

Abstract
Differentiation of smooth muscle cells is accompanied by the transcriptional activation of an array of muscle-specific genes controlled by serum response factor (SRF). Myocardin is a cardiac and smooth muscle-specific expressed transcriptional coactivator of SRF and is sufficient and necessary for smooth muscle gene expression. Here, we show that myocardin induces the acetylation of nucleosomal histones surrounding SRF-binding sites in the control regions of smooth muscle genes. The promyogenic activity of myocardin is enhanced by p300, a histone acetyltransferase that associates with the transcription activation domain of myocardin. Conversely, class II histone deacetylases interact with a domain of myocardin distinct from the p300-binding domain and suppress smooth muscle gene activation by myocardin. These findings point to myocardin as a nexus for positive and negative regulation of smooth muscle gene expression by changes in chromatin acetylation.
Smooth muscle cells (SMCs) are essential for the formation and function of the cardiovascular, respiratory, genitourinary, and digestive systems. Differentiation of SMCs is accompanied by the transcriptional activation of a set of genes whose products mediate the unique contractile, structural, and metabolic properties of the smooth musculature. Nearly every smooth muscle gene analyzed to date is controlled by one or more binding sites for serum response factor (SRF), a widely expressed MADS (MCM1, Agamous, Deficiens, SRF) box transcription factor (reviewed in reference 31). SRF binds as a homodimer to a DNA consensus sequence known as a CArG box [CC(A/T)6GG], which is found in the control regions of muscle-specific genes, as well as genes that are induced by growth factor signaling (34, 38). The specific genes activated by SRF are determined by the availability of positive and negative cofactors, as well as intracellular signals (42). The related MADS box factor myocyte enhancer factor 2 acts in a similar manner to control muscle- and signal-inducible gene expression (3).
Myocardin is an SRF cofactor expressed specifically in smooth and cardiac muscle lineages throughout embryonic development and adulthood (44; reviewed in reference 45). Myocardin belongs to the SAP (SAF-A/B, Acinus, PIAS) domain family of transcription factors, which has been implicated in chromatin dynamics (2), and stimulates SRF-dependent transcription by interacting with the MADS box of SRF and providing its strong transcription activation domain (TAD) (44). Remarkably, forced expression of myocardin in fibroblasts is sufficient to activate the smooth muscle differentiation program (8a, 10, 48, 52). Consistent with its potential role as a dominant activator of smooth muscle gene expression, knockout mice lacking myocardin die during mid-embryogenesis from a lack of differentiated vascular SMCs and consequent lethal cardiovascular abnormalities (25). Myocardin also regulates cardiac gene promoters in an SRF-dependent manner, and expression of a dominant negative mutant form of myocardin in Xenopus embryos can prevent heart formation (44). However, forced expression of myocardin in fibroblasts in culture does not result in activation of endogenous cardiac genes, suggesting that other factors are required for cardiac gene expression or that endogenous inhibitors suppress cardiogenic activity in this assay. In addition to myocardin, other SRF cofactors, including CRP1 and CRP2, appear to play important roles in regulating CArG-dependent smooth muscle gene expression (6).
Previous studies have suggested the involvement of chromatin acetylation-deacetylation in the control of smooth muscle gene expression (20, 28, 36). Histone acetylation, which is catalyzed by histone acetyltransferases (HATs), promotes gene transcription by destabilizing chromatin structure and facilitating access of transcriptional complexes to their target genes (reviewed in reference 19). Among the best-characterized HATs are p300 and CREB-binding protein (CBP), which act as bridging factors between transcription factors and other components of the transcriptional machinery and as targets for several intracellular signaling pathways that stimulate transcription (reviewed in references 5 and 13). While some transcription factors rely on the HAT activity provided by p300/CBP (and other HATs) to activate transcription, others possess intrinsic HAT activity.
The gene-activating function of HATs is antagonized by the activity of histone deacetylases (HDACs), which deacetylate nucleosomal histones, thereby promoting chromatin condensation (43). Mammalian HDACs can be categorized into three classes based on homology to yeast HDACs. Class I HDACs, which include HDAC1, -2, and -3, are widely expressed and contain simply a catalytic domain. Class II HDACs (HDAC4, -5, -7, and -9) exhibit tissue-restricted expression patterns and contain a bipartite structure with a C-terminal catalytic domain and an N-terminal extension that mediates associations with a variety of positive and negative cofactors. Class III HDACs are NAD dependent and related to yeast Sir2.
The ability of myocardin to activate smooth muscle genes that are otherwise silent in non-muscle cells suggests that myocardin is able to convert smooth muscle genes from a repressed to a transcriptionally active state. Here, we show that myocardin induces the acetylation of nucleosomal histones surrounding SRF-binding sites in the promoters of smooth muscle genes. Acetylation of smooth muscle genes is mediated by the association of myocardin with p300, which stimulates myocardin activity. The promyogenic activity of myocardin is counteracted by class II HDACs, which also interact with myocardin and repress its activity. These opposing influences of HATs and HDACs on myocardin activity provide a mechanism for modulation of the smooth muscle differentiation program and point to myocardin as a regulator of chromatin configurations of smooth muscle genes and an integrator of positive and negative influences on smooth muscle differentiation.
MATERIALS AND METHODS
Cell culture and transfection assays.
Transfection of COS and 10T1/2 cells and luciferase assays were performed as previously described (26, 44). Unless otherwise indicated, 100 ng of reporter plasmid and 100 ng of each activator plasmid were used. The total amount of DNA per well was kept constant by adding the corresponding amount of expression vector without a cDNA insert. All of the transfection experiments were repeated at least twice in duplicate. Values are expressed as means ± standard deviations.
Myocardin and HDAC expression vectors have been previously described (26, 44). The p300 expression vectors were obtained from Cheol Yong Choi (Sungkyunkwan University, Seoul, South Korea), Tso-Pang Yao (Duke University), and Wei Gu (Columbia University). The E1A expression plasmid was a gift of Tso-Pang Yao and Lishan Su (University of North Carolina). Myocardin and HDAC5 deletion mutant proteins were generated through PCR-based mutagenesis with the QuikChange kit from Stratagene. All mutations were confirmed by DNA sequencing. The SM22-luciferase reporter contained the 1,434-bp promoter (24), and the ANF-luciferase reporter contained the 638-bp promoter of the atrial natriuretic factor (ANF) gene (41). The 4 × SM22-CArG-luciferase reporter has been previously described (7). CMV-lacZ was included as an internal control for variations in transfection efficiency. All of the other reporters used have been described previously.
GST protein-binding assays.
A plasmid encoding a glutathione S-transferase (GST) fusion protein was transformed into BL21-codon plus cells (Stratagene). The cells were grown at 37°C in 2× YT medium to an optical density of 1.0. Isopropyl-β-d-thiogalactopyranoside (IPTG; 50 μM) was then added to the culture to induce protein expression. After being shaken at room temperature for 4 to 6 h, the cells were harvested and the GST protein was purified with glutathione beads in accordance with the Amersham procedure.
Proteins translated in vitro were labeled with [35S]methionine with a TNT T7 reticulocyte lysate system (Promega). Glutathione beads conjugated with 1 μg of protein were incubated with 10 μl of TNT product at 4°C for 2 h in 500 μl of GST-binding buffer (20 mM Tris [pH 7.3], 150 mM NaCl, 0.5% NP-40, protease inhibitor cocktail from Roche, 1 mM phenylmethylsulfonyl fluoride [PMSF]). The beads were washed three times with GST-binding buffer. Fifty microliters of sodium dodecyl sulfate (SDS) loading buffer was then added to the beads. After boiling, 20 μl was loaded onto an SDS-polyacrylamide gel electrophoresis (PAGE) gel and analyzed by autoradiography.
Coimmunoprecipitation assays.
COS cells were transiently transfected with plasmids encoding the epitope-tagged myocardin, HDAC4, HDAC5, and p300 proteins as indicated in the figure legends with FuGENE 6 reagent (Roche Molecular Biochemicals). Cells were harvested 48 h after transfection in lysis buffer composed of phosphate-buffered saline (PBS) containing 0.5% Triton X-100, 1 mM EDTA, 1 mM PMSF, and complete protease inhibitors (Roche Molecular Biochemicals). Following a brief sonication and removal of cellular debris by centrifugation, epitope-tagged proteins were precipitated with antibodies as indicated and protein A/G beads (Santa Cruz). The bound proteins were washed five times with lysis buffer, resolved by SDS-PAGE, and transferred to polyvinylidene difluoride membranes (Bio-Rad). Membranes were immunoblotted with antibodies as indicated, and proteins were visualized with a chemiluminescence detection system (Santa Cruz).
Immunostaining.
Immunostaining was performed as previously described (30). To determine the cellular localization of myocardin and HDAC5, COS cells were transfected with FLAG-tagged myocardin and Myc-tagged HDAC5 and stained with anti-FLAG (mouse monoclonal M2; Sigma) and anti-Myc (rabbit polyclonal; Santa Cruz) antibodies. Myogenic conversion assays with 10T1/2 cells were performed as previously described (26, 48) except that Lipofectamine reagent (Invitrogen) was used for transfection. Mouse anti-SM α-actin monoclonal antibody (1A4; Sigma) was used to monitor smooth muscle gene induction.
We determined the efficiency of myogenic conversion by myocardin by cotransfecting a CMV-lacZ expression vector with the FLAG-myocardin expression vector. Eighteen percent of lacZ-positive cells expressing myocardin also express smooth muscle proteins at a detectable level. By comparison, 26% of lacZ-positive cells expressing MyoD in transfection assays also express skeletal muscle proteins at a detectable level. We do not know whether the cells that express myocardin but not smooth muscle proteins express myocardin at a level too low to activate the smooth muscle differentiation program or, alternatively, whether smooth muscle proteins are expressed in cells below the level of detection by immunostaining.
Histone acetylation assays and HAT activity assays.
COS cells were transiently transfected with expression vectors encoding the FLAG-tagged myocardin deletion mutant proteins as described above. Forty-eight hours later, cells were harvested and proteins were immunoprecipitated with M2 anti-FLAG antibody-agarose (Sigma). Histone acetylation was assayed as previously described (35). Protein A-agarose beads were washed twice with HAT buffer (50 mM Tris [pH 8.0], 10% glycerol, 1 mM PMSF), followed by incubation in 30 μl HAT buffer containing 1 mM dithiothreitol, 100 ng of trichostatin A (TSA) per μl, 2 to 4 μl of 3H-labeled acetyl coenzyme A (5.3 μl/mmol; Amersham), and 10 μg of core histones from Roche. After 1 h of incubation at 30°C, 2× SDS loading dye was added to the reaction and the tubes were boiled at 95°C for 5 min. The proteins were separated by SDS-PAGE. The gel was stained with Coomassie blue, destained, and then incubated in Amplify solution (Amersham NAMP100) for 30 min at room temperature. After being dried, the gel was exposed to Amersham Hyperfilm MP.
ChIP assays.
Chromatin immunoprecipitation (ChIP) assays were carried out with the ChIP assay kit from Upstate Biotech. Briefly, cellular proteins were cross-linked with 1% formaldehyde at 37°C for 10 min. After being washed with cold PBS twice, cells were scraped in 500 μl of PBS and centrifuged at 5,000 rpm in an Eppendorf 547C centrifuge. The pellets were then resuspended in 300 μl of SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl [pH 8.1], protease inhibitors). The lysate was sonicated three times for 10 s each to shear the DNA into segments between 200 and 1,000 bp in length. After being centrifuged at 14,000 rpm in an Eppendorf 547C centrifuge for 10 min, the supernatants were diluted 10 times with ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl [pH 8.1], 167 mM NaCl, protease inhibitors). One milliliter of the diluted lysate was then cleared with 10 μg of salmon sperm DNA and 20 μl of protein A-agarose beads (Santa Cruz) at 4°C for 2 h. After brief centrifugation to pellet the beads, the supernatant was incubated with 2 μg of specific antibodies overnight at 4°C. The next day, 10 μg of salmon sperm DNA and 40 μl of protein A-agarose beads were added to the supernatant and the mixture was incubated for 2 h. The beads were then washed sequentially with low-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl [pH 8], 150 mM NaCl), high-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl [pH 8], 500 mM NaCl), and LiCl wash buffer (0.25 mM LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl [pH 8.1]) for 5 min each. The beads were then washed three times with Tris-EDTA, and the precipitates were then eluted twice with 250 μl of elution buffer (1% SDS, 0.1 M NaHCO3). Twenty microliters of 5 M NaCl was added to each eluate, and the mixture was incubated at 65°C for 4 h to reverse cross-linking. DNA fragments were then purified with a QIAquick spin column. The M2 anti-FLAG antibody used was from Sigma, and the anti-acetyl histone H3 antibody used was from Upstate. The primers for the SM α-actin promoter were AGCAGAACAGAGGAATGCAGTGGAAGAGAC and CCTCCCACTCGCCTCCCAAACAAGGAGC. The primers for the SM22 promoter were GGTCCTGCCCATAAAAGGTTT and TGCCCATGGAAGTCTGCTTGG. Both products contain two CArG boxes in the promoter regions. The primers for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) promoter were GCTCTCTGCTCCTCCCTGTT and CAATCTCCACTTTGCCACTGC.
Detection of transcripts by reverse transcription (RT)-PCR.
Total RNA was isolated from cells with Trizol reagent and used as the template for RT with primers specific for each transcript (Invitrogen) in accordance with the manufacturer's instructions. The sequences of the primers used are available upon request.
LacZ staining of mouse embryos.
Embryos heterozygous for a lacZ knockin allele into the HDAC5 locus (8) were fixed in formalin buffered with PBS, embedded in paraffin, and sectioned at 5 μm for histological examination. Sections were stained with hematoxylin and eosin and photographed under normal bright-field microscopic conditions. LacZ staining was performed as previously described (33). The expression pattern of the lacZ knockin allele faithfully reflects the pattern of expression of the endogenous HDAC5 gene (8).
RESULTS
Myocardin induces histone acetylation at smooth muscle gene promoters.
Myocardin is a potent transcriptional activator that activates smooth muscle genes by tethering to CArG boxes with SRF (44, 46). To determine the impact of myocardin on the acetylation state of nucleosomal histones associated with smooth muscle gene promoters, we used ChIP assays to monitor histone H3 acetylation in the vicinity of essential CArG boxes in the SM α-actin and SM22 promoters in 10T1/2 cells. In naïve 10T1/2 cells, histone H3 was not acetylated on these promoters. However, expression of myocardin, which activates these and other smooth muscle genes, induced histone H3 acetylation at these promoters (Fig. 1A). The gene for GAPDH, which is expressed constitutively, did not show a change in acetylation in response to myocardin. Thus, activation of endogenous smooth muscle target genes by myocardin is accompanied by the acetylation of histones on the corresponding gene promoters.
FIG. 1.

Acetylation of histones associated with smooth muscle gene promoters in response to myocardin and modulation of myocardin myogenic activity by p300 and HDAC5. (A) 10T1/2 cells were transiently transfected with a FLAG-tagged myocardin expression vector (+) or a control vector (−), and ChIP assays were performed as described in Materials and Methods with primers for sequences associated with the genes for SM α-actin, SM22, and GAPDH. The amount of DNA in each sample (input) is shown at the left. Immunoprecipitations were performed without primary antibody (no Ab) as a control and with anti-FLAG antibody and anti-acetyl histone H3 antibody, as indicated. (B) 10T1/2 cells were transiently transfected with expression vectors encoding myocardin (0.1 μg), p300 (0.5 and 1 μg), or HDAC5 (0.2 and 0.5 μg), as indicated, and SM α-actin protein expression was detected by immunostaining as described in Materials and Methods. Values are expressed as the relative number of positive cells compared to the number in cultures transfected with myocardin alone, which was assigned a value of 100.
Modulation of myocardin activity by p300.
The above findings suggested that myocardin itself might be a HAT or that a HAT might associate with myocardin. To test whether myocardin possesses intrinsic HAT activity, we expressed it in bacteria and assayed the purified protein for its potential ability to stimulate histone acetylation in vitro. Myocardin showed no detectable HAT activity in this assay (data not shown). We therefore tested whether myocardin activity might be modulated by p300, a ubiquitous transcriptional coactivator with intrinsic HAT activity. As reported previously (48), expression of myocardin in 10T1/2 fibroblasts was sufficient to activate smooth muscle gene expression measured by immunostaining for SM α-actin-positive cells. The promyogenic activity of myocardin was augmented by p300, whereas p300 alone had no effect on smooth muscle gene expression (Fig. 1B).
Cotransfection of expression plasmids encoding myocardin and p300 into COS cells also resulted in synergistic and dose-dependent activation of luciferase reporters linked to the SM22 promoter, as well as the cardiac muscle-specific ANF promoter (Fig. 2A), both of which are controlled by pairs of CArG boxes (18, 21, 23). Similarly, p300 enhanced the ability of myocardin to activate a luciferase reporter linked to four tandem copies of a CArG box (Fig. 2A), but not a mutant CArG box that cannot bind SRF (data not shown), demonstrating that the stimulatory effect of p300 was mediated by the SRF-binding site rather than sites for other transcription factors in the SM22 and ANF promoters. In the absence of myocardin, p300 did not stimulate expression of CArG box-dependent reporters, suggesting that its effects were directed at myocardin, not SRF, which is expressed endogenously in COS cells (Fig. 2A).
FIG. 2.

Modulation of myocardin transcriptional activity by p300 and HDAC5. (A) COS cells were transiently transfected with expression vectors encoding myocardin (0.1 μg) and p300 (0.5 and 1 μg) and the indicated luciferase reporters as described in Materials and Methods. (B) COS cells were transiently transfected with expression vectors encoding the GAL4 DNA-binding domain fused to amino acids 1 to 935 (GAL4-Myo) of myocardin (left graph) or 713 to 935 (GAL4-Myo-TAD) of myocardin, which encompasses the TAD (right graph) and p300 (1 μg) and a UAS-luciferase reporter. (C) COS cells were transiently transfected with expression vectors encoding myocardin and HDACs (0.1, 0.2, and 0.5 μg) and the indicated luciferase reporters. (D) COS cells were transiently transfected with expression vectors encoding the GAL4 DNA-binding domain fused to amino acids 1 to 935 (GAL4-Myo) of myocardin, HDAC5 (0.1 and 0.5 μg), or HDAC1 (0.1 and 0.5 μg) and a UAS-luciferase reporter. (E) COS cells were transiently transfected with expression vectors encoding myocardin (0.2 μg), p300 (1 μg), HDAC5 (0.5 μg), and increasing amounts of p300 (0.5, 1, 2, and 4 μg) and HDAC5 (0.5, 1, and 2 μg), respectively, as indicated, and the SM22-luciferase reporter.
Because myocardin requires SRF to activate smooth and cardiac muscle gene expression (44, 46) and SRF has been reported to associate with the p300-related HAT CBP (37), we sought to determine whether p300 could enhance the activity of a GAL4-myocardin fusion protein, which functions independently of SRF. As shown in Fig. 2B, p300 augmented the transcriptional activity of GAL4-myocardin on a reporter controlled by the GAL4 binding site, whereas p300 alone had no effect on this reporter. The TAD of myocardin is located near the C terminus, between residues 713 and 935, and its activity is enhanced when it is removed from the remainder of the protein (44). The transcriptional activity of the TAD was also enhanced by p300 (Fig. 2B). Together, these data suggest that p300 acts through the TAD of myocardin to stimulate transcription of CArG box-dependent cardiac and smooth muscle genes.
Class II HDACs repress the transcriptional activity of myocardin.
To further investigate the potential involvement of histone acetylation-deacetylation in the activation of smooth muscle gene expression by myocardin, we tested whether HDACs affect the transcriptional activity of myocardin. Indeed, HDAC5, a class II HDAC, potently suppressed the ability of myocardin to activate endogenous smooth muscle gene expression in transfected 10T1/2 cells (Fig. 1B). HDAC4, another class II HDAC, had an effect comparable to that of HDAC5, whereas the class I HDACs HDAC1 and -3 had no effect on myocardin activity in this assay (data not shown; see below). HDAC5 also suppressed the expression of reporters linked to the SM22 promoter and the multimerized CArG box in the presence of myocardin, whereas HDAC1 did not affect the activation of these reporters by myocardin (Fig. 2C).
To determine whether the repression of myocardin-dependent transcription by HDAC5 was mediated directly by repression of myocardin activity or indirectly by repression of SRF or another factor, we tested if HDAC5 could repress the transcriptional activity of myocardin fused to the GAL4 DNA-binding domain (Fig. 2D). HDAC5 strongly suppressed activation of the GAL4-dependent reporter by myocardin fused to GAL4. As in the other assays, HDAC1 did not repress the transcriptional activity of myocardin (Fig. 2D). The transcriptional activity of myocardin was dependent on the relative levels of p300 and HDAC5 such that increasing the relative level of p300 could counteract the repressive effect of HDAC5 and vice versa (Fig. 2E). We conclude that p300 and HDAC5 exert opposing influences on smooth muscle gene expression, at least in part by stimulating and suppressing, respectively, the activity of myocardin.
Expression of HDACs in SMCs.
To determine which class II HDACs are expressed in A7r5 SMCs, we performed RT-PCR assays with primers specific for each HDAC transcript. As shown in Fig. 3A, multiple class I and II HDACs were expressed in A7r5 cells. The class II HDACs HDAC4, -5, and -7 were expressed at comparable levels.
FIG. 3.

HDAC expression in SMCs. (A) Transcripts for the indicated HDACs were detected by RT-PCR with RNA isolated from A7r5 cells. Transcripts for calponin and ANF were used as markers of smooth and cardiac muscles, respectively, and GAPDH was detected as an internal control for RNA integrity and loading. (B) Embryonic day 12.5 mouse embryos heterozygous for a targeted HDAC5 allele in which lacZ was inserted in frame with the HDAC5 coding region were stained for lacZ expression. Histological sections were cut and visualized by bright-field microscopy; LacZ staining is indicated in pink. a, atrium; ao, aorta; e, esophagus; t, trachea.
We also examined the expression of HDAC5 in mouse embryos at embryonic day 12.5 by monitoring the expression of a lacZ reporter introduced into the gene for HDAC5 by homologous recombination (8). Expression of the gene for HDAC5 was readily detectable in the smooth muscle layer of the dorsal aorta, as well as in the esophagus and trachea, at this stage (Fig. 3B). The level of expression of the lacZ marker in these smooth muscle tissues was comparable to that in the myocardial layer of the heart (Fig. 3B), where HDAC5 has been shown to regulate cardiac growth and maturation (8).
Interaction of myocardin and p300.
In order to determine whether the stimulation of myocardin activity by p300 resulted from a direct interaction between the proteins, we performed coimmunoprecipitation assays with transiently transfected COS cells with epitope-tagged proteins. As shown in Fig. 4, an interaction between myocardin and p300 was readily detected. With a series of myocardin deletion mutant proteins, we mapped the domains responsible for this protein-protein interaction (Fig. 4A and B).
FIG. 4.

Mapping of the region of myocardin that interacts with p300. (A) Schematic diagram of myocardin and the mutant forms used to map the p300-binding domain. NT, not tested. (B) Coimmunoprecipitation assays. COS cells were transiently transfected with expression vectors encoding FLAG-tagged myocardin deletion mutant proteins and hemagglutinin (HA)-tagged p300. Myocardin proteins were immunoprecipitated (IP) from cell lysates with a monoclonal anti-FLAG antibody, and coimmunoprecipitating HA-tagged p300 was detected by immunoblotting (IB) with a polyclonal anti-HA antibody (top part). The expression of myocardin proteins was revealed by an anti-FLAG antibody (bottom part). NS, nonspecific background. (C) COS cells were transiently transfected with expression vectors encoding the indicated myocardin deletion mutant proteins, and immunoprecipitates were assayed for HAT activity in the presence of purified histones and 3H-labeled acetyl coenzyme A, followed by SDS-PAGE, as described in Materials and Methods.
Myocardin interacts with SRF through a basic and glutamine-rich domain near the N terminus (44, 47). The SAP domain of myocardin is not required for interaction with SRF or for transcriptional activity, but it appears to discriminate among different target genes since its deletion abolishes the responsiveness of some genes, but not others, to myocardin (44). Deletion of N-terminal sequences of myocardin up to amino acid 713, removing the conserved N-terminal, basic, glutamine-rich, and SAP domains, did not affect interaction with p300. However, deletion of sequences between amino acid 713 and the C terminus abolished this interaction. Therefore, the TAD of myocardin, which was sufficient to confer transcriptional responsiveness to p300, was necessary and sufficient for interaction with p300.
We further examined the association of myocardin with HAT activity by immunoprecipitating myocardin from transfected cells and assaying for HAT activity in vitro with purified histones as substrates. As shown in Fig. 4C, HAT activity was associated with myocardin. With a series of myocardin deletion mutant proteins, we found that residues 129 to 713 were devoid of activity, whereas the C-terminal TAD (residues 552 to 935 or 713 to 935) was associated with HAT activity (Fig. 4C). Given that p300 is ubiquitous and interacts with this region of myocardin, it is likely to account, at least in part, for the associated HAT activity, although other HATs could also be involved. Together, these findings suggest that myocardin recruits p300 and its HAT activity to the chromosomal location of smooth muscle genes through a direct protein-protein interaction.
Mapping of the myocardin-binding domain of p300.
To define the region of p300 that interacts with myocardin, we performed GST fusion protein pull-down assays. GST fused to the myocardin TAD (residues 713 to 935) was immobilized on glutathione-agarose beads and incubated with portions of p300 translated in vitro. As shown in Fig. 5, the amino-terminal region of p300 (residues 1 to 670) specifically interacted with myocardin, whereas no interaction was detected between other regions of p300 (residues 671 to 1194 or 1135 to 2414) and myocardin. As a negative control, GST alone or the amino terminus of myocardin (residues 128 to 513) fused to GST was used in the pull-down assays; no p300 binding to either of these proteins was detected (Fig. 5 and data not shown), indicating that the interaction between p300 and the C-terminal TAD of myocardin was specific. Further mapping experiments demonstrated that a region between residues 451 and 670 of p300, which includes the previously identified CREB-binding domain (5), specifically interacted with myocardin (Fig. 5). Together, these data demonstrate that myocardin and p300 directly interact in vivo and in vitro and that this interaction is mediated by the TAD of myocardin and the CREB-binding domain of p300.
FIG. 5.

Mapping of the region of p300 that interacts with myocardin. (A) Schematic diagram of p300 and mutant forms used to map the myocardin-binding domain. CH1, CH2, CH3, cysteine- and histidine-rich domains; BD, bromodomain; NHR, nuclear hormone receptor binding region; GRR, glutamine-rich region. (B) GST pull-down assays. GST-myocardin (amino acids 713 to 935) fusion protein or GST alone was incubated with [35S]methionine-labeled portions of p300 as described in Materials and Methods. Associated proteins were resolved by SDS-PAGE and analyzed by autoradiography (top and middle parts). In the bottom part, 10% of the [35S]methionine-labeled p300 protein was applied directly to the gel to control for input.
E1A blocks the effects of p300 on myocardin-dependent transcription.
The above data demonstrated that p300 was able to stimulate myocardin transcriptional activity through direct protein-protein interaction. In order to test if p300 activity was required for myocardin-dependent gene activation, we used E1A, a p300 antagonist, to block p300 activity. The adenovirus E1A protein can specifically bind to the C-terminal region of p300 and repress its transcriptional activity (1, 4, 17). To investigate whether E1A could disrupt the stimulatory effect of p300 on myocardin-dependent target genes, we performed reporter assays as described above in the presence and absence of an E1A expression plasmid. As shown in Fig. 6A, E1A interfered with myocardin's ability to activate the SM22 promoter, and it blocked activity of myocardin fused to the GAL4 DNA-binding domain. E1A also blocked activation of endogenous smooth muscle genes in 10T1/2 cells by myocardin (Fig. 6B). Similarly, expression of E1A in the A7r5 and PAC-1 SMC lines extinguished the expression of smooth muscle genes (data not shown). These findings support the conclusion that p300 is critical to the promyogenic activity of myocardin.
FIG. 6.

Blockade of myocardin-induced smooth muscle gene expression by E1A. (A) 10T1/2 cells were transiently transfected with expression vectors encoding myocardin and E1A and an SM22-luciferase reporter (left part) or GAL4-myocardin and E1A and a UAS-luciferase reporter (right part), as indicated. (B) 10T1/2 cells were transiently transfected with expression vectors encoding myocardin and E1A, as indicated, and cells were stained for expression of SM α-actin. A representative cell expressing SM α-actin is shown (left part), as is blockade of myocardin-induced SM α-actin expression by E1A (right part). Values are expressed as the relative number of positive cells compared to the number in cultures transfected with myocardin alone, which was assigned a value of 100.
Direct interaction of myocardin and HDAC5 and mapping of the domains that mediate their interaction.
We next investigated whether myocardin and class II HDAC proteins interact by coimmunoprecipitation assays with transfected COS cells. As shown in Fig. 7, myocardin specifically interacted with HDAC4 and HDAC5, whereas another transcriptional repressor, COOH-terminal binding protein (CtBP), did not interact with myocardin (Fig. 7B). Myocardin also did not interact with HDAC1 (data not shown), consistent with the failure of HDAC1 to suppress myocardin activity. Myocardin and HDAC5 colocalized precisely to the same intranuclear regions in transfected COS cells (Fig. 7C), consistent with the possible direct interaction of these two proteins in vivo.
FIG. 7.

Mapping of the region of myocardin that interacts with HDAC5. (A) Schematic diagram of myocardin and the mutant forms used to map the HDAC5-binding domain. (B) Coimmunoprecipitation assays. COS cells were transiently transfected with expression vectors encoding FLAG-tagged myocardin and Myc-tagged HDAC4, HDAC5, and CtBP1. Myc-tagged proteins were immunoprecipitated (IP) from cell lysates with a polyclonal anti-Myc antibody, and coimmunoprecipitating myocardin was detected by immunoblotting (IB) with a monoclonal anti-FLAG antibody (top part). The membrane was reprobed with anti-Myc antibody to reveal the total amount of Myc-tagged proteins (bottom part). (C) Immunostaining of myocardin and HDAC5. COS cells were transiently transfected with expression vectors for FLAG-tagged myocardin and Myc-tagged HDAC5, and the subcellular distribution of the proteins was determined by immunostaining as described in Materials and Methods (top and middle parts). Overlay of the individual staining patterns reveals a precise correlation between the intranuclear distribution of myocardin and that of HDAC5 (bottom part). (D) Coimmunoprecipitation assays. COS cells were transiently transfected with expression vectors encoding FLAG-tagged deletion mutant myocardin proteins and Myc-tagged HDAC5. Myc-tagged HDAC5 was immunoprecipitated from cell lysates with a polyclonal anti-Myc antibody, and coimmunoprecipitating myocardin proteins were detected by immunoblotting with a monoclonal anti-FLAG antibody (top part). The expression levels of HDAC5 (middle part) and myocardin mutant proteins (lower part) were revealed by Western blot assays with the indicated antibodies.
The domains of myocardin and HDAC5 that mediate their interaction were mapped by coimmunoprecipitation assays with a series of myocardin and HDAC5 deletion mutant proteins (Fig. 7A and D). Myocardin deletion mutant proteins lacking N-terminal residues up to amino acid 268 interacted with HDAC5, whereas a mutant protein lacking the first 328 amino acids failed to interact with HDAC5. Deletion mutant proteins lacking residues C terminal to amino acid 348 also retained the ability to interact with HDAC5. Thus, in contrast to p300, which interacted with the TAD of myocardin, HDAC5 interacted with a distinct region near the N terminus (between amino acids 268 and 328). Because the HDAC5-binding region of myocardin overlapped the SRF-binding region, we considered the possibility that HDAC5 might repress myocardin activity by displacing it from SRF. However, HDAC5 did not disrupt the formation of a ternary complex between SRF and myocardin in gel mobility shift assays (data not shown). The ability of HDAC5 to inhibit the activity of GAL4-myocardin on an upstream activating sequence (UAS)-luciferase reporter also discounts such a potential displacement mechanism for repression by HDAC5.
With a series of HDAC5 deletion mutant proteins, we found that mutant proteins lacking sequences N terminal to amino acid 153 retained the ability to interact with myocardin (Fig. 8A and B). However, further deletion to amino acid 201 completely abolished this interaction, suggesting that the region between residues 153 and 201 is essential for HDAC5 to interact with myocardin. Deletions from the C terminus revealed that residues downstream of amino acid 300 are not required for interaction with myocardin (Fig. 8A and B). We conclude that a myocardin-binding domain lies between amino acids 153 and 201 of HDAC5.
FIG. 8.

Mapping of the regions of HDAC5 that interact with myocardin. (A) Schematic diagram of HDAC5 and the mutant forms used to map the myocardin-binding domain. (B) Coimmunoprecipitation assays. COS cells were transiently transfected with expression vectors encoding Myc-tagged HDAC5 deletion mutant proteins and FLAG-tagged myocardin. Myc-tagged HDAC5 deletion mutant proteins were immunoprecipitated (IP) from cell lysates with a polyclonal anti-Myc antibody, and coimmunoprecipitating myocardin was detected by immunoblotting (IB) with a monoclonal anti-FLAG antibody (top parts). The membrane was reprobed with anti-Myc antibody to reveal the total amount of Myc-tagged HDAC5 proteins (bottom parts). (C) COS cells were transiently transfected with expression vectors encoding myocardin and HDAC5 deletion mutant proteins and the SM22 luciferase reporter.
There was a direct correlation between the abilities of HDAC5 deletion mutant proteins to interact with myocardin and repress its activity. Thus, only those HDAC5 deletion mutant proteins that contained amino acids 153 to 201 were able to interact with and repress myocardin. It is notable that the C-terminal region of HDAC5, which contains the catalytic domain, was not required for repression of myocardin activity (Fig. 8C).
Simultaneous interaction of p300 and HDAC5 with myocardin.
The finding that p300 and HDAC5 interact with separate regions of myocardin suggested the possibility that they might simultaneously associate with myocardin. Indeed, in coimmunoprecipitation experiments with FLAG-tagged myocardin, HA-tagged p300, and Myc-tagged HDAC5, we found that both p300 and HDAC5 could be coimmunoprecipitated in the presence, but not in the absence, of myocardin (Fig. 9). Thus, the balance between the stimulatory and repressive influences of p300 and HDAC5, respectively, is likely to dictate the transcriptional activity of myocardin.
FIG. 9.

Simultaneous interaction of myocardin with p300 and HDAC5. COS cells were transiently transfected with expression vectors encoding hemagglutinin (HA)-tagged p300, FLAG-tagged myocardin, and Myc-tagged HDAC5. FLAG-tagged myocardin was immunoprecipitated (IP) from cell lysates with a monoclonal anti-FLAG antibody, and coimmunoprecipitating p300 was detected by immunoblotting (IB) with an anti-HA antibody (top part). The membrane was then stripped and reprobed with anti-Myc antibody to reveal coimmunoprecipitating Myc-tagged HDAC5 proteins (second part from the top). Cell extracts were probed with antibodies against HA, FLAG, and Myc to detect p300, myocardin, and HDAC5, respectively (bottom three parts).
Effects of TSA on myocardin activity.
To further explore the involvement of histone acetylation-deacetylation in the control of myocardin activity, we tested the effect of the HDAC inhibitor TSA on the transcriptional activity of myocardin. As shown in Fig. 10A, TSA enhanced the transcriptional activity of the full-length myocardin protein fused to GAL4. In contrast, TSA treatment did not further enhance the activity of the myocardin TAD (data not shown). TSA also enhanced the expression of the SM22 promoter in the presence of myocardin to a level similar to that of myocardin plus p300 (Fig. 10B). Furthermore, cotransfecting TSA-treated cells with p300 did not increase myocardin activity further (Fig. 10B). Together, these data suggest that the primary function of p300 on myocardin is to relieve the repressive effect of HDAC5 on myocardin. Because the catalytic domain of HDAC5 was not required for repression but HDAC inhibition with TSA stimulated myocardin activity, we conclude that the effects of TSA are likely to be directed at class I HDACs, which can associate with HDAC5 (12, 43).
FIG. 10.

Effects of TSA on myocardin activity. (A) COS cells were transiently transfected with expression vectors encoding GAL4-myocardin fusion protein and the UAS-luciferase reporter. Cells were treated with 0.1 μM TSA after transfection and harvested 48 h later for determination of luciferase activity. (B) Effects of TSA on myocardin and p300 transcriptional activity. COS cells were transiently transfected with expression vectors encoding myocardin, p300, and the SM22-luciferase reporter. Cells were treated with 0.1 μM TSA after transfection and harvested 48 h later for determination of luciferase activity.
Repression of smooth muscle differentiation by HDAC5.
To further investigate the potential involvement of class II HDACs in the suppression of smooth muscle gene expression, we examined the effect of HDAC5 on the expression of smooth muscle genes in the A7r5 and PAC-1 SMC lines. As shown in Fig. 11, HDAC5 reduced the expression of smooth muscle genes in these cell lines. Mutant forms of HDAC5 that were unable to interact with myocardin had no effect on smooth muscle gene expression (data not shown). We conclude that HDAC5 suppresses smooth muscle gene expression by its association with myocardin on smooth muscle target genes.
FIG. 11.
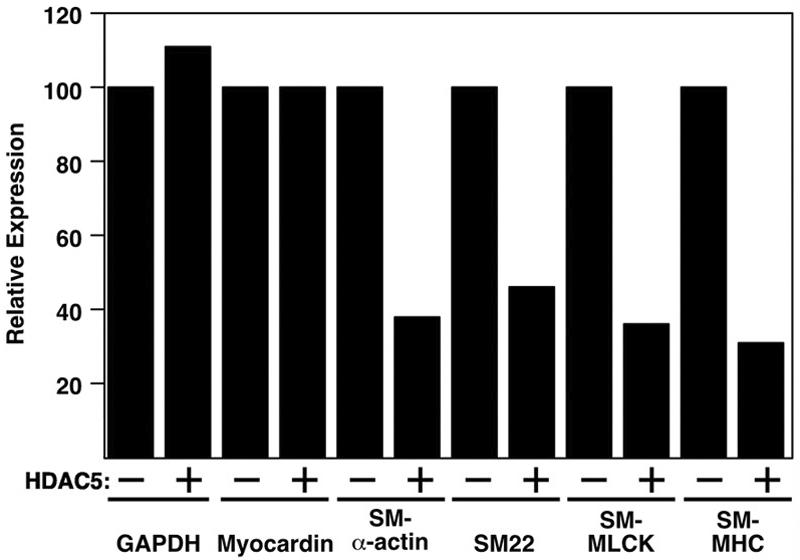
Suppression of smooth muscle gene expression in A7r5 and PAC-1 cells by HDAC5. A7r5 cells were infected with adenovirus encoding LacZ (−) or HDAC5 (+), and smooth muscle gene expression was assayed by RT-PCR. PCR products were quantified by phosphorimager analysis, and values were expressed relative to the level of expression of each transcript in lacZ virus-infected cells. SM-MLCK, SM-myosin light chain kinase; SM-MHC, SM-myosin heavy chain.
DISCUSSION
Differentiation of SMCs is accompanied by acetylation of histones associated with smooth muscle gene promoters (28, 36). However, the mechanisms and transcriptional targets of chromatin-modifying enzymes that influence smooth muscle gene expression have remained elusive. The results of this study demonstrate that myocardin induces the acetylation of histones associated with smooth muscle target genes and that p300 and HDAC5 stimulate and suppress, respectively, the promyogenic transcriptional activity of myocardin. These findings suggest that myocardin modulates the expression of its target genes, at least in part, by recruiting HATs and HDACs with consequent alterations in chromatin acetylation.
Stimulation of myocardin activity by p300.
Our results show that p300 associates specifically with the TAD of myocardin and enhances myocardin activity. Although the closely related HAT CBP has also been shown to interact with SRF to activate c-fos gene expression (37), this interaction is apparently insufficient to activate endogenous smooth muscle genes since CBP is expressed in a wide range of cell types in which smooth muscle genes are not expressed. Similarly, overexpression of p300 in 10T1/2 cells did not result in smooth muscle gene expression in the absence of myocardin, suggesting that additional factors are required. It is also important to note that both SRF and p300/CBP are ubiquitous, but smooth muscle promoters are unacetylated in non-SMCs, which further demonstrates that the potential interaction of SRF and p300/CBP is unable to modify chromatin associated with smooth muscle genes to a transcriptionally permissive state. In contrast, myocardin is able to induce the acetylation of nucleosomal histones associated with its target genes, which presumably is mediated by its recruitment of p300. We speculate that activation of the smooth muscle gene program may require a precise configuration of transcriptional activators on the corresponding promoters and that myocardin is required to orchestrate such a configuration. Given that myocardin preferentially activates promoters with two or more CArG boxes (44) and can form homodimers or heterodimers with myocardin-related transcription factors (MRTFs) (48) through its leucine zipper domain, perhaps it selectively activates smooth muscle target genes by bridging SRF bound at multiple sites.
p300 has been shown to stimulate transcription through various mechanisms, including the bridging of sequence-specific transcriptional activators to the basal transcription machinery, the recruitment of accessory factors with HAT activity, and the direct acetylation of histones and/or other transcription factors via its intrinsic HAT activity (5, 13). Any of these mechanisms could contribute to the synergistic activation of smooth muscle gene expression by myocardin and p300.
p300 associates with other transcriptional regulators primarily through three domains (5, 13). These include the amino-terminal zinc finger domain CH1, which interacts with TBP, RXR, STAT2, and others; the CREB-binding domain in the middle of the protein, which facilitates the interaction with CBP, YY1, c-Jun, BRCA1, and others; and the carboxyl-terminal zinc finger domain CH3, which mediates the interaction between p300 and GATA1, GATA4, MyoD, E1A, and others. Our results identify the CREB-binding domain of p300 as the region that interacts with myocardin. It will be of interest to determine whether other p300-associated proteins, such as CBP and YY-1, compete with myocardin for interaction with this domain. It is notable in this regard that YY-1, which functions as a transcriptional activator or repressor, binds to the CArG box (14, 22, 32).
Although it is not sufficient to activate muscle gene expression by itself, p300 is essential for embryogenesis and cardiac muscle differentiation (51). Because of an early embryonic lethality, the involvement of p300 in smooth and skeletal muscle development in vivo remains to be determined. Interestingly, E1A, which specifically inhibits p300, has been shown to suppress skeletal muscle differentiation by perturbing the activity of the MyoD family of myogenic transcription factors (49). Our results show that E1A represses myocardin activity and smooth muscle gene expression, further implicating p300 in smooth muscle gene activation. The domain of p300 that associates with E1A is distinct from the myocardin-binding region, suggesting that E1A does not disrupt the smooth muscle gene program by displacing myocardin from p300. E1A has previously been shown to block p300 function by inhibiting its HAT activity (1, 4, 17), suggesting that the HAT activity of p300 is directly involved in the regulation of myocardin transcriptional activity.
Repression of myocardin activity and smooth muscle differentiation by HDAC5.
In contrast to the interaction between myocardin and p300, which stimulates myocardin activity, the association of myocardin and class II HDACs strongly inhibits myocardin activity. Suppression requires interaction of HDAC5 with myocardin, which is mediated by a segment in the N-terminal extension of HDAC5 that tethers it to the N-terminal region of myocardin. Surprisingly, however, the catalytic domain of HDAC5 is not required for repression of myocardin activity. The N-terminal domain of class II HDACs has also been shown to repress myocyte enhancer factor 2 activity independently of the catalytic domain (12, 40, 55, 56). This may be attributable to the interaction of the N-terminal region with a variety of corepressors, such as CtBP (56), as well as class I HDACs (12, 43).
The TAD of myocardin (residues 713 to 935) is approximately 50-fold more active than the full-length protein when fused with a GAL4 DNA-binding domain (44), suggesting the presence of inhibitory factors associated with the N terminus of myocardin. The association of HDAC5 with the N terminus of myocardin may contribute to the repressive influence of the N-terminal sequences on the activity of the myocardin TAD.
The MADS box of SRF has recently been reported to interact with HDAC4, with resultant repression of SRF activity (9). While such an interaction could contribute to the repressive effect of HDAC5 on myocardin activity, the ability of HDAC5 to repress transcriptional activity of a GAL4-myocardin protein demonstrates that SRF is not required for repression by HDAC5. The facts that HDAC5 and -4 directly interact with myocardin and such direct association is required for the repression of myocardin further support our conclusion that HDAC5-mediated repression of smooth muscle gene expression is the result of direct repression of myocardin transcriptional activity, not through repression of other factors. Moreover, we believe that the interaction between HDAC4 and SRF is substantially weaker than that between HDAC4 and myocardin because in our experiments it was undetectable under conditions in which HDAC4-myocardin interactions were readily detectable (data not shown).
Recently, MyoD, a master transcriptional regulator of skeletal muscle differentiation, was shown to associate with p300 and HDAC1 (11, 27, 39, 53). MyoD apparently associates with HDAC1 in undifferentiated myoblasts, where it is kept in a silent state. However, when myoblasts begin to differentiate, MyoD dissociates from HDAC1 and eventually partners with HAT/pCAF to activate skeletal myogenesis (27). Our findings suggest that similar mechanisms operate during smooth muscle differentiation, with myocardin rather than MyoD serving as the target of positive and negative chromatin remodeling enzymes.
Expression of HDACs in smooth muscle.
Consistent with their potential involvement in SMC differentiation, multiple class I and II HDACs are expressed in the A7r5 SMC line. Our results also document robust expression of HDAC5 in the smooth muscle layer of the developing aorta during mouse embryogenesis. Because myocardin target genes are expressed in A7r5 cells and in the developing aorta at a time when HDAC5 is also expressed, the repressive influence of HDAC activity on myocardin must be overcome by counterregulatory mechanisms in these settings. Given the sensitivity of class II HDACs to calcium signals (29), it is interesting to speculate that such signaling systems may be activated in A7r5 cells and in the developing aorta, thereby favoring the promyogenic influence of p300 (or other HATs) on myocardin.
It is also notable that HDAC5 null mice are viable and show no overt SMC phenotype (8). Thus, we favor the interpretation that HDAC5 may influence the onset of SMC differentiation and/or participate in phenotypic remodeling of SMCs, but its repressive influence seems unlikely to be essential for homeostasis of SMCs. It is also conceivable that other class II HDACs mask potential functions of HDAC5.
Modularity of myocardin.
The antagonistic effects of p300 and HDAC5 on myocardin activity do not appear to result from direct competition between those proteins for myocardin interaction because p300 and HDAC5 bind to myocardin at different regions (Fig. 12). This modular structure potentially enables myocardin to recruit HDAC5 and/or p300 to CArG box-containing promoters while being tethered to SRF on DNA. Once in the vicinity of these control regions, HDAC5 and p300 can potentially exert negative or positive effects, respectively, on gene expression. Modulation of the activities of class II HDACs and p300 in response to intracellular signals may provide additional specificity to their effects on myocardin target genes.
FIG. 12.

Schematic diagram of protein interaction domains of myocardin. Domains in myocardin for SRF, HDACs, and p300 interactions are indicated. NTD, N-terminal domain; ++, basic domain; Q, glutamine-rich domain; LZ, leucine zipper.
Interestingly, the inhibitory effects of HDAC5 on myocardin appear to be dominant, as overexpression of p300 cannot completely overcome the repressive influence of HDAC5 on myocardin activity. We propose that the HDAC-mediated repression is probably the default pathway, whereas p300-mediated activation may be used only when target genes need to be activated.
Recently, we demonstrated that the ability of myocardin to activate smooth muscle gene expression can be regulated through competition between myocardin and Elk-1, a ternary complex factor that interacts with the same region of SRF as myocardin but represses SRF-dependent transcription (47). Unlike the inhibition by HDAC5, which did not affect myocardin/SRF association, Elk-1 represses myocardin-dependent smooth muscle gene expression by preventing the association of myocardin with SRF.
Given the fact that myocardin and the other two MRTFs, MRTF-A and -B, have the same structural organization as the conserved N-terminal, basic, Q-rich, and SAP domains, as well as the C-terminal TAD (46), it is likely that MRTFs are also targets for p300 and HDACs. However, unlike myocardin, MRTF-A and -B are ubiquitously expressed during embryogenesis and adulthood. The specificity and biological significance underlying the regulation of their transcriptional activity by p300 and HDACs remains to be determined.
Implications for cardiovascular development and disease.
The modulation of myocardin activity by HATs and HDACs is likely to play important roles in regulating smooth and cardiac muscle gene expression during disease. For example, numerous disease states, such as atherosclerosis, asthma, hypertension, and restenosis following angioplasty, are accompanied by the dedifferentiation and inappropriate proliferation of SMCs. In addition to its role in smooth muscle gene expression, myocardin has been implicated in cardiac gene expression on the basis of the ability of a dominant negative mutant form of myocardin to disrupt heart formation in Xenopus embryos (44). Myocardin is also a potent activator of the ANF promoter, which is activated in the heart in response to stress signals.
Several recent studies have demonstrated the importance of p300 and class II HDACs for hypertrophic growth of cardiac myocytes, which is accompanied by activation of a fetal gene program (8, 15, 16, 50, 54). The calcium-responsive phosphatase calcineurin and calcium/calmodulin-dependent protein kinase are powerful transducers of hypertrophic stimuli in cardiac myocytes and act by stimulating the activity of p300 and repressing the activity of class II HDACs, with consequent activation of stress-responsive cardiac genes such as ANF, a myocardin target gene (reviewed in reference 29). Thus, it will be of interest to determine whether p300 and class II HDACs influence cardiac gene expression via their influences on myocardin activity during cardiac development and disease.
Acknowledgments
We thank Cheol Yong Choi, Tso-Pang Yao, Lishan Su, and Wei Gu for reagents.
E.N.O. was supported by grants from the National Institutes of Health, the Donald W. Reynolds Foundation, and the Texas Advanced Technology Program. D.-Z.W. is a Basil O'Connor Scholar of the March of Dimes Birth Defects Foundation and was supported by grants from the National Institutes of Health, the Muscular Dystrophy Association, and the American Heart Association.
REFERENCES
- 1.Arany, Z., W. R. Sellers, D. M. Livingston, and R. Eckner. 1994. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell 77:799-800. [DOI] [PubMed] [Google Scholar]
- 2.Aravind, L., and E. V. Koonin. 2000. SAP—a putative DNA-binding motif involved in chromosomal organization. Trends Biochem. Sci. 25:112-114. [DOI] [PubMed] [Google Scholar]
- 3.Black, B. L., and E. N. Olson. 1998. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu. Rev. Cell Dev. Biol. 14:167-196. [DOI] [PubMed] [Google Scholar]
- 4.Chakravarti, D., V. Ogryzko, H. Y. Kao, A. Nash, H. Chen, Y. Nakatani, and R. M. Evans. 1999. A viral mechanism for inhibition of p300 and PCAF acetyltransferase activity. Cell 96:393-403. [DOI] [PubMed] [Google Scholar]
- 5.Chan, H. M., and N. B. La Thangue. 2001. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 114:2363-2373. [DOI] [PubMed] [Google Scholar]
- 6.Chang, D. F., N. S. Belaguli, D. Iyer, W. B. Roberts, S. P. Wu, X. R. Dong, J. G. Marx, M. S. Moore, M. C. Beckerle, M. W. Majesky, and R. J. Schwartz. 2003. Cysteine-rich LIM-only proteins CRP1 and CRP2 are potent smooth muscle differentiation cofactors. Dev. Cell 4:107-118. [DOI] [PubMed] [Google Scholar]
- 7.Chang, P. S., L. Li, J. McAnally, and E. N. Olson. 2001. Muscle specificity encoded by specific serum response factor-binding sites. J. Biol. Chem. 276:17206-17212. [DOI] [PubMed] [Google Scholar]
- 8.Chang, S., T. A. McKinsey, C. L. Zhang, J. A. Richardson, J. Hill, and E. N. Olson. 2004. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol. Cell. Biol. 24:8467-8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8a.Chen, J., C. M. Kitchen, J. W. Streb, and J. M. Miano. 2002. Myocardin: a component of a molecular swith for smooth muscle cell differentiation. J. Mol. Cell. Cardiol. 34:1345-1356. [DOI] [PubMed] [Google Scholar]
- 9.Davis, F. J., M. Gupta, B. Camoretti-Mercado, R. J. Schwartz, and M. P. Gupta. 2003. Calcium/calmodulin-dependent protein kinase activates serum response factor transcription activity by its dissociation from histone deacetylase, HDAC4. Implications in cardiac muscle gene regulation during hypertrophy. J. Biol. Chem. 278:20047-20058. [DOI] [PubMed] [Google Scholar]
- 10.Du, K. L., H. S. Ip, J. Li, M. Chen, F. Dandre, W. Yu, M. M. Lu, G. K. Owens, and M. S. Parmacek. 2003. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol. Cell. Biol. 23:2425-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eckner, R., T. P. Yao, E. Oldread, and D. M. Livingston. 1996. Interaction and functional collaboration of p300/CBP and bHLH proteins in muscle and B-cell differentiation. Genes Dev. 10:2478-2490. [DOI] [PubMed] [Google Scholar]
- 12.Fischle, W., F. Dequiedt, M. J. Hendzel, M. G. Guenther, M. A. Lazar, W. Voelter, and E. Verdin. 2002. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 9:45-57. [DOI] [PubMed] [Google Scholar]
- 13.Goodman, R. H., and S. Smolik. 2000. CBP/p300 in cell growth, transformation, and development. Genes Dev. 14:1553-1577. [PubMed] [Google Scholar]
- 14.Gualberto, A., D. LePage, G. Pons, S. L. Mader, K. Park, M. L. Atchison, and K. Walsh. 1992. Functional antagonism between YY1 and the serum response factor. Mol. Cell. Biol. 12:4209-4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gusterson, R., B. Brar, D. Faulkes, A. Giordano, J. Chrivia, and D. Latchman. 2002. The transcriptional co-activators CBP and p300 are activated via phenylephrine through the p42/p44 MAPK cascade. J. Biol. Chem. 277:2517-2524. [DOI] [PubMed] [Google Scholar]
- 16.Gusterson, R. J., E. Jazrawi, I. M. Adcock, and D. S. Latchman. 2003. The transcriptional co-activators CREB-binding protein (CBP) and p300 play a critical role in cardiac hypertrophy that is dependent on their histone acetyltransferase activity. J. Biol. Chem. 278:6838-6847. [DOI] [PubMed] [Google Scholar]
- 17.Hamamori, Y., V. Sartorelli, V. Ogryzko, P. L. Puri, H. Y. Wu, J. Y. Wang, Y. Nakatani, and L. Kedes. 1999. Regulation of histone acetyltransferases p300 and PCAF by the bHLH protein twist and adenoviral oncoprotein E1A. Cell 96:405-413. [DOI] [PubMed] [Google Scholar]
- 18.Hines, W. A., J. Thorburn, and A. Thorburn. 1999. A low-affinity serum response element allows other transcription factors to activate inducible gene expression in cardiac myocytes. Mol. Cell. Biol. 19:1841-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jenuwein, T., and C. D. Allis. 2001. Translating the histone code. Science 293:1074-1080. [DOI] [PubMed] [Google Scholar]
- 20.Kawahara, K., S. Watanabe, T. Ohshima, Y. Soejima, T. Oishi, S. Aratani, M. Nakata, M. Shibata, K. Inoue, T. Amano, R. Fujii, K. Yanai, M. Hagiwara, A. Fukamizu, I. Maruyama, and T. Nakajima. 1999. Hypernuclear acetylation in atherosclerotic lesions and activated vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 266:417-424. [DOI] [PubMed] [Google Scholar]
- 21.Kim, S., H. S. Ip, M. M. Lu, C. Clendenin, and M. S. Parmacek. 1997. A serum response factor-dependent transcriptional regulatory program identifies distinct smooth muscle cell sublineages. Mol. Cell. Biol. 17:2266-2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee, T. C., Y. Shi, and R. J. Schwartz. 1992. Displacement of BrdUrd-induced YY1 by serum response factor activates skeletal alpha-actin transcription in embryonic myoblasts. Proc. Natl. Acad. Sci. USA 89:9814-9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li, L., Z. Liu, B. Mercer, P. Overbeek, and E. N. Olson. 1997. Evidence for serum response factor-mediated regulatory networks governing SM22α transcription in smooth, skeletal, and cardiac muscle cells. Dev. Biol. 187:311-321. [DOI] [PubMed] [Google Scholar]
- 24.Li, L., J. M. Miano, B. Mercer, and E. N. Olson. 1996. Expression of the SM22α promoter in transgenic mice provides evidence for distinct transcriptional regulatory programs in vascular and visceral smooth muscle cells. J. Cell Biol. 132:849-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li, S., D. Wang, Z. Wang, J. Richardson, and E. N. Olson. 2003. The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc. Natl. Acad. Sci. USA 100:9366-9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu, J., T. A. McKinsey, C. L. Zhang, and E. N. Olson. 2000. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol. Cell 6:233-244. [DOI] [PubMed] [Google Scholar]
- 27.Mal, A., and M. L. Harter. 2003. MyoD is functionally linked to the silencing of a muscle-specific regulatory gene prior to skeletal myogenesis. Proc. Natl. Acad. Sci. USA 100:1735-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manabe, I., and G. K. Owens. 2001. Recruitment of serum response factor and hyperacetylation of histones at smooth muscle-specific regulatory regions during differentiation of a novel P19-derived in vitro smooth muscle differentiation system. Circ. Res. 88:1127-1134. [DOI] [PubMed] [Google Scholar]
- 29.McKinsey, T. A., and E. N. Olson. 2004. Cardiac histone acetylation—therapeutic opportunities abound. Trends Genet. 20:206-213. [DOI] [PubMed] [Google Scholar]
- 30.McKinsey, T. A., C. L. Zhang, J. Lu, and E. N. Olson. 2000. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408:106-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miano, J. M. 2003. Serum response factor: toggling between disparate programs of gene expression. J. Mol. Cell. Cardiol. 35:577-593. [DOI] [PubMed] [Google Scholar]
- 32.Natesan, S., and M. Gilman. 1995. YY1 facilitates the association of serum response factor with the c-fos serum response element. Mol. Cell. Biol. 15:5975-5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naya, F. J., C. Wu, J. A. Richardson, P. Overbeek, E. N. Olson. 1999. Transcriptional activity of MEF2 during mouse embryogenesis monitored with a MEF2-dependent transgene. Development 126:2045-2052. [DOI] [PubMed] [Google Scholar]
- 34.Norman, C., M. Runswick, R. Pollock, and R. Treisman. 1988. Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell 55:989-1003. [DOI] [PubMed] [Google Scholar]
- 35.Ogryzko, V. V., R. L. Schiltz, V. Russanova, B. H. Howard, and Y. Nakatani. 1996. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87:953-959. [DOI] [PubMed] [Google Scholar]
- 36.Qiu, P., and L. Li. 2002. Histone acetylation and recruitment of serum responsive factor and CREB-binding protein onto SM22 promoter during SM22 gene expression. Circ. Res. 90:858-865. [DOI] [PubMed] [Google Scholar]
- 37.Ramirez, S., S. Ait-Si-Ali, P. Robin, D. Trouche, A. Harel-Bellan, and S. Ait Si Ali. 1997. The CREB-binding protein (CBP) cooperates with the serum response factor for transactivation of the c-fos serum response element. J. Biol. Chem. 272:31016-31021. [DOI] [PubMed] [Google Scholar]
- 38.Reecy, J., N. S. Belaguli., and R. J. Schwartz. 1998. SRF/homeobox protein interactions, p. 273-290. In R. Harvey and N. Rosenthal (ed.), Heart development. Academic Press, San Diego, Calif.
- 39.Sartorelli, V., J. Huang, Y. Hamamori, and L. Kedes. 1997. Molecular mechanisms of myogenic coactivation by p300: direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol. Cell. Biol. 17:1010-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sparrow, D. B., E. A. Miska, E. Langley, S. Reynaud-Deonauth, S. Kotecha, N. Towers, G. Spohr, T. Kouzarides, and T. J. Mohun. 1999. MEF-2 function is modified by a novel co-repressor, MITR. EMBO J. 18:5085-5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sprenkle, A. B., S. F. Murray, and C. C. Glembotski. 1995. Involvement of multiple cis elements in basal- and alpha-adrenergic agonist-inducible atrial natriuretic factor transcription. Roles for serum response elements and an SP-1-like element. Circ. Res. 77:1060-1069. [DOI] [PubMed] [Google Scholar]
- 42.Treisman, R. 1994. Ternary complex factors: growth factor regulated transcriptional activators. Curr. Opin. Genet. Dev. 4:96-101. [DOI] [PubMed] [Google Scholar]
- 43.Verdin, E., F. Dequiedt, and H. G. Kasler. 2003. Class II histone deacetylases: versatile regulators. Trends Genet. 19:286-293. [DOI] [PubMed] [Google Scholar]
- 44.Wang, D., P. S. Chang, Z. Wang, L. Sutherland, J. A. Richardson, E. Small, P. A. Krieg, and E. N. Olson. 2001. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105:851-862. [DOI] [PubMed] [Google Scholar]
- 45.Wang, D. Z., and E. N. Olson. 2004. Control of smooth muscle development by the myocardin family of transcriptional coactivators. Curr. Opin. Genet. Dev. 14:558-566. [DOI] [PubMed] [Google Scholar]
- 46.Wang, D. Z., S. Li, D. Hockemeyer, L. Sutherland, Z. Wang, G. Schratt, J. A. Richardson, A. Nordheim, and E. N. Olson. 2002. Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc. Natl. Acad. Sci. USA 99:14855-14860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang, Z., D. Z. Wang, D. Hockemeyer, J. McAnally, A. Nordheim, and E. N. Olson. 2004. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 428:185-189. [DOI] [PubMed] [Google Scholar]
- 48.Wang, Z., D. Z. Wang, G. C. Pipes, and E. N. Olson. 2003. Myocardin is a master regulator of smooth muscle gene expression. Proc. Natl. Acad. Sci. USA 100:7129-7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Webster, K. A., G. E. Muscat, and L. Kedes. 1988. Adenovirus E1A products suppress myogenic differentiation and inhibit transcription from muscle-specific promoters. Nature 332:553-557. [DOI] [PubMed] [Google Scholar]
- 50.Yanazume, T., K. Hasegawa, T. Morimoto, T. Kawamura, H. Wada, A. Matsumori, Y. Kawase, M. Hirai, and T. Kita. 2003. Cardiac p300 is involved in myocyte growth with decompensated heart failure. Mol. Cell. Biol. 23:3593-3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yao, T. P., S. P. Oh, M. Fuchs, N. D. Zhou, L. E. Ch'ng, D. Newsome, R. T. Bronson, E. Li, D. M. Livingston, and R. Eckner. 1998. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 93:361-372. [DOI] [PubMed] [Google Scholar]
- 52.Yoshida, T., S. Sinha, F. Dandre, B. R. Wamhoff, M. H. Hoofnagle, B. E. Kremer, D. Z. Wang, E. N. Olson, and G. K. Owens. 2003. Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ. Res. 92:856-864. [DOI] [PubMed] [Google Scholar]
- 53.Yuan, W., G. Condorelli, M. Caruso, A. Felsani, and A. Giordano. 1996. Human p300 protein is a coactivator for the transcription factor MyoD. J. Biol. Chem. 271:9009-9013. [DOI] [PubMed] [Google Scholar]
- 54.Zhang, C. L., T. A. McKinsey, S. Chang, C. L. Antos, J. A. Hill, and E. N. Olson. 2002. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110:479-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang, C. L., T. A. McKinsey, J. R. Lu, and E. N. Olson. 2001. Association of COOH-terminal binding protein (CtBP) and MEF2-interacting transcription repressor (MITR) contributes to transcriptional repression of the MEF2 transcription factor. J. Biol. Chem. 276:35-39. [DOI] [PubMed] [Google Scholar]
- 56.Zhang, C. L., T. A. McKinsey, and E. N. Olson. 2001. The transcriptional corepressor MITR is a signal-responsive inhibitor of myogenesis. Proc. Natl. Acad. Sci. USA 98:7354-7359. [DOI] [PMC free article] [PubMed] [Google Scholar]