

Abstract
Concentrated seabed deposits of polymetallic nodules, which are rich in economically valuable metals (e.g., copper, nickel, cobalt, manganese), occur over vast areas of the abyssal Pacific Ocean floor. Little is currently known about the diversity of microorganisms inhabiting abyssal habitats. In this study, sediment, nodule, and water column samples were collected from the Clarion‐Clipperton Zone of the Eastern North Pacific. The diversities of prokaryote and microeukaryote communities associated with these habitats were examined. Microbial community composition and diversity varied with habitat type, water column depth, and sediment horizon. Thaumarchaeota were relatively enriched in the sediments and nodules compared to the water column, whereas Gammaproteobacteria were the most abundant sequences associated with nodules. Among the Eukaryota, rRNA genes belonging to the Cryptomonadales were relatively most abundant among organisms associated with nodules, whereas rRNA gene sequences deriving from members of the Alveolata were relatively enriched in sediments and the water column. Nine operational taxonomic unit (OTU)s were identified that occur in all nodules in this dataset, as well as all nodules found in a study 3000–9000 km from our site. Microbial communities in the sediments had the highest diversity, followed by nodules, and then by the water column with <1/3 the number of OTUs as in the sediments.
Keywords: 16S rRNA, 18S rRNA, diversity, microbial ecology, polymetallic nodules, sediment
1. Introduction
Polymetallic (i.e., manganese) nodules occur over vast areas of the abyssal ocean floor (Ghosh & Mukhopadhyay, 2000) and are enriched in commercially valuable minerals such as manganese (Mn), cobalt (Co), copper (Cu), nickel (Ni), and rare‐earth elements (Wegorzewski & Kuhn, 2014). Although nodules are estimated to have very slow growth rates of <1 nm year−1 (Kerr, 1984), rising global demand for these metals has renewed interests in commercial mining of deep‐sea nodule deposits. The Clarion‐Clipperton Zone (CCZ) in the Pacific Ocean is of particular interest for mining due to nodule concentrations up to 10–15 kg m−2 (Smith, Levin, Koslow, Tyler, & Glover, 2008). As of August 2015, the International Seabed Authority had granted 16 exploration licenses for nodule mining within the CCZ (Wedding et al., 2015). As currently conceived, mining operations would (1) remove nodules and seafloor sediments, and create near‐bottom sediment plumes that could spread over thousands of square kilometers, (2) pump nodules and associated sediments from the seabed to the ocean's surface, and (3) release mining tailings composed of nodule‐free sediments back into the water column, thereby potentially impacting much larger areas than due to the deep water plume (Rolinski, Segschneider, & Sundermann, 2001), and significantly disturbing large areas of the abyssal seafloor and overlying water column (Smith et al., 2008). To fully understand potential impacts on microbial processes that may be disturbed by mining activities, it is important to characterize the microbial communities of the abyssal seafloor and the overlying water column since both will likely be impacted by mining.
Although there is limited information on microbial communities inhabiting abyssal sediments in regions where polymetallic nodules are abundant, studies of abyssal sediments lacking nodules indicate that the resident prokaryotic communities are mainly composed of Proteobacteria, Acidobacteria, Actinomycetes, Chloroflexi, Planctomycetes, and Crenarchaeota (Durbin & Teske, 2010; Schauer, Bienhold, Ramette, & Harder, 2010), reviewed in Corinaldesi (2015). In the CCZ, the low particulate organic‐carbon flux from the overlying waters compared to continental margins results in oxygenated sediments to depths of 2–3 m (Mewes et al., 2014; Smith, De Leo, Bernardino, Sweetman, & Arbizu, 2008). The abyssal plain can be a relatively stable environment over a period of many years, and benthic production can be low as it depends on input of organic particles from the euphotic zone. The physical and chemical structure of polymetallic nodules potentially provides a unique niche for bacterial, archaeal, and microeukaryotic communities to colonize. Little is known about nodule formation, although it has been hypothesized to be an abiotic process (Kerr, 1984). However, a recent study posited a microbially mediated mechanism for nodule initiation because X‐ray and microscopy studies indicated high concentrations of bacteria in Mn‐rich micronodules (Wang, Schlossmacher, Wiens, Schroeder, & Mueller, 2009). Indeed, an early study of polymetallic nodules using light and scanning electron microscopy revealed biofilms and filamentous microorganisms associated with nodule surfaces (Burnett & Nealson, 1981). Three recent studies (Blothe et al., 2015; Tully & Heidelberg, 2013; Wu et al., 2013), relying on gene‐based surveys, identified unique bacterial and archaeal operational taxonomic unit (OTUs) specifically associated with nodules compared to surrounding sediments in the eastern North Pacific Subtropical Gyre (NPSG), the central South Pacific Gyre and the central and western NPSG. A fourth study investigated only bacterial diversity in the sediments of the CCZ but not from nodules (Wang et al., 2010). The nodule‐associated protistan community has thus far not been investigated beyond morphological identification of selected foraminifera (e.g., (Mullineaux, 1987; Veillette, Juniper, Gooday, & Sarrazin, 2007; Veillette, Sarrazin et al. 2007)).
Here we present findings of the bacterial, archaeal, and microeukaryotic communities associated with a polymetallic nodule field based on amplification and sequencing of rRNA genes. These analyses included 75 sediment samples, 24 water column samples, and 20 individual nodules from 11 stations randomly distributed over a 30 × 30 km stratum (AB‐01; Table 1, Figure 1a) within the United Kingdom's UK‐1 exploration claim area. Our results confirm that polymetallic nodules harbor diverse types of microorganisms that are distinct from both the surrounding sediments and overlying water, and that microbial diversity is substantially higher in the seafloor ecosystem than in the water column above. Our results reveal for the first time a diverse nodule eukaryotic community harboring OTUs assigned to the Sar, Opisthokonta, and Cryptophyceae lineages, as well as a core prokaryotic nodule community with OTUs found in nodules collected 3000 to >9000 km from our study site. These results suggest polymetallic nodules may harbor a persistent and stable microbiome.
Table 1.
Sampling site locations in the UK‐1 claim area, dates, and depths for this study
| Station | Location | Date (MM/DD/YY) | Station bottom depth (m) |
|---|---|---|---|
| A | (CTa) 13°52.90 N, 116°28.00 W | 10/08/13 | 4113 |
| B | (CT) 13°50.79 N, 116°37.59 W | 10/11/13 | 4025 |
| (BCb) 13°50.99 N, 116°38.70 W | 10/09/13 | 4108 | |
| (MCb) 13°50.79 N, 116°27.59 W | 10/10/13 | 4079 | |
| C | (BC) 13°47.60 N, 116°37.19 W | 10/11/13 | 4081 |
| (MC) 13°47.62 N, 116°42.19 W | 10/11/13 | 4078 | |
| D | (CT) 13°57.80 N, 116°34.10 W | 10/13/13 | 4025 |
| (MC) 13°57.80 N, 116°34.09 W | 10/13/13 | 4084 | |
| E | (BC) 13°49.45 N, 116°32.06 W | 10/14/13 | 4054 |
| (MC) 13°49.45 N, 116°32.06 W | 10/15/13 | 4054 | |
| F | (BC/MC)c 13°48.70 N, 116°42.60 W | 10/16/13 | 4076 |
| G | (MC) 13°45.71 N, 116°27.60 W | 10/18/13 | 4111 |
| H | (BC/MC)c 13°53.30 N, 116°41.40 W | 10/19/13 | 4150 |
| I | (BC) 13°45.00 N, 116°30.80 W | 10/18/13 | 4036 |
| (MC) 13°45.70 N, 116°27.62 W | 10/18/13 | 4111 | |
| J | (BC) 13°54.11 N, 116°35.40 W | 10/21/13 | 4163 |
| (MC) 13°54.10 N, 116°35.40 W | 10/21/13 | 4166 | |
| K | (BC) 13°51.78 N, 116°32.93 W | 10/20/13 | 4050 |
| (MC) 13°51.80 N, 116°32.80 W | 10/21/13 | 4053 |
CT, niskin rosette on the conductivity‐temperature‐depth device.
BC, Boxcore; MC, Megacore.
Ship remained on station; boxcore and megacore deployments occurred sequentially.
Figure 1.

Study location and design. (a) Location of AB‐01 stratum within the Clarion‐Clipperton Zone of the Pacific Ocean, where sampling occurred. Locations of sampling stations are indicated by dots and labeled with letters as in Table 1. (b) Example of a boxcore from which nodules (black globular structure) were sampled. In this example, the topwater has been drained from the box corer; box is 50 cm on a side. (Photo credit: Craig Smith.) (c) Example of a megacoring device after recovery with core tubes containing abyssal sediments during AB‐01
2. Experimental Procedures
2.1. Sample collection
Abyssal sediments, polymetallic nodules, and seawater samples were collected as part of the MV1313 research cruise (October 2013) aboard the R/V Melville to a 30 × 30 km stratum centered at 13°49′N, 116°36′W within the UK‐1 claim area in the Clarion‐Clipperton Zone. Samples were collected from stations randomly located within this 30 × 30 km area, designated AB‐01 or Stratum A (Table 1, Figure 1); 11 stations were sampled for sediments and nodules and three stations for seawater. Seawater samples were collected from eight discrete depths within the water column (5, 150, 300, 500, 1000, 2000, 3000 m, and near‐bottom waters) using a conductivity‐temperature‐depth (CTD; SBE 911plus; Sea‐Bird Electronics) rosette sampler equipped with 24 10 L sampling bottles. The rosette sensor package also included a fluorometer (Seapoint Chlorophyll Fluorometer; Seapoint Sensors, Inc.) and dissolved oxygen (O2) sensor (SBE 43; Sea‐Bird Electronics). Seawater (2 L each from 5, 150, 300, and 500 m and 8 L from 1000, 2000, 3000 m, and near‐bottom waters) was subsampled from the rosette bottles into polycarbonate carboys (4.5 L carboys for 2 L samples and 10 L carboys for 8 L samples) and immediately filtered using a peristaltic pump onto in‐line 25 mm diameter, 0.2 μm pore‐sized, Supor filters. Filtration times varied from 40 min to 2.5 hr depending on the volume. Filters were flash‐frozen in liquid nitrogen and stored at −80°C until shore‐based laboratory processing. Water samples (1.5 ml) for subsequent flow cytometric analyses of picoplankton abundance were fixed with 0.22 μm‐filtered formaldehyde (2% final concentration), incubated at 4°C for 15 min, flash‐frozen in liquid nitrogen, and stored at −80°C until shore‐based analyses.
Nodules and sediments were aseptically sampled from 0.25 m2 box cores (nodules) or 80 cm2 megacore tubes (nodules and sediments) (see (Glover, Dahlgren, Wiklund, Mohrbeck, & Smith, 2016) for box‐coring and megacoring equipment and sampling protocols). Nodules were mostly found within the 0–5 cm fraction of the box core and megacore, although some were recovered from our maximum sampling depth of 10 cm. All nodules found in megacore tubes designated for microbiology (usually 2 per deployment) were collected, as were a random subset of nodules from boxcores. From this collection of megacore and boxcore nodules, a random subset were selected for subsequent DNA extraction, and amplification and sequencing of 16S and 18S rRNA genes. In total, extracts from 20 nodules were used for 16S rRNA amplification and sequencing, with 18 of these nodule‐derived DNA extracts also used for 18S rRNA gene amplification and sequencing. Subcores of sediments were obtained using sterile 20 mL syringes with the tip ends cutoff, in each of four sediment horizons: 0–5 cm below seafloor (cmbf), 5–6 cmbf, 6–8 cmbf, and 8–10 cmbf. Sediment subcores were stored in sterile Whirl‐Pak bags (Nasco, Fort Atkinson, Wisconsin) at −80°C. Nodules were rinsed with 0.2 μm‐filtered ambient bottom water to remove sediment adhering to the surface and stored whole in sterile Whirl‐Pak bags at −80°C.
2.2. DNA extraction
In the shore‐based laboratory, genomic DNA was extracted from seawater samples using a DNeasy Plant Mini Kit (Qiagen) following a modified protocol (Paerl, Foster, Jenkins, Montoya, & Zehr, 2008). Briefly, the filters were subjected to chemical and physical (bead‐beating step using both 0.1 and 0.5 mm beads) means for cell disruption. Total lysates were purified using the DNeasy Mini spin column procedure (Qiagen) following the manufacturer's recommendations.
Under sterile laboratory conditions, the polymetallic nodules were rinsed with 0.2 μm‐filtered, autoclaved, bottom (~4000 m) seawater, returned to Whirl‐Pak bags, and broken while still in the bag using an autoclaved mortar and pestle. Two ~500 mg pieces from the interior of each nodule were subjected to DNA extraction. Extraction of DNA from nodules and sediments was performed using the FastDNA Spin Kit for Soil (MP Biomedicals, USA) following the manufacturer's protocol, modified as follows: homogenization was performed in a Mini‐Beadbeater‐16 (Biospec Products, Bartlesville, Oklahoma) and centrifugation following homogenization was extended to 15 min. An extraction blank (FastDNA Spin Kit for Soil spin column with no sample added) was processed alongside samples. DNA concentrations were determined from 4 μl of each sample using the Qubit 2.0 Fluorometer and the Qubit dsDNA High Sensitivity Assay kit (Life Technologies). Extracts with DNA concentrations >0.1 ng/μl were purified and concentrated using the Zymo Clean & Concentrator‐5 (2:1 DNA Binding Buffer) kit with the resulting DNA eluted in sterile, DNase‐free water.
2.3. PCR amplification and Illumina sequencing of 16S and 18S rRNA genes
The V4 region of the 16S rRNA gene was amplified by the polymerase chain reaction (PCR) using the oligonucleotide primer pair 515f/806r, which include the Illumina flowcell adapter sequences and a sample‐specific barcode exactly as described in Caporaso et al., (2011, 2012). Of the 20 nodules sampled for 16S rRNA genes, most were sampled in duplicate (i.e., two separate ~500 mg pieces from the same nodules were subjected to DNA extraction, PCR amplification, and sequencing). Initial 16S rRNA gene results from the duplicate samples generated from each nodule sample revealed a high degrees of similarity between profiles (Figure S3). To conserve the extracted genomic material, in most cases the duplicate extracts were pooled and only a single 18S rRNA gene amplification was performed from each nodule. The V9 region of the 18S rRNA gene was PCR amplified using the oligonucleotide primer pair 1391f/EukBr (Amaral‐Zettler, McCliment, Ducklow, & Huse, 2009) exactly as described in the Earth Microbiome Project 18S rRNA Amplification Protocol (Gilbert, Jansson, & Knight, 2014); this region (V9) of the 18S rRNA gene has previously been used in a large‐scale biodiversity assessment of marine eukaryotic communities (de Vargas et al., 2015). Sequencing of the 16S and 18S rRNA gene amplification products, including extraction blanks, was performed in separate runs on an Illumina MiSeq at the Hawaii Institute of Marine Biology Genetics Core Facility (Kaneohe, HI).
2.4. Bioinformatic analyses of sequences
Illumina paired‐end 16S rRNA gene reads were joined using the bioinformatic software fastq‐join (Aronesty, 2013) and sequences were processed, including an initial quality filtering and sequence sample‐mapping by barcode, using QIIME version 1.8.0 (Caporaso, Kuczynski, et al. 2010). Potentially chimeric sequences were identified using the UCHIME algorithm within the USEARCH package (Edgar, 2010) and removed from further analysis. Open reference‐based OTU picking was performed using the UCLUST algorithm (Edgar, 2010), one of the principal clustering algorithms in the QIIME package, at a 97% sequence similarity cutoff against the Greengenes rRNA gene database release 13_8 (DeSantis et al., 2006). OTUs that occurred as absolute singletons or were observed in the extraction and/or PCR blanks were filtered from the experimental samples. Taxonomy was assigned based on the Greengenes taxonomy (McDonald et al., 2012; Werner et al., 2012) using a UCLUST‐based consensus taxonomy assigner (Bokulich et al., 2015). A total of 13,835,715 high‐quality sequences were generated, with an average of 101,133 sequences/sample (minimum sequences/sample = 16,753; maximum sequences/sample = 236,702). These data were normalized to 16,000 reads/sample to account for uneven sampling depth using the script single_rarefaction.py, which randomly subsamples the input OTU table without replacement, and this normalized OTU table was used in subsequent analyses unless otherwise specified. The script summarize_otu_by_cat.py in the QIIME package was used to collapse this OTU table by sample type and/or depth when necessary. The only exceptions were the differential abundance analysis in which the full dataset was used, and alpha diversity analyses in which samples were collapsed by sample type (water column, nodules, or sediments) and the dataset was subsampled randomly multiple times at different depths, with a maximum depth of 2,401,000 sequences in order to take maximum advantage of this large dataset.
Illumina 5′ 18S rRNA gene reads were processed similarly to 16S rRNA reads, except reference‐based OTU picking was performed against the SILVA 119 rRNA gene database (Quast et al., 2013). Taxonomy was assigned based on the SILVA 119 taxonomy (Yilmaz et al., 2014) using BLAST (Altschul, Gish, Miller, Myers, & Lipman, 1990). The resulting OTUs were filtered to exclude 38,523 bacterial OTUs, 23,733 archaeal OTUs, and 3,126 OTUs that could not be identified at the domain level. A total of 54,819 Eukaryota OTUs comprised of 5,353,354 high‐quality sequences remained, with an average of 45,367 sequences/sample (minimum sequences/sample = 5450; maximum sequences/sample = 154,747). These data were normalized to 5,400 reads/sample to account for uneven sampling depth, and either this normalized OTU table, or a table normalized to relative abundance, was used in subsequent analyses, except the differential abundance analysis in which the full dataset was used and alpha diversity analyses in which samples were collapsed by sample type (water column, nodules, or sediments) and the dataset was subsampled randomly multiple times at different depths, with a maximum depth of 100,100 sequences. Joined, quality filtered 16S fastq files and 5′, quality filtered 18S fastq files have been deposited in the NCBI's Sequence Read Archive under BioProject ID PRJNA281530, SRA ID SRP057408.
The nodule prokaryotic core microbiome was computed using the script compute_core_microbiome.py within the QIIME package. The Wu et al. dataset was downloaded from NCBI and OTUs were picked and taxonomy assigned as described for our dataset. OTUs within our core microbiome that hit to Greengenes were compared to the newly created Wu et al. OTU table in order to identify reference‐based OTUs that were present in both datasets.
2.5. Statistical methodologies
Principal Coordinates Analysis (PCoA) was used to visualize patterns in microbial community structure based on sample type within the CCZ. Analysis of similarities (ANOSIM; (Chapman & Underwood, 1999) was performed on weighted UniFrac distance measurements of both 16S and 18S gene sequences, and implemented using the compare_categories.py script within the QIIME package. Briefly, UniFrac calculates a distance measure based on the fraction of branch length shared between two communities within a phylogenetic tree; weighted UniFrac additionally takes into account the differences in relative abundances of taxa within each community (Lozupone, Lladser, Knights, Stombaugh, & Knight, 2011). The prokaryotic phylogenetic tree used for UniFrac was built using FastTree (Price, Dehal, & Arkin, 2010) from representative sequences aligned with PyNAST (Caporaso, Desantis, et al., 2010), as implemented in the pick_open_reference_otus.py workflow, and is available as Figure S8; sequences which failed to align were omitted from both the tree and the OTU table. The eukaryotic phylogenetic tree was created similarly from representative sequences aligned with Infernal (Nawrocki, Kolbe, & Eddy, 2009) and is available as Figure S9. A heatmap (Figure 5) was created using the function heatmap.2 in the R package gplots (R Core Team, 2015; Warnes et al., 2016). A Bray–Curtis dissimilarity matrix was created from an OTU table containing the 10 most abundant OTUs in each habitat, average linkage hierarchical clustering was performed and a dendrogram was created using the R package vegan (Oksanen et al., 2016 ). Colors came from the R package RColorBrewer (Neuwirth, 2014). Average linkage hierarchical clustering was also done on the full dataset and the results were similar, that is, sediments, nodules, and the water column each formed groups (Figure S10). To create Figure 9, a differential analysis of count data using shrinkage estimation (DESeq2, (Love, Huber, & Anders, 2014)) was implemented on the full dataset (not rarefied) within the phyloseq package (McMurdie & Holmes, 2014). Differential OTUs which had a base mean of ≥100 (prokaryotes) or ≥10 (eukaryotes) were reported and visualized using the R package ggplot2 (Wickham, 2009).
Figure 5.
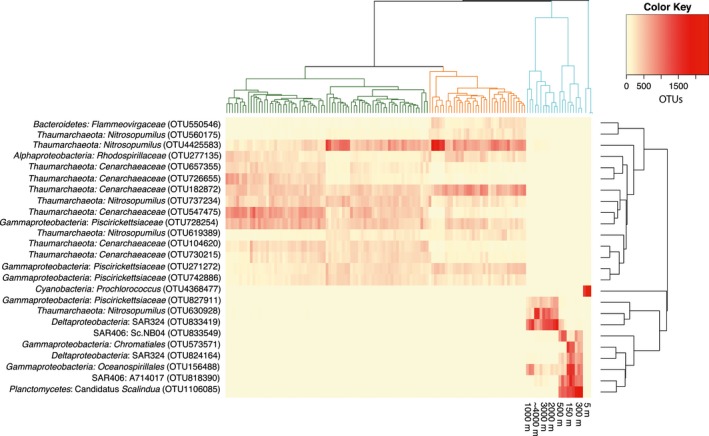
Heatmap of top 10 most abundant prokaryotic rRNA gene OTUs from each sample type. In some cases, the most abundant rRNA gene OTUs from sediments and nodules were the same, so total number of OTUs depicted is 25. Dendrograms were created via average linkage hierarchical clustering on a Bray–Curtis dissimilarity matrix of the selected dataset. Dendrogram on the Y‐axis is color‐coded by sample type as throughout the manuscript (blue = water, green = sediments, orange = nodules). Dendrogram on the X‐axis clusters OTUs that occur most frequently together. Heatmap color represents the number of OTUs found in each sample after normalization to 16,000 reads/sample. OTU, operational taxonomic unit
Figure 9.
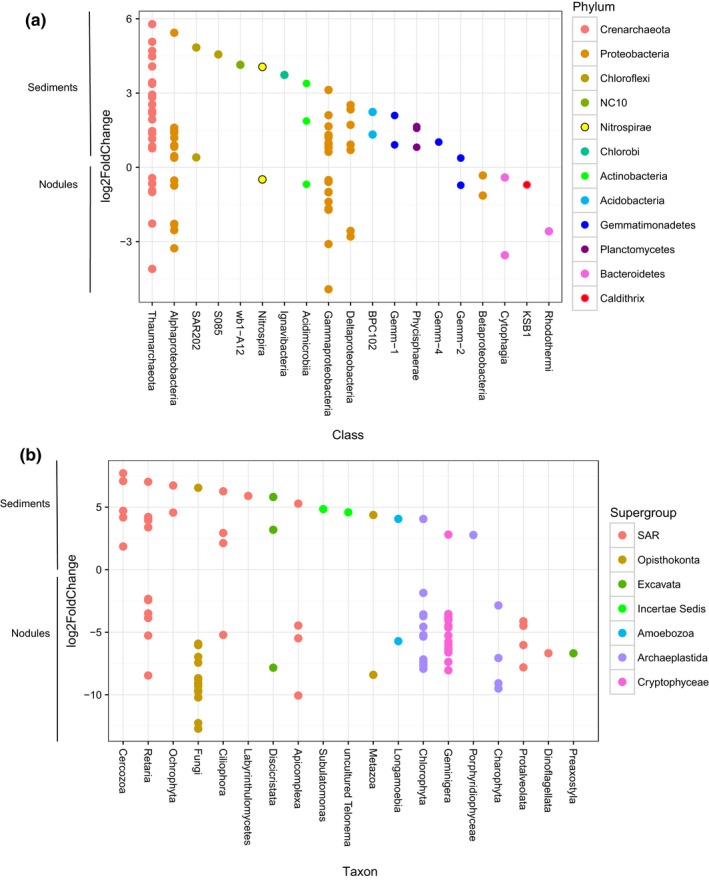
Plot depicting differentially abundant operational taxonomic units as the log2(fold change) of sediment versus nodule samples for (a) prokaryotic and (b) eukaryotic communities. OTUs that are more abundant in sediments are above the zero line, whereas those that are more abundant in nodules are below the zero line. OTUs are colored by phylum (prokaryotes) or supergroup (eukaryotes). OTU, operational taxonomic unit
2.6. Flow cytometric cell abundances
Seawater samples for flow cytometric analyses were thawed and 250 μl aliquots were transferred to 96‐well plates and stained with SYBR Green I (final concentration of 1X). Abundances of picoplanktonic cells were determined using an Attune Acoustic Focusing Cytometer (Life Technologies, Carlsbad, CA) at a flow rate of 100 μl min−1, using an excitation of 488 nm and detected using a 530/30 bandpass filter and side scatter.
2.7. Seawater nutrient and DOC concentrations
Seawater samples for subsequent analysis of phosphate (PO4 3‐), nitrate plus nitrite (NO3 − + NO2 −), silicate, and dissolved organic carbon (DOC) were collected into clean, acid washed 125 ml polyethylene bottles and frozen upright at −20°C. In the shore‐based laboratory, nutrient concentrations were measured colorimetrically using a Bran+Luebbe Autoanalyzer III (Karl et al., 2001). DOC analyses relied on high‐temperature combustion using a Shimadzu TOC‐V (DOM Analytical Lab, Santa Barbara, CA; (Carlson et al., 2010)).
3. Results and Discussion
3.1. Chemical characterization of UK‐1 claim area
Seawater nutrient concentrations and microbial cell abundances were sampled from vertical profiles of the water column overlying the abyssal seabed. Nutrient concentrations were typical for this region, with low (<0.2 μmol L−1) concentrations of nitrate + nitrite (N+N) in the near‐surface waters, increasing rapidly with depth, reaching concentrations (~45 μmol L−1) typical for the deep waters of the Eastern Tropical North Pacific (Garcia et al., 2014) (Table 2). One of the most prominent features was the presence of a large, relatively shallow, oxygen minimum zone (OMZ), where dissolved oxygen concentrations declined to <10 μmol L−1 (< 0.2 ml/L) between ~50 m to ~1000 m (Figure S1).
Table 2.
Mean concentrations of seawater nutrients, DOC, and picoplankton cell abundances from those depths where seawater samples were collected for rRNA gene analyses
| Depth (m) | PO4 3−(μmol L−1) | N+N (μmol L−1) | SiO4 (μmol L−1) | DOC (μmol L−1) | Picoplankton abundances (cells ml−1) |
|---|---|---|---|---|---|
| 5 | 0.18 ± 0.03 | 0.17 ± 0.01 | 1.89 ± 0.42 | 76.37 ± 0.54 | 5.68 × 105 ± 8.13 × 104 |
| 150 | 2.59 ± 0.02 | 26.29 ± 0.51 | 32.54 ± 0.50 | 46.35 ± 0.17 | 3.05 × 105 ± 2.37 × 104 |
| 300 | 2.82 ± 0.04 | 29.14 ± 0.69 | 41.51 ± 1.47 | 43.29 ± 0.71 | 1.55 × 105 ± 6.89 × 103 |
| 500 | 3.14 ± 0.00 | 37.03 ± 0.13 | 63.72 ± 0.31 | 39.98 ± 1.46 | 7.92 × 104 ± 4.34 × 103 |
| 1000 | 3.35 ± 0.01 | 46.63 ± 0.13 | 105.64 ± 0.03 | 39.06 ± 1.30 | 4.98 × 104 ± 4.41 × 103 |
| 2000 | 2.85 ± 0.02 | 41.46 ± 0.14 | 152.16 ± 0.50 | 37.49 ± 0.39 | 3.67 × 104 ± 1.78 × 103 |
| 3000 | 2.67 ± 0.01 | 39.34 ± 0.10 | 160.81 ± 0.12 | 37.08 ± 1.21 | 4.40 × 104 ± 7.27 × 103 |
| Near‐bottom waters (4013 –4103) | 2.59 ± 0.08 | 38.07 ± 1.35 | 156.98 ± 2.93 | 38.94 ± 0.64 | 3.86 × 104 ± 7.77 × 102 |
DOC, dissolved organic carbon; PO4 3–, phosphate; N+N, nitrate+nitrite; SiO4, silicic acid.
Shown are mean ± SD concentrations of the four water column stations sampled in the UK‐1 claim region.
3.2. General characteristics of the UK‐1 rRNA gene phylogenetic profiles
In total, after rarefaction, 112,926 distinct prokaryotic OTUs (defined as 16S rRNA gene sequences sharing ≥97% sequence identity) were recovered from the three types of samples (nodules, sediment, and seawater), with 74% of these amplicons deriving from bacteria, 24% from archaea, and an additional 2% that could not be confidently assigned at the Domain level. Within this dataset, a large proportion of OTUs were taxonomically assigned to the Proteobacteria (45% of sequences), including Gammaproteobacteria (21%), Alphaproteobacteria (14%), Deltaproteobacteria (9%), and Betaproteobacteria (1%). OTUs taxonomically assigned to the Thaumarchaeota also comprised a relatively large proportion of this dataset (22% of the sequences). Among the eukaryote 18S rRNA genes, 26,011 distinct OTUs (defined as sequences sharing ≥97% sequence identity) were recovered from the three sample types. The greatest proportion (51%) of these eukaryotic sequences fell into OTUs assigned to the Sar (formal taxon name derived from Stramenopiles, Alveolata, and Rhizaria; (Adl et al., 2012)) supergroup, whereas lower proportions grouped among the Opisthokonta (14%), Cryptophyceae (14%), Excavata (8%), Archaeplastida (8%), and Amoebozoa (2%). Sar was the dominant supergroup in all three habitats (seawater, sediment, and nodules). However, the Cryptomonadales, an order within the Cryptophyceae, represented the greatest relative abundance (25%) of classified sequences on nodules, more than any individual order within the Sar. In contrast, in the water column and sediments, the lineage Alveolata within the Sar supergroup dominated relative sequence abundances (43% and 30%, respectively).
3.3. Seawater communities
The distributions and relative abundances of prokaryote 16S rRNA genes in the water column of this region appeared typical of open‐ocean habitats, with notable exceptions observed in the low oxygen waters of the mesopelagic zone (Figure S2). OTUs identified as belonging to the candidate phylum Marine Group A (MGA) (formerly known as SAR406; (Fuhrman & Davis, 1997)), specifically the AB16 gene lineage, were the dominant lineage within the OMZ, comprising relative abundances of 24% and 32% at 300 m and 500 m, respectively (Figure 2j, and S2a). There are currently no cultivated representatives of the MGA, but these organisms appear abundant and diverse in OMZ waters (Allers et al., 2013; Fuchs, Woebken, Zubkov, Burkill, & Amann, 2005; Stevens & Ulloa, 2008), and recent metagenomic analyses from seawater samples collected in the North Pacific suggest these organisms are involved in oxidation of various reduced sulfur substrates (Wright et al., 2014). OTUs clustering among the Planctomycetes were also enriched in the oxygen minimum zone (averaging 12% relative abundance across all stations investigated, Figure S2a), and these OTUs demonstrated low relative abundances (averaging 2%) at other depths. This enrichment in Planctomycetes OTUs was driven by sequences deriving from members of the ‘Candidatus Scalindua’ (Figure 2g), a genus capable of catalyzing anaerobic ammonium oxidation (anammox; (Kuypers et al., 2003)). The anammox reaction is known to be a major loss for fixed nitrogen in various regions of the ocean (Kuypers et al., 2005; Ulloa, Canfield, DeLong, Letelier, & Stewart, 2012).
Figure 2.
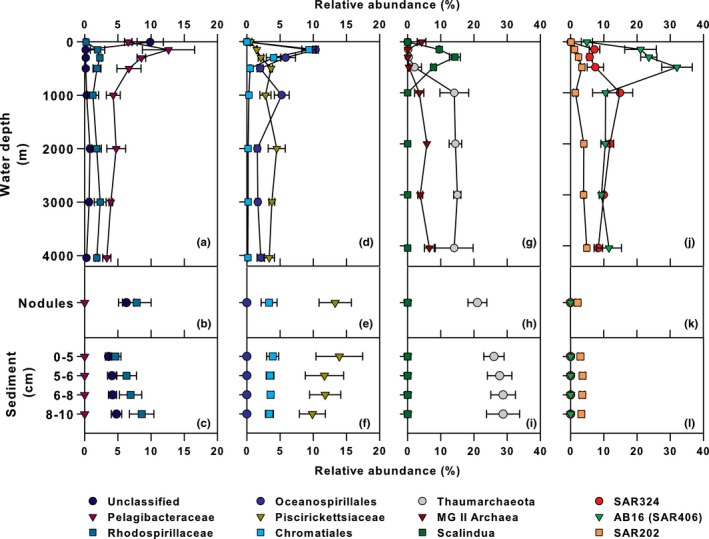
Vertical profiles depicting relative abundances of selected prokaryote rRNA gene lineages from the water column (a, d, g, & j), sediments (b, e, h, & k), and nodules (c, f, i, & l). Depicted are mean and standard deviation (error bars) of samples collected during AB‐01 from various locations in UK‐1 study region
The water column microeukaryote community was dominated by Protalveolata (Sar: Alveolata; 35%), Discicristata (Excavata: Discoba; 20%), and Retaria (Sar: Rhizaria; 15%); the relative abundances of these lineages varying with depth (Figure S2b). Nearly all the Protalveolata sequences clustered among the Syndiniales, a presumably exclusively endoparasitic group common in culture‐independent surveys of marine environments (Guillou et al., 2008; de Vargas et al., 2015). Group II Syndiniales dominated the anoxic and suboxic OMZ, whereas Groups I and II coexisted in the photic and oxygenated bathypelagic zones (Figure 3d). The Discicristata were solely Euglenozoa, mostly Diplonemea, a group of heterotrophic flagellates with major uncultured clades distributed throughout the deep‐sea (Lara, Moreira, Vereshchaka, & Lopez‐Garcia, 2009). This was reflected in our data, as the Diplonemea (Discicristata) were nearly absent from the euphotic zone samples and increased in relative abundance with depth (Figure 3j and S2b). The Retaria were represented by Radiolaria, mainly the Acantharia (4%), Polycystinea (6%), and members of radiolarian sequence group RAD B (5%). It has been hypothesized that one or more of these Radiolarian groups may serve as a host for the parasitic Syndiniales (Not, Gausling, Azam, Heidelberg, & Worden, 2007). Within our dataset, OTUs assigned to the Acantharia (Figure 3a) and Polycystinea (data not shown) had similar depth‐distribution patterns to OTUs assigned to Syndinales Group II (Figure 3d), a finding consistent with the Not et al. (2007) hypothesis.
Figure 3.
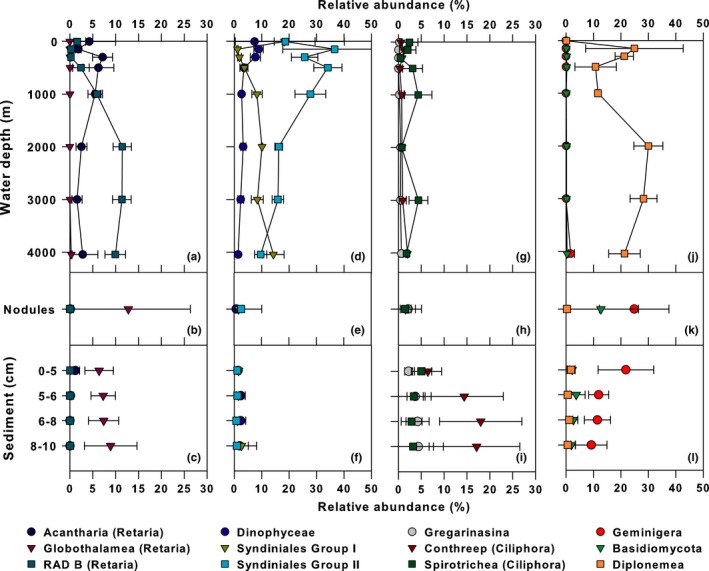
Vertical profiles depicting relative abundances of selected eukaryote rRNA gene lineages from the water column (a, d, g, & j), sediments (b, e, h, & k), and nodules (c, f, i, & l). Depicted are mean and standard deviation (error bars) of samples collected during AB‐01 from various locations in UK‐1 study region
3.4. Seabed‐associated communities
The relative abundance of nodule‐associated 16S rRNA gene sequences was dominated by members of the Gammaproteobacteria (23%), Thaumarchaeota (21%), and Alphaproteobacteria (18%; Figure S3), consistent with previous studies examining nodule‐associated archaeal and bacterial communities (Tully & Heidelberg, 2013; Wu et al., 2013). The same three major groups of prokaryotes were also dominant in the sediment samples (Figure 4): Thaumarchaeota (28%), Gammaproteobacteria (20%), Alphaproteobacteria (12%). Within the Thaumarchaeota, the genus Nitrosopumilus was prevalent in both the sediments and nodules (12% and 10%, respectively). The most abundant OTUs classified as Nitrosopumilus in the sediments and nodules were distinct from the abundant Nitrosopumilus OTU found in the bottom waters, and indeed, throughout the bathypelagic zone in AB‐01 (Figure 5), implying niche specialization between the sediment/nodules and water column. The genus Nitrosopumilus contains the ammonia‐oxidizing chemolithoautotrophic archaeal nitrifiers (Konneke et al., 2005); the relatively large proportion of these archaea in sediment and nodule samples could reflect increased importance of reduced substrates such as ammonia in the benthic environment as compared with the water column (Orcutt, Sylvan, Knab, & Edwards, 2011). In addition to the Thaumarchaeota, the class Nitrospira, containing potential chemolithoautotrophs, comprised a minor but measurable (1%) portion of the sediment community (Figure. 4).
Figure 4.
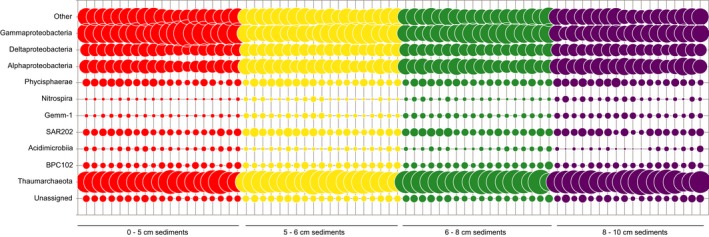
Major class‐level lineages of prokaryotic sediment taxa present at ≥2% relative rRNA gene abundances in at least two samples. Vertical lines represent the individual samples collected from each sediment horizon. Category “Other” represents all named taxa that did not reach the ≥2% relative abundance cutoff
Among the Gammaproteobacteria, sequences clustering among unclassified genera within the family Piscirickettsiaceae demonstrated the greatest relative abundance in both the sediments and nodules (12% and 13% relative abundances, respectively), with relative abundances decreasing with depth in the sediment. The Piscirickettsiaceae are a family of aerobic, aquatic bacteria, and a recent study indicated these organisms were enriched in seawater microcosms treated with cadmium (Wang et al., 2015). Generalized resistance to metal toxicity may explain their relatively high abundances within the nodule field. Additionally, sequences classified as belonging to the order Chromatiales comprised 2–5% of the total 16S rRNA genes throughout the sediments. Previous studies in the central and western Pacific have recovered rRNA genes belonging to the Chromatiales from sediments and nodules (Wu et al., 2013), as well as sediments associated with cobalt‐rich sediment crusts (Liao et al., 2011).
The dominant Alphaproteobacteria in both nodules and sediments were an unclassified genus within the family Rhodospirillaceae, occurring at 8% and 7% relative abundance, respectively, which was not present in the water column (Figure 5). Although Rhodospirillaceae are often found in anaerobic environments, this family contains the genus Magnetospirillum, a microaerophilic heterotroph with relatives known from sediments previously collected in the Pacific Nodule Province (Xu, Wang, Meng, & Xiao, 2007).
All the major Eukaryotic supergroups were represented in the nodule and sediment datasets (Figure S4, 6 and 3b–l). On nodules, the groups Geminigera (25%; a genus of cryptophytes), Fungi (14%), and Retaria (13%; a clade within Rhizaria) demonstrated the greatest relative abundances (Fig. S4). Fungi were almost exclusively comprised of Basidiomycota (13% of total sequences), a broad phylum that contains both yeasts and filamentous fungi. Sequences clustering among the Ciliophora (20%), Geminigera (13%), and Retaria (13%) were abundant in the sediments, although the relative abundances of these organisms varied with depth (Figure 3c, i and l). Ciliophora are relatively well‐studied ciliated, heterotrophic eukaryotes (Pierce & Turner, 1992; Verni & Gualtieri, 1997); however, the ecology and biogeochemical role of deep‐sea groups of these organisms remains unknown. Within our dataset, the Conthreep and Spirotrichea (Ciliophora) lineages were most abundant, with Conthreep OTUs increasing below the 0–5 cm sediment horizon, whereas the Spirotrichea remained at low relative abundance throughout the sediment (Figure 3i). The 18S rRNA profiles in the both the sediments and nodules also contained OTUs from the Archaeplastida and the Cryptophyceae, two groups of organisms most often associated with plastid‐containing photosynthetic organisms. In total, the sediment and nodule communities contained 1002 and 629 Archaeplastida OTUs and 1327 and 759 Cryptophyceae OTUs, respectively. Such results are consistent with previous 18S rRNA gene surveys in deep waters of the Gulf of California, and the Arctic and Southern Oceans (Edgcomb, Kysela, Teske, Gomez, & Sogin, 2002; Pawlowski et al., 2011), a finding attributed to sinking of surface water organisms and subsequent preservation in the sediment (Pawlowski et al., 2011).
Figure 6.
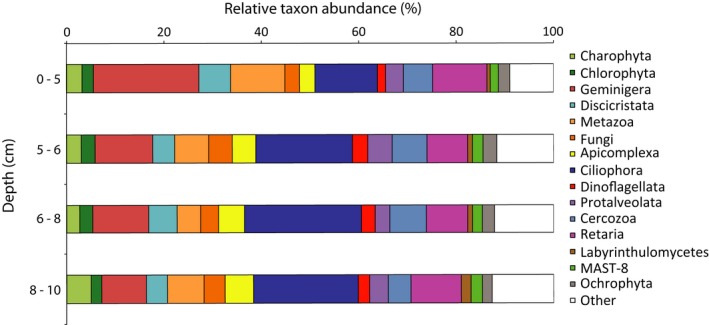
Eukaryotic sediment taxa from the 0–5 cm, 5–6 cm, 6–8 cm, and 8–10 cm horizons, averaged across all samples taken at a given depth; rRNA gene taxa present ≥2% relative abundance (on average) in at least one horizon are depicted. Category “Other” represents all named taxa that did not reach the ≥2% relative abundance cutoff
3.5. Alpha diversity of Clarion‐Clipperton Zone microbial communities
After pooling by habitat and rarefaction to 2,401,000 sequences per habitat in order to account for differences in sequencing depth while still utilizing a large portion of the dataset, 33,732 prokaryotic OTUs were found in the water column, 93,790 prokaryotic OTUs were found in the nodules, and 111,413 prokaryotic OTUs were found in the sediments (Table S1). Species accumulation curves indicate that OTUs were still accumulating in the sediments and nodules at this depth of sequencing, whereas sequence diversity in the water column appeared to plateau (Figure 7). Chao1 predicts a species richness of 35,083 prokaryotic OTUs in the water column, 118,552 prokaryotic OTUs in the nodules, and 184,335 prokaryotic OTUs in the sediments (Table S1). For eukaryotes, at a sampling depth of 100,100 sequences (pooled by habitat), the accumulation curves do not appear to approach an asymptote for nodules, the water column, or the sediments, indicating undersampling of all three habitats (Figure 7). There were 6,704 eukaryotic observed OTUs in the water column, 4,744 eukaryotic OTUs in the nodules, and 9004 eukaryotic OTUs in the sediments, with Chao1 richness estimates of 13,373, 8831, and 15,344 OTUs, respectively (Table S1).
Figure 7.
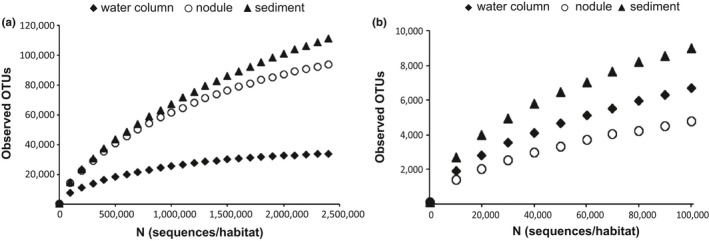
Rarefaction analyses of observed operational taxonomic units among nodule, sediment, and water column (a) prokaryotic and (b) eukaryotic communities
Based on the three different measures of diversity assessed in this study (Chao1, exponential of Shannon's, and observed OTUs), nodule and sediment prokaryotic communities harbored greater alpha diversity than water column communities when pooled by habitat (Table S1). This held true when the alpha diversity of individual samples within these habitats were considered as well (rarefied to 16,000 sequences/sample, nonparametric t‐test, observed OTUs, p = .003 for both nodule versus water column and sediment vs. water column; Chao1, p = .003 for both nodule versus water column and sediment vs. water column; exponential of Shannon's, p = .003 for both nodules vs. water column and sediment vs. water column). Additionally, on average, prokaryotic sediment communities demonstrated greater alpha diversity than nodule communities (nonparametric t‐test, observed OTUs, p = .006).
3.6. Beta and gamma diversity of Clarion Clipperton Zone microbial communities
Principal Coordinates Analysis (PCoA) of weighted UniFrac (Lozupone & Knight, 2005) distances of the 16S and 18S rRNA gene amplicon communities, revealed that the seawater, sediments, and nodules each harbored distinct prokaryotic (Figure S5a) and eukaryotic (Figure S5b) assemblages and this was supported by ANOSIM (R statistic = .8203, p = .001 and R statistic = .7996, p = .001, respectively). PCoA analysis of the nodules and different sediments layers was conducted, and both nodules and 0–5 cm sediments harbored prokaryotic and eukaryotic communities distinct from each other and from the deeper sediments (Figure 8). Microbial prokaryote and eukaryote assemblages within the sediments were further shown to vary with depth via ANOSIM (R statistic = .3105, p = 0.001 and R statistic = .3357, p = 0.001, respectively). However, there were no significant differences in microbial community structure among the different stations sampled within the 30 × 30 km stratum of the study site (ANOSIM; prokaryotes: R statistic = .1126, p = .002; eukaryotes: R statistic = .0432, p = .079), suggesting that these assemblages are widespread within the ~900 km2 sampling area. Repeating the PCoA analyses with abundance weighted Jaccard distance matrices gave similar results (Figure S6 & S7).
Figure 8.
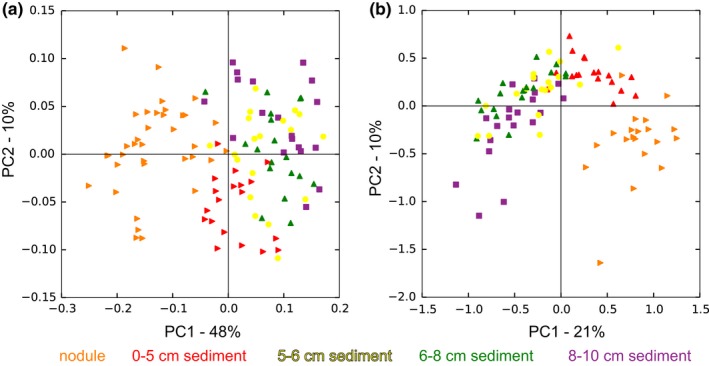
Principal Component Analysis (PCoA) plots based on weighted UniFrac distance measurements comparing: (a) prokaryotic communities; (b) eukaryotic communities associated with different layers of sediment and nodules in the study area
3.7. Trends in microbes differentially represented in sediments and nodules
A differential analysis of count data (Love et al., 2014) was performed to determine whether specific OTUs discriminated between nodules and sediment samples. Ninety‐three prokaryotic OTUs with assigned taxonomy were identified as differentiating the 75 sediment samples from the 36 nodule samples (Figure 9a). Ninety eukaryotic OTUs differentiated the 74 sediment samples from the 20 pooled nodule samples (Figure 9b). Although the prokaryotic OTUs that differentiated the sediment and nodule samples came from diverse phyla, some general trends emerged. All the differential Chloroflexi OTUs recovered (4 OTUs; 4% of differential OTUs) were more abundant in sediment samples than nodule samples (Figure 9a). This agrees with data from the German mining claim area, to the west of our study site, where Chloroflexi were found in sediments but were not associated with nodules (Blothe et al., 2015). Fifteen (16%) of the differentially abundant OTUs were Alphaproteobacteria (Figure 9a). Of those Alphaproteobacteria more highly represented on the nodules, two fell into the family Hyphomicrobiaceae within the Rhizobiales, a group that contains members known to be involved in manganese cycling (Larsen, Sly, & McEwan, 1999). The Alphaproteobacterial OTUs identified as more abundant in the sediments than nodules were either Rhodospirillales or unclassified beyond the class level; none of them were classified as Rhizobiales, potentially indicating a unique niche for Rhizobiales on the nodules. Five of the seven deltaproteobacterial OTUs were overrepresented in the sediments relative to the nodules. Deltaproteobacteria are ubiquitous members of the deep‐sea surface sediment community (Kouridaki, Polymenakou, Tselepides, Mandalakis, & Smith, 2010; Schauer et al., 2010), including within polymetallic nodule fields (Wang et al., 2010; Xu, Wang, Wang, & Xiao, 2005).
Eukaryotic communities in both the various sediment samples and on collected nodules demonstrated considerable sample‐to‐sample heterogeneity (Figure S4), but differential abundance analysis identified several taxa that appear to prefer either sediments or nodules (Figure 9b). For example, three of the four Ciliophora OTUs identified as differentially abundant were more abundant in the sediments, and differential OTUs from the clade Cercozoa, a diverse group of heterotrophic protists, were found solely in the sediments (Figure 9b). Fifteen (23%) of the OTUs that were more abundant on the nodules derived from the Opisthokonta, the supergroup containing both the Fungi and the Metazoa (Figure 9b). This included one metazoan OTU unique to this study classified as Enoplea (a nematode), and 14 fungal OTUs, three of which were related to the yeast‐like fungus Pseudozyma.
3.8. Comparisons to other polymetallic nodule datasets
In addition to describing the microbial communities associated with the sediments, nodules, and overlying waters, we compared our results to previously published studies from geographically diverse sites within the Pacific Ocean to place our observations in a basin‐scale context of known nodule‐associated microbial communities. The large number of nodules collected in this study allowed us to compute a “nodule core microbiome” for the AB‐01 region in order to compare it to nodules across the CCZ. In total, 196 prokaryotic OTUs were present in 100% of our nodules sampled; 168 of these fell into OTUs with representative sequences within the Greengenes database whereas 28 were novel (Table S2). In order to look at connectivity across the CCZ, we compared the 168 reference‐based OTUs to OTUs identified in the three nodules sampled from two sites at distances ~3000 km and >9000 km from AB‐01, within the North Pacific (Wu et al., 2013). Forty‐seven (28%) of these OTUs were also retrieved in at least one of the nodules studied by Wu et al.; nine of these OTUs were found in all three nodules studied by Wu et al. (Table 3). Many of these core OTUs fell into taxa that our differential abundance analyses revealed to prefer nodules rather than sediments, such as the Cytophagia and the Rhizobiales (Table 3, Figure 9a). These results indicate that certain stable and consistent associations between microbes and nodules exist over thousands of kilometers of abyssal ocean.
Table 3.
Core nodule OTUs found in all nodules in this study and all nodules in Wu et al., at 3000 – 9000 km distance from the AB‐01 site
| Greengenes OTU ID | Taxonomy |
|---|---|
| 1054 | Archaea; Crenarchaeota; Thaumarchaeota; Cenarchaeales; Cenarchaeaceae |
| 182872 | Archaea; Crenarchaeota; Thaumarchaeota; Cenarchaeales; Cenarchaeaceae |
| 196754 | Archaea; Crenarchaeota; Thaumarchaeota; Cenarchaeales; Cenarchaeaceae |
| 540244 | Archaea; Crenarchaeota; Thaumarchaeota; Cenarchaeales; Cenarchaeaceae |
| 737234 | Archaea; Crenarchaeota; Thaumarchaeota; Cenarchaeales; Cenarchaeaceae; Nitrosopumilus |
| 550546 | Bacteria; Bacteroidetes; Cytophagia; Cytophagales; Flammeovirgaceae |
| 704361 | Bacteria; Planctomycetes; Phycisphaerae; C86 |
| 567782 | Bacteria; Proteobacteria; Alphaproteobacteria |
| 260794 | Bacteria; Proteobacteria; Alphaproteobacteria; Rhizobiales; Hyphomicrobiaceae |
A recent study of the microbes associated with two polymetallic nodules in the east German license area, ~300 km from our study site, found that members of the gammaproteobacterial genera Colwellia and Shewanella made up 30–50% of the bacterial sequences (n = 75) in clone libraries and 66–84% of gammaproteobacterial sequences derived from pyrosequencing (Blothe et al., 2015). Similarly, a study of three nodules collected in the South Pacific Gyre found that Colwellia comprised ~40–55% of the prokaryotic community on one of three nodules, but were absent or nearly absent (0.3%) on the other two nodules (Tully & Heidelberg, 2013). Our analysis of 20 nodules from 8 stations, sampled by two different apparatus (megacore and boxcore), recovered no sequences classified as Colwellia and a minute fraction (<0.0006%) classified as Shewanella. One possible explanation for the high amounts of Colwellia and Shewanella in the Tully and Heidelberg and Blothe et al. studies could be sediment contamination, since our methodology employed a stringent double wash of the exterior of the nodule samples prior to DNA extraction and amplification. However, we find vanishingly small amounts of Colwellia (0.00008%) and Shewanella (0.002%) even in our sediment samples. Blothe et al. considered this possibility as well and performed group‐specific PCR of sediment samples to ensure that the Shewanella signal they found truly came from the nodules rather than sediment contamination. Additionally, in the Tully and Heidelberg study, the Colwellia OTUs were predominantly retrieved from the inner portion of the nodule sample (42–55%) rather than the outer portion (6%). Hence, we suspect sediment contamination is likely not the explanation for differences between our study and these other studies. Colwellia and Shewanella are generally considered copiotriophs, demonstrating rapid growth rates and hence are found in regions where concentrations of organic and inorganic nutrients are elevated (Lauro et al., 2009), and Colwellia and Shewanella are both known to colonize marine invertebrates (Gillan, Speksnijder, Zwart, & De Ridder, 1998; Ivanova et al., 2004). The greater abundances of these Alteromondales in various nodules from other studies and complete absence in this study (as well as absence from individual nodules in the Tully and Heidelberg (2013) study) may reflect regional differences within the CCZ in the colonization of nodules by invertebrates and their associated microflora, or simply a high degree of endemism at the abyssal seafloor when examined over larger spatial scales (>300 km) (Bienhold, Zinger, Boetius, & Ramette, 2016). In addition, differences among these studies could reflect biases associated with the choice of PCR primers; for this study, the forward (515f) and reverse (805r) PCR primers we relied on for prokaryotic identification can yield single‐nucleotide mismatches to the 16S rRNA genes of members of the Thaumarcheota Marine Group I (Parada, Needham, & Fuhrman, 2015) and the SAR11 clade (Apprill, McNally, Parsons, & Weber, 2015), respectively. However, this would not explain the lack of the Gammaproteobacteria Colwellia and Shewanella associated with nodules sampled in our study. We further checked these primers using Silva TestPrime 1.0 and found that the primer pair 515f/805r had coverage of 92% within the Shewanella and 89% within the Colwellia when no mismatches were allowed. When one mismatch was allowed, this coverage rose to 96 and 94%, respectively. Therefore, it seems unlikely that the primers used for amplification in our study are biased against these genera. We utilized the same primers for all the samples collected for this study, facilitating comparative assessment of prokaryotic diversity across the different types of habitats (seawater, sediment, and nodules) and stations sampled. Finally, there may have been differences in the physical and/or chemical structure of the nodules within the German claim area and those nodules sampled from the UK claim area, which might promote differences in the microbiota observed between these studies.
4. Conclusions
We provide strong evidence supporting the growing view that polymetallic nodules, surrounding sediments, and the overlying water column constitute distinct microbial habitats with characteristic microbial assemblages. Microbial assemblages differ with depth into the sediment such that surface sediment removal and/or accelerated burial by resettling from a near‐bottom sediment plume are likely to fundamentally alter microbial community structure. Sediments and nodules are major reservoirs of microbial diversity distinct from even the deep water column, suggesting that large‐scale removal of nodules and sediments might alter local, and even regional, patterns of microbial diversity and ultimately modify specific ecosystem functions. Over the ~30 km scales of our study, each of these habitats appears to harbor similar microbial assemblages. Additional work is needed to determine if microbial assemblages in the sediments and nodules vary over the ~6 million square km expanse of the Clarion‐Clipperton Zone, as suggested by the lack of Colwellia and Shewanella sequences in our samples versus within the German claim area ~300 km away. However, our current study provides the first evidence of a widespread core nodule microbial community across large regions of the Pacific Ocean. Additionally, many of the prominent prokaryotic genera retrieved from the sediment and nodules in our study and others suggest an important role for chemoautotrophy within and above the nodule field. The energy sources sustaining such metabolisms remain unknown. Finally, future work is needed to understand the stability and resilience of these microbial ecosystems to perturbations such as those likely to result from nodule‐mining operations.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Acknowledgments
We thank all members of the ABYSSLINE science party for logistical help at sea and on land, particularly D. Amon, as well as the captain, officers, and crew of the R/V Melville for their assistance on MV1313. We are grateful to S. Curless, L. Fujieki, and F. Santiago‐Mandujano for their assistance with nutrient analyses and CTD data processing; E. DeLong for computational resources; and B. Pedler and S. Goldberg for guidance on the flow cytometric analyses. We also thank three anonymous reviewers for their helpful and thorough comments and suggestions, which improved the manuscript. Funding for this research derived from a contract from UK Seabed Resources, LTD. (UKSR) to C.R. Smith and M. Church of the University of Hawaii. UKSR had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Shulse, C. N. , Maillot, B ., Smith, C. R. and Church, M. J. Polymetallic nodules, sediments, and deep waters in the equatorial North Pacific exhibit highly diverse and distinct bacterial, archaeal, and microeukaryotic communities. MicrobiologyOpen. 2017;6:e00428 https://doi.org/10.1002/mbo3.428
References
- Adl, S. M. , Simpson, A. G. B. , Lane, C. E. , Lukes, J. , Bass, D. , Bowser, S. S. , Brown, M. W. , Burki, F. , Dunthorn, M. , Hampl, V. , Heiss, A. , Hoppenrath, M. , Lara, E. , … Spiegel, F. W . (2012). The revised classification of eukaryotes. Journal of Eukaryotic Microbiology, 59, 429–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allers, E. , Wright, J. J. , Konwar, K. M. , Howes, C. G. , Beneze, E. , Hallam, S. J. , & Sullivan, M. B. (2013). Diversity and population structure of Marine Group A bacteria in the Northeast subarctic Pacific Ocean. ISME Journal, 7, 256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S. F. , Gish, W. , Miller, W. , Myers, E. W. , & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Amaral‐Zettler, L. A. , McCliment, E. A. , Ducklow, H. W. , & Huse, S. M. (2009). A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small‐subunit ribosomal RNA genes. PLoS ONE, 4, e6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apprill, A. , McNally, S. , Parsons, R. , & Weber, L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquatic Microbial Ecology, 75, 129–137. [Google Scholar]
- Aronesty, E. (2013). Comparison of sequencing utility programs. The Open Bioinformatics Journal, 7, 1–8. [Google Scholar]
- Bienhold, C. , Zinger, L. , Boetius, A. , & Ramette, A. (2016). Diversity and biogeography of bathyal and abyssal seafloor bacteria. PLoS ONE, 11, e0148016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blothe, M. , Wegorzewski, A. V. , Muller, C. , Simon, F. , Kuhn, T. , & Schippers, A. (2015). Manganese‐cycling microbial communities inside deep‐sea manganese nodules. Environmental Science and Technology, 49, 7692–7700. [DOI] [PubMed] [Google Scholar]
- Bokulich, N. A. , Rideout, J. R. , Kopylova, E. , Bolyen, E. , Patnode, J. , Ellett, Z. , … Caporaso, J. G . (2015). A standardized, extensible framework for optimizing classification improves marker‐gene taxonomic assignments. PeerJ PrePrints, 3, e1502. [Google Scholar]
- Burnett, B. R. , & Nealson, K. H. (1981). Organic films and microorganisms associated with manganese nodules. Deep‐Sea Research. Part I, Oceanographic Research Papers, 28, 637–645. [Google Scholar]
- Caporaso, J. G. , Bittinger, K. , Bushman, F. D. , DeSantis, T. Z. , Andersen, G. L. , & Knight, R. (2010). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics, 26, 266–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso, J. G. , Kuczynski, J. , Stombaugh, J. , Bittinger, K. , Bushman, F. D. , Costello, E. K. , … Knight, R . (2010). QIIME allows analysis of high‐throughput community sequencing data. Nature Methods, 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso, J. G. , Lauber, C. L. , Walters, W. A. , Berg‐Lyons, D. , Huntley, J. , Fierer, N. , … Knight, R . (2012). Ultra‐high‐throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME Journal, 6, 1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso, J. G. , Lauber, C. L. , Walters, W. A. , Berg‐Lyons, D. , Lozupone, C. A. , Turnbaugh, P. J. , … Knight, R . (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences of the United States of America, 108, 4516–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson, C. A. , Hansell, D. A. , Nelson, N. B. , Siegel, D. A. , Smethie, W. M. , Khatiwala, S. , … Halewood, E. (2010). Dissolved organic carbon export and subsequent remineralization in the mesopelagic and bathypelagic realms of the North Atlantic basin. Deep Sea Research. Part II Top Study Oceanography, 57, 1433–1445. [Google Scholar]
- Chapman, M. G. , & Underwood, A. J. (1999). Ecological patterns in multivariate assemblages: information and interpretation of negative values in ANOSIM tests. Marine Ecology Progress Series, 180, 257–265. [Google Scholar]
- Corinaldesi, C. (2015). New perspectives in benthic deep‐sea microbial ecology. Frontiers in March Science, 2, 17. [Google Scholar]
- DeSantis, T. Z. , Hugenholtz, P. , Larsen, N. , Rojas, M. , Brodie, E. L. , Keller, K. , … Andersen, G. L. (2006). Greengenes, a chimera‐checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environment Microbiology, 72, 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin, A. M. , & Teske, A. (2010). Sediment‐associated microdiversity within the Marine Group I Crenarchaeota. Environmental Microbiology Reports, 2, 693–703. [DOI] [PubMed] [Google Scholar]
- Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26, 2460–2461. [DOI] [PubMed] [Google Scholar]
- Edgcomb, V. P. , Kysela, D. T. , Teske, A. , Gomez, A. D. , & Sogin, M. L. (2002). Benthic eukaryotic diversity in the Guaymas Basin hydrothermal vent environment. Proceedings of the National Academy of Sciences of the United States of America, 99, 7658–7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs, B. M. , Woebken, D. , Zubkov, M. V. , Burkill, P. , & Amann, R. (2005). Molecular identification of picoplankton populations in contrasting waters of the Arabian Sea. Aquatic Microbial Ecology, 39, 145–157. [Google Scholar]
- Fuhrman, J. A. , & Davis, A. A. (1997). Widespread archaea and novel bacteria from the deep sea as shown by 16S rRNA gene sequences. Marine Ecology Progress Series, 150, 275–285. [Google Scholar]
- Garcia, H. E. , Locarnini, R. A. , Boyer, T. P. , Antonov, J. I. , Baranova, O. K. , & Zweng, M. M . (2014) World Ocean Atlas 2013, Vol 4: Dissolved Inorganic Nutrients (phosphate, nitrate, silicate) In Levitus S. (Ed.) NOAA Atlas NESDIS 76, (p. 25). U.S. Department of Commerce, Silver Spring, MD. [Google Scholar]
- Ghosh, A. K. , & Mukhopadhyay, R. (2000). Mineral Wealth of the Ocean. New Delhi: Oxford & IBH Pub. Co. [Google Scholar]
- Gilbert, J. A. , Jansson, J. K. , & Knight, R. (2014). The Earth Microbiome project: successes and aspirations. BMC Biology, 12, 000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillan, D. C. , Speksnijder, A. , Zwart, G. , & De Ridder, C. (1998). Genetic diversity of the biofilm covering Montacuta ferruginosa (Mollusca, Bivalvia) as evaluated by denaturing gradient gel electrophoresis analysis and cloning of PCR‐Amplified gene fragments coding for 16S rRNA. Applied and Environment Microbiology, 64, 3464–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover, A. G. , Dahlgren, T. G. , Wiklund, H. , Mohrbeck, I. , & Smith, C. R. (2016). An end‐to‐end DNA taxonomy method for benthic biodiversity survey in the clarion‐clipperton zone, Central Pacific Abyss. Journal of Marine Science and Engineering, 4, 2–34. [Google Scholar]
- Guillou, L. , Viprey, M. , Chambouvet, A. , Welsh, R. M. , Kirkham, A. R. , Massana, R. , … Worden, A. Z. (2008). Widespread occurrence and genetic diversity of marine parasitoids belonging to Syndiniales (Alveolata). Environmental Microbiology, 10, 3349–3365. [DOI] [PubMed] [Google Scholar]
- Ivanova, E. P. , Nedashkovskaya, O. I. , Sawabe, T. , Zhukova, N. V. , Frolova, G. M. , Nicolau, D. V. , … Bowman, J. P. (2004). Shewanella affinis sp nov., isolated from marine invertebrates. International Journal of Systematic and Evolutionary Microbiology, 54, 1089–1093. [DOI] [PubMed] [Google Scholar]
- Karl, D. M. , Bjorkman, K. M. , Dore, J. E. , Fujieki, L. , Hebel, D. V. , Houlihan, T. , … Tupas, L. M. (2001). Ecological nitrogen‐to‐phosphorus stoichiometry at station ALOHA. Deep Sea Research. Part II Top Study Oceanography, 48, 1529–1566. [Google Scholar]
- Kerr, R. A. (1984). Manganese nodules grow by rain from above. Science, 223, 576–577. [DOI] [PubMed] [Google Scholar]
- Konneke, M. , Bernhard, A. E. , de la Torre, J. R. , Walker, C. B. , Waterbury, J. B. , & Stahl, D. A. (2005). Isolation of an autotrophic ammonia‐oxidizing marine archaeon. Nature, 437, 543–546. [DOI] [PubMed] [Google Scholar]
- Kouridaki, I. , Polymenakou, P. N. , Tselepides, A. , Mandalakis, M. , & Smith, K. L. Jr (2010). Phylogenetic diversity of sediment bacteria from the deep Northeastern Pacific Ocean: a comparison with the deep Eastern Mediterranean Sea. International Journal of Microbiology, 13, 143–150. [DOI] [PubMed] [Google Scholar]
- Kuypers, M. M. M. , Lavik, G. , Woebken, D. , Schmid, M. , Fuchs, B. M. , Amann, R. , … Jetten, M. S. M. (2005). Massive nitrogen loss from the Benguela upwelling system through anaerobic ammonium oxidation. Proceedings of the National Academy of Sciences of the United States of America, 102, 6478–6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuypers, M. M. M. , Sliekers, A. O. , Lavik, G. , Schmid, M. , Jorgensen, B. B. , Kuenen, J. G. , … Jetten, M. S. M. (2003). Anaerobic ammonium oxidation by anammox bacteria in the Black Sea. Nature, 422, 608–611. [DOI] [PubMed] [Google Scholar]
- Lara, E. , Moreira, D. , Vereshchaka, A. , & Lopez‐Garcia, P. (2009). Pan‐oceanic distribution of new highly diverse clades of deep‐sea diplonemids. Environmental Microbiology, 11, 47–55. [DOI] [PubMed] [Google Scholar]
- Larsen, E. I. , Sly, L. I. , & McEwan, A. G. (1999). Manganese(II) adsorption and oxidation by whole cells and a membrane fraction of Pedomicrobium sp. ACM 3067. Archives of Microbiology, 171, 257–264. [Google Scholar]
- Lauro, F. M. , McDougald, D. , Thomas, T. , Williams, T. J. , Egan, S. , Rice, S. , … Cavicchioli, R. (2009). The genomic basis of trophic strategy in marine bacteria. Proceedings of the National Academy of Sciences of the United States of America, 106, 15527–15533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, L. , Xu, X.‐W. , Jiang, X.‐W. , Wang, C.‐S. , Zhang, D.‐S. , Ni, J.‐Y. , & Wu, M. (2011). Microbial diversity in deep‐sea sediment from the cobalt‐rich crust deposit region in the Pacific Ocean. FEMS Microbiology Ecology, 78, 565–585. [DOI] [PubMed] [Google Scholar]
- Love, M. I. , Huber, W. , & Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15, 000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone, C. , & Knight, R. (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Applied and Environment Microbiology, 71, 8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone, C. , Lladser, M. E. , Knights, D. , Stombaugh, J. , & Knight, R. (2011). UniFrac: an effective distance metric for microbial community comparison. ISME Journal, 5, 169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, D. , Price, M. N. , Goodrich, J. , Nawrocki, E. P. , DeSantis, T. Z. , Probst, A. , … Hugenholtz, P. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME Journal, 6, 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie, P. J. , & Holmes, S. (2014). Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comptational Biology, 10, 000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewes, K. , Mogollon, J. M. , Picard, A. , Ruehlemann, C. , Kuhn, T. , Noethen, K. , & Kasten, S. (2014). Impact of depositional and biogeochemical processes on small scale variations in nodule abundance in the Clarion‐Clipperton Fracture Zone. Deep‐Sea Research. Part I, Oceanographic Research Papers, 91, 125–141. [Google Scholar]
- Mullineaux, L. S. (1987). Organisms living on manganese nodules and crusts: distribution and abundance at three North Pacific sites. Deep‐Sea Research. Part I, Oceanographic Research Papers, 34, 165–184. [Google Scholar]
- Nawrocki, E. P. , Kolbe, D. L. , & Eddy, S. R. (2009). Infernal 1.0: inference of RNA alignments. Bioinformatics, 25, 1335–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwirth, E . (2014) RColorBrewer: ColorBrewer Palettes. R package version 1.1‐2. Retrieved from: http://CRAN.R-project.org/package=RColorBrewer. [Google Scholar]
- Not, F. , Gausling, R. , Azam, F. , Heidelberg, J. F. , & Worden, A. Z. (2007). Vertical distribution of picoeukaryotic diversity in the Sargasso Sea. Environmental Microbiology, 9, 1233–1252. [DOI] [PubMed] [Google Scholar]
- Oksanen, J. , Blanchet, F. G. , Friendly, M. , Kindt, R. , Legendre, P. , & McGlinn, D . … Wagner, H. (2016) vegan: Community Ecology Package. R package version 2.4‐1. Retrieved from http://CRAN.R-project.org/package=vegan.
- Orcutt, B. N. , Sylvan, J. B. , Knab, N. J. , & Edwards, K. J. (2011). Microbial Ecology of the Dark Ocean above, at, and below the Seafloor. Microbiology and Molecular Biology Reviews, 75, 361–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paerl, R. W. , Foster, R. A. , Jenkins, B. D. , Montoya, J. P. , & Zehr, J. P. (2008). Phylogenetic diversity of cyanobacterial narB genes from various marine habitats. Environmental Microbiology, 10, 3377–3387. [DOI] [PubMed] [Google Scholar]
- Parada, A. , Needham, D. M. , & Fuhrman, J. A. (2015). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time‐series and global field samples. Environmental Microbiology, 18, 1403–1414. [DOI] [PubMed] [Google Scholar]
- Pawlowski, J. , Christen, R. , Lecroq, B. , Bachar, D. , Shahbazkia, H. R. , Amaral‐Zettler, L. , & Guillou, L. (2011). Eukaryotic richness in the abyss: insights from pyrotag sequencing. PLoS ONE, 6, e18169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce, R. W. , & Turner, J. T. (1992). Ecology of planktonic ciliates in marine food webs. Reviews in Aquatic Sciences, 6, 139–181. [Google Scholar]
- Price, M. N. , Dehal, P. S. , & Arkin, A. P. (2010). FastTree 2‐approximately maximum‐likelihood trees for large alignments. PLoS ONE, 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , Yarza, P. , … Glockner, F. O. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic Acids Research, 41, D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2015) R: A language and environment for statistical computing. In. Vienna, Austria: R Foundation for Statistical Computing.
- Rolinski, S. , Segschneider, J. , & Sundermann, J. (2001). Long‐term propagation of tailings from deep‐sea mining under variable conditions by means of numerical simulations. Deep Sea Research. Part II Top Study Oceanography, 48, 3469–3485. [Google Scholar]
- Schauer, R. , Bienhold, C. , Ramette, A. , & Harder, J. (2010). Bacterial diversity and biogeography in deep‐sea surface sediments of the South Atlantic Ocean. ISME Journal, 4, 159–170. [DOI] [PubMed] [Google Scholar]
- Smith, C. R. , De Leo, F. C. , Bernardino, A. F. , Sweetman, A. K. , & Arbizu, P. M. (2008). Abyssal food limitation, ecosystem structure and climate change. Trends in Ecology & Evolution, 23, 518–528. [DOI] [PubMed] [Google Scholar]
- Smith, C. R. , Levin, L. A. , Koslow, P. A. , Tyler, P. A. , & Glover, A. G . (2008) The near future of deep seafloor ecosystems In Polunin N., (Ed) Aquatic ecosystems: Trends and global prospects (pp. 334–351).Cambridge, UK; New York: Cambridge University Press. [Google Scholar]
- Stevens, H. , & Ulloa, O. (2008). Bacterial diversity in the oxygen minimum zone of the eastern tropical South Pacific. Environmental Microbiology, 10, 1244–1259. [DOI] [PubMed] [Google Scholar]
- Tully, B. J. , & Heidelberg, J. F. (2013). Microbial communities associated with ferromanganese nodules and the surrounding sediments. Frontiers in Microbiology, 4, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulloa, O. , Canfield, D. E. , DeLong, E. F. , Letelier, R. M. , & Stewart, F. J. (2012). Microbial oceanography of anoxic oxygen minimum zones. Proceedings of the National Academy of Sciences of the United States of America, 109, 15996–16003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vargas, C. , Audic, S. , Henry, N. , Decelle, J. , Mahe, F. , Logares, R. , … Tara Oceans Coordinators . (2015). Eukaryotic plankton diversity in the sunlit ocean. Science, 348, 1261605. [DOI] [PubMed] [Google Scholar]
- Veillette, J. , Juniper, S. K. , Gooday, A. J. , & Sarrazin, J. (2007). Influence of surface texture and microhabitat heterogeneity in structuring nodule faunal communities. Deep‐Sea Research. Part I, Oceanographic Research Papers, 54, 1936–1943. [Google Scholar]
- Veillette, J. , Sarrazin, J. , Gooday, A. J. , Galeron, J. , Caprais, J.‐C. , Vangriesheim, A. , … Juniper, S. K. (2007). Ferromanganese nodule fauna in the Tropical North Pacific Ocean: species richness, faunal cover and spatial distribution. Deep‐Sea Research. Part I, Oceanographic Research Papers, 54, 1912–1935. [Google Scholar]
- Verni, F. , & Gualtieri, P. (1997). Feeding behaviour in ciliated protists. Micron, 28, 487–504. [Google Scholar]
- Wang, C. S. , Liao, L. , Xu, H. X. , Xu, X. W. , Wu, M. , & Zhu, L. Z. (2010). Bacterial diversity in the sediment from polymetallic nodule fields of the Clarion‐Clipperton Fracture Zone. Journal of Microbiology, 48, 573–585. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Schlossmacher, U. , Wiens, M. , Schroeder, H. C. , & Mueller, W. E. G. (2009). Biogenic origin of polymetallic nodules from the Clarion‐Clipperton Zone in the Eastern Pacific Ocean: electron microscopic and EDX evidence. Marine Biotechnology, 11, 99–108. [DOI] [PubMed] [Google Scholar]
- Wang, K. , Zhang, D. , Xiong, J. , Chen, X. , Zheng, J. , Hu, C. , … Zhu, J. (2015). Response of bacterioplankton communities to cadmium exposure in coastal water microcosms with high temporal variability. Applied and Environment Microbiology, 81, 231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes, G. R. , Bolker, B. , Bonebakker, L. , Gentleman, R. , Huber, W. , & Liaw, A . … Venables, B. (2016) gplots: Various R Programming Tools for Plotting Data. In. R package version 3.0.1.Retrieved from: http://CRAN.R-project.org/package=gplots.
- Wedding, L. M. , Reiter, S. M. , Smith, C. R. , Gjerde, K. M. , Kittinger, J. N. , Friedlander, A. M. , … Crowder, L. B. (2015). Managing mining of the deep seabed. Science, 349, 144–145. [DOI] [PubMed] [Google Scholar]
- Wegorzewski, A. V. , & Kuhn, T. (2014). The influence of suboxic diagenesis on the formation of manganese nodules in the Clarion Clipperton nodule belt of the Pacific Ocean. Marine Geology, 357, 123–138. [Google Scholar]
- Werner, J. J. , Koren, O. , Hugenholtz, P. , DeSantis, T. Z. , Walters, W. A. , Caporaso, J. G. , … Ley, R. E. (2012). Impact of training sets on classification of high‐throughput bacterial 16S rRNA gene surveys. ISME Journal, 6, 94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, H. (2009). ggplot2: elegant graphics for data analysis. New York: Springer‐Verlag. [Google Scholar]
- Wright, J. J. , Mewis, K. , Hanson, N. W. , Konwar, K. M. , Maas, K. R. , & Hallam, S. J. (2014). Genomic properties of Marine Group A bacteria indicate a role in the marine sulfur cycle. ISME Journal, 8, 455–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y.‐H. , Liao, L. , Wang, C.‐S. , Ma, W.‐L. , Meng, F.‐X. , Wu, M. , & Xu, X.‐W. (2013). A comparison of microbial communities in deep‐sea polymetallic nodules and the surrounding sediments in the Pacific Ocean. Deep‐Sea Research. Part I, Oceanographic Research Papers, 79, 40–49. [Google Scholar]
- Xu, M. , Wang, F. , Meng, J. , & Xiao, X. (2007). Construction and preliminary analysis of a metagenomic library from a deep‐sea sediment of east Pacific Nodule Province. FEMS Microbiology Ecology, 62, 233–241. [DOI] [PubMed] [Google Scholar]
- Xu, M. X. , Wang, P. , Wang, F. P. , & Xiao, X. (2005). Microbial diversity at a deep‐sea station of the Pacific nodule province. Biodiversity and Conservation, 14, 3363–3380. [Google Scholar]
- Yilmaz, P. , Parfrey, L. W. , Yarza, P. , Gerken, J. , Pruesse, E. , Quast, C. , … Glockner, F. O. (2014). The SILVA and “All‐species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Research, 42, D643–D648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials