

Summary
Circular RNAs (circRNAs) are abundant and evolutionarily conserved RNAs of largely unknown function. Here, we show that a subset of circRNAs is translated in vivo. By performing ribosome footprinting from fly heads, we demonstrate that a group of circRNAs is associated with translating ribosomes. Many of these ribo-circRNAs use the start codon of the hosting mRNA, are bound by membrane-associated ribosomes, and have evolutionarily conserved termination codons. In addition, we found that a circRNA generated from the muscleblind locus encodes a protein, which we detected in fly head extracts by mass spectrometry. Next, by performing in vivo and in vitro translation assays, we show that UTRs of ribo-circRNAs (cUTRs) allow cap-independent translation. Moreover, we found that starvation and FOXO likely regulate the translation of a circMbl isoform. Altogether, our study provides strong evidence for translation of circRNAs, revealing the existence of an unexplored layer of gene activity.
Keywords: circRNA, translation, muscleblind, cap-independent
Graphical Abstract
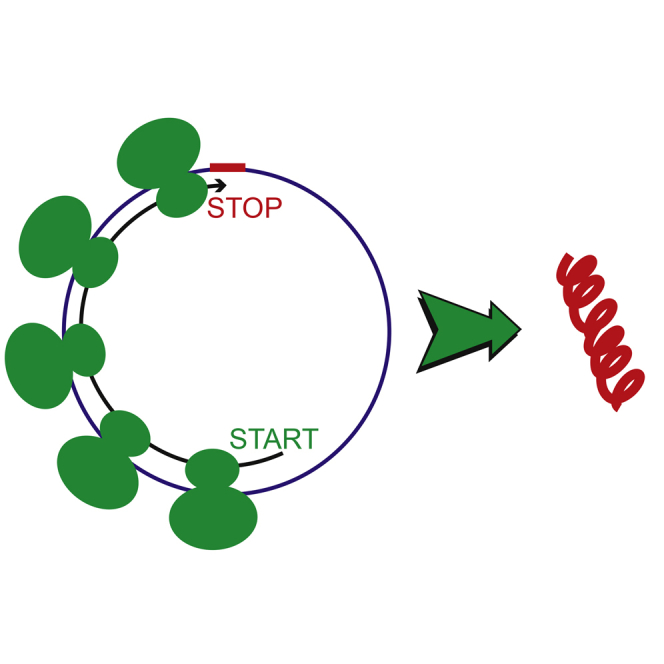
Highlights
-
•
Specific circRNAs are associated with translating ribosomes
-
•
Ribosome footprinting reads match termination codon signature for circMbl
-
•
circMbl3-derived protein is detected by mass spectrometry
-
•
circRNAs are translated in vitro and in vivo in a cap-independent manner
Pamudurti et al. show that a subset of circRNAs is translated. These circRNAs generally share the start codon with the hosting RNA, encode proteins with specific protein domains, and are translated in a cap-independent manner.
Introduction
Circular RNAs (circRNAs) in animals are a large class of particularly stable RNAs produced by circularization of specific exons (Jeck and Sharpless, 2014, Wang et al., 2014). In vivo, circRNAs are generated by the spliceosome via backsplicing: the 3′ end of an exon is covalently linked to the 5′ end of an upstream exon (Ashwal-Fluss et al., 2014, Starke et al., 2015). Most circRNAs originate from protein-coding genes and contain complete exons. Particularly in the fly brain (Westholm et al., 2014) and mammalian neuronal and muscle tissues (Rybak-Wolf et al., 2015), circRNAs are highly abundant and evolutionarily conserved.
The function of only a few circRNAs has been elucidated. At the molecular level, CDR1as acts as a microRNA (miRNA) sponge (Hansen et al., 2013, Memczak et al., 2013). Other circRNAs can regulate the function of RNA-binding proteins (Ashwal-Fluss et al., 2014) or the transcription rate of its host gene (Li et al., 2015). However, the functions of thousands of described circRNAs (Glažar et al., 2014) remain unknown.
Although it has been shown that in principle circRNAs can be translated in vitro and in vivo (Abe et al., 2015, Chen and Sarnow, 1995, Li and Lytton, 1999, Wang and Wang, 2015), the possibility that circRNAs are endogenously translated has so far been only indirectly tested (Guo et al., 2014, Jeck and Sharpless, 2014). In eukaryotes, the canonical translation process begins with the binding of the open pre-initiation complex, which contains the small ribosomal subunit (Aitken and Lorsch, 2012, Sonenberg and Hinnebusch, 2009). Translation-competent mRNAs are effectively circularized because of interaction between proteins that bind the cap and the polyA regions (Aitken and Lorsch, 2012, Sonenberg and Hinnebusch, 2009). The small ribosomal subunit then scans the mRNA until encountering a start codon. This leads to the recruitment of the 60S ribosomal subunit. In addition ribosomes can be recruited to an internal start codon by a mechanism that depends on an internal ribosome entry site (IRES). Translation of several viral proteins is dependent on IRES, and cellular IRESs have been identified in many organisms, including humans, although their mechanism of action remains controversial (Jackson, 2013, Weingarten-Gabbay et al., 2016).
Results
A Subset of Drosophila CircRNAs Is Associated with Translating Ribosomes
To determine the specific coding potential of Drosophila circRNAs, we identified those with open reading frames (ORFs) across the backspliced junction. We found that most circRNAs have coding potential (see list in Table S1). However, we obtained similar results with a set of control exons (data not shown).
After annotating the Drosophila circRNA ORFs (cORFs), we searched for evidence of their translation. We utilized previously published ribosome footprinting (RFP) datasets (Aspden et al., 2014, Dunn et al., 2013, Kronja et al., 2014, Miettinen and Björklund, 2015) and searched for reads across the circRNA-specific junctions. Using a database of backsplice and canonical junctions (Figure 1A), we identified 37 circRNAs (referred hereafter as ribo-circRNAs) with at least one specific RFP read (Figure 1B; Table S2). The association of these ribo-circRNAs with the ribosomes seems to be more than a chance event because: (1) ribo-circRNAs contain circRNAs expressed at various levels (Figure 1C); and (2) despite the low RFP counts, some ribo-circRNAs are detected on the same scale as their hosting mRNA (Figure S1A; Table S2), suggesting comparable translational rates. Moreover, for a few circRNAs, we found more RFP reads covering the exons containing the circRNAs than other exons (Figures 1D and 1E). In this experiment, the polysomes were separated according to size before the footprinting assay. Indeed, the enrichment of RFP reads in specific circRNA-containing exons was prominent only in RNAs associated with light polysomal fractions. This was highly significant for circTai (7.2-fold enrichment, p < 1 × 10−54; Figure 1D) and circCt (2.6-fold enrichment, p < 1 × 10−31; Figure 1E).
Figure 1.

A Subset of CircRNAs Is Associated with Translating Ribosomes
(A) Approach used to detect circRNA reads in RFP data.
(B) Datasets utilized and ribo-circRNAs detected.
(C) circRNA expression was plotted against their ranking in these datasets. Red dots indicate ribo-circRNAs.
(D and E) Integrated genome browser (IGV) snapshots of small and large RFP datasets for tai (D) and ct (E) show that more RFP reads are in the circularized exons, mainly in the lighter polysomes (red).
See Table S1.
Minigenes of Ribo-CircRNAs Produce Proteins
We then generated intron-exon-intron minigenes for expression of specific circRNAs in Drosophila S2 cells. We focused on the ribo-circRNAs circMbl, circCdi, and circPde8 and used as negative controls circRNAs for which we did not observe RFP reads in this cell type: circHaspin and circCamK1. We engineered the minigenes to express a V5-tagged protein in case of a circularization event (Figure 2A). Cells transfected with circMblV5, circCdiV5, or circPde8V5 minigenes produced V5-tagged proteins, whereas those transfected with circHaspinV5 or circCamK1V5 minigenes failed to express a V5-tagged protein (Figure 2B). The two V5-tagged protein products in the cells transfected with the circPde8V5 minigene are due to the translation of two proteins generated from the circRNA with or without an internal intron. We detected a non-specific band above the circCdi protein (Figure 2B).
Figure 2.

CircRNAs Can Produce Proteins as Tagged Minigenes
(A) Ribo-circRNA tagging strategy. MT, metallothionein promoter.
(B) circMbl V5, Pde8 V5, and Cdi V5 minigenes produce proteins of their expected size (red arrows). GFP was co-transfected.
(C) circMbl minigenes produce both circMbl and linear concatemers when transfected into Drosophila S2 cells. Left: EtBr staining. Right: northern blot using a probe directed against the circMbl backsplice junction.
(D) Top: scheme of the minigene expressing a split Cherry molecule under the control of the circMbl cUTR. Bottom: representative picture of Drosophila S2 cells transfected with this minigene.
(E) Western blot from Drosophila S2 cells transfected with the specified circMbl V5 minigenes.
(F) Northern blot using a probe directed against the circMbl backsplice junction. Samples were prepared from heads of control (actin-gal4/+) or circMbl overexpression (OE) flies (actin-gal4; UAS-circMbl). Left: EtBr staining. Right: northern blot. The asterisk indicates an unknown RNA species that is detected by the probe, and it is resistant to RNaseR.
(G) RT-PCR analysis of control and circMbl OE flies. Gene expression was normalized to rp49 and 28S RNAs. Mean ± SD (n = 2 for control and n = 3 for circMbl samples).
(H) Western blot of control or circMbl OE flies utilizing the anti-MBL or anti-tubulin antibodies.
See Figures S1 and S2.
A protein of the expected size was also observed when we utilized an anti-MBL antibody (see “Input” in Figure S1B). Moreover, the MBL-positive and V5-positive western signals originate from the same protein (Figure S1B). We detected stronger anti-MBL immunoreactivity in cells that expressed the circMbl minigenes without the V5-tag sequence, despite similar amounts of circMbl RNA (data not shown). This result suggests that several RNA isoforms could originate from the circMblV5 minigene. Therefore, we visualized the RNA by northern blot (Figure S1C). Indeed, we found that circMbl and circMblV5 minigenes generate linear concatemers that can also potentially produce the observed peptides (Figures 2C, S1D, and S1E). The circMbl minigene produces more protein than the circMblV5 minigene (see “Input” in Figure S1B) despite much lower levels of concatemers, suggesting that the circRNA is the main source of the detected protein. The circPde8V5 minigene construct also produces substantial amounts of linear concatemers (Figures S1F and S1G). Interestingly, other minigenes (i.e., circCamK1V5 and circCdiV5; Figures S1G–S1I) produce lower levels or no concatemers.
To determine whether the produced proteins are generated from the circRNA, we determined whether minigene-derived circRNA molecules are associated with translating ribosomes. We co-transfected Drosophila S2 cells with the circMblV5, circCdiV5, and circCamKIV5 minigenes, a plasmid driving expression of GFP, and a plasmid driving expression of a circRNA in which a split Cherry protein is under the control of the circMbl cUTR (UTR of circMbl; all the sequences in circMbl that are not within the putative ORF; see Figure 2D). This Cherry protein construct contains the CamKI flanking intronic sequences that drive very efficient RNA circularization, with no detectable concatemers (Figures 2D and S1G–S1I). Cells transfected with the split Cherry constructs display strong expression of CHERRY protein (Figure 2D). We then performed sucrose density gradient centrifugation. Both GFP and an endogenous mRNA (rp49) were strongly associated with monosomes and polysomes (Figures S2A and S2B). Most circCamKI RNAs co-migrate with the lighter fractions, whereas most of the circMbl, circCdi, and circCherry RNA co-migrates with the monosome and polysomal fractions (Figure S2A and S2B). We then treated the RNA from the different polysome gradient fractions with RNaseR. This assay confirmed that the RNAs derived from the circMblV5, circCherry minigenes, and to a lesser extent, the circCdiV5 minigene are indeed circular (Figures S2C–S2E). RNaseR efficiently degraded rp49 mRNA (Figure S2F).
Importantly, the expression of the V5-tagged protein from the circMblV5 minigene depends on the start codon in the circMbl exon (Figure 2E). In addition, mutation of the splice site resulted in the disappearance of anti-V5 immunoreactivity and in the generation of a slightly longer protein because of a stop codon in the downstream intron (Figure 2E). Indeed, mutation of the 5′ splice site (5′ss) leads to the use of a cryptic 5′ splice site and the generation of a new type of circRNA molecule (circMblV5′ss; Figures S1D and S1E), which is likely the template for the longer protein product observed in Figure 2E. From these experiments we conclude that a substantial fraction of the V5-tagged proteins detected in Figure 2B originates from circRNA molecules. We also concluded that the intron-exon-intron minigenes should be used with caution.
To extend these findings to an in vivo system, we generated transgenic flies expressing a circMbl minigene. To minimize formation of concatemers, we did not tag the circMbl-encoded protein. We expressed this circMbl minigene with the actin-gal4 driver and analyzed muscleblind RNA and protein isoforms from fly heads. Indeed, expression of the minigene resulted in a 4-fold increase in the levels of circMbl (Figures 2F and 2G). In these flies, we did not observe linear concatemers, although we observed small amounts of a larger RNaseR-resistant RNA, likely a larger circMbl molecule (see asterisk in Figure 2F). Importantly, in these flies we consistently detected MBL-immunoreactive bands of approximately 10 kDa (Figure 2H), strongly suggesting that circMbl can also produce a protein in adult flies.
RFP from Fly Heads
To determine whether circRNAs are translated in a more relevant tissue, we set up the RFP methodology in fly heads. For doing so, we used two different extraction buffers, which contain none or standard detergent concentrations. As expected, we obtained strong coverage of the known ORFs and their 5′ UTRs (Figure 3A).
Figure 3.

Genome-wide Analysis of Translation in Fly Heads by RFP
(A) Meta-analysis of the RFP reads.
(B) “Pile ups” of the RFP reads at two different resolutions in the Arr2 locus.
(C) Meta-analysis of the RFP reads in the proximity of the start and stop codons of all annotated genes. The total frequency of RFP read 5′ end positions plotted against the relative distance (in nucleotides) to the start/stop codon (left). The same information, but subdivided into reads of different length, is shown, with frequency represented by color (middle). Relative enrichment of RFP reads around real start/stop codons over the background frequency for reads of such length in the vicinity of start/stop codons, expressed as log ratio of these frequencies (right). Positive scores (red) indicate consistency with the signature of start/stop codons, and negative scores (blue) consistency with the uniform background distribution.
(D) Main over-represented GO terms for the mRNAs significantly higher in the normal detergent library.
(E) Venn diagrams displaying the number of circRNAs found in the different libraries (number of backspliced reads). The detergent libraries were sequenced 3.4 times deeper.
(F) The x axis presents the number of backspliced reads, and the y axis presents the number of linear reads from the downstream junction of the circ-hosting gene. The asterisks (∗) indicate the position of the three circRNAs with higher number of RFP reads. We also indicated the number of RFP reads within the parentheses.
(G) Stop codon RFP pattern observed around the putative stop codon of circMbl. Top: considering genome-aligned as well as backspliced reads, the pattern is not consistent with a stop codon because of superimposed reads from mbl mRNA. Bottom: when extracting only backspliced reads, the RFP pattern is consistent with a stop codon at the inferred position in the circRNA.
RFP fragment sequencing experiments preferentially fall into particular positions with respect to the ORF. As expected, we observed phasing of the reads (Figure 3B), which indicates that our data can be exploited to assert translation in a candidate region (Calviello et al., 2016, Fields et al., 2015, Ingolia et al., 2009, Ji et al., 2015, Lareau et al., 2014).
We observed that reads of different length have different frame preferences, and furthermore, that these preferences depend on the extraction buffer conditions used in the experiment (Figures S3A and S3B). Next, we developed an algorithm that converts the frame preferences of RFP reads of different lengths into a scoring scheme. This scoring scheme is able to distinguish annotated ORFs, start and stop codons, from randomly selected AUGs or (out-of-frame) stop codons inside the same CDS, and it is specific to the actual read-frame preferences (Figures 3C, S3C, and S3D). The score is highly reproducible between biological replicates (Figure S3E).
Interestingly, when using different lysis buffers, we observed differences in the mRNAs covered by the RFP reads. A Gene Ontology (GO) enrichment analysis for the RNAs differentially expressed between the two types of RFP libraries revealed that mRNAs encoding membrane and transmembrane proteins are highly enriched in the samples lysed by the detergent-containing buffer (Figure 3D). Other enriched GO terms included post-synaptic membrane and proteins harboring typical neuronal transmembrane domains. These results strongly suggest that the no detergent samples are depleted for mRNAs translated by membrane-associated ribosomes (i.e., in the endoplasmic reticulum [ER] or synaptic spaces).
A Subset of CircRNAs Is Associated with Translating Ribosomes in Fly Heads
We therefore set out to investigate circRNAs for potential translation. Interestingly, we found a much larger number of RFP reads in the detergent-lysed samples (Figures 3E and S4A), suggesting that the potential translation of circRNAs happens by membrane-associated ribosomes or in subcellular compartments rich in membranes (i.e., synaptic space). The presence of backsplicing reads does not seem to be due to random binding, because we did not find reads for many highly abundant circRNAs and we found RFP reads originated from circRNAs of middle and low abundance (Figure S4B). In most cases the number of RFP reads encompassing circRNA junctions is in the same scale as the RFP exon-junction reads of the corresponding linear mRNA (Figures 3F and S4C; Table S2). The fly head RFP identified 122 ribo-circRNAs, 7 of which we previously identified in the S2 or Kc samples (Figure S4D). This is a notable overlap supporting specific ribosome association (p < 5 × 10−4).
We next validated the circularity of top fly head candidates. Indeed, seven of the eight candidates showed strong resistance to RNaseR treatment (Figure S4E). circSh showed sensitivity to RNaseR, suggesting that the assay detected some linear RNA species.
Only patterns in RFP reads encompassing the backsplicing junction can be unequivocally attributed to circRNA translation, which abrogates our capacity to look for AUG patterns in all cases and stop codons in most cases, and significantly diminishes the power for detecting ribosome phasing encompassing the ORF. Despite these limitations, we observed a high stop codon score for the most abundant ribo-circRNA, circMbl (score = 37; p < 0.007; Figure 3G, bottom panels). Thus, this result supports translation of circMbl in fly heads. Interestingly, the high score of this stop codon disappears when all the RFP reads (not only the ones containing the backspliced reads) are considered (Figure 3G, top panels). Therefore, this stop codon is used only in the context of the circRNA. In summary, the RFP pattern around the stop codon of circMbl strongly supports endogenous translation of this circRNA.
It could be that the RFP backspliced reads arise from a small, undetected fraction of linear, trans-spliced products rather than circRNA molecules. To test this possibility, we utilized a fly strain carrying a transgene for expressing EGFP-tagged ribosomes (Huang et al., 2013). We immunopurified the tagged ribosomes and analyzed the RNA bound to them. We measured the relative immunoprecipitation (IP) enrichment by comparing the enrichment (immunoprecipitate/input ratio) between the assayed target and an abundant pre-mRNA (pre-mbl RNA), which is not expected to be associated with ribosomes. Indeed, we found a strong enrichment for four ribo-circRNAs assayed, demonstrating their association with ribosomes (Figure 4A). To distinguish between associations of ribosomes with linear and circRNA molecules, we treated ribosome-bound RNAs with RNaseR. For doing so, we set up an approach to determine RNaseR sensitivity from low RNA amounts (see STAR Methods). Indeed, we found that the four assayed ribo-circRNAs are strongly resistant to RNaseR, demonstrating that they are bound to the ribosome as circRNA molecules (Figure 4B). Importantly, the RNaseR treatment resulted in complete degradation of a linear RNA in the same sample (rp49 mRNA; Figure 4C). In addition, we found that the ribosome-bound circSh fraction is strongly resistant to RNaseR treatment (Figure 4C, red bars), even when this circRNA displays sensitivity to RNaseR treatment in total RNA and in the input fraction (Figures 4C and S3E). This suggests the existence of two populations of transcripts carrying the backsplice junction (one linear and one circular). The circular transcript is bound to the translating ribosomes.
Figure 4.

A Subset of CircRNAs Is Associated with Translating Ribosomes
(A) Results of the translating ribosome affinity purification (TRAP) assay. Ribo-GFP strains: Actin-Gal4; UAS-EGFPL10a. Control strain: UAS-EGFPL10a. The number indicates the difference in the normalized ratio between the Ribo-GFP and control strains. n = 4.
(B). RNaseR validation of the ribosome-bound circRNAs. RNA from the input and IP TRAP samples was subjected to RNaseR treatment. Mouse RNA was added and quantified as spike-in for the normalization. The RNaseR resistance was assessed and normalized to the one observed in the input sample. n = 3.
(C) RNaseR validation of the ribosome-bound RNAs. Normalization was performed using the internal spike-in. n = 3.
(D) RNA expression in the mbl locus. Red track represents the expression of the different exons in fly heads. Hemi-circles indicate the most abundant circRNAs. Top inset: comparison of the expression levels in the mock and RNase-treated samples corroborates the high expression of circMbl3 and circMbl4. Right boxes indicate the size of the proteins produced by the annotated mbl mRNAs or circRNAs.
(E) Top: schematics of the circMbl3-encoded protein and the peptides generated by proteolysis.
(F) Northern blot assay showing the presence of circMbl3 in fly heads of yw and CS flies. In both strains this circRNA is resistant to RNaseR. Blotting was done using probes for the relevant circRNA junction.
In (A)–(C), error bars indicate SEM. See also Figure S4.
A CircRNA Generated from the mbl Locus Produces a Detectable Protein
The mbl locus produces several circRNAs, some of which are highly abundant (see Figure 4D; Ashwal-Fluss et al., 2014, Westholm et al., 2014). Most of those circRNAs include the second exon, which contains the start codon for the putative circRNA-encoded ORF and would also include the same 5′ UTR regulatory sequences. To determine the presence of any of these proteins in fly head extracts, we performed targeted mass spectrometry from a fly head MBL immunoprecipitate. We focused on the C terminus of the putative proteins, which is encoded across the circRNA junction. We utilized synthetic peptides to determine their characteristic spectra for which we then searched in the fly head immunoprecipitate. Unfortunately, the ionization capacity of the peptide designed to identify the circMbl1-encoded peptide was extremely low, precluding the efficient identification of our best candidate. However, we found a consistent and very high confidence hit for a peptide that can only be produced by circMbl3 (Figures 4D, 4E, and S4F). This circRNA encodes a 37.04 kDa protein and might either include or skip an alternative internal exon (exon 5 of mbl). To verify the existence of this circRNA in fly heads, we performed a Northern blot analysis using a specific circMbl3 probe. We detected two RNaseR-resistant bands of the predicted size due to alternative splicing of the fifth mbl exon in the assayed fly strains (yw and CantonS [CS]; Figure 4E). Importantly, circMbl3 was identified as resistant to RNaseR treatment in our published dataset (Ashwal-Fluss et al., 2014). Because the identified peptide could only have originated from the circMbl3 molecule and not from any of the other known MBL isoforms (Figure 4E), these data constitute very strong evidence for the translation of this circRNA.
Analysis of CircRNA Features
We utilized 151 ribo-circRNAs (Table S2) for further analysis. We compared these ribo-circRNAs with the set of control exons utilized for cORF identification, as well as the ORF contained within circRNAs for which we did not find evidence of translation (untranslated cORFs, or utcORFs). Interestingly, we found that the ribo-circRNAs have a strong bias toward 5′ UTRs (p < 0.0055; Figure 5A). In 40% of these circRNAs, translation is predicted to share the start codon with the host gene (Figure 5B). We observed a similar bias for both parameters for the utcORFs, suggesting the presence of false negatives (Figures 5A and 5B). Interestingly, ribo-circRNAs are significantly longer than the control groups (Figure S5A), allowing them to accommodate regulatory sequences. Indeed, 72% of ribo-circRNAs have cUTR sequences (sequence outside the cORF), contrasting with 58% in the control dataset (Figure 5C). Moreover, the mean length of the cUTR of the ribo-circRNAs is significantly longer than the control groups (Figure 5D).
Figure 5.

Features of Fly Ribo-CircRNAs
(A) Ribo-circRNAs are strongly enriched for 5′ UTR and CDS overlap (p < 0.0055, Fisher’s exact test).
(B) Percentage of control, utc-circRNAs, and ribo-circRNAs ORFs, which share their putative start codon with their hosting linear mRNA. The differences between ribo-circRNAs and control or utc-circRNAs are statistically significant (p < 2 × 10−9 and p < 0.05, respectively, Fisher’s exact test).
(C) Percentage of control, cORFs, and ribo-circRNAs that contain cUTR. The differences between ribo-circRNAs and control or utc-circRNAs are significant (p < 0.002 and p < 0.05, respectively, Fisher’s exact test).
(D) Average length of cUTR for control groups as well as for ribo-circRNAs (p < 0.019, double-sided Mann-Whitney U test).
(E) Excerpt of a multiple species alignment of circMbl. First codon position is in uppercase, and other codon positions are in lowercase. STP, stop codon. Arrows indicate the beginning (green) and end (red) of the exon.
(F) Ratio between the linear and circular forms of the second exon of mbl in different Drosophila species. n = 2.
(G) Boxplot showing the distribution of stop codon conservation scores for circRNAs composed of CDS and 5′ UTR exonic sequences. Negative controls are randomly selected stop codons inside the same 5′ UTR stretches. CDS denotes annotated stop codons from mRNAs sharing the circRNA exons. Red lines indicate the median, and blue dots indicate the mean. Boxes show interquartile range. Whiskers show 5th to 95th percentile range. Significantly different medians are indicated by asterisks: ∗∗∗∗p < 0.0001, two-sided Mann-Whitney U test.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. See also Figures S5 and S6 and Tables S3, S4, S5, and S6.
If the translation of a subset of circRNAs is functionally important, it is reasonable to expect that their translation potential should be evolutionary conserved. Indeed, we observed strong conservation of the coding potential and levels of circMbl, the most abundant Drosophila circRNA. We based our conclusion on the results of multiple species alignment, as well as on the validation and quantification of backsplice junctions by Sanger sequencing for five different Drosophila species (Figures 5E, 5F, and S5B).
To investigate whether cORFs are under negative selection, we built multiple species alignments of each circRNA candidate from up to 24 insect species (see STAR Methods; unpublished data). Figure 5E shows excerpts of such an alignment for circMbl (see Table S3 for the scores of all cORFs). Indeed, we observed that the cORFs show higher conservation of the stop codon compared with randomly selected stop codons in the same 5′ UTR (Figure 5G). These results are encouraging but should be taken with caution because of two limitations of the conservation assessments. First, cORF stop codons tend to fall close to the head-to-tail junction of the circRNA, and hence near the splice sites (the distribution of distances from the splice site is indeed to a good approximation geometric; see Figure S5C). However, splice sites are under additional evolutionary constraints because of the requirement for proper splice-site recognition. Second, the cORF stop codon could also be the stop of an upstream ORF (uORF), potentially starting in a preceding 5′ UTR exon. When accounting for these factors, we observe higher conservation of stop codons that are linked to a potential uORF in both circRNAs and controls (Figure 5G; Figure S5D). Thus, despite the higher degree of evolutionary conservation in comparison with the hosting 5′ UTR sequences, it is impossible to determine whether the observed conservation is due to the cORF, the uORF, or both.
As stated earlier, a large fraction of the ribo-circRNAs are in frame and share the start codon with the ORF of the hosting gene. Interestingly, we observed that many (31 out of 132) proteins encoded in ribo-circRNAs contain at least one identifiable protein domain (see Table S4), suggesting that they can carry out functions. We compared the protein domains present in the predicted cORFs with those present in random exons from the same hosting genes. Interestingly, we found a subset of domains, which were significantly enriched in the cORF group (Figure S5E). Half of those domains (three of six) are among those encoded by exons contained in ribo-circRNAs. We did not observe RFP reads for exons containing any of the domains enriched in our random dataset, strongly suggesting that ribo-circRNAs contain specific types of domains.
Ribo-CircRNAs in Mouse and Rat
To extend our observation to a mammalian system, we sought relevant RFP datasets (Ribo-seq) from mammalian cells and tissues (in which also most circRNAs have cORFs; Figure S6A). We utilized a Ribo-seq dataset from rat brain and liver, and one from mouse C2C12 cells (de Klerk et al., 2015, Ori et al., 2015). We found unequivocal RFP reads for 34 and 158 circRNAs in the rat and mouse samples, respectively (Tables S5 and S6). As in Drosophila, these ribo-circRNAs are not necessarily among the most abundant circRNAs in the assayed tissue (Figures S6B and S6C). However, the numbers of backsplice junction mapping RFP reads were too low to assess phasing or make predictions on the rate of translation. This finding agrees with the number of spliced reads spanning normal mRNA junction, suggesting that levels of circRNA translation may be comparable with those of the hosting RNAs (Figures S6D and S6E). As observed in Drosophila, we found that the ribo-circRNAs are strongly enriched for 5′ UTR overlap and tend to share the start codon with linear RNAs (Figures S6F–S6H).
Ribo-CircRNAs Contain Sequences that Can Promote Cap-Independent Translation
Because circRNAs do not contain a 5′ cap, their translation should be dependent on the presence of a translation regulatory sequence. Cap-dependent translation can be efficiently inhibited by overexpression of the 4E-BP protein (Marr et al., 2007, Olson et al., 2013). We examined whether overexpression of Drosophila 4E-BP inhibits translation of the V5-tagged circRNA minigenes. Although the levels of GFP expressed from a linear mRNA molecule were severely diminished by co-expression of 4E-BP, expression of the circMbl or circCdi V5-tagged proteins was not affected (Figure 6A). Because the main protein product of the Pde8 minigene co-migrates with the 4E-BP protein (which is also V5 tagged), it was not possible to distinguish between these proteins. However, we found that the smaller (14.5 kDa) isoform was insensitive to 4E-BP expression, suggesting that sequences in the cUTR of this circRNA can drive cap-independent translation (Figure S7A). We observed a similar phenomenon with our circCherry artificial minigene (Figures 2F and 6B).
Figure 6.

Characterization of CircRNAs Putative IRESs
(A) Proteins expressed by the circMbl and circCdi minigenes are resistant to the activity of 4E-BP.
(B) Quantification of the fluorescent signal from cells transfected with the Cherry minigene and GFP in the presence or absence of 4E-BP, detected by high-throughput microscopy. n = 3. In each experiment more than 3,000 cells were measured.
(C) Relative firefly luciferase levels of the indicated bicistronic reporters. To normalize between experiments, we divided the luciferase reads to the ones obtained with the HCV IRES construct. n = 6. Error bars represent SEM.
(D) Drosophila S2 cells were transfected with a bicistronic IRES reporter and with or without a plasmid expressing 4E-BP. The results show the ratio between the firefly/Renilla coefficients in presence or absence of 4E-BP. Error bars indicate SEM. n = 6.
(E) Expression of the G-capped RLuc reporter bearing mbl 5′ UTR (m7Gppp-circMbl-RLuc) was compared with the analogous A-capped reporter (Appp-circMbl-RLuc) and bicistronic reporter (m7Gppp-FLuc-circMbl-RLuc) in a cell-free Drosophila translation system. RLuc activity was normalized to FLuc activity; expression of m7Gppp-circMbl-RLuc was taken for 100%, and the rest was expressed accordingly.
(F) Linear reporter containing the A-capped CrPV IRES (CrPV-RL) was used as a positive control, and circular reporters with mbl 5′ UTR in a reverse orientation (circMblRevcUTR-RLuc) or deleted mbl 5′ UTR (circMblcUTR-RLuc) were used as negative controls in a cell-free Drosophila translation system. Each reaction was assembled in the presence or absence of m7GpppG.
See Figure S7.
To validate these results, we cloned the circMbl, circCdi, circPde8, and circTai cUTRs in the straight or inverted orientation into a bicistronic reporter (Figure 6C, top; Olson et al., 2013). Remarkably, we found that the circMbl, circCdi, circTai, and circPde8 cUTRs can drive higher expression of the luciferase ORF from this bicistronic construct (Figures 6C and S7B). Interestingly, we noticed that the putative regulatory elements seem to act at least partially by increasing RNA stability (Figure S7C). We then measured firefly and Renilla luciferase levels in the presence of 4E-BP. As previously reported, overexpression of 4E-BP resulted in an increase of the firefly/Renilla ratio of the Hepatitis C virus (HCV) reporter by approximately 40% (Figure 6D; Olson et al., 2013). We observed a more than 4-fold increase in the firefly luciferase/Renilla luciferase ratio for all the cUTR reporters after co-transfection with a 4E-BP expression plasmid (Figure 6D). The constructs containing the inverted cUTR sequences displayed much lower resistance to 4E-BP overexpression with the exception of the reverse Pde8 sequence, which displays luciferase levels barely above background (Figures 6C, 6D, and S7B).
CircMbl Can Be Translated In Vitro in a Cap-Independent Way
To study the mbl 5′ UTR in a more controlled system, we performed an in vitro translation assay. We generated RNA reporters that contain a Renilla luciferase ORF downstream of the mbl 5′ UTR and used them in a Drosophila cell-free translation system (Castagnetti et al., 2000, Chekulaeva et al., 2006). We first compared translation of two reporters: one bearing m7Gppp-cap (cap-circMbl-RLuc) and another containing an Appp-cap analog (circMbl-RLuc). Substitution of m7Gppp-cap with Appp-cap analog reduced mbl reporter translation 8-fold (Figure 6E). Similarly, bicistronic reporter with mbl 5′ UTR was translated 10-fold less efficiently than m7Gppp-capped monocistronic mbl reporter (cap-Fluc-circMbl-RLuc; Figure 6E). These results show that in this system, the linear version of the mbl mRNA is translated mainly via cap-dependent initiation. However, because circRNAs represent a unique case of RNAs in which there is no cap, we reasoned that even 10% activity might still be relevant. Therefore, we generated a circRNA reporter that contains the circMbl cUTR (circMblcUTR; Figures S6D and S6E). Additionally, we generated circRNA reporters with cUTR in a reverse orientation (RevcUTR) and a reporter from which most of the cUTR was deleted (ΔcUTR). In the context of the circRNA, circMbl cUTR was able to drive Renilla translation two times less efficiently than CrPV IRES (Figure 6F). Deletion of most of the cUTR abolished this activity, and the cUTR activity was reduced 2-fold when this element was inserted in a reverse orientation. Addition of a cap analog in trans was used as a further control to demonstrate that the CrPV IRES and circMbl reporters do not carry m7Gppp-cap and, therefore, cannot be translated in a cap-dependent manner (Figure 6F). In summary, our experiments demonstrate that the circMbl cUTR sequences can drive translation from a circRNA in a cap-independent manner, although this mechanism is not prevailing for a linear mbl reporter, at least in vitro. Interestingly, the inverted circMbl sequence can also promote circRNA translation, perhaps suggesting that the functional element is of a structural nature.
A Putative CircMbl1-Encoded Protein Is Enriched in Synaptosomes and Modulated by Starvation and FOXO
Using the available antibody against MBL, we could not always detect an endogenous band for putative circMbl-encoded proteins in fly head extracts. Therefore, we determined whether we could detect bands of the expected size in synaptosomal preparations from fly heads (Depner et al., 2014). Indeed, following subcellular fractionation, we could observe a band of the expected size (slightly lighter than 10 kDa for circMbl1) from the fractions P2 and LP1, which are strongly enriched for synaptosomes. We also observed a band of the expected size (37 kDa) for the protein product produced from circMbl3 in the cytoplasmic fractions. However, we cannot rule out that this band is not the endogenous MBL isoform of 32 kDa. In addition, circMbl1 and circMbl3 RNA were also present in the same synaptosomal preparation (Figure 7B).
Figure 7.

MBL Isoforms Are Expressed in Different Cellular Fractions, and Their Levels Are Modulated by Starvation and FOXO
(A) Wild-type (CS) fly heads were subjected to centrifugation-based biochemical fractionation and western blot using antibodies against MBL and Brunchpilot (BRP). The fraction P2 contains synaptosomes, and the fraction LP1 contains the pre- and post-synaptic membranes.
(B) RT-PCR analysis examining circular, linear, and precursor transcript levels in the synaptosomal fractions. The mbl (2–3) and mbl (1–2) primer pairs are used to quantify different mbl mRNA isoforms. The y axis indicates the ratio between the signal in the synaptosomal (P2) and predominantly nuclear (P1) fractions. Data were normalized to spike-in and to the volume of the fraction. Mean ± SD. n = 2.
(C) Western blot analysis of Drosophila S2 cells stably transfected with copper-induced Foxo. Cells were harvested at the indicated time after copper induction.
(D) Adult flies were starved (or not) for 12 hr, harvested, and their head extracts were subjected to western blot analysis. The arrow indicates the position of the putative circMbl-1-encoded protein.
(E) RT-PCR analysis examining circular and mbl mRNA levels in response to starvation. Mean ± SD (n = 2). Data were normalized to rp49 and 28S rRNA. Actin-gal4 flies were used for the experiment.
(F) Western blot analysis showing the MBL 10 kDa isoform expression upon induction of FOXO expression (elav-GS; UAS-Foxo fly line). The experiment was performed by triplicate.
See Table S7.
We next sought to identify signals or factors that could modulate the level of translation from circRNAs. Those factors theoretically should also increase translation from mbl mRNAs, because those RNAs share the start codon and most of the 5′ regulatory regions with the circMbl isoforms. To test this, we utilized a Drosophila S2 cell line carrying a plasmid allowing conditional expression of FOXO (Puig et al., 2003). FOXO expression strongly inhibits cap-dependent translation (Puig et al., 2003). Induction of FOXO resulted in a strong accumulation of at least two MBL protein isoforms (Figure 7C). The accumulation of these isoforms does not correlate with higher levels of either mbl mRNA or pre-mRNA, suggesting that it is due to stimulation of their specific protein synthesis (Figure S7F). Those isoforms are not likely originated from circRNAs, because the levels of circMbl isoforms are very low in Drosophila S2 cells.
To extend this observation to a more relevant system, we determined the effect of starvation, which activates cap-independent translation (Bar-Peled and Sabatini, 2014), on the levels of the ∼10 kDa MBL-immunoreactive band observed in Figure 7A. As a positive control, we utilized a fly extract of the fly strain overexpressing circMbl (see earlier). Overexpression of circMbl leads to the presence of at least one clear ∼10 kDa band (which in some cases was accompanied by a slightly heavier MBL-immunoreactive band; see Figure 7A). However, following 12 hr starvation, we observed a clear band of this size also in wild-type flies (Figure 7D), indicating that this protein is either produced and/or stabilized following starvation. Moreover, inducible expression of FOXO in the fly brain for 16 hr (by the use of the CNS-specific GeneSwitch transgene) resulted in the presence of the ∼10 kDa band (Figure 7F). Importantly, neither starvation nor FOXO overexpression resulted in any changes in the levels of circMbl1 or the tested mbl mRNA isoforms (Figure 7E), demonstrating that the effect is post-transcriptional.
Discussion
In this manuscript, we present multiple lines of evidence that strongly support circRNAs translation: (1) specific association of circRNAs with translating ribosomes; (2) capacity to generate proteins from circRNA minigenes; (3) finding of RFP reads supporting a stop codon in circMbl; (4) presence of sequences able to promote cap-independent translation on several circRNAs; (5) finding of a peptide matching circMbl3, but no known linear isoform; (6) detection of putative circMbl-encoded proteins by western blot; and (7) determination that physiological factors can regulate the levels of these proteins.
Analyzing circRNA data is difficult because their identification requires exclusive focus on backspliced reads, which cover only a small part of the circRNA molecule. Even for mRNA, the low number of spliced RFP reads prohibits isoform-specific analyses of phasing. The same difficulty arises in identifying circRNA-produced peptides by mass spectrometry. Similarly, evolutionary analysis of cORFs needs to focus on the stop codon, which even if it resides in 5′ UTR sequence may well be shared with a putative uORF. Thus, none of the sequence or sequencing-based analyses alone can without ambiguity support circRNA translation. However, taken together and viewed in light of the additional experimental validation presented here, we provide strong evidence of endogenous circRNAs translation. In this context, although it is impossible to quantitatively estimate translation efficiencies of circRNAs from the ratio of backspliced and linear reads, the situation is comparable with mRNA isoforms when restricted to a single, diagnostic junction. Thus, it appears that, once initialized, translation of circRNAs is not much different from translation of mRNAs.
It was recently reported that artificially, strongly overexpressed circRNAs can be translated independently of IRES sequences (Abe et al., 2015). However, our results suggest the existence of a specific sequence to enable translation initiation in a natural, endogenous context. Moreover, the fact that we detected circRNAs with very different abundance associated to ribosomes and that some of our minigenes did not generate proteins strongly suggest that translation of a subset of circRNAs is not by chance, but a specific effect. Importantly, the results obtained using the bicistronic constructs, as well as the in vitro assay, do not necessary mean that the assayed cUTRs have conventional IRES activity. This is because the results obtained using the bicistronic constructs could be caused by the presence of cryptic promoters and/or cleavage and polyadenylation signals, and because circRNAs in the in vitro assay could potentially be cleaved in the extract. However, the fact that in both assays the luciferase signal obtained from constructs carrying the cUTRs is resistant to inhibition of cap-dependent translation demonstrates the capacity of these sequences to promote translation in a cap-independent way, and hence from circular molecules. Further research is necessary to uncover how these sequences promote translation.
Many of our observations suggest that circRNA translation might be important in the brain. First, circMbl1 and the putative circMbl1-encoded peptide are present in synaptosome fractions. Moreover, ribo-circRNAs are predominantly bound to membrane-associated ribosomes. Importantly, the proteins encoded by the mbl and circMbl molecules do not contain identifiable peptide signal sequences, raising the possibility that circMbl and maybe some mbl mRNA isoforms are translated in synapses. Indeed, a recent report demonstrated that acute fasting regulates synaptic function by a mechanism dependent on 4E-BP and FOXO, both of which can regulate translation from the circMbl cUTR.
In summary, here we provide evidence for translation of circRNAs, an abundant and widespread type of RNA that is distinct from, but shares sequence with, mRNA. Our findings thus indicate the presence of a repertoire of protein isoforms and likely uncharacterized modes of regulation of gene and protein expression.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-MBL | Lab of Prof. Darren Monckton PMID:17309604 | N/A |
| Mouse monoclonal Anti-V5 | Sigma-Aldrich | CAT# V8012; RRID: AB_261888 |
| Rabbit polyclonal anti-GFP | Abcam | CAT# Ab290; RRID: AB_303395 |
| mouse monoclonal DM1A anti α-tubulin | Sigma-Aldrich | CAT# T6199; RRID: AB_477583 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Synthetic peptide RAADTTDMFPLIM | SpikeTides, JPT | N/A |
| Critical Commercial Assays | ||
| Dual Luciferase Assay kit | Promega | CAT# E1960 |
| Deposited Data | ||
| Drosophila RFP RNA-seq data | This paper | GSE79626 |
| C2C12 RFP RNA-seq data | de Klerk et al., 2015 | PRJEB7207 |
| rat RFP RNA-seq data | Ori et al., 2015 | GSE66715 |
| Kc167 cells RFP RNA-seq data | Miettinen and Björklund, 2015 | PRJEB5938 |
| S2 cells (dataset2) and embryo RFP RNA-seq data | Dunn et al., 2013 | GSE49197 |
| eggs and oocyte RFP RNA-seq data | Kronja et al., 2014 | GSE52799 |
| S2 cells RFP RNA-seq data | Aspden et al., 2014 | GSE60384 |
| Experimental Models: Cell Lines | ||
| D. melanogaster: Cell line S2: S2-DRSC | N/A | N/A |
| FOXO stable cell line | Laboratory of Michael T. Marr and Puig et al., 2003. PMID:12893776 | N/A |
| Experimental Models: Organisms/Strains | ||
| D. melanogaster: w1118 | Bloomington Drosophila stock center | BDSC: 5905 FlyBase:FBal0018186 |
| D. melanogaster: yw | Bloomington Drosophila stock center | BDSC: 1495 FlyBase:FBal0018607 |
| D. melanogaster: Canton S | Bloomington Drosophila stock center | BDSC: 6366 |
| D. melanogaster: Actin-Gal4: w[1118]; P{w[+mC] = AyGAL4}25/CyO | Bloomington Drosophila stock center | BDSC: 3953 FlyBase:FBti0012290 |
| D. melanogaster: UAS-EGFPL10a (III) | Huang et al., 2013 PMID:24348200 | N/A |
| D. melanogaster: UAS-foxo: y[1] w[∗]; P{w[+mC] = UAS-foxo.P}2 | Bloomington Drosophila stock center | BDSC: 9575 FlyBase: FBti0076467 |
| D. melanogaster: Elav-Gal GeneSwitch | Lab of Dr. Minoru Saitoe and S. Naganos et al., 2012 PMID:22342328 | N/A |
| D. melanogaster: UAS-circMbl | This paper | N/A |
| Oligonucleotides | ||
| Oligos for cloning, qPCR, and Northern Blotting see Table S7 | This paper | N/A |
| Recombinant DNA | ||
| Mbl | Ashwal-Fluss et al., 2014 PMID:25242144 | N/A |
| Haspin V5 | This paper | N/A |
| Camk1 V5 | This paper | N/A |
| Mbl V5 | This paper | N/A |
| CircMblV5 5′ss | This paper | N/A |
| CircMblV5 ΔATG | This paper | N/A |
| Cdi V5 | This paper | N/A |
| Pde8 V5 | This paper | N/A |
| Camk1-mbl-cherry-camk1 | This paper | N/A |
| circMbl IRES | This paper | N/A |
| circPde8 IRES | This paper | N/A |
| circCdi IRES | This paper | N/A |
| circTai IRES | This paper | N/A |
| circMbl IRES reverse | This paper | N/A |
| circPde8 IRES reverse | This paper | N/A |
| circCdi IRES reverse | This paper | N/A |
| circTai IRES reverse | This paper | N/A |
| UAS-circMbl OE | This paper | N/A |
| Software and Algorithms | ||
| Find_circ.py | Memczak et al. 2013 | http://circbase.org/cgi-bin/downloads.cgi |
| cORF_prediction_pipeline.py (prediction of ORFs from circRNA sequences) | This paper | https://github.com/kadenerlab/cORF_pipeline |
| SRCP.py (short read circRNA pipeline for detection of back-splice reads in RFP RNA-seq) | This paper | To be published, can be provided upon request |
| Toptat2 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| HTSeq | Anders et al., 2015 | http://www-huber.embl.de/HTSeq |
| DESeq | Anders and Huber, 2010 | http://bioconductor.org/packages/release/bioc/html/DESeq.html |
| DAVID | Huang da et al., 2009 | https://david.ncifcrf.gov/home.jsp |
Contact for Reagents and Resource Sharing
Further information and request of reagents should be addressed to the Lead Contact, Dr. Sebastian Kadener (skadener@mail.huji.ac.il).
Method Details
Fly strains and Reagents
Plasmids
The minigenes constructs for Drosophila S2 cells were generated in pMT-V5 (Invitrogen). HCV IRES, MARL bicistronic reporter and 4E-BP plasmids were kindly provided by Mike Marr and previously described (Olson et al., 2013). The pAc-GFP plasmid was previously described (Weiss et al., 2014). Custom DNA fragments synthesis (gBlocks®, Integrated DNA Technologies) was used to generate split cherry exon plasmid construct with circCamk1 flanking introns. The primers used for cloning are described in Table S7.
Generation of UAS-circMbl flies
To generate circMbl overexpression flies we utilized a variant of the pUAS-attB plasmid (Addgene) in which we eliminated the SV40 small intron and replaced the SV40 3′ UTR with the one present in the pMT plasmid (Invitrogen). We followed by cloning the circMbl minigene (Ashwal-Fluss et al., 2014) into this plasmid by standard cloning techniques. This plasmid was sent to injection to BestGene Inc (CA, USA).
Fly Strains
WT flies used in this study are w118, yw and CantonS (CS, CSIso3H) strain (Bloomington Stock Centre, Indiana). Drosophila yakuba, Drosophila simulans, Drosophila virilis, and Drosophila pseudoobscura were obtained from the Drosophila Species Stock Center (DSSC) at the University of California, San Diego. Actin-gal4 and UAS-EGFPL10a (III) strains were previously described (Huang et al., 2013, Ito et al., 1997). The UAS-foxo (stock# 9575; (Puig et al., 2003)) was obtained from Bloomington stock center. Elav-Gal GeneSwitch (GS) were generously provided by Dr. Minoru Saitoe, Tokyo Metropolitan Institute of Medical Science, Japan.
Starvation Experiment
Flies were starved for 12 hr on 2% agar vials, flash frozen in liquid nitrogen and heads were separated using a sieve after mechanical decapitation.
Gene switch experiment
6 days old flies were starved for 12 hr and transferred to 5% sugar containing 400ug/ml RU486 food vials to induce the Gal4 expression for 24 hr, flash froze and separated the heads performed western blotting as described below.
Molecular Biology Methods
Cell culture and transfections
Drosophila S2 cells were maintained in 10% fetal bovine serum (Invitrogen) insect tissue culture medium (HyClone). Cells were seeded in a six-well/ twelve-well plate. Transfection was performed at 60%–80% confluence according to company recommendations: 6μl of TransIT 2020transfection reagent (Mirus Bio, MIR 5400A) and 2μg of total DNA. In the minigene experiments 1 μg of the minigene-expressing vector were used. 0.4μg of 4E-BP was used in all co-transfection experiment. When copper (Cu) induction was performed, 500μM of copper was added to the media and the cells were collected 48h after the induction. FOXO stable cell line was previously described (Puig et al., 2003).
Western blotting
Fly heads (20 heads per sample) were collected on dry ice. Heads were homogenized in RIPA lysis buffer (50 mM Tris-HCl at pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40 0.5% Sodium deoxycholate, and 0.1% sodium dodecyl sulfate (SDS), 1 mM DTT, supplemented by protease inhibitor cocktail and phosphatase inhibitors) using motorized pestle. Head lysates were then centrifuged at max speed for 10 min and the supernatant was saved. Cell samples were collected by centrifugation (500 g 5min), washed twice with PBS and homogenized in RIPA lysis buffer. All lysates were boiled with protein sample buffer (Bio-Rad) and resolved by Criterion XT Bis-Tris gels (Bio-Rad). Antibodies used in this study: Anti-MBL antibody was kindly provided by Prof. Darren Monckton (School of Life Sciences, University of Glasgow). Anti-V5 (Mouse monoclonal V8012, Sigma Aldrich) was used for Immunoprecipitation and immunoblotting. Rabbit polyclonal anti-GFP (ab290, Abcam) and anti α-tubulin (mouse monoclonal DM1A, Sigma, Aldrich) were used for western blotting. All western blot analysis represents at least three independent biological repeats done.
Northern Blotting
RNA was treated with RNaseR (3U/μg, 15 min 37°C) or mock treated and run on denaturing agarose gels (1% formaldehyde) at 90 V for 2:30 hr. The gels were stained with SYBR Gold to ensure equal loading. The RNA was blotted onto a Hybond N+ membrane (GE Healthcare) at 15 V for 1 hr, the membrane was dried and crosslinked (0.12 J/cm2), equilibrated and hybridized (68°C, o/n) in NorthernMax Hybridization Buffer (Ambion) with 0.1 nM DIG-labeled RNA-probe (max. 150 nt, DIG labeling mix - Roche). The Northern blots were washed stringently (twice 2xSSC, 0,1% SDS and twice 0.2xSSC, 0.1% SDS at 68°C) developed using an anti-Digoxigenin antibody (Roche), CDP star (Roche) and a LAS4000.
Polysome sucrose gradient-
S2 cells were co-transfected with the circRNA minigenes. 12 hr later the cells were stimulated with copper and collected for 36 hr. Polysome profiling and RNA isolation from each fraction were done as previously described (Aspden et al., 2014). RNA from the each fraction was mock/ RNaseR treated as described below. In cases in which RNA amounts were lower than 2 μg, an in vitro transcribed Luciferase transcript (see utilized oligonucleotides on Table S6) was added to the RNaseR/ mock reaction.
RNaseR treatment of total RNA
Total RNA was RNaseR treated as previously described (Memczak et al., 2013, Suzuki et al., 2006). 2 μg of total RNA were treated with 3U/μg RNaseR (Epicenter) for 15 min at 37°C or mock treated. The RNA was immediately transferred to ice, spiked with 10% mouse RNA, extracted with TRI Reagent (Sigma, Aldrich) according to the manufacturer’s protocol. The RNA concentration of the mock-treated sample was determined and 5μl of the mock treated RNA was used for reverse transcription (RT, using iScript and random primers, Bio-Rad), the same volume of the RNaseR treated RNA was used for reverse transcription. cDNA was used for qPCR quantification, whereas the spike-in was used for normalization.
Analysis of gene expression by quantitative real-time PCR
Total RNA was prepared from adult fly heads and Drosophila S2 cells using TRI Reagent (Sigma, Aldrich)) according to the manufacturer’s protocol. RNA was DNase treated (DNaseI, NEB) and cDNA derived from this RNA (using iScript and random primers, Bio-Rad) was utilized as a template for quantitative real-time PCR performed with the C1000 Thermal Cycler Bio-Rad. The PCR mixture contained Taq polymerase (SYBR green Bio-Rad). Cycling parameters were 95°C for 3 min, followed by 40 cycles of 95°C for 10 s, 55°C for 10 s, and 72°C for 30 s. fluorescence intensities were plotted versus the number of cycles by using an algorithm provided by the manufacturer. Primer efficiency was determined for all primers described in this study and incorporated into the relative expression calculation. All the primers used in this assay are detailed in Table S7.
Protein Immunoprecipitation (IP)
Drosophila S2 Cells (one 10cm plate per sample) were washed twice with cold PBS and lysed in 500μl of lysis buffer (30 mM HEPES KOH at pH 7.4, 100 mM KAcetate, 2 mM MgAcetate, 5% glycerol, 0.1% Triton X-100, 1 mM EGTA, 5 mM DTT, protease inhibitors [Complete mini, Roche)). The lysates were incubated for 10 min on ice and centrifuged at 14,000 rpm for 12 min at 4°C. 1/10 of the supernatant was used as input. 9/10 of the supernatant was incubated with either anti-v5 or anti-MBL antibodies at 4°C with rotation. 2 hr later, 50μl of protein G-plus-Agarose beads (Santa Cruz, sc-2002; previously washed with lysis buffer) were added and incubated one additional hour. After incubation, beads were washed five times for 5 min at 4°C in lysis buffer, before the addition of protein sample buffer and heating of the sample at 95°C for 5 min. As a negative control for the antibody specificity we included in each IP experiment a sample incubated with a normal IgG corresponding to the host species of the primary antibodies used (sheep IgG for α-MBL, sc-2717 and mouse IgG for α-V5, Jackson ImmunoResearch).
RFP
For “no detergent” samples, 200 frozen (in liquid N2) heads fly heads (head1 and head2) were homogenized in homogenization buffer (20mM HEPES at pH 7.3, 150mM KCL, 5mM MgCL2, protease inhibitors (Complete mini, Roche), 0.5mM DTT, RNasin 10ul, 100μg/ml CHX). Heads were homogenized immediately using a motorized pestle and centrifuged at 20,000g for 20 min at 4°C. The supernatant was carefully transferred to a new 1.5 mL tube and stored in −80°C until further use. For the “detergent samples” (Heads 3 and Head4), 200 heads frozen headsand lysed in 500ul of lysis buffer (30 mM HEPES KOH at pH 7.4, 100 mM KAcetate, 2 mM MgAcetate, 5% glycerol, 0.1% Triton X-100, 1 mM EGTA, 5 mM DTT, 0.4 U/μL murine RNase inhibitor (NEB), protease inhibitors (Complete mini, Roche), and 100ug/ml CHX). The lysates were incubated for 10 min on ice and centrifuged at 14,000 rpm for 12 min at 4°C. The supernatant was carefully transferred to new 1.5ml eppendorf tube, flash frozen in liquid nitrogen and stored in −80°C. For isolation of ribosome-protected RNA fragments we followed (Calviello et al., 2016). Briefly, 120ul of the lysate was digested with 3 μL of RNase I (Life Technologies, AM2294) for 45 min at room temperature with rotation. Digestion was stopped by the addition of 4 μL of Superase-In (Life Technologies, AM2694). Meanwhile, MicroSpin S-400 HR columns (GE Healthcare, 27-5140-01) were equilibrated with 3ml of mammalian polysome buffer by gravity flow and emptied by centrifugation at 600g for 4 min. We then immediately loaded 100 μl of the digested lysate on the column and eluted the column by centrifugation at 600g for 2 min. RNA was extracted from the flow-through (approximately 125μl) using Trizol LS (Life Technologies, 10296-010). Ribosomal RNA fragments were removed using the RiboZero Kit (Illumina, MRZH11124) and separated on a 17% denaturing urea-PAGE gel (National Diagnostics, EC-829). The 27-33nt RNA fragments were cut eluted from the gel and used to generate small RNA libraries. The oligonucleotides utilized are listed on Table S7.
Ribosome IP (TRAP)
Ribo IP (TRAP) was performed as previously described (Huang et al., 2013). The RNaseR treatment was performed in the following way: 100ng of RNA from the immunoprecipitated or input fraction was combined with 1.9 μg of in vitro transcribed 70nt RNAs from a luciferase template using the Megascript T7 Kit (ambion AM1334) according to the manufacturers protocol (see utilized oligonucleotides on Table S7). After performing the mock or RNaseR treatment, we added 100ng of mouse RNA which was used for normalization purposes and proceeded to RT-PCR as described above.
Fluorescence reporter expression analysis in S2 cells
24hs after transfection, cells were transferred to optic-suitable 384-well plates and visualized the fluorescence using ScanˆR high-throughput fluorescent microscope (Olympus). The intensity from the transfection control reporter (Cherry for circMbl IRES and GFP for the cap-dependent activity experiments), cell size and roundness were evaluated. Cherry or/and GFP levels were assessed only for cell populations that met intensity and cell morphology criteria previously described (Lerner et al., 2015).
Dual luciferase assay
Thirty hours of post transfection, cells were assayed using the Dual Luciferase Assay kit (Promega) following the manufacturer’s instructions.
In vitro translation
All capped mRNA templates for in vitro translation were generated as previously described (Chekulaeva et al., 2006). For the circMbl-RLuc RNA production, we fused either the complete circMbl UTR region in the reverse / straight orientation / only the 20 most distal nucleotides with the Renilla luciferase or coding sequence by PCR. The resulting PCR fragment was used as a template for in vitro transcription. Transcription, circularization and purification of the circularized RNA by TBE-Urea PAAG electrophoresis were performed according to (Chen and Sarnow, 1995). Preparation of Drosophila embryo extract and in vitro translation assays was done as previously described (Castagnetti et al., 2000, Gebauer et al., 1999). Translation reactions contained 1.5 nM exogenous mRNA or circRNA.
Synaptoneurosome preparation
Synaptosome extraction was carried out as previously described (Depner et al., 2014). Briefly, 5ml of fly heads were first crushed into a fine powder with a pre-chilled mortar and ground. The powder was re-suspended in ice-cold homogenization buffer (320 mM sucrose and 4 mM HEPES, pH 7.4) and homogenized with a Polytron 3000 Kinematica homogenizer in 10 pulses. This homogenate was centrifuged (2,700 rpm, 10 min in a SS34 rotor). The supernatant was further centrifuged (11,000 rpm, 15 min in a SS34 rotor). The pellet contains a soft synaptosome layer, which was collected, and a hard mitochondria core. Samples were taken from all steps for RNA purification.
Mass spectrometry
Collection of the reference spectra
The synthetic heavy labeled RAADTTDMFPLIM (SpikeTides, JPT Inc.) was resuspended in 50% acetonitrile, 0.1% formic acid. The spectra were recorded using a Q-Exactive plus mass spectrometer (Thermo Scientific) using higher energy collision dissociation method (HCD) with a mass resolution of 70000 for the MS and 35000 for the MS/MS scans. The recorded spectra were analyzed using the MaxQuant software package (version 1.5.8) using a custom made database, with carbamidomethylation of cysteins as a fixed and oxidation of methionines as a variable modification. For peptides and proteins a FDR of 1% was applied.
SRM-Mass Spectrometry
Based on the fragmentation pattern of the RAADTTDMFPLIM peptide in the Q-Exactive plus mass spectrometer a SRM method for a Q-TRAP 6500 was developed monitoring the b8, b9 and b11 fragment ions.
Proteins bound on the antibody resin were released from the beads by a treatment with 1% formic acid (Merck) for 15 min at RT. The extract was then neutralized at pH = 8.0 with 1M ammonium bicarbonate (Sigma, Aldrich). Disulfide bonds were first reduced with 10 mM TCEP (tris(2-carboxyethyl)phosphine) (Sigma, Aldrich) for 20 min and then cysteine groups were alkylated with 55 mM 2-chloroacetamide (Sigma, Aldrich) for 30 min on an automated setup (Kanashova et al., 2015). The proteins were digested with endopeptidase Lys-C (Wako). Peptides were extracted, desalted and stored on reversed-phase (C18) StageTips (Rappsilber et al., 2007). After elution, the peptides were lyophilized and resuspended in 3% TFA and 5% acetonitrile in preparation for LC-MS analysis. The heavy-labeled RAADTTDMFPLIM internal standard peptide (SpikeTides, JPT Inc.) was spiked into the solution at a concentration of 200 fmol per microliter of sample. Peptides were then separated on an in-house packed 20 cm reversed-phase column (75 μM inner diameter, 3 μm Reprosil C18-beads, Dr. Maisch) on a gradient from 3% to 32% acetonitrile in 90 min, and detected by a Q-Trap 6500 (AB Sciex). SRM signals were analyzed using the Analyst 1.6 software package (AB Sciex).
List of the transitions monitored for the identification of the peptide RAADTTDMFPLIM
| PRECURSOR MASS | TRANSITION MASS | DWELL TIME | FRAGMENTATION ID | COLLISION ENERGY | RETENTION TIME |
|---|---|---|---|---|---|
| 744.85 | 1219.57 | 30 | b11_heavy | 37.736 | 49 min |
| 1009.43 | 30 | b9_heavy | |||
| 862.36 | 30 | b8_heavy | |||
| 741.35 | 1219.57 | 200 | b11 | 37.604 | 49 min |
| 1009.43 | 200 | b9 | |||
| 862.36 | 200 | b8 |
Computational Methods
ORFs Prediction
To predict the cORFs we utilized circRNAs dataset generated from flies (Ashwal-Fluss et al., 2014), mouse and human (obtained from circBase.org; Glažar et al., 2014)). Analysis was performed with only annotated circRNA (in which both junctions of the circRNA are matched to known annotated exons). Each circRNA sequence, excluding intronic sequences, was multiplied four times, and the longest ORF spanning the circRNA junction was selected for each one of the three frames (minimum cORF length threshold was 20aa). In the ribo-circRNA analysis we used the longest non-infinity (“moebius”) cORF detected in any frame. We generated the control dataset by randomly selecting junctions from genes not hosting circRNAs and correcting to obtain a similar 5′ bias to the circRNA data.
cORF selection
We scored all possible ORFs in a circRNA by the following function:
KNOWN_START is 1 if the start codon coincides with the annotated start of the mRNA ORF
HEAD_TO_TAIL is 1 if the cORF spans the head-to-tail junction
MOEBIUS_ORF is 1 if the cORF has no stop codon and thus theoretically never terminates
The length of the ORF is used to break ties, with longer cORFs preferred over shorter, and cORFs in frame with the known ORF preferred over out-of-frame cORFs (for 5′UTR overlapping cORFs this implies KNOWN_START), and HEAD_TO_TAIL cORFs preferred over non-head-to-tail cORFs.
Expression of circRNA in total-RNA seq data
In order to assess the ribo-circRNA expression levels in total RNA-seq data we applied the find_circ pipeline (Memczak et al., 2013) on the relevant RNA-seq datasets (for C2C12 and Kc167 cells, see STAR Methods table)or used an available circRNA expression data. For fly and S2 cells we used our recently published data (Ashwal-Fluss et al., 2014). For embryo we used data published in (Westholm et al., 2014).
Start/Stop scoring
From this, for each experiment, we compute a scoring-matrix for the vicinity of start and stop codons as follows:
with r being the relative position, l the length of the read, and R the number of positions taken into accound. Thus we compare the specific occurrence frequency at each position to a mean, uniform background frequency, for each read length.
An observed pattern of reads is then scored as follows:
where O_rl is the observation matrix, containing the number of observed reads of length l at relative position r to the start or stop codon.
ORF-scoring
The ORF score is slightly more complicated, because each read can not only support translation in one or more frames, but can also reduce support for translation in other frames. As a simple example, most RFP experiments show a clear preference of 28nt reads to occur in frame1 relative to the start codon. Therefore, if we observed many such reads in frame 2 of a candidate region, this would lend strong support that the scored region is actually translated in frame2 and weaken any support for frames 1 and 3. Thus, we need to properly sum up all evidence contributed by each read, even if occurring in an adjacent frame. First, the scoring matrix is computed completely analogous to Q_rl with the frame replacing the realtive position (also denoted by r). The same is true for the observation matrix O_rl, where we take the modulo of each RFP 5′position in the candidate region with three to derive the frame r. Then, we compute one aggregate score for each frame of the candidate region:
(the ORF-score matrix plots show everything on the right before being summed over l, i.e. the contribution of each length to translation in frame 1,2,3).
The highest score determines the most likely frame in which the region is translated:
We asserted that the vast majority of known ORFs with at least 10 RFP reads is assigned the correct frame1 relative to the CDS (not shown).
Negative controls and significance estimate
We implemented two different negative controls. First, we randomly permuted the scoring matrix for each ORF that was scanned. This strongly abrogated the scores assigned to known Start/Stop codons or ORFs. However, since the scoring matrix still contains the same values, rare, large scores can be observed. Second, we randomly assign reads to a frame, ignoring its actual position relative to the candidate region, but keeping the length frequency distribution and the scoring-matrix intact.
This shuffling is very fast and can be used to compute an empirical P value as the fraction of shuffled scores that reaches or exceeds the observed, non-shuffled score.
Backspliced reads
When scoring a putative circRNA start/stop codon or ORF, we extract the RFP alignments to the back-splice junction of the circRNA in question and convert the coordinates of each reads 5′ end to relative coordinates in the ORF. Then, scoring proceeds as explained above. Optionally, scoring can be performed exclusively for back-spliced alignments, excluding any possible interference with mRNA-derived RFP reads.
Multiple species alignments
We downloaded the 24way Drosophila alignment and 60 way mouse MAF-blocks from UCSC (Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC (Blanchette et al., 2004, Kent et al., 2002). Next we obtained the full genomic sequences of all used insect genomes. We then used a combination of indexing and custom scripts (to be published elsewhere) to extract and splice together the orthologous sequences to the exons in a dm6 Drosophila melanogaster or mm10 Mus musculus transcript model. The set of all obtained sequences was run through MUSCLE (Edgar, 2004) to rebuild a full multiple sequence alignment.
Stop codon scoring
We used our multiple-species alignment code to extract specifically the stop codon sequences, aligned to all known stop codons in the ENSEMBL gene annotation for dm6, and, separately mm10, downloaded from UCSC (Karolchik et al., 2004). We repeated this analysis for all stop codon sequences occurring in internal (not first) 5′UTR exons from the same transcript catalog, recording their distance from the 5′ splice site. From this data we computed for each aligned species the frequency with which the given stop codon in the dm6 reference (one of UAA, UAG, or UGA) is replaced by another, or a non-stop triplet. Thus, the 5′UTR stops yield position-dependent background models for the evolution of 5′UTR exonic sequence that is intrinsically tuned to evolutionary branch length and species-specific changes in sequence composition. Comparing these background frequencies to the observed substitution matrices for annotated stop codons yields a scoring matrix (the log-ratios), analogous to our approach for scoring RFP read patterns. We build separate matrices for splice-site distances up to 10nt. After this distance we accumulated all data to build a model for stop codons 11nt or further away from the 5′ splice site. Next, we selected from our circRNA stes only those with cORFs composed of coding and 5′UTR exonic sequence, where the stop codon resides in 5′UTR sequence, such that our model would be applicable. We then ran a script that uses these scoring matrices to score all stop codons of cORFs and the various controls we generated. For each species, the observed substitution or conservation is scored with the log-ratio matrix. Positive log-ratios indicate that the sequence fits the evolution of annotated stop codons, negative scores indicate that the sequence fits better to the 5′UTR background model. Scores are added for all species for which there is an alignable orthologous sequence.
Domain Analysis
We used ScanProsite tool (http://prosite.expasy.org/scanprosite) to find domains in the predicted cORF sequences. We used only cORF that contains STOP codon. As a control set we used ORF predicted from randomly selected exons obtained from the same hosting genes. We count the number of occurrence of each domain in the two datasets and select the ones enriched in the cORF dataset comparing to the random control (found at least 4 times more in the cORF).
Quantification and Statistical Analysis
The statistical analysis as well as the number of repeats are stated in the figure legends as well as in the STAR method sections. For the meta-analysis described in this paper (in Figures 3, 5, S3, and S5) we used Fisher’s exact test or Mann-Whitney U test as indicated in the figure legends.
Data and Software Availability
All raw sequencing data are available at NCBI GEO: GSE79626. The cORF prediction algorithm has been deposited in https://github.com/kadenerlab/cORF_pipeline. The SRCP algorithm to detect circRNAs from RFP reads can be provided upon request.
Author Contributions
N.R.P. and O.B. performed most of the experiments. M.J., R.A.-F., S.S., and M.S. performed the computational analysis. C.S. performed the northern blots. L.R. and M.C. performed the in vitro translation. M.H. performed the synaptosomal purifications. E.W. and M.L. set up the RFP assays. D.P.-H., E.R., and G.D. performed the mass spectrometry. N.R. supervised most of the computational experiments. S.K. wrote the manuscript and designed and supervised the project.
Acknowledgments
We thank M. Marr for reagents and Y. Arava for help. S.K. is funded by the ERC Consolidator Grant (ERC Grant 647989). M.C., N.R., and L.R. are supported by the Einstein Foundation grant “Single molecule RNA network.” E.R. was supported by the Berlin School of Integrative Oncology (grant GSC 1091). M.J. received a fellowship from DFG (GRK 1772). C.S. is funded by BIF. N.R.P. is funded by a JBC fellowship.
Published: March 23, 2017
Footnotes
Supplemental Information includes seven figures and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2017.02.021.
Supplemental Information
References
- Abe N., Matsumoto K., Nishihara M., Nakano Y., Shibata A., Maruyama H., Shuto S., Matsuda A., Yoshida M., Ito Y., Abe H. Rolling circle translation of circular RNA in living human cells. Sci. Rep. 2015;5:16435. doi: 10.1038/srep16435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken C.E., Lorsch J.R. A mechanistic overview of translation initiation in eukaryotes. Nat. Struct. Mol. Biol. 2012;19:568–576. doi: 10.1038/nsmb.2303. [DOI] [PubMed] [Google Scholar]
- Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwal-Fluss R., Meyer M., Pamudurti N.R., Ivanov A., Bartok O., Hanan M., Evantal N., Memczak S., Rajewsky N., Kadener S. circRNA biogenesis competes with pre-mRNA splicing. Mol. Cell. 2014;56:55–66. doi: 10.1016/j.molcel.2014.08.019. [DOI] [PubMed] [Google Scholar]
- Aspden J.L., Eyre-Walker Y.C., Phillips R.J., Amin U., Mumtaz M.A., Brocard M., Couso J.P. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. eLife. 2014;3:e03528. doi: 10.7554/eLife.03528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L., Sabatini D.M. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014;24:400–406. doi: 10.1016/j.tcb.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M., Kent W.J., Riemer C., Elnitski L., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calviello L., Mukherjee N., Wyler E., Zauber H., Hirsekorn A., Selbach M., Landthaler M., Obermayer B., Ohler U. Detecting actively translated open reading frames in ribosome profiling data. Nat. Methods. 2016;13:165–170. doi: 10.1038/nmeth.3688. [DOI] [PubMed] [Google Scholar]
- Castagnetti S., Hentze M.W., Ephrussi A., Gebauer F. Control of oskar mRNA translation by Bruno in a novel cell-free system from Drosophila ovaries. Development. 2000;127:1063–1068. doi: 10.1242/dev.127.5.1063. [DOI] [PubMed] [Google Scholar]
- Chekulaeva M., Hentze M.W., Ephrussi A. Bruno acts as a dual repressor of oskar translation, promoting mRNA oligomerization and formation of silencing particles. Cell. 2006;124:521–533. doi: 10.1016/j.cell.2006.01.031. [DOI] [PubMed] [Google Scholar]
- Chen C.Y., Sarnow P. Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science. 1995;268:415–417. doi: 10.1126/science.7536344. [DOI] [PubMed] [Google Scholar]
- de Klerk E., Fokkema I.F., Thiadens K.A., Goeman J.J., Palmblad M., den Dunnen J.T., von Lindern M., ’t Hoen P.A. Assessing the translational landscape of myogenic differentiation by ribosome profiling. Nucleic Acids Res. 2015;43:4408–4428. doi: 10.1093/nar/gkv281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depner H., Lützkendorf J., Babkir H.A., Sigrist S.J., Holt M.G. Differential centrifugation-based biochemical fractionation of the Drosophila adult CNS. Nat. Protoc. 2014;9:2796–2808. doi: 10.1038/nprot.2014.192. [DOI] [PubMed] [Google Scholar]
- Dunn J.G., Foo C.K., Belletier N.G., Gavis E.R., Weissman J.S. Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. eLife. 2013;2:e01179. doi: 10.7554/eLife.01179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields A.P., Rodriguez E.H., Jovanovic M., Stern-Ginossar N., Haas B.J., Mertins P., Raychowdhury R., Hacohen N., Carr S.A., Ingolia N.T. A regression-based analysis of ribosome-profiling data reveals a conserved complexity to mammalian translation. Mol. Cell. 2015;60:816–827. doi: 10.1016/j.molcel.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer F., Corona D.F., Preiss T., Becker P.B., Hentze M.W. Translational control of dosage compensation in Drosophila by Sex-lethal: cooperative silencing via the 5′ and 3′ UTRs of msl-2 mRNA is independent of the poly(A) tail. EMBO J. 1999;18:6146–6154. doi: 10.1093/emboj/18.21.6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glažar P., Papavasileiou P., Rajewsky N. circBase: a database for circular RNAs. RNA. 2014;20:1666–1670. doi: 10.1261/rna.043687.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J.U., Agarwal V., Guo H., Bartel D.P. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014;15:409. doi: 10.1186/s13059-014-0409-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen T.B., Jensen T.I., Clausen B.H., Bramsen J.B., Finsen B., Damgaard C.K., Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang Y., Ainsley J.A., Reijmers L.G., Jackson F.R. Translational profiling of clock cells reveals circadianly synchronized protein synthesis. PLoS Biol. 2013;11:e1001703. doi: 10.1371/journal.pbio.1001703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Awano W., Suzuki K., Hiromi Y., Yamamoto D. The Drosophila mushroom body is a quadruple structure of clonal units each of which contains a virtually identical set of neurones and glial cells. Development. 1997;124:761–771. doi: 10.1242/dev.124.4.761. [DOI] [PubMed] [Google Scholar]
- Jackson R.J. The current status of vertebrate cellular mRNA IRESs. Cold Spring Harb. Perspect. Biol. 2013;5:a011569. doi: 10.1101/cshperspect.a011569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeck W.R., Sharpless N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014;32:453–461. doi: 10.1038/nbt.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z., Song R., Regev A., Struhl K. Many lncRNAs, 5′UTRs, and pseudogenes are translated and some are likely to express functional proteins. eLife. 2015;4:e08890. doi: 10.7554/eLife.08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanashova T., Popp O., Orasche J., Karg E., Harndorf H., Stengel B., Sklorz M., Streibel T., Zimmermann R., Dittmar G. Differential proteomic analysis of mouse macrophages exposed to adsorbate-loaded heavy fuel oil derived combustion particles using an automated sample-preparation workflow. Anal. Bioanal. Chem. 2015;407:5965–5976. doi: 10.1007/s00216-015-8595-4. [DOI] [PubMed] [Google Scholar]
- Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronja I., Yuan B., Eichhorn S.W., Dzeyk K., Krijgsveld J., Bartel D.P., Orr-Weaver T.L. Widespread changes in the posttranscriptional landscape at the Drosophila oocyte-to-embryo transition. Cell Rep. 2014;7:1495–1508. doi: 10.1016/j.celrep.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau L.F., Hite D.H., Hogan G.J., Brown P.O. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. eLife. 2014;3:e01257. doi: 10.7554/eLife.01257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner I., Bartok O., Wolfson V., Menet J.S., Weissbein U., Afik S., Haimovich D., Gafni C., Friedman N., Rosbash M., Kadener S. Clk post-transcriptional control denoises circadian transcription both temporally and spatially. Nat. Commun. 2015;6:7056. doi: 10.1038/ncomms8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.F., Lytton J. A circularized sodium-calcium exchanger exon 2 transcript. J. Biol. Chem. 1999;274:8153–8160. doi: 10.1074/jbc.274.12.8153. [DOI] [PubMed] [Google Scholar]
- Li Z., Huang C., Bao C., Chen L., Lin M., Wang X., Zhong G., Yu B., Hu W., Dai L. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015;22:256–264. doi: 10.1038/nsmb.2959. [DOI] [PubMed] [Google Scholar]
- Marr M.T., 2nd, D’Alessio J.A., Puig O., Tjian R. IRES-mediated functional coupling of transcription and translation amplifies insulin receptor feedback. Genes Dev. 2007;21:175–183. doi: 10.1101/gad.1506407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S.D., Gregersen L.H., Munschauer M. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- Miettinen T.P., Björklund M. Modified ribosome profiling reveals high abundance of ribosome protected mRNA fragments derived from 3′ untranslated regions. Nucleic Acids Res. 2015;43:1019–1034. doi: 10.1093/nar/gku1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganos S., Horiuchi J., Saitoe M. Mutations in the Drosophila insulin receptor substrate, CHICO, impair olfactory associative learning. Neurosci. Res. 2012;73:49–55. doi: 10.1016/j.neures.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Olson C.M., Donovan M.R., Spellberg M.J., Marr M.T., 2nd The insulin receptor cellular IRES confers resistance to eIF4A inhibition. eLife. 2013;2:e00542. doi: 10.7554/eLife.00542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori A., Toyama B.H., Harris M.S., Bock T., Iskar M., Bork P., Ingolia N.T., Hetzer M.W., Beck M. Integrated Transcriptome and Proteome Analyses Reveal Organ-Specific Proteome Deterioration in Old Rats. Cell Syst. 2015;1:224–237. doi: 10.1016/j.cels.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig O., Marr M.T., Ruhf M.L., Tjian R. Control of cell number by Drosophila FOXO: downstream and feedback regulation of the insulin receptor pathway. Genes Dev. 2003;17:2006–2020. doi: 10.1101/gad.1098703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J., Mann M., Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- Rybak-Wolf A., Stottmeister C., Glažar P., Jens M., Pino N., Giusti S., Hanan M., Behm M., Bartok O., Ashwal-Fluss R. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cell. 2015;58:870–885. doi: 10.1016/j.molcel.2015.03.027. [DOI] [PubMed] [Google Scholar]
- Sonenberg N., Hinnebusch A.G. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starke S., Jost I., Rossbach O., Schneider T., Schreiner S., Hung L.H., Bindereif A. Exon circularization requires canonical splice signals. Cell Rep. 2015;10:103–111. doi: 10.1016/j.celrep.2014.12.002. [DOI] [PubMed] [Google Scholar]
- Suzuki H., Zuo Y., Wang J., Zhang M.Q., Malhotra A., Mayeda A. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 2006;34:e63. doi: 10.1093/nar/gkl151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Wang Z. Efficient backsplicing produces translatable circular mRNAs. RNA. 2015;21:172–179. doi: 10.1261/rna.048272.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.L., Bao Y., Yee M.C., Barrett S.P., Hogan G.J., Olsen M.N., Dinneny J.R., Brown P.O., Salzman J. Circular RNA is expressed across the eukaryotic tree of life. PLoS ONE. 2014;9:e90859. doi: 10.1371/journal.pone.0090859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarten-Gabbay S., Elias-Kirma S., Nir R., Gritsenko A.A., Stern-Ginossar N., Yakhini Z., Weinberger A., Segal E. Comparative genetics. Systematic discovery of cap-independent translation sequences in human and viral genomes. Science. 2016;351 doi: 10.1126/science.aad4939. [DOI] [PubMed] [Google Scholar]
- Weiss R., Bartok O., Mezan S., Malka Y., Kadener S. Synergistic interactions between the molecular and neuronal circadian networks drive robust behavioral circadian rhythms in Drosophila melanogaster. PLoS Genet. 2014;10:e1004252. doi: 10.1371/journal.pgen.1004252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westholm J.O., Miura P., Olson S., Shenker S., Joseph B., Sanfilippo P., Celniker S.E., Graveley B.R., Lai E.C. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 2014;9:1966–1980. doi: 10.1016/j.celrep.2014.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.