette (1477 bp) flanked by 1005 bp of 5′UTR (−1006 to −1) and 1006 bp of 3′UTR (+6438 to +7444) sequences of LkGLT1. This module (3488 bp) was amplified by overlapped extension PCR with deoxyoligonucleotides 111 and 114 using a template built up by three independent modules: (1) the LkGLT1 5′UTR amplified using 111 and 112 deoxyoligonucleotides, (2) the natMx4 module flanked by homologous regions of the 5′UTR and 3′UTR of the LkGLT1 gene, which was amplified from p4339 plasmid using deoxyoligonucleotides 109 and 110, and (3) the LkGLT1 3′UTR amplified using deoxyoligonucleotides 113 and 114. The PCR product was transformed into the Lk156‐1 strain. Transformants were selected for nourseothricin resistance (100 μg ml−1). Deoxyoligonucleotides 115 and 116 were used to verify the construction Lkglt1Δ::natMx4, these deoxyoligonucleotides generated a module of 1012 bp (+875 of natMx4 to +1887 of 3′UTR of LkGLT1) using genomic DNA of the nourseothricin‐resistant transformants as a template.
To generate the L. kluyveri (Lk156‐4) double‐mutant strain, the above described Lkgdh1Δ::kanMx4 cassette was transformed into the Lk156‐3 strain. Transformants were selected for G418 (geneticin) (200 μg ml−1) and nourseothricin resistance (100 μg ml−1). Deoxyoligonucleotides 108‐1 and 108‐2 were used to verify the construction as described above for the Lk156‐2 mutant strain using genomic DNA of the G418 and nourseothricin‐resistant transformants as a template.
K. lactis Klgdh1Δ (KlWM37‐2) mutant strain was obtained by replacing the ORF of KlGDH1 with the selectable marker natMx4 . The KlGDH1 gene was replaced by homologous recombination using a module containing the natMx4 cassette (1578 bp) flanked by 1000 bp of 5′UTR (−1000 to −1) and 997 bp of 3′UTR (+1386 to +2383) sequences of KlGDH1. This module (3575 bp) was amplified by overlapped extension PCR with deoxyoligonucleotides 119 and 122 using a template built up by three independent modules: (1) the KlGDH1 5′UTR amplified using 119 and 120 deoxyoligonucleotides, (2) the natMx4 module flanked by homologous regions of the 5′UTR and 3′UTR of the KlGDH1 gene, which was amplified from p4339 plasmid using deoxyoligonucleotides 117 and 118, and (3) the KlGDH1 3′UTR amplified using deoxyoligonucleotides 121 and 122. The PCR product was transformed into the KlWM37‐1 strain. Transformants were selected for nourseothricin resistance (100 μg ml−1). Deoxyoligonucleotides 123 and 124 were used to verify the construction Klgdh1Δ::natMx4, these deoxyoligonucleotides generated a module of 2030 bp (−277 of 5′UTR of KlGDH1 to +1670 of 3′UTR of KlGDH1) using genomic DNA of the nourseothricin‐resistant transformants as a template.
To generate the K. lactis Klgdh1Δ Klglt1Δ double mutant strain (KlWM37‐4), the above described Klgdh1Δ::natMx4 cassette was transformed into the KlWM37‐3 strain (Valenzuela et al., 1995). Transformants were selected for G418 (geneticin) (200 μg ml−1) and nourseothricin resistance (100 μg ml−1). Deoxyoligonucleotides 123 and 124 were used to verify the construction as described above for the KlWM37‐2 mutant strain using genomic DNA of G418 and nourseothricin‐resistant transformants as a template.
2.2. Growth conditions
Strains were routinely grown on minimal medium (MM) containing salts, trace elements, and vitamins following the formula of yeast nitrogen base (Difco). Sterilized glucose (2%, w/v) or ethanol (2%, w/v) was used as a carbon source. A quantity of 40 mmol/L ammonium sulfate or 5 mmol/L glutamate was used as a nitrogen source. Supplements needed to satisfy auxotrophic requirements were added at 0.1 mg ml−1. Cells were incubated at 30°C with shaking (250 rpm). Growth was monitored by measuring optical density at 600 nm (Thermo Fisher Scientific, Genesys 20 model 4001/4 spectrophotometer).
2.3. Construction of Plasmids Bearing the ScGDH1, ScGDH3, LkGDH1, or KlGDH1 Genes
All standard molecular biology techniques were followed as previously described (Sambrook, Fritsch, & Maniatis, 1989). The ScGDH1, ScGDH3, LkGDH1, and KlGDH1 genes were PCR amplified together with their 5′ promoter sequence and cloned into either the pRS416 (CEN6‐ARSH4‐URA3), pLk‐EE (CEN6‐ARSH4‐LkURA3) (provided by Dr. Lina Riego‐Ruiz), or YEpKD352 (pKD1 ori‐KlURA3), respectively (Colon et al., 2011; Wach, Brachat, Pohlmann, & Philippsen, 1994). Cloning into the pRS416 plasmid was made as follows: for the ScGDH1 gene, a 2514 bp region between −850 bp upstream the start codon and +275 bp downstream the stop codon was amplified with deoxyoligonucleotides 133 and 134 using genomic DNA from the S. cerevisiae (CLA1) WT strain as a template; for the ScGDH3 gene, a 2466 bp region between −780 bp upstream the start codon and +288 bp downstream the stop codon was amplified with deoxyoligonucleotides 135 and 136 using genomic DNA from the S. cerevisiae (CLA1) WT strain as a template; for the LkGDH1 gene, a 2611 bp region between −920 bp upstream the start codon and +281 bp downstream the stop codon was amplified with deoxyoligonucleotides 137 and 138 using genomic DNA from the L. kluyveri (Lk156‐1) WT strain as a template; and for KlGDH1, a 2532 bp region between −874 bp upstream the start codon and +248 bp downstream the stop codon was amplified with deoxyoligonucleotides 139 and 140 using genomic DNA from the K. lactis (KlWM37‐1) WT strain as a template. The PCR products and pRS416 plasmid were digested with restriction enzymes (BamHI/XhoI for ScGDH1 and ScGDH3, BamHI/SacI for LkGDH1 and BamHI/XbaI for KlGDH1) and after gel purification were ligated.
Cloning into the pLk‐EE plasmid was made following the same strategy: the ScGDH1, ScGDH3, LkGDH1, and KlGDH1 genes products were (BamHI/XhoI, BamHI/SacI, BamHI/XbaI, and BamHI/SmaI, respectively) digested and after gel purification were ligated. And for cloning into the YEpKD352 plasmid, the ScGDH1, ScGDH3, LkGDH1 and KlGDH1 genes products were (BamHI/SacI, BamHI/SmaI, BamHI/XbaI and BamHI/XhoI, respectively) digested and after gel purification were ligated. The cloned genes were sequenced to check ORF integrity and after were transformed into the CLA2, CLA5, Lk156‐2, and KlWM37‐2 strains as indicated in Table 1. Yeast strains (S. cerevisiae, L. kluyveri, and K. lactis) were transformed following a previously described method (Ito, Fukuda, Murata, & Kimura, 1983). Transformants were selected for uracil prototrophy on MM.
2.4. NADP‐GDH purification
2.4.1. Cloning and expression
The ScGDH1 and ScGDH3 genes were PCR amplified using the deoxyoligonucleotides pairs 125/126 and 127/128, respectively, using genomic DNA of the CLA1 WT strain as a template. PCR products and the pET‐28a(+) plasmid were NheI/XhoI digested and after gel purification were ligated. The LkGDH1 gene was amplified with the deoxyoligonucleotides 129 and 130 using genomic DNA of the Lk156‐1 WT strain as a template. PCR product and the pET‐28a(+) plasmid were NdeI/BamHI digested and after gel purification were ligated. The KlGDH1 gene was amplified with the deoxyoligonucleotides 131 and 132 using genomic DNA of the KlWM37‐1 WT strain as a template. PCR product and the pET‐28a(+) plasmid were NheI/BamHI digested and after gel purification ligated.
Ligations were transformed into the DH5α E. coli strain. After plasmid purification, correct cloning was verified by sequencing. For heterologous expression, the BL21 E. coli strain was transformed. Selected clones were grown in LB medium supplemented with 30 μg ml−1 of kanamycin incubated at 37°C with shaking (250 rpm). When the cultures reached an OD of 0.6 at 600 nm, the expression of the proteins was induced with 100 μmol/L of IPTG (Iso‐Propil‐Tio‐Galactoside), incubated 4 hr at 30°C with shaking (250 rpm), harvested by centrifugation at 1100g for 15 min, and the cellular pellet was stored at −70°C until used.
2.5. Whole cell soluble protein extract
Cells were thawed and resuspended in 20 ml of 30 mmol/L imidazol, 1 mmol/L EDTA, 1 mmol/L dithiothreitol, 1 mmol/L phenylmethylsulfonylfluoride (PMSF). Protein extracts were obtained by sonication (Ultrasonic Processor Model: VCX 130) with a tip sonicator keeping the tubes on ice; five cycles (60% amplitude, one second on and one second off for 1 min) with 1 min of incubation on ice between each cycle. After centrifugation at 1100g for 20 min at 4°C, the supernatant was stored at −20°C until used.
2.6. Affinity Chromatography
To purify the NADP‐GDH proteins, the supernatant was loaded on an equilibrated nickel column (Ni‐NTA Agarose 100, Thermo Fisher Scientific), which was then washed 10 times with 30 mmol/L imidazol. The protein was eluted with 500 mmol/L imidazol and stored at −20°C until used. Homogeneity of proteins was verified with a polyacrylamide gel electrophoresis 12% (SDS‐PAGE) stained with Coomassie Blue (Fig. S2).
2.7. Enzyme assay and protein determination
Whole yeast cell soluble protein extracts were prepared by sonication lysis of cell pellets harvested during exponential growth. The NADP‐GDH activity was assayed by the method of Doherty (Doherty, 1970). Protein was measured by the method of Lowry (Lowry, Rosebrough, Farr, & Randall, 1951), using bovine serum albumin as a standard.
2.8. Enzyme kinetics and analysis of kinetic data
NADP‐GDH activity was assayed for the reductive amination reaction at different concentrations of α‐KG (0.02–12 mmol/L), NADPH (5–500 μmol/L), or ammonium chloride (1–100 mmol/L) and at saturating concentrations of the remaining substrates (8 mmol/L α‐KG, 250 μmol/L NADPH, and 100 mmol/L ammonium chloride). The progress of the reaction was always kept below 5% conversion of the initial substrate. Measurements were made in 100 mmol/L Tris at pH 7.5 for ScGdh1, ScGdh3, and LkGdh1 or 0.1 mol/L potassium phosphate at pH 7.5 for KlGdh1. For experiments in which pH was 5.8, 25 mmol/L acetic acid, 25 mmol/L MES, and 50 mmol/L of TRIS or potassium phosphate at pH 5.8 was used as buffer. Kinetic data were analyzed by nonlinear regression using the program GraphPad Prism 5.00 (Software Inc.). All assays were performed at 340 nm, 30°C in a Varian Cary 50 spectrophotometer.
2.9. Glutamic inhibition
To study glutamic inhibition, were prepared protein extracts of S. cerevisiae WT, Scgdh1Δ, L. kluyveri WT y K. lactis WT strains grown on MM with ammonium sulfate as a nitrogen source and 2% glucose or ethanol as carbon source. Saturation curves were determined at the following glutamic concentrations: 0, 3, 5, 10, 15, 20, 50, 100, 200, 300, 400, 500, 600, 700, 800, 900, and 1000 mmol/L for every strain. At every glutamic concentration, the α‐KG, ammonium chloride and NADPH were fixed (8 μmol/L, 100 mmol/L, and 250 μmol/L, respectively). In order to select the inhibition model, the data were fitted to different models, with the program Dynafit. IC50 results were globally obtained with program GraphPad Prism 7.00 (Software Inc.).
2.10. Northern blot analysis
Northern blot analysis was carried out as previously described Struhl K. and Davis RW (1981). Total yeast RNA was prepared from 100 ml aliquots of cultures grown to an OD 600 nm of 0.6 in MM with ammonium sulfate as a nitrogen source and 2% glucose or ethanol as carbon source. PCR products were used as probes. For ScGDH1, a 645 bp product was amplified with deoxyoligonucleotides 141 and 142; for ScGDH3, a 1156 bp PCR product was amplified with deoxyoligonucleotides 143 and 144; 1200 bp fragment amplified using deoxyoligonucleotides 145 and 146 of ScACT1 was used as internal loading standard; for LkGDH1, a 1180 bp product was amplified with deoxyoligonucleotides 147 and 148; 477 bp fragment amplified using deoxyoligonucleotides 149 and 150 of Lk18s was used as internal loading standard; for KlGDH1, a 1386 bp product was amplified with deoxyoligonucleotides 151 and 152 and 477 bp fragment amplified using deoxyoligonucleotides 153 and 154 of Kl18s was used as internal loading standard. Blots were scanned with ImageQuant 5.2 program (Molecular Dynamics).
2.11. Nucleosome scanning assay (NuSA)
The nucleosome scanning assay was made to see the chromatin organization ScGDH1, ScGDH3, LkGDH1 y KlGDH1 promoter, and the procedure to the study of the positioning of nucleosomes on promoters was made as described by Infante et al. 2011. When the cultures reached an OD of 0.6 at 600 nm, genetic DNA was obtained of Cla1, Cla2, KlWM37‐1, and Lk156‐1 strains grown on minimal medium with ammonium sulfate as nitrogen source and 2% glucose or ethanol as carbon source. Cells were treated with formaldehyde (1% final concentration) for 20 min at 37°C and then glycine (125 mmol/L final concentration) for 5 min at 37°C. Formaldehyde‐treated cells were harvested by centrifugation, washed with Tris‐buffered saline, and then incubated in Buffer Z2 (1 mol/L Sorbitol, 50 mmol/L Tris‐Cl at pH 7.4, 10 mmol/L β‐mercaptoethanol) containing 2.5 mg of zymolase 20T for 20 min at 30°C on rocker platform. Spheroplast were pelleted by centrifugation at 3000g, and resuspended in 1.5 ml of NPS buffer (0.5 mmol/L Spermidine, 0.075% NP‐40, 50 mmol/L NaCl, 10 mmol/L Tris pH 7.4, 5 mmol/L MgCl2, 1 mmol/L CaCl2, 1 mmol/L β‐mercaptoethanol). Samples were divided into three 500 μl aliquots that were then digested with 22.5 U of MNase (Nuclease S7 from Roche) at 50 min at 37°C. Digestions were stopped with 12 μl of Stop buffer (50 mmol/L EDTA and 1% SDS) and were treated with 100 μg of proteinase K at 65°C over night. DNA was extracted twice by phenol/chloroform and precipitated with 20 μl of 5 mol/L NaCl and equal volume of isopropanol for 30 min at −20°C. Precipitates were resuspended in 40 μl of TE and incubated with 20 μg RNase A for 1 hr at 37°C. DNA digestions were separated by gel electrophoresis from a 1.5% agarose gel. Monosomal bands (150 bp) were cut and purified by Wizard SV Gel Clean‐Up System Kit (Promega, REF A9282). DNA samples were diluted 1:30 and used in quantitative polymerase chain reactions (qPCR) to quantify the relative MNase protection of each ScGDH1, ScGDH3, LkGDH1 y KlGDH1 template. qPCR analysis was performed using a Corbett Life Science Rotor Gene 6000 machine. The detection dye used was SYBR Green (2× KAPA SYBR FAST qBioline and Platinum SYBR Green from Invitrogen). Real‐time PCR was carried out as follows: 94° for 5 min (1 cycle), 94° for 15 s, 58° for 20 s, and 72° for 20 s (35 cycles). Relative protection was calculated as a ratio to the control ScVCX1, LkVCX1, and KlVCX1 template found within a well‐positioned nucleosome in +250 bp of the ORFs. The PCR primers amplify from around −950 to +250 bp (Table S3) of ScGDH1, ScGDH3, LkGDH1 y KlGDH1 locus whose coordinates are given relative to the ATG (+1).
2.12. Metabolite extraction and analysis
Cell extracts were prepared from exponentially growing cultures. Samples used for intracellular amino acid determination were treated as previously described (Quezada et al., 2008).
2.13. Phylogenetic analysis
A total of 26 taxa were used in the analysis, including two ascomycetes as outgroup (Table S2). Glutamate dehydrogenase sequences were obtained from YGOB (http://ygob.ucd.ie) (Byrne & Wolfe, 2005) and ESEMBLFungi (http://fungi.ensembl.org/index.html) (Kersey et al., 2016) databases using ScGdh1 sequence as query.
The bootstrap neighbor‐joining tree (500 replicates) was constructed with MEGA version 6 software (http://www.megasoftware.net/) (Tamura, Stecher, Peterson, Filipski, & Kumar, 2013), based on the sequence alignment constructed with the multi‐alignment program Muscle. Alternatively, phylogenetic analysis was also conducted with Maximum Likelihood method, in order to improve the accuracy of the phylogenetic analysis.
3. Results
3.1. NADP‐GDH is the main glutamate‐producing pathway in S. cerevisiae, L. kluyveri, and K. lactis
To analyze the relative contribution of glutamate dehydrogenases (NADP‐GDH) and glutamate synthase (GOGAT) to glutamate biosynthesis, mutant strains were constructed in which the genes encoding for the NADP‐GDH (GDH1/GDH3) or GOGAT (GLT1) were inactivated (Table 1). Growth rates of these mutants were determined on minimal media with glucose or ethanol as carbon sources and ammonium as nitrogen source (Table 2). In the three yeast species, inactivation of the NADP‐GDH‐encoding genes resulted in a strong reduction in growth rate on both carbon sources (from 60% to 80% relative to the corresponding WT strains) indicating that the proteins ScGdh1/ScGdh3 in S. cerevisiae, LkGdh1 in L. kluyveri and KlGdh1 in K. lactis, are the main contributors to glutamate production under the conditions studied. The glutamine synthetase‐GOGAT pathway in S. cerevisiae made a marginal contribution to glutamate production under the conditions studied because the glt1Δ mutant strain grew as well as the wild‐type strain (Table 2). However, in L. klyveri and K. lactis, the GOGAT pathway made a significant contribution since inactivation of the GLT1 genes, resulted in reduction of growth rates ranging from 25% to 60% (Table 2). As expected, the mutants lacking NADP‐GDH and GOGAT‐encoding genes were full glutamate auxotrophs (Table 2). In the pre‐WGD species, only one gene is responsible for the NADP‐GDH activity because inactivation of either LkGDH1 or KlGDH1 resulted in complete lack of this activity (Table 2). In agreement with previous reports (DeLuna et al., 2001), the contribution of ScGdh3 was evident on ethanol but not on glucose.
Table 2.
Growth rates and NADP‐GDH specific activities
| Strains | Growth rates | Specific activities | ||||||
|---|---|---|---|---|---|---|---|---|
| Glucose | Glucose +Glu | Ethanol | Ethanol +Glu | Glucose | Glucose +Glu | Ethanol | Ethanol +Glu | |
| S. cerevisiae | ||||||||
| WT ScGDH1 ScGDH3 ScGLT1 | 100 | 100 | 100 | 100 | 0.741 (0.05) | 0.710 (0.05) | 0.818 (0.05) | 0.801 (0.06) |
| Scgdh1Δ ScGDH3 ScGLT1 | 37 | 98 | 60 | 95 | 0.048 (0.02) | 0.046 (0.07) | 0.459 (0.04) | 0.435 (0.04) |
| ScGDH1 Scgdh3Δ ScGLT1 | 91 | 93 | 80 | 91 | 0.749 (0.06) | 0.698 (0.03) | 0.938 (0.03) | 0.704 (0.04) |
| Scgdh1Δ Scgdh3Δ ScGLT1 | 20 | 96 | 35 | 95 | ND | ND | ND | ND |
| ScGDH1 ScGDH3 Scglt1Δ | 97 | 94 | 94 | 92 | 0.654 (0.03) | 0.694 (0.08) | 0.901 (0.06) | 0.781 (0.02) |
| Scgdh1Δ Scgdh3Δ Scglt1Δ | ND | 74 | ND | 78 | ND | ND | ND | ND |
| L. kluyveri | ||||||||
| WT LkGDH1 LkGLT1 | 100 | 100 | 100 | 100 | 0.252 (0.02) | 0.279 (0.07) | 0.465 (0.03) | 0.424 (0.02) |
| Lkgdh1Δ LkGLT1 | 17 | 96 | 20 | 95 | ND | ND | ND | ND |
| LkGDH1 Lkglt1Δ | 70 | 81 | 59 | 77 | 0.229 (0.03) | 0.253 (0.03) | 0.418 (0.05) | 0.451 (0.08) |
| Lkgdh1Δ Lkglt1Δ | ND | 72 | ND | 73 | ND | ND | ND | ND |
| K. lactis | ||||||||
| WT KlGDH1 KlGLT1 | 100 | 100 | 100 | 100 | 0.431 (0.03) | 0.489 (0.08) | 0.645 (0.04) | 0.682 (0.05) |
| Klgdh1Δ KlGLT1 | 43 | 98 | 32 | 97 | ND | ND | ND | ND |
| KlGDH1 Klglt1Δ | 75 | 76 | 64 | 62 | 0.446 (0.04) | 0.434 (0.04) | 0.593 (0.02) | 0.587 (0.08) |
| Klgdh1Δ Klglt1Δ | ND | 78 | ND | 65 | ND | ND | ND | ND |
Glu, glutamate. ND, not detected. Numbers in parentheses are standard deviations.
Growth rates values are shown relative to the WT strains: for S. cerevisiae 0.258 hr−1 and 0.156 hr−1; for L. kluyveri, 0.179 hr−1 and 0.073 hr−1; for K. lactis, 0.322 hr−1 and 0.264 hr−1 on glucose and ethanol, respectively, in the absence of glutamate. However, in the presence of this amino acid, growth rates of the WT strains were for S. cerevisiae 0.261 hr−1 and 0.154 hr−1; for L. kluyveri, 0.182 hr−1 and 0.075 hr−1; for K. lactis, 0.325 hr−1 and 0.268 hr−1 on glucose and ethanol, respectively. For the growth rates, the standard deviations of at least three independent cultures were less than 5%. Specific activity values are shown in μmol min−1 mg−1.
When glutamate was supplemented to the growth media, gdhΔ mutant strains recovered wild‐type growth (Table 2). However, this was not the case for the L. kluyveri and K. lactis glt1Δ mutant strains, which did not recover wild‐type growth rate by glutamate addition. As previously reported, in addition to glutamate biosynthesis, GOGAT plays other role, which has been found to be critical for the maintenance of the redox balance and cytosolic NADH homeostasis (Guillamon et al., 2001).
3.2. Glutamate is not a negative regulator of the S. cerevisiae, L. kluyveri and K. lactis, NADP‐GDHs
To further analyze the regulation of the NADP‐GDH enzymes, specific activities in the presence of glutamate were determined. The use of 5 mmol/L glutamate as nitrogen source did not result on a reduction in NADP‐GDH specific activities as compared to those observed when ammonium sulfate was used (Table 2). Furthermore, when clarified extracts from the WT strains were analyzed in the presence of increasing glutamate concentrations, the half inhibitory concentrations were in the range of 376–681 mmol/L (Fig. S1); this range is much higher than the estimated cytosolic glutamate concentration, 9–46 mmol/L (Table 4). These results indicate that glutamate does not trigger strong negative regulatory mechanisms (e.g., repression of transcription or feedback inhibition) of glutamate biosynthesis under the conditions studied.
Table 4.
Intracellular metabolite pools
| α‐KG | Glutamate | |||||||
|---|---|---|---|---|---|---|---|---|
| Glucose | Ethanol | Glucose | Ethanol | |||||
| (nmol×108 cells) | (mmol/L) | (nmol×108 cells) | (mmol/L) | (nmol×108 cells) | (mmol/L) | (nmol×108 cells) | (mmol/L) | |
| S. cerevisiae | 1.0 (0.05) | 0.5 | 2.4 (0.21) | 1.1 | 100 (4) | 46 | 81 (14) | 37 |
| L. kluyveri | 2.1 (0.12) | 1.0 | 0.2 (0.03) | 0.1 | 20 (1) | 9 | 98 (8) | 45 |
| K. lactis | 1.5 (0.23) | 0.7 | 1.4 (0.13) | 0.7 | 25 (1) | 11 | 40 (5) | 18 |
Concentrations in mmol/L represent the cytosolic pool and were estimated considering a cell volume of 29 μm3 of which 75% of corresponded to cytosol (Kitamoto et al., 1988). Numbers in parentheses are standard deviations.
3.3. S. cerevisiae, L. kluyveri, and K. lactis showed different patterns of carbon source‐dependent transcriptional regulation of NADP‐GDH encoding genes
In order to deepen the studies with regard to carbon source‐dependent regulation of the NADP‐GDH enzymes, specific activities, transcript levels of the corresponding genes and nucleosome positioning on the promoter regions were analyzed. As previously reported, transcription of the ScGDH3 gene was higher on ethanol as carbon source compared to that observed on glucose (Figure 1a) (Avendano et al., 2005 and Riego et al., 2002). This was accompanied by nucleosome clearance on the −754 bp to −128 bp ScGDH3 promoter region (Figure 1c) (Avendano et al., 2005). Transcript levels of the ScGDH1 gene and nucleosome positioning on the promoter region were similar on both carbon sources (Figure 1a and b). Albeit in S. cerevisiae, the overall activity was similar on both carbon sources (Table 2), and the relative contribution of the ScGdh3 isoform was higher on ethanol than on glucose. When the mutant strain Scgdh1Δ was grown on ethanol, the ScGdh3‐specific activity was 10‐fold increased (Table 2). This is in accordance with previous results (DeLuna et al., 2001) demonstrating the observed differential contributions of each enzyme to growth rates (Table 2). When L. kluyveri and K. lactis were grown on ethanol as carbon source, activities were increased 80% and 50%, respectively, compared to those observed on glucose (Table 2). In L. kluyveri, transcription of the LkGDH1 gene was slightly increased on ethanol as carbon source concomitantly with nucleosome clearance on the −738 bp to −336 bp promoter region (Figure 2a and b), whereas that transcriptional levels and nucleosome positioning of KlGDH1 gene no change in carbon sources studied (Figure 2c and d).
Figure 1.
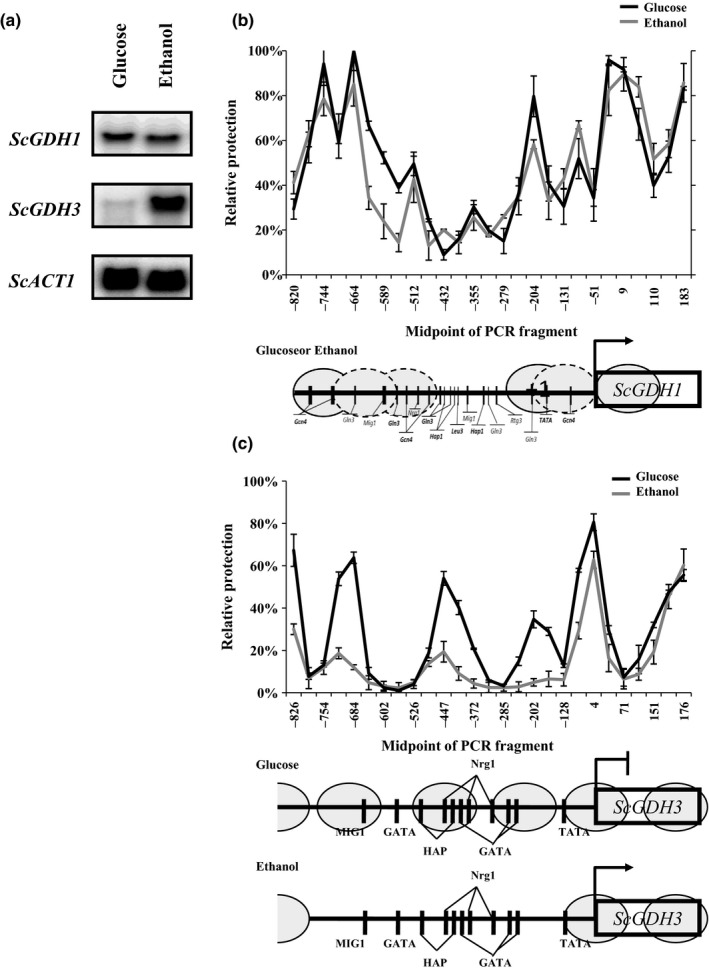
ScGDH1 and ScGDH3 NuSA and expression profile. (a) Northern blot analysis shown expression profile of ScGDH1 gene, ScGDH3 gene, and ScACT1 gene as control in glucose or ethanol as carbon source. (b) Nucleosome Scanning Assay (NuSA) ScGDH1 gene promoter in glucose (black line) or ethanol (gray line); and (c) Nucleosome Scanning Assay (NuSA) ScGDH3 gene promoter in glucose (black line) or ethanol (gray line); nucleosomes are shown in gray ovals and black vertical lines shown DNA‐binding sites. Error bars represent the standard deviations
Figure 2.
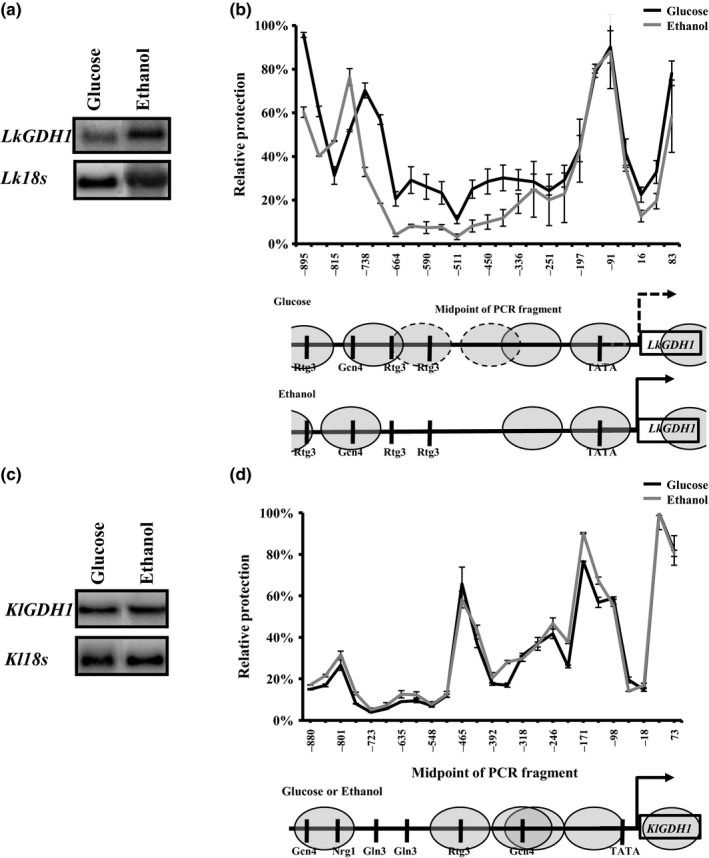
LkGDH1 and KlGDH1 NuSA and expression profile. (a) Northern blot analysis shown expression profile of LkGDH1 gene and (c) Northern blot analysis shown expression profile KlGDH3 gene, in both cases Lk18s or Kl18s ribosomal gene was used as control in glucose or ethanol as carbon source. (b) Nucleosome scanning assay (NuSA) LkGDH1 gene promoter in glucose (black line) or ethanol (gray line); and (d) Nucleosome Scanning Assay (NuSA) KlGDH1 gene promoter in glucose (black line) or ethanol (gray line); nucleosomes are shown in gray ovals and black vertical lines shown DNA binding sites. Error bars represent the standard deviations
3.4. ScGDH1/LkGDH1 and ScGDH3 / KlGDH1 gene pairs showed distinctive heterologous complementation patterns
To determine to what extent the various NADP‐GDHs were specialized to the metabolic peculiarities of the species they belong to, heterologous complementation tests were made. To this end, ScGDH1, ScGDH3, LkGDH1, and KlGDH1 were cloned in the low copy‐number plasmids pRS416, pLk‐EE, and YEpKD352 for expression in S. cerevisiae, L. kluyveri or K. lactis, respectively, under the transcriptional control of their native promoters as described in Experimental procedures. Mutant strains lacking NADP‐GDH activity were selectively transformed with the pertinent plasmids and their growth rates were compared (Figure 3).
Figure 3.
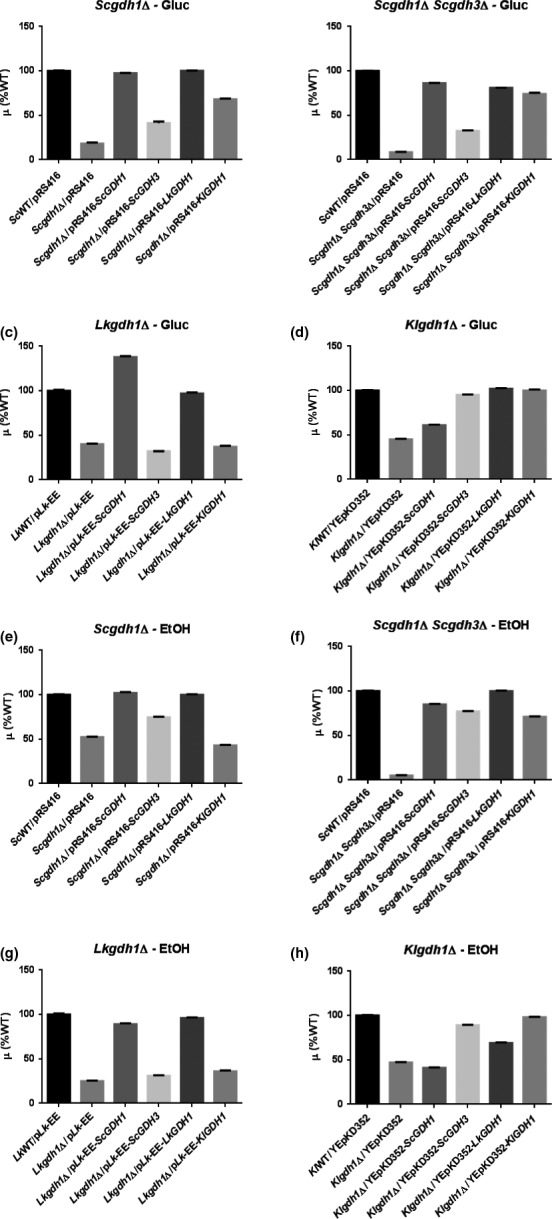
Complementation tests. Growth rates values are shown relative to the WT strains carrying the empty plasmid: for S. cerevisiae 0.23 hr−1 and 0.15 hr−1; for L. kluyveri, 0.12 hr−1 and 0.07 hr−1; for K. lactis, 0.35 hr−1 and 0.14 hr−1 on glucose and ethanol, respectively. In all cases, standard deviations of at least three independent cultures were less than three percent. For ectopic expression, the plasmid pRS416 was used for S. cerevisiae, the pL k‐EE for L. kluyveri and the YEpKD352 for K. lactis as described in Experimental procedures
As expected, homologous expression of ScGDH1 in the S. cerevisiae Scgdh1Δ mutant strain (third bar in Figure 3a and e), of LkGDH1 in the L. kluyveri Lkgdh1Δ mutant strain (fifth bar in Figure 3c and g) and of KlGDH1 in the K. lactis Klgdh1Δ mutant strain (last bar in Figure 3d and h), restored wild‐type growth when strains were grown on either glucose or ethanol. Homologous expression of the S. cerevisiae ScGDH3 gene resulted in a discrete but significant complementation of the gdh1Δ mutant strain on glucose (fourth bar in Figure 3a and b) and in almost full complementation on ethanol as carbon source (fourth bar in Figure 3e and f). These last results are in agreement with the proposed specialized role of ScGdh3 during respiratory conditions and with the carbon source‐dependent regulation of ScGDH3 gene (Avendano et al., 2005; DeLuna et al., 2001).
Heterologous expression of the ScGDH1 and LkGDH1genes resulted in similar patterns of complementation in S. cerevisiae and L. kluyveri where expression of these genes restored growth rates close to those of the WT reference strains (third bar in Figure 3c and g, and fifth bar in a, b, e and f). This effect, however, was not observed upon expression in K. lactis (Figure 3d and h). In this yeast, expression of the L. kluyveri LkGDH1 gene resulted in full complementation of the Klgdh1Δ mutant strain on glucose and in partial complementation on ethanol (fifth bars in Figure 3d vs. h), whereas expression of the S. cerevisiae ScGDH1 gene resulted in poor growth on either glucose or ethanol (third bars in Figure 3d and h). Interestingly, expression of the ScGDH1 and LkGDH1 genes also showed a similar trend when expressed on K. lactis grown on ethanol. In this case, however, the corresponding growth rates were significantly lower than those of the WT strain (third and fifth bars in Figure 3h), which showed the opposite trend to that observed upon expression on S. cerevisiae or L. kluyveri.
Ectopic expression of the ScGDH3 and KlGDH1 genes also showed a similar trend in the complementation experiments. When expressed on L. kluyveri, they did not improve growth of the Lkgdh1Δ mutant strain (fourth and sixth bars in Figure 3c and g). When expressed on S. cerevisiae, they showed significant complementation but the growth rates were still lower than those of the corresponding WT strains (fourth and sixth bars in Figure 3a, b, e and f). Transcriptional repression of the ScGDH3 gene on glucose (Avendano et al., 2005), may have contributed to the low levels of complementation of the ScGDH3 gene in the S. cerevisiae mutant strain grown on glucose (fourth and sixth bars in Figure 3a and b). Furthermore, when the ScGDH3 gene was expressed in K. lactis, it fully complemented growth of the Klgdh1Δ mutant strain at levels similar to those of the endogenous KlGDH1 gene (fourth and sixth bars in Figure 3d and h).
These results suggest that peculiar transcriptional and/or kinetic regulatory mechanisms cluster the ScGDH1 and LkGDH1 genes, or their encoded proteins, in a separate group from the ScGDH3 and KlGDH1 genes. To determine to what extent the amount of active enzyme is responsible of this effect, the NADP‐GDH specific activities were determined (Figure 4). As expected, the non‐complemented Scgdh1Δ Scgdh3Δ double‐mutant strain, as well as the Lkgdh1Δ and Klgdh1Δ single mutants did not show detectable activity (lack of a second bar in Figure 4b, c, d, f, g and h). Growth of these mutant strains, which are devoid of NADP‐GDH activity, may have involved the GOGAT‐dependent glutamate‐producing pathway (Magasanik, 2003). In the Scgdh1Δ single mutant strain, however, presence of the ScGDH3 gene was responsible of a very low activity on glucose and a significant activity on ethanol (second bars in Figure 4a and e), this was consistent with the induction of the ScGDH3 gene under respiratory conditions (Figure 1a). In general, a clear correlation between growth rate and NADP‐GDH activity was observed (Figure 3 vs. 4) indicating that regulatory mechanisms determining synthesis of NADP‐GDH (e.g., transcriptional and translational) are similar between KlGDH1 and ScGDH3 and between LkGDH1 and ScGDH1. However, in the K. lactis‐mutant strain grown on glucose, expression of the LkGDH1 gene restored growth rate and the specific activity to levels similar to those observed when the homologous KlGDH1gene was expressed (fifth and sixth bars in Figures 3d and 4d) and a significant complementation was also observed on ethanol (fifth and sixth bars in Figures 3h and 4h) suggesting that, in K. lactis, expression of the LkGDH1 gene is similar to that of the homologous KlGDH1gene. Reciprocal expression, however, resulted in poor growth and in absent or very low specific activity (sixth bars in Figures 3c, g and 4c, g), indicating that the contribution to the heterologous complementation capacity of LkGDH1 and KlGDH1 genes is mainly determined by the levels of active enzyme and not by their kinetic properties. Additionally, the lack of reciprocal complementation between these genes indicates that mechanisms determining synthesis of the NADP‐GDHs are regulated differently in K. lactis and L. kluyvery in spite of the close phylogenetic relationship between these species Figure 6).
Figure 4.
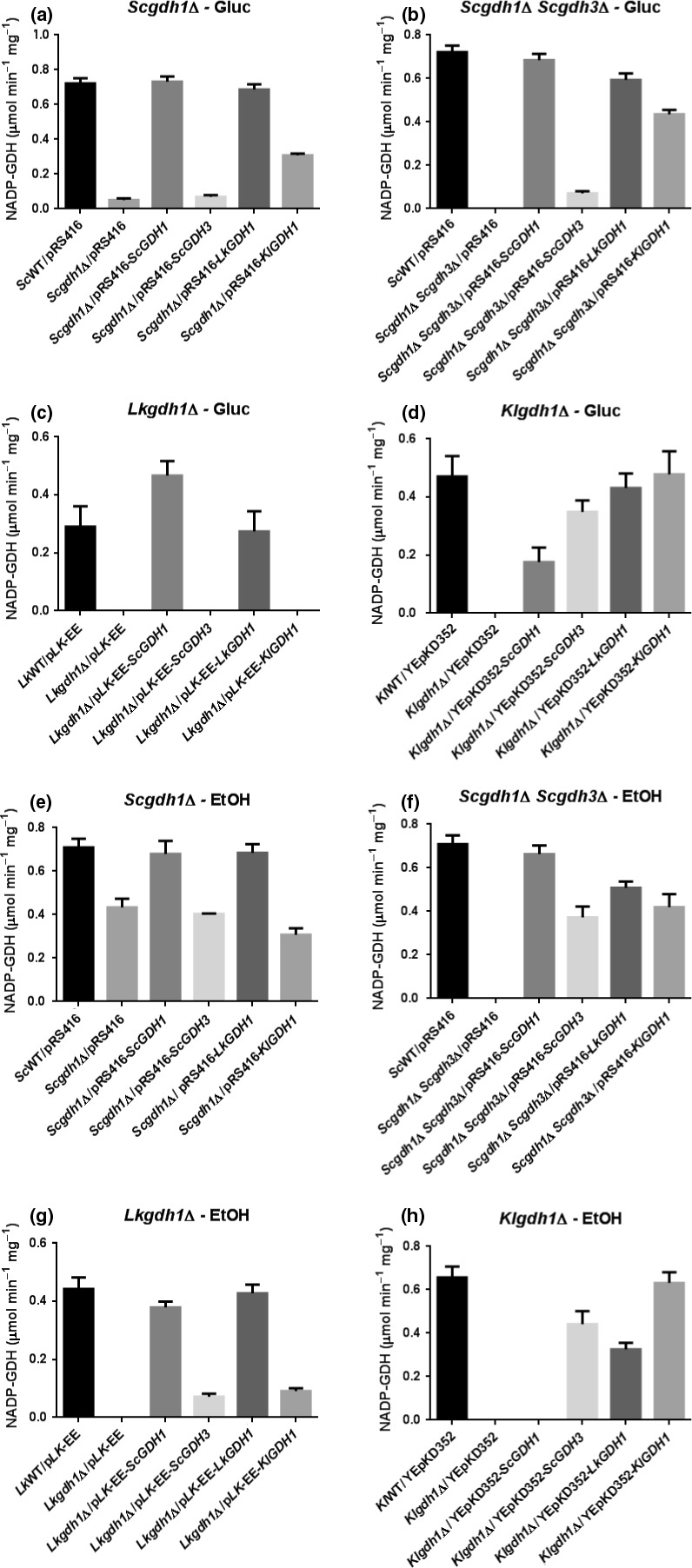
NADP‐GDH specific activities in complemented strains. NADP‐GDH specific activities values are shown in μmol min−1 mg−1. ND, not detected. Numbers in parentheses are standard deviations of at least three independent cultures
Figure 6.
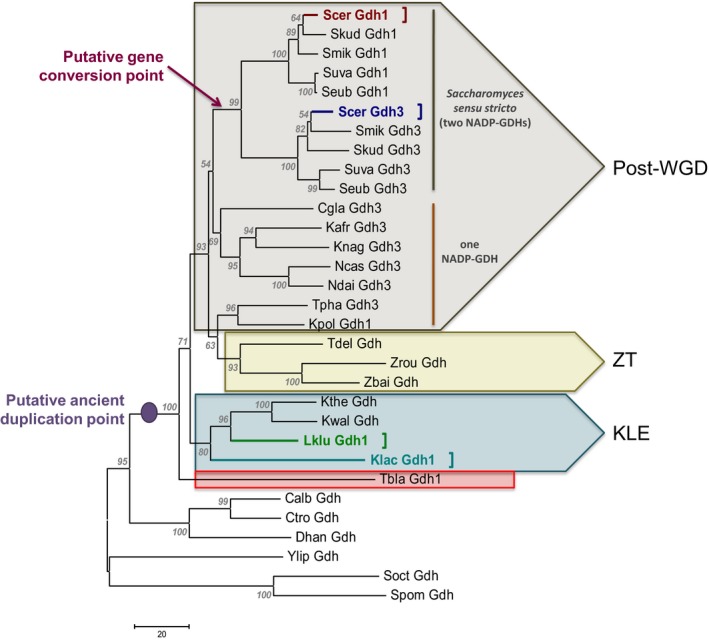
Evolutionary relationships of NADP‐GDHs from yeasts. The phylogeny was constructed using the neighbor‐joining method (Saitou & Nei, 1987). The optimal tree with the sum of branch length = 1043.39892578 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown next to the branches (Felsenstein, 1985). The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the number of differences method (Nei & Kumar, 2000) and are in the units of the number of amino acid differences per sequence. The analysis involved 31 amino acid sequences. All ambiguous positions were removed for each sequence pair. There were a total of 484 positions in the final dataset. Evolutionary analyses were conducted in MEGA6 (Tamura et al., 2013). Scer Gdh1, Saccharomyces cerevisiae ScGdh1 (red letters and square bracket); Scer Gdh3, Saccharomyces cerevisiae ScGdh3 (dark blue letters and square bracket); Lklu Gdh1, Lachancea kluyveri LkGdh1 (green letters and square bracket); Klac Gdh1, Kluyveromyces lactis KlGdh1 (light blue letters and square bracket). Post‐WGD, post‐whole genome duplicacion clade (light brown group); ZT, Zygosaccharomyces‐Torulaspora clade (light yellow group); KLE, Kluyveromyces‐Lachancea‐Eremothecium clade (light blue group). Tbla Gdh1, Torulaspora blattae Gdh1 (red square). NADP‐GDH sequence accession numbers, taxa used and their corresponding abbreviations are included in Table S2
Unexpectedly, NADP‐GDH activity was not detected in three complemented strains: the Lkgdh1Δ mutant strain grown on glucose bearing the ScGDH3 and KlGDH1 genes (lack of fourth and sixth bars in Figure 4C), and the Klgdh1Δ mutant strain grown on ethanol bearing the ScGDH1 gene (lack of a third bar in Figure 4h). This suggested that expression of the heterologous genes in these strains was very low and below the detection limit. Interestingly, heterologous complementation of K. lactis with the S. cerevisiae ScGDH1 and ScGDH3 genes resulted in higher activities of ScGdh3 than those observed for ScGdh1 (third and fourth bars in Figure 4d and h) and this effect was similar to the relative contribution of the two isoforms observed in the WT strain grown on ethanol (Table 2).
3.5. K. lactis KlGdh1 and S. cerevisiae ScGdh3 isoforms showed cooperativity for α‐KG utilization, whereas L. kluyveri LkGdh1 and S. cerevisiae ScGdh1 isoforms showed hyperbolic kinetics
In order to analyze the NADP‐GDHs biochemical characteristics and find additional elements that could contribute to better understand the results obtained in the complementation tests, substrate utilization kinetics was studied. The His‐tagged ScGdh1, ScGdh3, LkGdh1, and KlGdh1 enzymes were purified to electrophoretic homogeneity after heterologous expression in E. coli (Fig. S2). Apparent molecular masses of the ScGdh1, ScGdh3, LkGdh1, and KlGdh1 monomers, respectively, were: 53 kD, 55 kD, 54 kD, and 54 kD. In all cases, values are close to those expected. Initial velocity measurements were made at different substrate concentrations (α‐KG, NADPH and ammonium chloride); when the amount of one substrate was varied, the other two were kept at saturating concentrations. The reaction was assayed as previously reported (DeLuna et al., 2001).
The responses to increasing substrate concentrations were heterogeneous: they varied from hyperbolic for the three substrates in the S. cerevisiae isoform ScGdh1 and the L. kluyveri enzyme LkGdh1, to sigmoidal for the three substrates in the K. lactis enzyme KlGdh1 (Figure 5, S3 and S4). The S. cerevisiae isoform ScGdh3, however, showed hyperbolic response for the three substrates at pH 7.5, but sigmoidal for α‐KG at pH 5.8, in agreement with a previous study in which the nontagged homologous purified protein was used (DeLuna et al., 2001). This pH‐dependent difference in the shape of the α‐KG saturation curve was only observed for the ScGdh3 isoform (Figure 5, S3 and S4). Experimental data from the hyperbolic or sigmoidal responses were fitted to the Michaelis–Menten or the Hill equations, respectively, and the resulting kinetic parameters are shown in Table 3. At pH 7.5, the four enzymes showed similar turnover numbers, which indicate the catalytic events per unit of time (k cat), and similar affinities for NADPH and ammonium (Table 3). The ScGdh1, ScGdh3, and LkGdh1 isoforms also showed similar affinities for α‐KG, showing similar K m‐αKG values. However, KlGdh1 showed lower affinity and strong cooperativity for α‐KG utilization, its affinity constant (S 0.5) for α‐KG was about eight times higher than the K m‐αKG of the other enzymes (Table 3) and the fitted Hill number (n H‐α‐KG) resulted to be 4.4. These KlGdh1 characteristics were also observed at pH 5.8 and, at this pH value, were also shared by the ScGdh3 isoform. At the acidic pH, the k cat value of the K. lactis enzyme resulted to be three times lower than that observed at pH 7.5 (Table 3). Enzymatic activities of the LkGdh1 and KlGdh1 enzymes were assayed at different pH values ranging from 5 to 9. Maximum activities were observed at pH 7.0 (data not shown), which is close to the 6.8 value reported for the S. cerevisiae isoforms (DeLuna et al., 2001).
Figure 5.
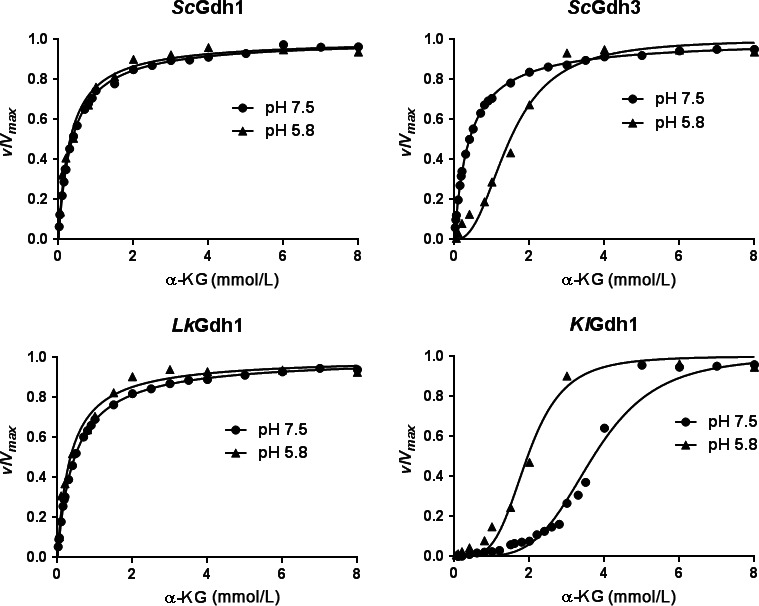
Effect of pH on the NADP‐GDH kinetic responses to α‐KG saturation. Initial velocities are shown as fractions of the corresponding V max at different α‐KG concentrations. The reductive amination reaction was measured in pure recombinant proteins from S. cerevisiae (ScGdh1 and ScGdh3), L. kluyveri (LkGdh1), and K. lactis (KlGdh1). The corresponding kinetic parameters are shown in Table 3
Table 3.
Kinetic parameters of the studied NADP‐GDHs
| Enzyme | pH 7.5 | pH 5.8 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| k cat (s−1) | K m‐α‐KG (mmol/L) | n H‐ α‐KG | K m‐NADPH (μmol/L) | n H‐NADPH | K m‐NH4+ (mmol/L) | n H‐NH4+ | k cat (s−1) | K m‐ α‐KG (mmol/L) | n H‐ α‐KG | |
| ScGdh1 | 13 | 0.37 | 45 | 8.6 | 12 | 0.31 | ||||
| ScGdh3 | 14 | 0.4 | 42 | 6.9 | 13 | S 0.5=1.48 | 2.4 | |||
| LkGdh1 | 20 | 0.46 | 46 | 7.4 | 10 | 0.34 | ||||
| KlGdh1 | 21 | S 0.5=3.61 | 4.4 | S 0.5=39 | 1.8 | S 0.5=21.4 | 2.7 | 6 | S 0.5=1.95 | 4 |
Data were fitted to the Michaelis–Menten equation except for the ScGdh3 isoform at pH 5.8 and the KlGdh1 enzyme which were fitted to the Hill equation. Standard errors for the fitted parameters at pH 7.5 were lower than 8% and, at pH 5.8, lower than 16%. For all the fitting analyses, the R 2 value was higher than 0.982.
In order to get some insight into the in vivo kinetic regulation of the NADP‐GDHs, the α‐KG and glutamate intracellular pools were determined (Table 4). Similar α‐KG values were detected in the three yeast species, except for L. kluyveri grown on ethanol which showed much lower α‐KG content, which was probably associated with the very low growth rate observed for this yeast (Table 2). The estimated α‐KG cytosolic concentrations in S. cerevisiae and L. kluyveri were in the range of 0.1–1.14 mmol/L which is close to 0.4–1.76 mmol/L, the reported values for S. cerevisiae (Cueto‐Rojas, Maleki Seifar, Ten Pierick, Heijnen, & Wahl, 2016; Hans, Heinzle, & Wittmann, 2003). To estimate these concentrations, a cell volume of 29 μm3 of which 75% of corresponded to cytosol was used (Kitamoto, Yoshizawa, Ohsumi, & Anraku, 1988), and to compare them with the reported values, a cell volume of 1.7 ml/g cell dry weight was used (Zhang et al., 2015). The estimated α‐KG physiological concentrations correspond to 0.3–3 K m‐α‐KG (Tables 3 and 4). At these concentrations, the catalytic rates of the ScGdh1, ScGdh3, and LkGdh1 enzymes were highly responsive to changes in substrate concentration (Figure 3). In K. lactis, the α‐KG estimated concentration was 0.7 mmol/L, which is around one‐fourth of the S 0.5‐α‐KG (Tables 3 and 4). At this concentration, and because of its strong cooperativity, the KlGdh1 enzyme catalytic rate was not highly responsive to changes in substrate concentration (Figure 3).
The intracellular glutamate content observed on ethanol as carbon source was similar to that observed on glucose in S. cerevisiae and K. lactis, although the pools in the former were higher than in the latter (Table 4). In L. kluyveri, however, the intracellular glutamate pool was much higher on ethanol than on glucose. This could be the results of the high‐affinity LkGdh1 has for α‐KG, driving intermediate flux to glutamate biosynthesis, which could result in decreased growth rate due to a restriction of carbon flow through Krebs Cycle and limited energy production resulting in low α‐KG intracellular concentration (Table 4). The estimated glutamate intracellular concentrations was found in the range of 9–46 mmol/L (Table 4), which are near to the 30–80 mmol/L, reported values for S. cerevisiae (Cueto‐Rojas et al., 2016; Hans et al., 2003). At these concentrations, glutamate does not exert a significant inhibitory effect over the NADP‐GDH activity as indicated by the high IC50 values, which were detected (Fig. S1).
3.6. Phylogenetic analysis of NADP‐GDH‐sequences revealed that the evolutionary origin of these proteins does not correlate with their kinetic properties
To analyze whether the observed ScGdh1, ScGdh3, LkGdh1, and KlGdh1 regulatory and kinetic properties correlated with the evolutionary origin of these proteins, a phylogenetic tree was constructed with NADP‐dependent Gdh sequences from fungal representatives of the different taxonomical classes (Figure 6). Overall, GDH phylogenies resembled the taxonomical classifications; however, three major aspects should be highlighted. 1) ScGdh1 (Scer Gdh1) and ScGdh3 (Scer Gdh3) were grouped in a separated clade together with Gdh1 and Gdh3 sequences from Saccharomyces sensu stricto species but not with other expected orthologs identified by synteny at the YGOB database (e.g., Scer Gdh1 with Lklu Gdh1 or Scer Gdh3 with Cgla Gdh3). These results suggest that gene conversion between the ancestral GDH duplicated copies of the S. cerevisiae lineage occurred after the whole genome duplication event (Figure 6, red arrow: putative gene conversion point). 2) Aside the Saccharomyces sensu stricto clade, all post‐WGD yeasts have conserved only one of the Gdh copies (in almost all of the cases the ScGdh3 orthologous counterpart) (Figure 6, orange line). 3) KlGdh1 (Klac Gdh1) and LkGdh1 (Lklu Gdh1) grouped together and might have been originated from a GDH clade which was generated through an ancient genome duplication event that was the consequence of an hybridization process which preceded the divergence between Saccharomyces and a clade containing the genera Kluyveromyces, Lachancea and Eremothecium (Figure 6, KLE group), as has been recently proposed by Marcet‐Houben and Gabaldón (2015). This observation is also supported by the fact that Tetrapisispora blattae Gdh sequence (Tbla Gdh1) (Figure 6, red rectangle) grouped outside even of the KLE clade and not with the other post‐WGD species Tetrapisispora phaffii (Tpha Gdh1) and Vanderwaltozyma polyspora (Vpol Gdh1) Gdh sequences as has been previously established (Marcet‐Houben & Gabaldón, 2015).
4. Discussion
This study addressed the question of whether there is a correlation between the regulation of the NADP‐GDHs activity and the evolutionary history of the corresponding genes in three Saccharomycetales species showing different levels of adaptation to the fermentative lifestyle. To this end, we compared the results of phylogenetic, functional, and kinetic analyses. Presented results show that kinetic properties and heterologous complementation capacities of the S. cerevisiae ScGdh1 isoform are closer to those of the L. kluyveri LkGdh1 enzyme than to those of its ScGdh3 paralogous isoform. While kinetics and heterologous complementation abilities of the ScGdh3 enzyme, resemble those of the KlGdh1, K. lactis enzyme.
4.1. Functional characterization was necessary to group the NADP‐GDHs into similar pairs
Carbon source‐dependent transcriptional regulation of the ScGDH1 gene was similar to that of the KlGDH1 gene because transcript levels and nucleosome positioning did not change with the nature of the carbon source (Figures 1a, b and 2c and d). Furthermore, transcriptional regulation of the ScGDH3 and LkGDH1 genes resulted in higher transcript levels on ethanol than on glucose and this was accompanied by chromatin remodeling (Figures 1a, c and 2a and b). However, kinetic characterization of the NADP‐GDHs and heterologous complementation patterns of the corresponding genes resulted in opposite relationships. Thus, transcriptional responses did not give a complete portrait of Gdh isozymes function. Yet, the close correlation between the NADP‐GDH specific activities (Figure 4) and growth rates (Figure 3) suggest that the ScGDH1 and the LkGDH1 genes could be expressed at similar levels in S. cerevisiae and in L. kluyveri. Similarly, ScGDH3 and KlGDH1 genes in S. cerevisiae and K. lactis, could share similar functions and biological properties. However, further research is necessary to determine the contribution of transcriptional regulation to the heterologous complementation similarities, and to identify the transcriptional regulators involved.
4.2. Kinetic properties comparisons allow grouping ScGdh1 and LkGdh1 enzymes in a peculiar group, different from ScGdh3 and KlGdh1
Hyperbolic kinetics and high affinity for α‐KG are shared properties of the ScGdh1 and LkGdh1 enzymes, while cooperativity for α‐KG utilization is a shared property of the ScGdh3 isoform with the KlGdh1 enzyme (Table 3, Figure 5). However, the physiological significance of ScGdh3 cooperativity is not clear since it has been only observed at pH 5.8 (Table 3) or lower (DeLuna et al., 2001). Intracellular pH during exponential growth has been reported to be close to neutrality: 7.2 on glucose and 6.8 on a mix of 2% ethanol and 2% glycerol (Orij, Postmus, Ter Beek, Brul, & Smits, 2009). In non‐growing glucose‐starved cells, however, it drops to 5.5–6.0 (Orij et al., 2009). These last conditions may reflect the stationary phase context in which ScGdh3 plays a significant role (Lee et al., 2012). It is possible that ScGdh3 cooperativity is a reminiscent feature of the ancestor protein without a true physiological and metabolic role in vivo; however, it can also be the case that an unknown allosteric effector induces cooperativity during exponential growth on ethanol, whose effect may be mimicked by acidic pH “in vitro”. The similarities in complementation patterns of the KlGdh1 and ScGdh3 enzymes suggest that cooperativity of ScGdh3 may be important “in vivo”.
Cooperativity for the utilization of the three substrates was observed in the K. lactis enzyme KlGdh1, most notably for α‐KG and ammonium utilization for which the Hill numbers were 4.4 and 2.7, respectively (Table 3). The estimated α‐KG physiological concentration in this yeast was 0.7 mmol/L (Table 4) and at this concentration, the KlGdh1 catalytic rate is not highly responsive to changes in α‐KG (Figure 3). This suggests that α‐KG availability does not determine glutamate synthesis in K. lactis. However, the low catalytic rate observed for the KlGdh1 enzyme at 0.7 mmol/L (Figure 3), may not be compatible with the fast growth observed for K. lactis (Table 2). It is possible that unknown activators contribute to modulation of the K. lactis enzyme in vivo. Interestingly, the S. cerevisiae and L. kluyveri enzymes are highly responsive to changes in α‐KG at the physiological concentrations (Table 4 and Figure 3) this suggests that the rate of glutamate synthesis is highly influenced by α‐KG availability as was previously proposed (Quezada et al., 2013). Worth of mention is the fact that there is growing evidence indicating that α‐KG plays a role in metabolic regulation. Thus, modulation of the intracellular α‐KG levels could constitute important mechanisms of metabolic control. In this regard, it has been proposed that in Caenorhabditis elegans, α‐KG is a key metabolite mediating longevity by dietary restriction (Chin et al., 2014). Intracellular α‐KG/succinate levels can contribute to the maintenance of cellular identity and have a mechanistic role in the transcriptional and epigenetic state of mouse stem cells (Carey, Finley, Cross, Allis, & Thompson, 2015). Most interestingly, recent studies of Gdh1 function has revealed that gdh1 mutants show enhanced N‐terminal histone H3 proteolisis, suggesting that α−KG has a key regulatory role in telomere silencing in S. cerevisiae (Su & Pillus, 2016).
Reported NADPH intracellular concentration is around 286 μmol/L (Zhang et al., 2015; authors considered a cellular volume of 1.7 ml/g cell dry weight), which corresponds to 6–7 K m‐NADPH or S 0.5‐NADPH (Table 3). At this concentration, the activities of the herein studied NADP‐GDH enzymes were not responsive to changes on the NADPH concentration (Fig. S4). This indicates that the physiological concentration of this substrate is close to saturation and does not determine the NADP‐GDH activity in vivo. By contrast, the reported intracellular ammonium concentration is 2.2 mmol/L (Cueto‐Rojas et al., 2016; considering a cellular volume of 1.7 ml/g cell dry weight as in Zhang et al., 2015). This value is well below the K m‐NH4+ or S 0.5‐NH4+ shown in Table 3, which indicates that the ammonium availability modulates the NADP‐GDH activity in vivo. Thus, glutamate synthesis by NADP‐GDH seems to be mainly determined by α‐KG and ammonium availability and not by product inhibition by glutamate.
Most interesting was the fact that, the kinetic behavior of the enzymes present in the two yeast species which show significant fermentative capacity when grown on high glucose media (ScGdh1 in S. cerevisiae and LkGdh1 in L. kluyveri) was hyperbolic, showing high affinity for α‐KG (K m‐α‐KG ≈ 0.4 mmol/L). The enzyme present in the yeast with a predominantly respiratory metabolism, KlGdh1 from K. lactis, and the ScGdh3 isoform whose contribution to glutamate synthesis increases during respiratory metabolism in S. cerevisiae (Table 2), were cooperative and showed low affinity for α‐KG (S 0.5KlGdh1, pH7.5 = 3.61 mmol/L and S 0.5ScGdh3, pH5.8 = 1.95 mmol/L). Assuming that during S. cerevisiae evolution, L. kluyveri and K. lactis, selective pressures drove changes in the NADP‐GDHs, these enzymes could have changed from cooperative to hyperbolic. As cooperativity was observed in the NADP‐GDH from K. lactis and in the ScGdh3 isoform, it seems possible that the common ancestor of the three yeast species had a cooperative NADP‐GDH and that this property was lost two times: one in the L. kluyveri lineage after divergence of the S. cerevisiae and L. kluyveri branches, and the other after the WGD event which resulted in the conservation of the hyperbolic ScGdh1 and the cooperative ScGdh3. This suggests that NADP‐GDH kinetics may be related to adaptation to the fermentative or respiratory lifestyles, and further research in various yeast species is needed to explore this possibility.
Conflict of Interest
None declared.
Supporting information
Acknowledgments
This study was performed in partial fulfillment of the requirements for the PhD degree in Biomedical Sciences of José Carlos Campero‐Basaldua at the Programa de Doctorado en Ciencias Biomédicas de la Universidad Nacional Autónoma de México, which he carried with a CONACYT doctoral fellowship. We thank Juan Pablo Pardo and Diego Hartasánchez Frenk for illuminating discussions during the course of this work, Nicolás Gómez Hernández, Javier Montalvo‐Arredondo, Mirelle Citlali Flores‐Villegas, Edson E Robles‐Gómez, Ximena Martínez de la Escalera and Beatriz Aguirre‐López, for helpful technical assistance, and Rocío Romualdo Martínez for helpful secretarial assistance. This study was funded by Dirección General de Asuntos del Personal Académico, UNAM, grant IN201015 (http://dgapa.unam.mx); Consejo Nacional de Ciencia y Tecnología (CONACYT grant CB‐2014‐239492‐B). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Campero‐Basaldua, C. , Quezada, H. , Riego‐Ruíz, L. , Márquez, D. , Rojas, E. , González, J. , El‐Hafidi, M. and González, A. Diversification of the kinetic properties of yeast NADP‐glutamate‐dehydrogenase isozymes proceeds independently of their evolutionary origin. MicrobiologyOpen. 2017;6:e00419 https://doi.org/10.1002/mbo3.419
References
- Alba‐Lois, L. , Segal, C. , Rodarte, B. , Valdes‐Lopez, V. , DeLuna, A. , & Cardenas, R. (2004). NADP‐glutamate dehydrogenase activity is increased under hyperosmotic conditions in the halotolerant yeast Debaryomyces hansenii . Current Microbiology, 48, 68–72. [DOI] [PubMed] [Google Scholar]
- Avendano, A. , Deluna, A. , Olivera, H. , Valenzuela, L. , & Gonzalez, A. (1997). GDH3 encodes a glutamate dehydrogenase isozyme, a previously unrecognized route for glutamate biosynthesis in Saccharomyces cerevisiae . Journal of Bacteriology, 179, 5594–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avendano, A. , Riego, L. , DeLuna, A. , Aranda, C. , Romero, G., Ishida, C. , … González, A. (2005). Swi/SNF‐GCN5‐dependent chromatin remodelling determines induced expression of GDH3, one of the paralogous genes responsible for ammonium assimilation and glutamate biosynthesis in Saccharomyces cerevisiae . Molecular Microbiology, 57, 291–305. [DOI] [PubMed] [Google Scholar]
- Breunig, K. D. , Bolotin‐Fukuhara, M. , Bianchi, M. M. , Bourgarel, D. , Falcone, C. , Ferrero, I. I. … Zeeman, A. M. (2000). Regulation of primary carbon metabolism in Kluyveromyces lactis . Enyzme and Microbial Technology, 26, 771–780. [DOI] [PubMed] [Google Scholar]
- Byrne, K. P. , & Wolfe, K. H. (2005). The Yeast Gene Order Browser: Combining Curated Homology and Syntenic Context Reveals Gene Fate in Polyploid Species. Genome Research, 15, 1456–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey, B. W. , Finley, S. L. W. , Cross, R. J. , Allis, D. C. , & Thompson, B. C. (2015). Intracellular α‐ketoglutarate maintains the pluripotency of embryonic stem cells. Nature, 518, 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, M. R. , Fu, X. , Pai, Y. M. , Vergnes, L. , Hwang, H. , Deng, G. … Meisheng, J. (2014). The metabolite α‐ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature, 510(7505), 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury, R. , & Punekar, N. S. (2009). Aspergillus terreus NADP‐glutamate dehydrogenase is kinetically distinct from the allosteric enzyme of other Aspergilli. Mycological Research, 113, 1121–1126. [DOI] [PubMed] [Google Scholar]
- Colon, M. , Hernandez, F. , Lopez, K. , Quezada, H. , Gonzalez, J. , Lopez, G. … González, A. (2011). Saccharomyces cerevisiae Bat1 and Bat2 aminotransferases have functionally diverged from the ancestral‐like Kluyveromyces lactis orthologous enzyme. PLoS ONE, 6, e16099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cueto‐Rojas, H. F. , Maleki Seifar, R. , Ten Pierick, A. , Heijnen, S. J. , & Wahl, A. (2016). Accurate measurement of the in vivo ammonium concentration in Saccharomyces cerevisiae . Metabolites, 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuna, A. , Avendano, A. , Riego, L. , & Gonzalez, A. (2001). NADP‐glutamate dehydrogenase isoenzymes of Saccharomyces cerevisiae. Purification, kinetic properties, and physiological roles. Journal of Biological Chemistry, 276, 43775–43783. [DOI] [PubMed] [Google Scholar]
- Doherty, D. (1970). L‐glutamate dehydrogenases (yeast). Methods in Enzymology, 17, 850–856. [Google Scholar]
- Felsenstein, J. (1985). Confidence limits on phylogenies: An approach using the bootstrap. Evolution, 39, 783–791. [DOI] [PubMed] [Google Scholar]
- Fincham, J. R. (1951). The occurrence of glutamic dehydrogenase in Neurospora an its apparent absence in certain mutant strains. Journal of General Microbiology, 5, 793–806. [DOI] [PubMed] [Google Scholar]
- Gancedo, J. M. (1998). Yeast carbon catabolite repression. Microbiology and Molecular Biology Reviews, 62, 334–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillamon, J. M. , van Riel, N. A. , Giuseppin, M. L. , & Verrips, C. T. (2001). The glutamate synthase (GOGAT) of Saccharomyces cerevisiae plays an important role in central nitrogen metabolism. FEMS Yeast Research, 1, 169–175. [DOI] [PubMed] [Google Scholar]
- Hagman, A. , Säll, T. , & Piskur, J. (2014). Analysisof the yeast short‐term Cabtree effect and its origin. FEBS Journal, 281, 4805–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hans, M. A. , Heinzle, E. , & Wittmann, C. (2003). Free intracellular amino acid pools duringautonomous oscillations in Saccharomyces cerevisiae . Biotechnology and Bioengineering, 82, 143–151. [DOI] [PubMed] [Google Scholar]
- Holmes, A. R. , Collings, A. , Farnden, K. J. , & Shepherd, M. G. (1989). Ammonium assimilation by Candida albicans and other yeasts: Evidence for activity of glutamate synthase. Journal of General Microbiology, 135, 1423–1430. [DOI] [PubMed] [Google Scholar]
- Infante, J. J. , Glynn, L. , & Elton, Y. (2011). Chromatin Remodelingand Protocols. Methods in Molecular Biology, 833, 63–87. [Google Scholar]
- Ito, H. , Fukuda, Y. , Murata, K. , & Kimura, A. (1983). Transformation of intact yeast cells treated with alkali cations. Journal of Bacteriology, 153, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersey, J. P. , Allen, E. J. , Armean, I. , Boddu, S. , Bolt, J. B. , Carvalho‐Silva, D. … Staines, DM. (2016). Ensembl Genomes 2016: More genomes, more complexity. Nucleic Acids Research, 44(D1), D574–D580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamoto, K. , Yoshizawa, K. , Ohsumi, Y. , & Anraku, Y. (1988). Dynamic aspects of vacuolar andcytosolic amino acid pools of Saccharomyces cerevisiae . Journal of Bacteriology, 170, 2683–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y. J. , Kim, K. J. , Kang, H. Y. , Kim, H. R. , & Maeng, P. J. (2012). Involvement of GDH3‐encoded NADP+‐dependent glutamate dehydrogenase in yeast cell resistance to stress‐induced apoptosis in stationary phase cells. Journal of Biological Chemistry, 287, 44221–44233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry, O. H. , Rosebrough, N. J. , Farr, A. L. , & Randall, R. J. (1951). Protein measurement with the Folin phenol reagent. Journal of Biological Chemistry, 193, 265–275. [PubMed] [Google Scholar]
- Macheda, M. L. , Hynes, M. J. , & Davis, M. A. (1999). The Aspergillus nidulans gltA gene encoding glutamate synthase is required for ammonium assimilation in the absence of NADP‐glutamate dehydrogenase. Current Genetics, 34, 467–471. [DOI] [PubMed] [Google Scholar]
- Magasanik, B. (2003). Ammonia assimilation by Saccharomyces cerevisiae . Eukaryotic Cell, 2, 827–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcet‐Houben, M. , & Gabaldón, T. (2015). Beyond the Whole‐Genome Duplication: Phylogenetic Evidence for an Ancient Interspecies Hybridization in the Baker′s Yeast Lineage. Plos Biology, 13(8), 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, S. M. , & Magasanik, B. (1990). Role of NAD‐linked glutamate dehydrogenase in nitrogenmetabolism in Saccharomyces cerevisiae . Journal of Bacteriology, 172, 4927–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller, K. , Christensen, B. , Forster, J. , Piskur, J. , Nielsen, J. , & Olsson, L. (2002). Aerobic glucose metabolism of Saccharomyces kluyveri: Growth, metabolite production, and quantification of metabolic fluxes. Biotechnology and Bioengineering, 77, 186–193. [DOI] [PubMed] [Google Scholar]
- Moller, K. , Olsson, L. , & Piskur, J. (2001). Ability for anaerobic growth is not sufficient for development of the petite phenotype in Saccharomyces kluyveri . Journal of Bacteriology, 183, 2485–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalvo‐Arredondo, J. , Jímenez‐Benítez, A. , Colón‐González, M. , González‐Flores, J. , Flores‐Villegas, M. , González, A. , & Riego‐Ruíz, L. (2015). Funtional roles of a predicted branched chain aminotransferase encoded by the LkBAT1 gene of the yeast Lachancea kluiyveri . Fungal Genetics and Biology, 85, 71–82. [DOI] [PubMed] [Google Scholar]
- Nei, M. , & Kumar, S. (2000). Molecular Evolution and Phylogenetics. New York.: Oxford University Press. 352. [Google Scholar]
- Noor, S. , & Punekar, N. S. (2005). Allosteric NADP‐glutamate dehydrogenase from aspergilli: Purification, characterization and implications for metabolic regulation at the carbon‐nitrogen interface. Microbiology, 151, 1409–1419. [DOI] [PubMed] [Google Scholar]
- Orij, R. , Postmus, J. , Ter Beek, A. , Brul, S. , & Smits, G. J. (2009). In vivo measurement of cytosolic and mitochondrial pH using a pH‐sensitive GFP derivative in Saccharomyces cerevisiae reveals a relation between intracellular pH and growth. Microbiology, 155, 268–278. [DOI] [PubMed] [Google Scholar]
- Perysinakis, A. , Kinghorn, J. R. , & Drainas, C. (1994). Biochemical and genetical studies of NADP‐specific glutamate dehydrogenase in the fission yeast Schizosaccharomyces pombe . Current Genetics, 26, 315–320. [DOI] [PubMed] [Google Scholar]
- Pfeiffer, T. , & Morley, A. (2014). An evolutionary perspective on the Crabtree effect. Frontiers in Molecular Biosciences, 1, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quezada, H. , Aranda, C. , DeLuna, A. , Hernández, H. , Calcagno, M. , Marín‐Hernández, A. , & González, A. (2008). Specialization of the paralogue LYS21 determines lysine biosynthesis under respiratory metabolism in Saccharomyces cerevisiae . Microbiology, 154, 1656–1667. [DOI] [PubMed] [Google Scholar]
- Quezada, H. , Marín‐Hernández, A. , Arreguín‐Espinosa, R. , Rumjanek, F. D. , Moreno‐Sánchez, R. , & Saavedra, E. (2013). The 2‐oxoglutarate supply exerts significant control on the lysinesynthesis flux in Saccharomyces cerevisiae . FEBS Journal, 22, 5737–5749. [DOI] [PubMed] [Google Scholar]
- Riego, L. , Avendaño, A. , DeLuna, A. , Rodríguez, E. , & González, A. (2002). GDH1 expression isregulated by GLN3, GCN4, and HAP4 under respiratory growth. Biochemical and Biophysical ResearchCommunications, 293, 79–85. [DOI] [PubMed] [Google Scholar]
- Saitou, N. , & Nei, M. (1987). The neighbor‐joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4, 406–425. [DOI] [PubMed] [Google Scholar]
- Sambrook, J. , Fritsch, E. F. , & Maniatis, T . (1989). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory: New York. [Google Scholar]
- Seoighe, C. , & Wolfe, K. H. (1999). Updated map of duplicated regions in the yeast genome. Gene, 238, 253–261. [DOI] [PubMed] [Google Scholar]
- Struhl, K. , & Davis, R. W. (1981). Transcription of the HIS3 gene region in Saccharomyces cerevisiae . Journal of Molecular Biology, 152, 535–552. [DOI] [PubMed] [Google Scholar]
- Su, X. B. , & Pillus, L . (2016). Functions for diverse metabolic activities in heterochromatin. PNAS, 113, E1526–E1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Stecher, G. , Peterson, D. , Filipski, A. , & Kumar, S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0 . Molecular Biology and Evolution, 30, 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Y. , Sieg, A. , & Trotter, P. J. (2011). (1)(3)C‐metabolic enrichment of glutamate in glutamate dehydrogenase mutants of Saccharomyces cerevisiae . Microbiological Research, 166, 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela, L. , Guzman‐Leon, S. , Coria, R. , Ramirez, J. , Aranda, C. , & Gonzalez, A. (1995). A NADP‐glutamate dehydrogenase mutant of the petit‐negative yeast Kluyveromyces lactis uses the glutamine synthetase‐glutamate synthase pathway for glutamate biosynthesis. Microbiology, 141(Pt 10), 2443–2447. [DOI] [PubMed] [Google Scholar]
- Wach, A. , Brachat, A. , Pohlmann, R. , & Philippsen, P. (1994). New heterologous modules for classical or PCR‐based gene disruptions in Saccharomyces cerevisiae . Yeast, 10, 1793–1808. [DOI] [PubMed] [Google Scholar]
- Wolfe, K. H. , & Shields, D. C. (1997). Molecular evidence for an ancient duplication of the entire yeast genome. Nature, 387, 708–713. [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Ten Pierick, A. , van Rossum, H. M. , Seifar, R. M. , Ras, C. , Daran, J. M. , … Wahl, S. A. (2015). Determination of the cytosolic NADPH/NADP ratio in Saccharomyces cerevisiae using shikimate dehydrogenase as sensor reaction. Scientific Reports, 5, 12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials