

Abstract
Background
Hereditary proximal spinal muscular atrophy (SMA) is a severe neuromuscular disease of childhood caused by homozygous loss of function of the survival motor neuron (SMN) 1 gene. The presence of a second, nearly identical SMN gene (SMN2) in the human genome ensures production of residual levels of the ubiquitously expressed SMN protein. Alpha-motor neurons in the ventral horns of the spinal cord are most vulnerable to reduced SMN concentrations but the development or function of other tissues may also be affected, and cardiovascular abnormalities have frequently been reported both in patients and SMA mouse models.
Methods
We systematically reviewed reported cardiac pathology in relation to SMN deficiency. To investigate the relevance of the possible association in more detail, we used clinical classification systems to characterize structural cardiac defects and arrhythmias.
Conclusions
Seventy-two studies with a total of 264 SMA patients with reported cardiac pathology were identified, along with 14 publications on SMA mouse models with abnormalities of the heart. Structural cardiac pathology, mainly septal defects and abnormalities of the cardiac outflow tract, was reported predominantly in the most severely affected patients (i.e. SMA type 1). Cardiac rhythm disorders were most frequently reported in patients with milder SMA types (e.g. SMA type 3). All included studies lacked control groups and a standardized approach for cardiac evaluation.
The convergence to specific abnormalities of cardiac structure and function may indicate vulnerability of specific cell types or developmental processes relevant for cardiogenesis. Future studies would benefit from a controlled and standardized approach for cardiac evaluation in patients with SMA.
Electronic supplementary material
The online version of this article (doi:10.1186/s13023-017-0613-5) contains supplementary material, which is available to authorized users.
Keywords: Spinal muscular atrophy, SMA, Werdnig-Hoffmann, Kugelberg-Welander, Cardiac pathology, Cardiac abnormalities
Background
Hereditary proximal spinal muscular atrophy (SMA) is an important genetic cause of infantile mortality and childhood disability. Degeneration of α-motorneurons in the ventral horns of the spinal cord is the most salient feature but other organs, in particular the heart, may also be affected as suggested by numerous case reports [1, 2].
SMA is caused by deficiency of the survival motor neuron (SMN) protein due to homozygous loss of function of the SMN1 gene. The human SMN locus contains a second, nearly identical, SMN copy (SMN2) that contains a critical point mutation in exon 7, resulting in exclusion of exon 7 from most SMN2 mRNA transcripts. SMN2 therefore produces residual levels of full length SMN2 mRNA and functional SMN protein [3–7]. SMN protein is ubiquitously expressed and is part of multiprotein complexes that probably have both general and motor neuron specific functions, including small nuclear ribonucleic protein (snRNP) assembly, pre-mRNA splicing, post-transcriptional gene regulation, axonal mRNA transport, ubiquitination homeostasis, maintenance and neuronal differentiation of embryonic stem cells and embryonic organ development [6, 8–13]. Variation in SMN2 copy numbers, which partly explains differences in SMN protein levels between patients, is the most important modifier of SMA severity. The severity spectrum encompasses prenatal SMA (type 0), infantile onset severe SMA (type 1), an intermediate form (SMA type 2), childhood onset SMA (type 3), and adult onset SMA (type 4). Higher copy numbers are associated with milder forms of SMA [5, 6].
The identification of non-neuromuscular complications of severe SMA, including disorders of the heart and cardiovascular system, may help to elucidate pathogenic pathways and are furthermore of increasing clinical importance since therapies that aim to attenuate or reverse SMN deficiency may be introduced soon.
To study the evidence for an association of SMA with cardiac pathology in more detail, we performed a systematic review of the available clinical and experimental literature.
Methods
Search
We searched MEDLINE and Embase for articles on SMA and cardiac pathology published up to January 31st 2016, using a combination of the following terms: ‘spinal muscular atrophy’, ‘Werdnig Hoffmann’ and ‘Kugelberg Welander’, ‘heart’, ‘cardiac’, and ‘ECG’. Numerous word variations were included and specific types of cardiac abnormalities were added to the search, including ‘congenital heart disease’, ‘atrial septal defect’, ‘ventricular septal defect’, ‘cardiac malformations’, and ‘arrhythmias’, in order to identify as many relevant articles as possible. The query that retrieved the largest number of relevant results was used and is shown in Table 1.
Table 1.
Details on systematic search
| Search terms used for PubMed search | |
|---|---|
| Topic | Querya |
| SMA | (“spinal muscular atrophy” OR muscular atrophb OR “werdnig hoffmann” OR “kugelberg welander”) |
| Cardiac Pathology | AND (“heart” OR “cardiac” OR cardiolb OR ventricb OR “septum” OR “ECG”) |
aSimilar terms were used in all searches, tailored to the specific requirements of each search engine. The addition of specific cardiac abnormalities did not retrieve relevant additional results and were therefore excluded from our final query
b indicates that word variations of the search term were also searched
The MEDLINE database was searched using PubMed. In addition, Scopus, OvidSP, and Web of Science were used to obtain as many relevant original papers as possible. For OvidSP, the following resources were selected: ‘MEDLINE’, ‘OLD MEDLINE’ and ‘MEDLINE In-Process’. Similar terms were used for all searches, tailored to the specific requirements of each search engine. No field limitations or language restrictions were applied. We used indexed search terms, if applicable, to ensure inclusion of relevant related terms. MeSH-indexed terms were not used in order to prevent missing recently published articles that had not yet been MeSH-indexed at the time of our search. For articles possibly relevant to our search but unavailable online, we searched university library catalogues using ‘Picarta’ (http://www.picarta.org/) to check for offline availability, and contacted the author(s) of the original publications if e-mail or correspondence addresses were available.
Selection of relevant articles
Two of the authors [CAW, MS] independently conducted the search and selection processes. After screening title and abstract of all obtained articles, potentially relevant papers were screened full text using predefined inclusion and exclusion criteria (Table 2). Both clinical and experimental studies of patients and mouse models were included. We also systematically checked the references of all included papers and used Thomson Reuters’ ‘Web of Science’ for a cited references search and a related articles search, to ensure identification of all relevant literature. Details of the search and selection process are summarized in Fig. 1.
Table 2.
Criteria used for critical selection of papers retrieved from our search
| Applied criteria for the selection of relevant papers | |
|---|---|
| Inclusion criteria | Diagnosis of SMA types 1–4a or SMA mouse model; presence of cardiac abnormalities; original study with identifiable case(s). |
| Exclusion criteria | No diagnosis of SMA or substantial doubts about diagnosis; diagnosis of non 5q-SMA (e.g.: SMARD, distal SMA); no cardiac pathology present; SMA with additional chromosomal abnormalities associated with (congenital) heart disease (e.g. trisomy 21); cardiac abnormalities due to medication or in moribund patients (e.g. bradycardia); redundant publication of previously reported case(s); congress reports; mouse model of non 5q-SMA (e.g. IGHMBP2 model); animal research other than mouse models; only abstract available with unidentifiable cases. |
aThis also includes the SMA subtypes, e.g. ‘type 0’, ‘type 1a’, ‘type 1b’, ‘type 3a’ and ‘type 3b’. SMA spinal muscular atrophy; SMARD spinal muscular atrophy with respiratory distress; IGHMBP2 Immunoglobulin Mu Binding Protein 2
Fig. 1.
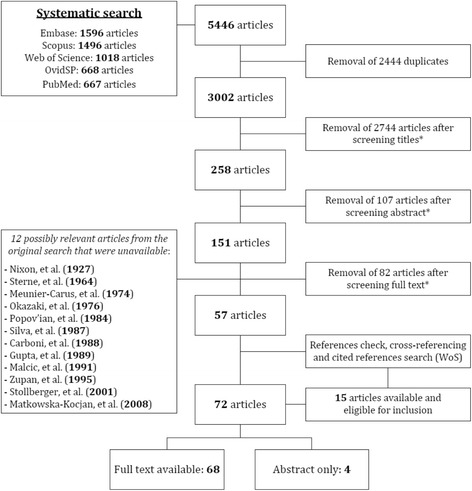
Flowchart of search and selection process. Summary of search and selection process of eligible articles for inclusion. *: predefined inclusion and exclusion criteria were applied, as shown in Table 2. WoS: ‘Web of Science’
Data extraction
After all relevant data was extracted from the selected papers [by CAW], two authors [CAW and ACB] independently categorized structural (congenital) cardiac defects using the 2012 version of the European Paediatric Cardiac Code (International Paediatric and Congenital Cardiac Code (IPCCC) Short List) [14, 15] that distinguishes 8 groups, based on affected anatomical areas of the heart (Table 3). We also classified abnormalities of cardiac rhythm using the system suggested by Korpas [16], which is based upon mechanisms of origin, i.e.: arrhythmias due to abnormal impulse initiation or abnormal impulse conduction. Impulse initiation disorders were further subdivided into 3 groups, based upon the area of the cardiac conduction system involved: sinoatrial (sinus) node, supraventricular, or ventricular (Table 5). Initial classification disagreements were resolved by consensus. A comprehensive overview of all retrieved cases of SMA patients with cardiac pathology is shown in Additional file 1: Tables S1–S3.
Table 3.
Classification of structural cardiac defects in SMA type 1 patients
| Diagnostic groups | Reported occurrence | |
|---|---|---|
| 1. | Abnormalities of position and connection of heart | 2x |
| 2. | Tetralogy of Fallot and variants | 1x |
| 3. | Abnormalities of great veins | – |
| 4. | Abnormalities of atria and atrial septum | 23x |
| 5. | Abnormalities of atrioventricular valves and atrioventricular septal defect | 3x |
| 6. | Abnormalities of ventricles and ventricular septum | 17x |
| 7. | Abnormalities of ventriculo-arterial valves and great arteries | 7x |
| 8. | Abnormalities of coronary arteries, arterial duct and pericardium | 6x |
Table 5.
Arrhythmias in SMA type 3
| Arrhythmias in patients with SMA type 3 | ||
|---|---|---|
| Arrhythmia type | Anatomical site of origin |
Reported ECG abnormalities
(number of times observed in patients with SMA type 3) |
| Impulse initiation disorder | Sinus/SA node initiation disorders | Sinus arrest (2) |
| Sinus dysfunction (1) | ||
| Sinus tachycardia (1) | ||
| Supraventricular initiation disorders | Atrial fibrillation (5) | |
| AV junctional rhythm (5) | ||
| Supraventricular extrasystoles (2) | ||
| Atrial flutter (2) | ||
| Atrial tachycardia (1) | ||
| Ventricular initiation disorders | Ventricular extrasystoles (2) | |
| Non-sustained ventricular tachycardias (2) | ||
| Impulse conduction disorders | n/a | Left anterior hemiblock (4) |
| AV-block (n.o.s.) (3) | ||
| Right bundle branch block (3) | ||
| 1st degree AV block (3) | ||
| Complete AV block (2) | ||
| 2nd degree AV block, Mobitz I (Wenckebach) (1) | ||
| Prolonged junctional recovery time (1) | ||
Reported arrhythmias in patients with SMA type 3 (n = 24). Some patients had more than one ECG abnormality. Also see Additional file 1: Table S3
Abbreviations: SA sinoatrial; AV atrioventricular; n.o.s. not otherwise specified; n/a not applicable
The small number of both patients and SMA model mice with histological abnormalities of cardiac tissue precluded the use of available classification systems, nor was it possible to classify cardiac abnormalities in SMA mouse models due to significant methodological differences between studies. A comprehensive overview of all included SMA mouse models is shown in Additional file 1: Table S5.
Results
We retrieved 3002 articles with our initial search. After selection, 72 articles met our predefined inclusion criteria, including 4 articles of which only the abstract was available [17–20]. These abstracts contained sufficient detailed information and were included for further analysis. We were unable to obtain full text or detailed abstracts of 15 possibly relevant articles. Twelve of these articles were identified in the original search, whilst the other 3 were found through the related articles search (Additional file 1: Table S4, [21–35]).
We identified a total of 264 published cases of SMA patients with cardiac pathology. Seven studies contained descriptions of patients with several SMA types, 28 studies of SMA type 1 only and 23 studies of SMA type 3 only. We found a total of 14 studies on cardiac pathology in SMA mouse models (Additional file 1: Tables S1–S3 and S5).
Cardiac pathology in patients with SMA type 1
We identified 77 patients with SMA type 1 (‘Werdnig-Hoffmann Disease’) and cardiac pathology [36–69]. Most studies used well defined clinical criteria for the diagnosis of SMA. Tests for homozygous SMN1 deletion were performed in 36 (47%) patients and confirmed in 31: five patients did not have a homozygous SMN1 deletion. It was not specified whether these patients had intragenic SMN1 point mutations, or a different neuromuscular disorder. The diagnosis in these 5 patients was based upon clinical characteristics combined with supportive information from muscle biopsies (n = 5), EMG (n = 4) and autopsy findings (n = 2) [44, 50, 54, 59].
Thirty-three out of 77 (43%) patients had electrocardiogram (ECG) abnormalities. ECGs of 15 patients (19%) showed severe symptomatic bradycardias, defined as a heart rate of less than 40 beats per minute. Baseline tremors were reported in the other 18 (23%) patients. These ECG baseline tremors were initially interpreted as abnormalities or even as being suggestive of cardiac pathology, but nowadays they are interpreted as artefacts due to peripheral muscle tremors. Two patients were diagnosed with a ‘late form of Werdnig-Hoffmann’ but it is unclear whether these two patients had SMA type 2 or 3 and were misclassified or had a relatively mild form of SMA type 1 (i.e. type 1c) with longer survival [70, 71]. The two children, aged 10 and 12, had echocardiographic abnormalities compatible with left ventricular hypertrophy (Additional file 1: Table S1, patients 8 and 9) [39].
The remaining 42 patients (55%) with SMA type 1 had structural cardiac defects. We used the IPCCC diagnostic classification system as outlined in Table 3 [14, 15].
Twenty-one of the 42 patients (50%) had a single structural abnormality of the heart. This included 11 (26%) patients with an atrial septal defect (ASD) and 5 (12%) with an isolated ventricular septal defect (VSD). The other 21 patients (50%) had multiple structural cardiac abnormalities. The combination of an ASD (IPCCC group 4) and VSD (IPCCC group 6) was reported relatively frequently (n = 5; 12%). Hypoplastic left heart syndrome was also found in 5 (12%) patients. Additional file 1: Table S1 includes details on all included patients. Figure 2 illustrates the reported structural cardiac pathology in SMA type 1.
Fig. 2.
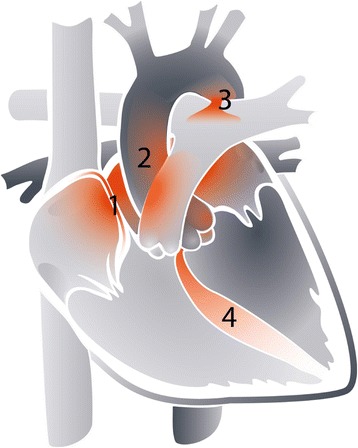
Structural cardiac pathology in SMA type 1. Areas of the heart frequently reported to show cardiac defects in patients with SMA type 1 (n = 42) are shown in color. Rarely affected and unaffected areas are shown in shades of grey. Numbers indicate 1: atrial septum; 2: cardiac outflow tract; 3: patent ductus arteriosus; 4: ventricular septum
We next assessed the relationship between SMN2 copy numbers and the severity of cardiac pathology. SMN2 copy numbers were available for only 13 of the 77 patients with SMA type 1 (17%). Ten patients had 1 SMN2 copy and 3 had 2 SMN2 copies (Table 4) [46, 53, 56, 63, 65–68].
Table 4.
SMN2 copy numbers in patients with SMA type 1 and cardiac abnormalities
| Reference | Patient no. | SMN2 copy number | Cardiac pathology | IPCCC diagnostic group(s) |
|---|---|---|---|---|
| Devriendt (1996) [46] | 1 | 1 copy | Small VSD | Group 6 |
| Macleod (1999) [53] | 2 | 1 copy χ | ASD | Group 4 |
| 3 | 1 copy χ | ASD and mitral hypoplasia | Groups 4 and 5 | |
| García-Cabezas (2004) [56] | 4 | 1 copy | ASD (secundum type) | Group 4 |
| Rüdnik-Schöneborn (2008) [63] | 5 | 1 copy | Large ASD (sinus venosus type), multiple VSDs and PDA | Groups 4, 6 and 8 |
| 6 | 1 copy | Common atrium (i.e. a very large ASD) and PDA | Groups 4 and 8 | |
| 7 | 1 copy | Large ASD (secundum type) and VSD | Groups 4 and 6 | |
| 8 | 2 copies | Small VSD and PDA | Groups 6 and 8 | |
| Lumaka (2009) [65] | 9 | 2 copies | ASD (secundum type) | Group 4 |
| Rüdnik-Schöneborn (2010) [66] | 10 | 1 copy | Large ASD, mild pulmonary and mild aortic stenosis | Groups 4 and 7 |
| 11 | 1 copy | AVSD | Group 5 | |
| Parra (2012) [67] | 12 | 1 copy | HLHS | Group 6 |
| Ekici (2012) [68] | 13 | 2 copies | Dextrocardia and Tetralogy of Fallot | Groups 1 and 2 |
The number of SMN2 copies of 13 patients with SMA type 1
Abbreviations: IPCCC International Paediatric and Congenital Cardiac Code; VSD ventricular septal defect; ASD atrial septal defect; PDA patent ductus arteriosus; AVSD atrioventricular septal defect; HLHS hypoplastic left heart syndrome. χ = SMN2 copy numbers were calculated based on the SMN:MPZ ratio provided by the authors in the original publication [53]
Cardiac pathology in patients with SMA type 2
We found 5 studies with a total of 63 patients with SMA type 2 and cardiac pathology [37, 40, 41, 49, 72]. No genetic tests to confirm the diagnosis were performed. Clinical characteristics, sometimes combined with documented muscle biopsy (n = 21) and EMG results (n = 8), were used for diagnosis. Many of the original publications mentioned the use of ancillary investigations to support the diagnosis (e.g. EMG, muscle biopsy, autopsy) but did not further specify the tests.
All 63 patients had ECG baseline tremors (Additional file 1: Table S2). A small number of patients also had one or more additional ECG abnormalities [72]. This included disorders of impulse initiation (n = 2; both with a sinus tachycardia), disorders of impulse conduction (n = 2; both with a right bundle branch block), signs of atrial enlargement (n = 3) or ventricular enlargement (n = 4), signs of myocardial damage (abnormal Q-waves n = 2, ST-changes n = 3), or signs of ventricular hypertrophy (n = 7).
Cardiac pathology in patients with SMA type 3
We identified 124 patients with SMA type 3 (‘Kugelberg-Welander Disease’, ‘Wolfhart-Kugelberg-Welander Disease’) and cardiac pathology [17–20, 36, 37, 40, 41, 47, 49, 72–91]. Genetic tests to confirm homozygous deletion of SMN1 were performed in 5 patients (4%) and confirmed in 4 [47, 86, 88, 89, 91]. It remains unclear whether this one patient might have had a hemizygous deletion in combination with an intragenic SMN1 point mutation, or ‘non-5q’ SMA [88]. SMN2 copy numbers were not available for any of the patients (Additional file 1: Table S3).
Sixty-seven (54%) of the 124 patients had tremors of the ECG baseline only. Six patients (5%) had both ECG abnormalities and structural cardiac pathology [80, 82]. One presented with mitral and tricuspid valve prolapse (IPCCC group 5), five patients had a prolonged QT time interval combined with one or more structural abnormalities: mitral valve prolapse (n = 3, IPCCC group 5), a hypertrophic interventricular septum (n = 2, IPCCC group 6) or a hypertrophic (n = 1) or atrophic (n = 1) posterior ventricular wall. One of these patients also had a diminished left ventricular end diastolic volume.
Two patients had structural abnormalities of the heart without abnormalities of the cardiac rhythm. One, with genetically confirmed SMA type 3a, had a complex cardiac malformation consisting of an ASD, L-transposition of the great arteries, functional single ventricle and a patent ductus arteriosus (IPCCC groups 2, 4, 6, and 8) [47]. The other patient had an ASD (ostium secundum type, IPCCC group 2) [90].
Thirty-nine (31%) of the 124 patients with SMA type 3 had abnormalities of the cardiac rhythm only. With details provided in the original publications we were able to further classify cardiac rhythm abnormalities of 24 patients (19%), using a classification system suggested previously (Table 5) [16].
Additionally, 9 patients (7%) were reported with ECG signs suggestive of myocardial damage (e.g. Q-waves or ST-changes) [72, 73, 76, 77, 80, 81], two patients had systolic pump function abnormalities [86, 88], and 2 others had ECG abnormalities that could not be classified further [20, 83].
Finally, information on histological cardiac abnormalities from autopsy (n = 5) or cardiac biopsy (n = 3) was available for 8 patients (6%). Myocardial fibrosis was the most frequent finding (n = 4) [19, 73, 75–78, 89]. Two studies contained detailed descriptions of the myocardial histology, including findings of deranged, atrophic and degenerated myocytes and myocardial fibers [75, 78]. Ultrastructural myocardial changes, such as focal degeneration of myocardial cells, were also noted [75].
Cardiac pathology in SMA mouse models
We found 14 studies that reported the presence of cardiac pathology in SMA mouse models (Additional file 1: Table S5) [10, 92–104]. Methods used to evaluate and classify cardiac involvement differed considerably between studies, complicating a comparison of reported outcomes. The most important macroscopic findings were decreased heart size including decreased left ventricular (LV) mass [10, 92–94, 96, 102, 104]. This finding could at least partially be explained by reduced body size and weight. Furthermore, reduced thickness of the LV wall and interventricular septum (IVS) were frequently reported [10, 94, 96, 99, 100, 102]. Abnormal cardiogenesis of the IVS, LV and arterial walls, which also accounts for thinning and partial flattening of the cardiac arterial walls, was suggested as a possible underlying cause in one study [94].
The most prominent microscopic finding was myocardial fibrosis [94, 96, 98, 100]. Other abnormalities included vascular remodelling, including decreased numbers of coronary capillaries, and ultrastructural changes, e.g. abnormal expression of postnatal cardiac development markers indicating loss of contractile components (Additional file 1: Table S5).
Microscopic abnormalities of the cardiac autonomic nervous system (ANS) were also reported, including reduced neuronal branching and presence of thinner cardiac sympathetic ANS nerves [93, 95]. Abnormalities of the cardiac ANS or cardiac rhythm were reported in most studies. Bradyarrhythmias were reported in almost all studies [92–97, 101, 103, 104]. Few studies included detailed information on murine cardiac rhythm, which precludes further classification. The available data suggest that both disorders of impulse initiation and impulse conduction can be found in mouse models of SMA.
The main finding indicating reduced cardiac function was a significant reduction in pumping efficiency, i.e. reduced stroke volume and cardiac output, mainly due to left ventricular dysfunction [92, 93, 104]. All findings are summarized in Additional file 1: Table S5.
Discussion
Vulnerability to SMN deficiency may not be confined to motor neurons. Cardiovascular abnormalities are among the most frequently reported non-neuromuscular complications in SMA [2]. In this systematic review, we identified 264 published possible cases of SMA with cardiac abnormalities and 14 studies reporting cardiac involvement in SMA mouse models. Structural cardiac pathology was almost exclusively reported in patients with SMA type 1, while acquired cardiac pathology, including arrhythmias and conduction abnormalities, were reported more frequently in less severely affected patients. Detailed classification of the reported abnormalities suggests convergence to specific pathologies in patients with SMA that may be linked to downstream effects of SMN deficiency. We did not identify large controlled studies that indicate the presence of cardiac pathology in SMA, preventing a definite conclusion as to whether the incidence of cardiac abnormalities is increased in SMA.
Structural cardiac abnormalities in SMA type 1 were almost exclusively defects of atrial and ventricular septa and/or defects of the cardiac outflow tract. Ventricular septal defects (VSD), pulmonary stenosis, a patent ductus arteriosus (PDA), and atrial septal defects (ASD) are, however, the most common structural cardiac abnormalities in newborns, with a reported incidence of approximately 1% [105–110]. Low SMN protein levels may increase the odds of abnormal cardiac development. This hypothesis is supported by several observations: interventricular septum abnormalities were also observed in animal models of severe SMA, and abnormal embryonic cardiogenesis induced by low SMN protein levels was identified as a possible underlying cause in one study [94]. Moreover, there was an over-representation of patients with SMA type 1 and cardiac defects who had only one SMN2 copy, which is associated with the lowest residual SMN protein levels that are compatible with life at birth [5]. The association between the lowest SMN2 copy number and occurrence of non-neuromuscular pathology, including cardiac abnormalities, has been suggested previously [63].
Disturbances of cardiac rhythm were a second abnormality reported across the spectrum of SMA severity, i.e. in SMA types 1–3. Leaving out baseline tremors, which are to be considered an artefact caused by the characteristic peripheral tremor in patients with SMA, impulse initiation disorders were the most common cardiac rhythm abnormalities. Taking into account the very low reported incidence of for example atrial flutter or atrial fibrillation in patients under the age of 50 years [111, 112], impulse initiation disorders occurred at a strikingly young age in patients with SMA (atrial flutter, n = 2, ages 24 and 49 years [77, 80]; atrial fibrillation, n = 4, reported ages ranging from 29 to 35 years [17, 19, 88]). This may suggest a developmental origin associated with SMN deficiency. In theory, both dysfunction of either the cardiac electrical conduction system or the ANS, which influences cardiac rhythm in vivo, may underlie cardiac arrhythmias [113]. Significant abnormalities of the cardiac ANS were also found in SMA mouse models [93, 95].
Myocardial fibrosis was reported in 8 patients and may contribute to arrhythmias in SMA [19, 73, 76, 89]. Fibrosis of the myocardium was also a frequent finding in both severe and intermediate SMA mouse models, in which arrhythmias were virtually omnipresent. Bradycardia was reported most often, due to delays in the cardiac electrical conduction system, causing various types of atrioventricular and bundle branch blocks. It should be noted that myocardial fibrosis is a hallmark of normal aging [114], and the limited number of patients precludes a definite conclusion whether impulse conduction disorders in SMA are caused by presenile cardiac fibrosis secondary to SMN deficiency.
There are several other possible explanations how SMN deficiency causes cardiac abnormalities, including specific mRNA-splicing defects that could interfere with normal cardiac development [11, 115]. Low SMN levels have already been shown to influence embryonic organ development in animal models, including cardiogenesis [2, 6, 116]. Furthermore, very low levels of SMN protein may predispose to dysfunction of specific cell types other than alpha-motor neurons, that are involved in cardiogenesis [117–119]. A potential candidate cell type is the neural crest cell (NCC), as a subset of NCCs migrate and differentiate into cardiac neural crest cells (cNCCs) that are involved in development of the musculoconnective tissue (tunica media) of the great vessels, cardiac outflow tract septa (dividing the conotruncus into the aorta and pulmonary trunk) and, to some extent, septation of the atria and ventricles [120–126]. SMN protein deficiency may alter the function of downstream signalling pathways that are important for the migratory process of NCCs [123]. Furthermore, although the cardiac electrical conduction system itself originates from cardiomyocytes [127], the cardiac ANS that contributes to arrhythmias, develops from NCCs [113].
Several limitations of this systematic review need to be addressed. First, we cannot exclude the possibility of publication bias towards cases with particular findings or with severe forms of SMA and heart disease. Published cases may, therefore, not be representative of all cardiac pathology in SMA and single cases could have been missed if they were not represented in the databases used. However, given the relatively large number of patients included, it is unlikely that these cases would have substantially influenced our overall findings. Furthermore, publications and reports differed significantly in clinical detail and the time of diagnosis. Many studies were published before genetic testing for homozygous SMN1 deletion became widely available (i.e.: cases before 1995) which leaves open the possibility of inclusion of disorders other than SMA, in particular for SMA types 2 and 3. The cases of patients with SMA and heart disease included in our work were published between the late 1960s [73] and 2015 [91]. During this period, significant modifications of diagnostic criteria and classifications of SMA types occurred [128, 129]. Although these changes are largely irrelevant with regard to observing a cardiac abnormality in a patient with SMA, we had to assume the correct diagnosis of SMA (in the absence of genetic confirmation of the diagnosis) and severity in some patients. With a view to addressing these issues, at least in part, we reviewed all available clinical data of included cases (Additional file 1: Tables S1–S3) in an attempt to maximise diagnostic accuracy. Finally, considerable differences in the diagnostic methodology for cardiac evaluation, ranging from a limited number of diagnostic tools to assess cardiac pathology, to a more comprehensive combination of ECG, radiographs, echocardiography, or autopsy, clearly results in differences in quality of observations between studies.
Conclusions
On the basis of the data available, if present, structural abnormalities of the heart are predominantly expected in the more severely affected SMA patients (i.e. SMA type 1), disturbances of the cardiac rhythm in the more mildly affected patients (i.e. SMA types 2 and 3). Future studies would benefit greatly from a controlled, standardized, uniform, and comprehensive protocol for cardiac work-up of genetically confirmed cases of SMA.
Acknowledgments
We thank Prof. Dr. Jeroen Bakkers, Professor of Molecular Cardiogenetics at the University Medical Center of Utrecht and the Hubrecht Institute of Developmental Biology & Stem Cell Research, for his helpful comments and contribution to our work.
Funding
SMA research at the University Medical Center of Utrecht has been supported by grants from the Prinses Beatrix Spierfonds (WAR08-24, WAR13-07, WAR14-26), and Stichting Spieren voor Spieren. The funders had no role in study design, data collection, data analysis or interpretation, writing the manuscript, or decisions in submitting this manuscript.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed for the writing of this review. The data collected from the original publications is available in the Additional file 1: Tables S1–S5.
Authors’ contributions
CAW conceived and designed the review, performed the systematic search, selected the papers for inclusion, extracted and analyzed the data, drafted the initial manuscript, and reviewed and revised the final manuscript. ACB analyzed the data, participated in drafting, reviewing and revising the manuscript. MS participated in the systematic search and selection process, data extraction, and reviewing the manuscript. RIW and LHB contributed in reviewing and revising the manuscript. WLP contributed in designing the review, analyzing the data, and reviewing and revising the manuscript. All authors approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ANS
Autonomic nervous system
- ASD
Atrial septal defect
- AV
Atrioventricular
- AVSD
Atrioventricular septal defect
- cNCC
Cardiac neural crest cell
- ECG
Electrocardiogram
- EMG
Electromyogram
- HLHS
Hypoplastic left heart syndrome
- IGHMBP2
Immunoglobulin Mu Binding Protein 2
- IPCCC
International Pediatric Cardiac Code
- IVS
Intraventricular septum
- LV
Left ventricular
- mRNA
Messenger-ribonucleic acid
- NCC
Neural crest cell
- PDA
Patent ductus arteriosus
- SA
Sinoatrial
- SMA
Spinal muscular atrophy
- SMARD
Spinal muscular atrophy with respiratory distress
- SMN
Survival motor neuron
- snRNP
Small nuclear ribonucleic protein
- VSD
Ventricular septal defect
Additional file
Tables S1, S2, and S3: provide an overview of all included studies and all individual cases of patients with SMA and cardiac pathology. Table S4: overview of articles (n = 12) possibly relevant to our systematic search of which no full text or detailed abstract was available. Table S5. Overview of SMA mouse models with cardiac pathology. (DOCX 153 kb)
Contributor Information
C. A. Wijngaarde, Email: C.A.Wijngaarde-2@umcutrecht.nl
A. C. Blank, Email: a.c.blank@umcutrecht.nl
M. Stam, Email: m.stam-4@umcutrecht.nl
R. I. Wadman, Email: r.i.wadman@umcutrecht.nl
L. H. van den Berg, Email: l.h.vandenberg@umcutrecht.nl
W. L. van der Pol, Email: W.L.vanderPol@umcutrecht.nl
References
- 1.Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120–2133. doi: 10.1016/S0140-6736(08)60921-6. [DOI] [PubMed] [Google Scholar]
- 2.Hamilton G, Gillingwater TH. Spinal muscular atrophy: going beyond the motor neuron. Trends Mol Med. 2013;19:40–50. doi: 10.1016/j.molmed.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 4.Coovert DD, Le TT, McAndrew PE, Strasswimmer J, Crawford TO, Mendell JR, Coulson SE, Androphy EJ, Prior TW, Burghes AH. The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet. 1997;6:1205–1214. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 5.Mailman MD, Heinz JW, Papp AC, Snyder PJ, Sedra MS, Wirth B, Burghes AH, Prior TW. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med. 2002;4:20–26. doi: 10.1097/00125817-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. 2009;10:597–609. doi: 10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lorson CL, Rindt H, Shababi M. Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum Mol Genet. 2010;19:R111–8. doi: 10.1093/hmg/ddq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vitte JM, Davoult B, Roblot N, Mayer M, Joshi V, Courageot S, Tronche F, Vadrot J, Moreau MH, Kemeny F, Melki J. Deletion of murine Smn exon 7 directed to liver leads to severe defect of liver development associated with iron overload. Am J Pathol. 2004;165:1731–41. doi: 10.1016/S0002-9440(10)63428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boyer JG, Bowerman M, Kothary R. The many faces of SMN: deciphering the function critical to spinal muscular atrophy pathogenesis. Future Neurol. 2010;5:873–890. doi: 10.2217/fnl.10.57. [DOI] [Google Scholar]
- 10.Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, Krainer AR. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123–6. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li DK, Tisdale S, Lotti F, Pellizzoni L. SMN control of RNP assembly: From post-transcriptional gene regulation to motor neuron disease. Semin Cell Dev Biol. 2014;32:22–29. doi: 10.1016/j.semcdb.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang W-F, Xu J, Chang C-C, Yang S-H, Li H-Y, Hsieh-Li HM, Tsai M-H, Wu S-C, Cheng WTK, Liu J-L, Sung L-Y. SMN is required for the maintenance of embryonic stem cells and neuronal differentiation in mice. Brain Struct Funct. 2015;220(3):1539–53. [DOI] [PubMed]
- 13.Maeda M, Harris AW, Kingham BF, Lumpkin CJ, Opdenaker LM, McCahan SM, Wang W, Butchbach MER. Transcriptome profiling of spinal muscular atrophy motor neurons derived from mouse embryonic stem cells. PLoS One. 2014;9:e106818. doi: 10.1371/journal.pone.0106818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Franklin RCG, Jacobs JP, Krogmann ON, Béland MJ, Aiello VD, Colan SD, Elliott MJ, William Gaynor J, Kurosawa H, Maruszewski B, Stellin G, Tchervenkov CI, Walters Iii HL, Weinberg P, Anderson RH. Nomenclature for congenital and paediatric cardiac disease: historical perspectives and The International Pediatric and Congenital Cardiac Code. Cardiol Young. 2008;18(Suppl 2):70–80. doi: 10.1017/S1047951108002795. [DOI] [PubMed] [Google Scholar]
- 15.The International Society for Nomenclature of Paediatric and Congenital Heart Disease (ISNPCHD). European Paediatric Cardiac Code – The Short List with ICD-9 & ICD-10 cross-mapping. Retrieved from: http://ipccc.net/ipccc-download-form/.
- 16.Korpas D. Implantable Cardiac Devices Technology. Boston: Springer US; 2013. [Google Scholar]
- 17.Sterz H, Harrer G, Marchet H, Kaserer HP, Schlamberger H, Samec H, Stark U. [Primary and neurogenic skeletal muscle diseases or paralysis with marked disturbances of cardiac rhythm] Z Kreislaufforsch. 1971;60:1–13. [PubMed] [Google Scholar]
- 18.Matsumoto K, Kakiuchi F, Kaklhana M. Kugelberg-Welander disease with cardiopathy of unknown etiology. Clinical report of a case. Respir Circ. 1971;19:863–870. [Google Scholar]
- 19.Sugimura F, Iijima M, Ozawa Y, Oki Y, Watanabe S. [2 cases of Kugelberg-Welander disease with cardiopathy] Rinsho Shinkeigaku. 1973;13:79–86. [PubMed] [Google Scholar]
- 20.Lagarde P, Bakouche P, Lamotte-Barrillon S. The Wohlfart-Kugelberg-Welander disease. A clinical case with nonobstructive myocardiopathy. Sem Hop. 1976;52:1017–21. [PubMed] [Google Scholar]
- 21.Nixon CE, Oliver J. Early infantile progressive muscular atrophy(Werdnig–Hoffman), a clinical and pathologic study of two cases. J Lab Clin Med. 1927;12:837–44. [Google Scholar]
- 22.Sterne J, Lavieuville M. CARDIAC MANIFESTATIONS DURING DISEASES OF THE NERVOUS AND MUSCULAR SYSTEMS. Gaz médicale Fr. 1964;71:1925–32. [PubMed] [Google Scholar]
- 23.Brandt S. [Werdnig-Hoffmann’s infantile progressive muscular atrophy; clinical aspects, pathology, heredity, and relation to Oppenheim’s amyotonia congenita and other morbid conditions with laxity of the joints or muscles in children]. Nord Med. 1950;44:1499. [PubMed]
- 24.Kohn R. Postmortem findings in a case of Wohlfart-Kugelberg-Welander disease. Confin Neurol. 1968;30:253–60. doi: 10.1159/000103537. [DOI] [PubMed] [Google Scholar]
- 25.Meunier-Carus J, Lonsdorfer J, Lampert E. Functional cardio-respiratory repercussions of neuromuscular diseases of the thoraco-abdominal wall. Poumon Coeur. 1974;30:13–9. [PubMed] [Google Scholar]
- 26.Okazaki K, Sakata S. Saito T. Masui. 1976;25:398–401. [PubMed] [Google Scholar]
- 27.Ikuta F, Ohama E, Nakanishi T, Mannen T, Toyokura Y. Postmortem findings in a case of KW disease; presence of underdeveloped schwann cells and axons. In: Foundation Japan Medical Research., editor. Amyotrophic Lateral Sclerosis. Tokyo: University of Tokyo Press; 1979. pp. 227–84. [Google Scholar]
- 28.Popov’ian MD, Dubinskaia EE, Proshina OV, Tul’skaia MP, Loginova LA. Myocardial lesions in several forms of progressive muscular atrophy. Zh Nevropatol Psikhiatr Im S S Korsakova. 1984;84:330–5. [PubMed] [Google Scholar]
- 29.Silva AC, Russo AK, Piçarro IC, Schmidt B, Gabbai A, Oliveira AS, Tarasantchi J. Cardiorespiratory responses to exercise in patients with spinal muscular atrophy and limb-girdle dystrophy. Braz J Med Biol Res. 1987;20:565–8. [PubMed] [Google Scholar]
- 30.Carboni P, Porro G. Usefulness of ECG in the early diagnosis of infantile spinal muscular dystrophy. Minerva Pediatr. 1988;40:321–5. [PubMed] [Google Scholar]
- 31.Gupta PC, Ahuja B, Dhamija K, Mullick DN. Spinal muscular atrophy: some easy clues to diagnosis. Indian Pediatr. 1989;26:977–80. [PubMed] [Google Scholar]
- 32.Malcić I, Barisić N, Brzović Z, Pazanin L, Senecić I. Cardiomyopathies in children with neuromuscular disorders. Neurol Croat. 1991;40:221–30. [PubMed] [Google Scholar]
- 33.Zupan A. Cardiovascular aberrations in patients with neuromuscular diseases. Acta Cardiomiologica. 1995;7:107–11. [Google Scholar]
- 34.Stöllberger C, Finsterer J. Echocardiography in storage and neuromuscular disorders. Wien Klin Wochenschr. 2001;113:408–15. [PubMed] [Google Scholar]
- 35.Matkowska-Kocjan A, Kuchar E, Szenborn L. Vaccinations in children’s palliative care - Our proposal for the algorithm. Onkol Pol. 2008;11(4):159–62. [Google Scholar]
- 36.Russman BS, Fredericks EJ. Use of the ECG in the diagnosis of childhood spinal muscular atrophy. Arch Neurol. 1979;36:317–8. doi: 10.1001/archneur.1979.00500410095017. [DOI] [PubMed] [Google Scholar]
- 37.Dawood A, Moosa A. Hand and ECG tremor in spinal muscular atrophy. Arch Dis Child. 1983;58:376–8. doi: 10.1136/adc.58.5.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Towfighi J, Young RS, Ward RM. Is Werdnig-Hoffmann disease a pure lower motor neuron disorder? Acta Neuropathol. 1985;65:270–80. doi: 10.1007/BF00687008. [DOI] [PubMed] [Google Scholar]
- 39.Lintermans JP. Echocardiography in neurological disorders. Eur J Pediatr. 1987;146:15–20. doi: 10.1007/BF00647275. [DOI] [PubMed] [Google Scholar]
- 40.Coletta C, Carboni P, Carunchio A, Porro G, Bacci V. Electrocardiographic abnormalities in childhood spinal muscular atrophy. Int J Cardiol. 1989;24:283–8. doi: 10.1016/0167-5273(89)90005-3. [DOI] [PubMed] [Google Scholar]
- 41.Moosa A, Dawood A. Spinal muscular atrophy in African children. Neuropediatrics. 1990;21:27–31. doi: 10.1055/s-2008-1071453. [DOI] [PubMed] [Google Scholar]
- 42.Møller P, Moe N, Saugstad OD, Skullerud K, Velken M, Berg K, Nitter-Hauge S, Børresen AL. Spinal muscular atrophy type I combined with atrial septal defect in three sibs. Clin Genet. 1990;38:81–3. doi: 10.1111/j.1399-0004.1990.tb03553.x. [DOI] [PubMed] [Google Scholar]
- 43.Kirkinen P, Ryynänen M, Haring P, Torkkeli H, Pääkkönen L, Martikainen A. Prenatal activity of a fetus with early-onset, severe spinal muscular atrophy. Prenat Diagn. 1994;14:1074–6. doi: 10.1002/pd.1970141111. [DOI] [PubMed] [Google Scholar]
- 44.Bürglen L, Spiegel R, Ignatius J, Cobben JM, Landrieu P, Lefebvre S, Munnich A, Melki J, Burglen L. SMN gene deletion in variant of infantile spinal muscular atrophy. Lancet. 1995;346:316–317. doi: 10.1016/S0140-6736(95)92206-7. [DOI] [PubMed] [Google Scholar]
- 45.Rudnik-Schöneborn S, Forkert R, Hahnen E, Wirth B, Zerres K. Clinical spectrum and diagnostic criteria of infantile spinal muscular atrophy: further delineation on the basis of SMN gene deletion findings. Neuropediatrics. 1996;27:8–15. doi: 10.1055/s-2007-973741. [DOI] [PubMed] [Google Scholar]
- 46.Devriendt K, Lammens M, Schollen E, Van Hole C, Dom R, Devlieger H, Cassiman JJ, Fryns JP, Matthijs G. Clinical and molecular genetic features of congenital spinal muscular atrophy. Ann Neurol. 1996;40:731–8. doi: 10.1002/ana.410400509. [DOI] [PubMed] [Google Scholar]
- 47.Mulleners WM, van Ravenswaay CM, Gabreëls FJ, Hamel BC, van Oort A, Sengers RC. Spinal muscular atrophy combined with congenital heart disease: a report of two cases. Neuropediatrics. 1996;27:333–4. doi: 10.1055/s-2007-973805. [DOI] [PubMed] [Google Scholar]
- 48.Bürglen L, Amiel J, Viollet L, Lefebvre S, Burlet P, Clermont O, Raclin V, Landrieu P, Verloes A, Munnich A, Melki J. Survival motor neuron gene deletion in the arthrogryposis multiplex congenita-spinal muscular atrophy association. J Clin Invest. 1996;98:1130–2. doi: 10.1172/JCI118895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang JJ, Jong YJ, Huang MY, Chiang CH, Huang TY. Electrocardiographic findings in children with spinal muscular atrophy. Jpn Heart J. 1996;37:239–42. doi: 10.1536/ihj.37.239. [DOI] [PubMed] [Google Scholar]
- 50.Bingham PM, Shen N, Rennert H, Rorke LB, Black AW, Marin-Padilla MM, Nordgren RE. Arthrogryposis due to infantile neuronal degeneration associated with deletion of the SMNT gene. Neurology. 1997;49:848–51. doi: 10.1212/WNL.49.3.848. [DOI] [PubMed] [Google Scholar]
- 51.Rijhsinghani A, Yankowitz J, Howser D, Williamson R. Sonographic and maternal serum screening abnormalities in fetuses affected by spinal muscular atrophy. Prenat Diagn. 1997;17:166–9. doi: 10.1002/(SICI)1097-0223(199702)17:2<166::AID-PD14>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 52.Jong YJ, Chang JG, Wu JR. Large-scale deletions in a Chinese infant associated with a variant form of Werdnig-Hoffmann disease. Neurology. 1998;51:878–9. doi: 10.1212/WNL.51.3.878. [DOI] [PubMed] [Google Scholar]
- 53.MacLeod MJ, Taylor JE, Lunt PW, Mathew CG, Robb SA. Prenatal onset spinal muscular atrophy. Eur J Paediatr Neurol. 1999;3:65–72. doi: 10.1016/S1090-3798(99)80015-4. [DOI] [PubMed] [Google Scholar]
- 54.Courtens W, Johansson A-B, Dachy B, Avni F, Telerman-Toppet N, Scheffer H. Infantile spinal muscular atrophy variant with congenital fractures in a female neonate: evidence for autosomal recessive inheritance. J Med Genet. 2002;39:74–77. doi: 10.1136/jmg.39.1.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.El-Matary W, Kotagiri S, Cameron D, Peart I. Spinal muscle atrophy type 1 (Werdnig-Hoffman disease) with complex cardiac malformation. Eur J Pediatr. 2004;163:331–2. doi: 10.1007/s00431-004-1437-6. [DOI] [PubMed] [Google Scholar]
- 56.García-Cabezas M a, García-Alix A, Martín Y, Gutiérrez M, Hernández C, Rodríguez JI, Morales C. Neonatal spinal muscular atrophy with multiple contractures, bone fractures, respiratory insufficiency and 5q13 deletion. Acta Neuropathol. 2004;107:475–8. doi: 10.1007/s00401-004-0825-3. [DOI] [PubMed] [Google Scholar]
- 57.Cook AL, Curzon CL, Milazzo AS. An infant with hypoplastic left heart syndrome and spinal muscular atrophy. Cardiol Young. 2006;16:78–80. doi: 10.1017/S1047951105002131. [DOI] [PubMed] [Google Scholar]
- 58.Menke L a, Poll-The BT, Rozemuller JM, LoaNjoe SM, Lemmink HH, Cobben JM. Hypoplastisch linkerhartsyndroom als uiting van een bijzondere vorm van de ziekte van Werdnig-Hoffmann. Tijdschr Kindergeneeskd. 2006;74:214–217. doi: 10.1007/BF03061635. [DOI] [Google Scholar]
- 59.Sarnat HB, Trevenen CL. Motor neuron degeneration in a 20-week male fetus: spinal muscular atrophy type 0. Can J Neurol Sci. 2007;34:215–20. doi: 10.1017/S0317167100006077. [DOI] [PubMed] [Google Scholar]
- 60.Vaidla E, Talvik I, Kulla A, Sibul H, Maasalu K, Metsvaht T, Piirsoo A, Talvik T. Neonatal spinal muscular atrophy type 1 with bone fractures and heart defect. J Child Neurol. 2007;22:67–70. doi: 10.1177/0883073807299954. [DOI] [PubMed] [Google Scholar]
- 61.Bach JR. Medical considerations of long-term survival of Werdnig-Hoffmann disease. Am J Phys Med Rehabil. 2007;86:349–55. doi: 10.1097/PHM.0b013e31804b1d66. [DOI] [PubMed] [Google Scholar]
- 62.Menke LA, Poll-The BT, Clur SA, Bilardo CM, van der Wal AC, Lemmink HH, Cobben JM. Congenital heart defects in spinal muscular atrophy type I: a clinical report of two siblings and a review of the literature. Am J Med Genet A. 2008;146A:740–744. doi: 10.1002/ajmg.a.32233. [DOI] [PubMed] [Google Scholar]
- 63.Rudnik-Schoneborn S, Heller R, Berg C, Betzler C, Grimm T, Eggermann T, Eggermann K, Wirth R, Wirth B, Zerres K, Rudnik-Schöneborn S. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J Med Genet. 2008;45:635–8. doi: 10.1136/jmg.2008.057950. [DOI] [PubMed] [Google Scholar]
- 64.Araujo A prufer de QCAA prufer de QC, Araujo M, Swoboda KJ, Araujo Ade Q. Vascular perfusion abnormalities in infants with spinal muscular atrophy. J Pediatr. 2009;155:292–4. [DOI] [PMC free article] [PubMed]
- 65.Lumaka A, Bone D, Lukoo R, Mujinga N, Senga I, Tady B, Matthijs G, Lukusa TP. Werdnig-Hoffmann disease: report of the first case clinically identified and genetically confirmed in central Africa (Kinshasa-Congo) Genet Couns. 2009;20:349–58. [PubMed] [Google Scholar]
- 66.Rudnik-Schöneborn S, Vogelgesang S, Armbrust S, Graul-Neumann L, Fusch C, Zerres K. Digital necroses and vascular thrombosis in severe spinal muscular atrophy. Muscle Nerve. 2010;42:144–7. doi: 10.1002/mus.21654. [DOI] [PubMed] [Google Scholar]
- 67.Parra J, Alias L, Also-Rallo E, Martínez-Hernández R, Senosiain R, Medina C, Alejos O, Rams N, Amenedo M, Ormo F, Jesús Barceló M, Calaf J, Baiget M, Bernal S, Tizzano EF. Evaluation of fetal nuchal translucency in 98 pregnancies at risk for severe spinal muscular atrophy: possible relevance of the SMN2 copy number. J Matern Fetal Neonatal Med. 2012;25:1246–9. doi: 10.3109/14767058.2011.636101. [DOI] [PubMed] [Google Scholar]
- 68.Ekici B, Demir IH, Ocak S, Yesil G, Tatli B, Celebi A, Yeşil G, Tatlı B, Çelebi A. An infant with spinal muscular atrophy and tetrology of Fallot. Clin Neurol Neurosurg. 2012;114:1033–1034. doi: 10.1016/j.clineuro.2012.01.049. [DOI] [PubMed] [Google Scholar]
- 69.Khera S, Ghuliani R. Type 0 spinal muscular atrophy with multisystem involvement. Indian Pediatr. 2014;51:923–924. doi: 10.1007/s13312-015-0557-6. [DOI] [PubMed] [Google Scholar]
- 70.Mercuri E, Bertini E, Iannaccone ST. Childhood spinal muscular atrophy: controversies and challenges. Lancet Neurol. 2012;11:443–452. doi: 10.1016/S1474-4422(12)70061-3. [DOI] [PubMed] [Google Scholar]
- 71.Wadman RI, Stam M, Gijzen M, Lemmink HH, Snoeck IN, Wijngaarde CA, Braun KPJ, SChoenmakers MAGC, Van den Berg LH, Dooijes D, Van der Pol W-L. Association of motor milestones and SMN2 copy and outcome in spinal muscular atrophy types 0-4. J Neurol Neurosurg Psychiatry. 2016. (in press) [DOI] [PubMed]
- 72.Carter GT, Abresch RT, Fowler WM, Johnson ER, Kilmer DD, McDonald CM. Profiles of neuromuscular diseases. Spinal muscular atrophy. Am J Phys Med Rehabil. 1995;74(5 Suppl):S150–9. doi: 10.1097/00002060-199509001-00009. [DOI] [PubMed] [Google Scholar]
- 73.Gardner-Medwin D, Hudgson P, Walton JN. Benign spinal muscular atrophy arising in childhood and adolescence. J Neurol Sci. 1967;5:121–58. doi: 10.1016/0022-510X(67)90013-5. [DOI] [PubMed] [Google Scholar]
- 74.Emery AE. Abnormalities of the electrocardiogram in hereditary myopathies. J Med Genet. 1972;9:8–12. doi: 10.1136/jmg.9.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nuruki K, Tanaka H, Nishi S, Uemura N, Toyama Y, Kanehisa T, Tanaka N, Ohshige K, Ohkatsu Y, Igata A. Electrophysiological and Histological Studies of the Heart in Myotonic Dystrophy and Kugelberg-Welander’s Disease. Proceeding of the 38th Annual Meeting. Jpn Circ J. 1974;38:644. [Google Scholar]
- 76.Tomlinson BE, Walton JN, Irving D. Spinal cord limb motor neurones in muscular dystrophy. J Neurol Sci. 1974;22:305–327. doi: 10.1016/0022-510X(74)90003-3. [DOI] [PubMed] [Google Scholar]
- 77.Tanaka H, Uemura N, Toyama Y, Kudo A, Ohkatsu Y, Kenehisa T. Cardiac involvement in the Kugelberg-Welander syndrome. Am J Cardiol. 1976;38:528–32. doi: 10.1016/0002-9149(76)90473-2. [DOI] [PubMed] [Google Scholar]
- 78.Tanaka H, Nishi S, Nuruki K, Tanaka N. Myocardial ultrastructural changes in Kugelberg-Welander syndrome. Br Heart J. 1977;39:1390–3. doi: 10.1136/hrt.39.12.1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kimura S, Yokota H, Tateda K, Miyamoto K, Yamamoto K, Shibata J. A case of the Kugelberg-Welander syndrome complicated with cardiac lesions. Jpn Heart J. 1980;21:417–22. doi: 10.1536/ihj.21.417. [DOI] [PubMed] [Google Scholar]
- 80.Ceroni M, Grandi A, Poloni M, Venco A. Association of cardiomyopathy with Kugelberg-Welander disease. Ital J Neurol Sci. 1982;3:143–7. doi: 10.1007/BF02043948. [DOI] [PubMed] [Google Scholar]
- 81.Hofstad H, Ohm OJ, Mørk SJ, Aarli JA. Heart disease in myasthenia gravis. Acta Neurol Scand. 1984;70:176–84. doi: 10.1111/j.1600-0404.1984.tb00817.x. [DOI] [PubMed] [Google Scholar]
- 82.Stegaru-Hellring B, Nitsche A, Struwe O, Berlit P, Lipinski CG, Brittinger WD. Kardiale Befunde bei verschiedenen Formen der Muskeldystrophie und neurogenen Muskelatrophie im Kindes- und Jugendalter. Aktuelle Neurol. 1988;15:102–106. doi: 10.1055/s-2007-1020637. [DOI] [Google Scholar]
- 83.Bataille J, Guillon F, Urtizberea A, Estournet B, Richard S, Barois A. [Pathological anatomy of the heart in myopathies and infantile muscular atrophies] Ann Med Interne (Paris) 1991;142:5–8. [PubMed] [Google Scholar]
- 84.Elkohen M, Vaksmann G, Elkohen MR, Francart C, Foucher C, Rey C. Cardiac involvement in Kugelberg-Welander disease. A prospective study of 8 cases. Arch Mal Coeur Vaiss. 1996;89:611–7. [PubMed] [Google Scholar]
- 85.Liu YB, Chen WJ, Lee YT. Atrial standstill in a case of Kugelberg-Welander syndrome with cardiac involvement: an electrophysiologic study. Int J Cardiol. 1999;70:207–10. doi: 10.1016/S0167-5273(99)00078-9. [DOI] [PubMed] [Google Scholar]
- 86.Yasuma F, Kuru S, Konagaya M. Dilated cardiomyopathy in Kugelberg-Welander disease: coexisting sleep disordered breathing and its treatment with continuous positive airway pressure. Intern Med. 2004;43:951–4. doi: 10.2169/internalmedicine.43.951. [DOI] [PubMed] [Google Scholar]
- 87.Takahashi N, Shimada T, Ishibashi Y, Sugamori T, Hirano Y, Oyake N, Murakami Y. Cardiac involvement in Kugelberg-Welander disease: a case report and review. Am J Med Sci. 2006;332:354–6. doi: 10.1097/00000441-200612000-00009. [DOI] [PubMed] [Google Scholar]
- 88.Roos M, Sarkozy A, Chierchia GB, De Wilde P, Schmedding E, Brugada P. Malignant ventricular arrhythmia in a case of adult onset of spinal muscular atrophy (Kugelberg-Welander disease) J Cardiovasc Electrophysiol. 2009;20:342–4. doi: 10.1111/j.1540-8167.2008.01327.x. [DOI] [PubMed] [Google Scholar]
- 89.Kuru S, Sakai M, Konagaya M, Yoshida M, Hashizume Y, Saito K. An autopsy case of spinal muscular atrophy type III (Kugelberg-Welander disease) Neuropathology. 2009;29:63–7. doi: 10.1111/j.1440-1789.2008.00910.x. [DOI] [PubMed] [Google Scholar]
- 90.Vilela H, Santos J, Colaço J, Oliveira E, Canas-da-Silva P. Reversal of neuromuscular blockade with sugammadex in a patient with spinal muscular atrophy type III (Kugelberg-Welander syndrome) J Anesth. 2012;26:306–7. doi: 10.1007/s00540-011-1308-z. [DOI] [PubMed] [Google Scholar]
- 91.Haliloglu G, Gungor M, Anlar B. The Role of Electrocardiography in the Diagnosis of Spinal Muscular Atrophy Type III. J Pediatr. 2015;166:1092. doi: 10.1016/j.jpeds.2014.12.056. [DOI] [PubMed] [Google Scholar]
- 92.Bevan AK, Hutchinson KR, Foust KD, Braun L, McGovern VL, Schmelzer L, Ward JG, Petruska JC, Lucchesi P a, Burghes AHM, Kaspar BK. Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum Mol Genet. 2010;19:3895–905. doi: 10.1093/hmg/ddq300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Heier CR, Satta R, Lutz C, DiDonato CJ. Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum Mol Genet. 2010;19:3906–18. doi: 10.1093/hmg/ddq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shababi M, Habibi J, Yang HT, Vale SM, Sewell WA, Lorson CL. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum Mol Genet. 2010;19:4059–71. doi: 10.1093/hmg/ddq329. [DOI] [PubMed] [Google Scholar]
- 95.Gogliotti RG, Quinlan K a, Barlow CB, Heier CR, Heckman CJ, Didonato CJ. Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not a cause, of motor neuron dysfunction. J Neurosci. 2012;32:3818–29. doi: 10.1523/JNEUROSCI.5775-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Biondi O, Lopes P, Desseille C, Branchu J, Chali F, Ben Salah A, Pariset C, Chanoine C, Charbonnier F. Physical exercise reduces cardiac defects in type 2 spinal muscular atrophy-like mice. J Physiol. 2012;590(Pt 22):5907–25. doi: 10.1113/jphysiol.2012.238196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Osborne M, Gomez D, Feng Z, McEwen C, Beltran J, Cirillo K, El-Khodor B, Lin M-Y, Li Y, Knowlton WM, McKemy DD, Bogdanik L, Butts-Dehm K, Martens K, Davis C, Doty R, Wardwell K, Ghavami A, Kobayashi D, Ko C-P, Ramboz S, Lutz C. Characterization of behavioral and neuromuscular junction phenotypes in a novel allelic series of SMA mouse models. Hum Mol Genet. 2012;21:4431–47. doi: 10.1093/hmg/dds285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shababi M, Habibi J, Ma L, Glascock JJ, Sowers JR, Lorson CL. Partial restoration of cardio-vascular defects in a rescued severe model of spinal muscular atrophy. J Mol Cell Cardiol. 2012;52:1074–82. doi: 10.1016/j.yjmcc.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schreml J, Riessland M, Paterno M, Garbes L, Rossbach K, Ackermann B, Kramer J, Somers E, Parson SH, Heller R, Berkessel A, Sterner-Kock A, Wirth B, Roßbach K, Krämer J. Severe SMA mice show organ impairment that cannot be rescued by therapy with the HDACi JNJ-26481585. Eur J Hum Genet. 2013;21:643–52. doi: 10.1038/ejhg.2012.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cobb MS, Rose FF, Rindt H, Glascock JJ, Shababi M, Miller MR, Osman EY, Yen P-F, Garcia ML, Martin BR, Wetz MJ, Mazzasette C, Feng Z, Ko C-P, Lorson CL. Development and characterization of an SMN2-based intermediate mouse model of Spinal Muscular Atrophy. Hum Mol Genet. 2013;22:1843–55. doi: 10.1093/hmg/ddt037. [DOI] [PubMed] [Google Scholar]
- 101.Keil JM, Seo J, Howell MD, Hsu WH, Singh RN, DiDonato CJ. A short antisense oligonucleotide ameliorates symptoms of severe mouse models of spinal muscular atrophy. Mol Ther Nucleic Acids. 2014;3:e174. doi: 10.1038/mtna.2014.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tsai L-K, Chen C-L, Ting C-H, Lin-Chao S, Hwu W-L, Dodge JC, Passini M a, Cheng SH. Systemic administration of a recombinant AAV1 vector encoding IGF-1 improves disease manifestations in SMA mice. Mol Ther. 2014;22:1450–9. doi: 10.1038/mt.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Heier CR, DiDonato CJ. ECG in neonate mice with spinal muscular atrophy allows assessment of drug efficacy. Front Biosci (Elite Ed) 2015;7:107–16. doi: 10.2741/e721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bogdanik LP, Osborne MA, Davis C, Martin WP, Austin A, Rigo F, Bennett CF, Lutz CM. Systemic, postsymptomatic antisense oligonucleotide rescues motor unit maturation delay in a new mouse model for type II/III spinal muscular atrophy. Proc Natl Acad Sci U S A. 2015;112:E5863–72. doi: 10.1073/pnas.1509758112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mitchell SC, Korones SB, Berendes HW. Congenital heart disease in 56,109 births. Incidence and natural history. Circulation. 1971;43:323–32. doi: 10.1161/01.CIR.43.3.323. [DOI] [PubMed] [Google Scholar]
- 106.Hoffman JI, Christianson R. Congenital heart disease in a cohort of 19,502 births with long-term follow-up. Am J Cardiol. 1978;42:641–7. doi: 10.1016/0002-9149(78)90635-5. [DOI] [PubMed] [Google Scholar]
- 107.Hoffman JIE, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–900. doi: 10.1016/S0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 108.Bernier P-L, Stefanescu A, Samoukovic G, Tchervenkov CI. The challenge of congenital heart disease worldwide: epidemiologic and demographic facts. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2010;13:26–34. doi: 10.1053/j.pcsu.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 109.van der Bom T, Zomer a C, Zwinderman AH, Meijboom FJ, Bouma BJ, Mulder BJM. The changing epidemiology of congenital heart disease. Nat Rev Cardiol. 2011;8:50–60. doi: 10.1038/nrcardio.2010.166. [DOI] [PubMed] [Google Scholar]
- 110.Brown DW, Fulton DR. Congenital Heart Disease in Children and Adolescents. In: Fuster V, Walsh RA, Harrington RA. Hurst’s The Heart, 13th edition. New York: McGraw-Hill; 2011.
- 111.Wilke T, Groth A, Mueller S, Pfannkuche M, Verheyen F, Linder R, Maywald U, Bauersachs R, Breithardt G. Incidence and prevalence of atrial fibrillation: an analysis based on 8.3 million patients. Europace. 2013;15:486–93. doi: 10.1093/europace/eus333. [DOI] [PubMed] [Google Scholar]
- 112.Granada J, Uribe W, Chyou PH, Maassen K, Vierkant R, Smith PN, Hayes J, Eaker E, Vidaillet H. Incidence and predictors of atrial flutter in the general population. J Am Coll Cardiol. 2000;36:2242–6. doi: 10.1016/S0735-1097(00)00982-7. [DOI] [PubMed] [Google Scholar]
- 113.Shen MJ, Zipes DP. Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ Res. 2014;114:1004–21. doi: 10.1161/CIRCRESAHA.113.302549. [DOI] [PubMed] [Google Scholar]
- 114.Biernacka A, Frangogiannis NG. Aging and Cardiac Fibrosis. Aging Dis. 2011;2:158–173. [PMC free article] [PubMed] [Google Scholar]
- 115.Lotti F, Imlach WL, Saieva L, Beck ES, Hao LT, Li DK, Jiao W, Mentis GZ, Beattie CE, McCabe BD, Pellizzoni L. An SMN-dependent U12 splicing event essential for motor circuit function. Cell. 2012;151:440–54. doi: 10.1016/j.cell.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shababi M, Lorson CL, Rudnik-Schöneborn SS. Spinal muscular atrophy: a motor neuron disorder or a multi-organ disease? J Anat. 2014;224:15–28. doi: 10.1111/joa.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bowerman M, Murray LM, Beauvais A, Pinheiro B, Kothary R. A critical smn threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromuscul Disord. 2012;22:262–276. doi: 10.1016/j.nmd.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 118.Kariya S, Park G-H, Maeno-Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, Landmesser LT, Monani UR. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:2552–2569. doi: 10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Park GH, Maeno-Hikichi Y, Awano T, Landmesser LT, Monani UR. Reduced survival of motor neuron (SMN) protein in motor neuronal progenitors functions cell autonomously to cause spinal muscular atrophy in model mice expressing the human centromeric (SMN2) gene. J Neurosci. 2010;30:12005–12019. doi: 10.1523/JNEUROSCI.2208-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Etchevers HC, Vincent C, Le Douarin NM, Couly GF. The cephalic neural crest provides pericytes and smooth muscle cells to all blood vessels of the face and forebrain. Development. 2001;128:1059–68. doi: 10.1242/dev.128.7.1059. [DOI] [PubMed] [Google Scholar]
- 121.Le Douarin NM, Creuzet S, Couly G, Dupin E. Neural crest cell plasticity and its limits. Development. 2004;131:4637–50. doi: 10.1242/dev.01350. [DOI] [PubMed] [Google Scholar]
- 122.Crane JF, Trainor P a. Neural crest stem and progenitor cells. Annu Rev Cell Dev Biol. 2006;22:267–86. doi: 10.1146/annurev.cellbio.22.010305.103814. [DOI] [PubMed] [Google Scholar]
- 123.Kirby ML, Hutson MR. Factors controlling cardiac neural crest cell migration. Cell Adh Migr. 2010;4:609–621. doi: 10.4161/cam.4.4.13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Brade T, Pane LS, Moretti A, Chien KR, Laugwitz K-L. Embryonic heart progenitors and cardiogenesis. Cold Spring Harb Perspect Med. 2013;3:a013847. doi: 10.1101/cshperspect.a013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vincentz, JW, Firulli AB. The Cardiac Neural Crest and Their Role in Development and Disease. In: Neural Crest Cells: Evolution, Development and Disease, 1st edition. London: Elsevier inc.; 2014.
- 126.Mayor R, Theveneau E. The role of the non-canonical Wnt-planar cell polarity pathway in neural crest migration. Biochem J. 2014;457:19–26. doi: 10.1042/BJ20131182. [DOI] [PubMed] [Google Scholar]
- 127.Mikawa T, Hurtado R. Development of the cardiac conduction system. Semin Cell Dev Biol. 2007;18:90–100. doi: 10.1016/j.semcdb.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 128.Kolb SJ, Kissel JT. Spinal muscular atrophy: a timely review. Arch Neurol. 2011;68:979–84. doi: 10.1001/archneurol.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dubowitz V. Ramblings in the history of spinal muscular atrophy. Neuromuscul Disord. 2009;19:69–73. doi: 10.1016/j.nmd.2008.10.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed for the writing of this review. The data collected from the original publications is available in the Additional file 1: Tables S1–S5.