

Abstract
Large-scale metabolic profiling requires the development of novel economical high-throughput analytical methods to facilitate characterization of systemic metabolic variation in population phenotypes. We report a fit-for-purpose direct infusion nanoelectrospray high-resolution mass spectrometry (DI-nESI-HRMS) method with time-of-flight detection for rapid targeted parallel analysis of over 40 urinary metabolites. The newly developed 2 min infusion method requires <10 μL of urine sample and generates high-resolution MS profiles in both positive and negative polarities, enabling further data mining and relative quantification of hundreds of metabolites. Here we present optimization of the DI-nESI-HRMS method in a detailed step-by-step guide and provide a workflow with rigorous quality assessment for large-scale studies. We demonstrate for the first time the application of the method for urinary metabolic profiling in human epidemiological investigations. Implementation of the presented DI-nESI-HRMS method enabled cost-efficient analysis of >10 000 24 h urine samples from the INTERMAP study in 12 weeks and >2200 spot urine samples from the ARIC study in <3 weeks with the required sensitivity and accuracy. We illustrate the application of the technique by characterizing the differences in metabolic phenotypes of the USA and Japanese population from the INTERMAP study.
Keywords: direct infusion mass spectrometry, metabolic profiling, molecular epidemiology, high-throughput analysis
1. Introduction
Technological advancements including automated sample handling, data processing, together with reduction in cost have seen an increase in the application of metabolic profiling approaches in molecular epidemiology.1,2 To achieve sufficient power to discover statistically significant associations at the population level, these studies require consecutive analysis of tens or hundreds thousands of samples. Analyses on a molecular epidemiology scale can deliver solutions for metabolome-wide association (MWAS) studies, exploring the statistical relationships between disease risk factors and metabolite concentrations in large human sample cohorts.3 One strategy for achieving a higher sample throughput in metabolic profiling is to completely omit the chromatographic separation phase by using direct infusion mass spectrometry (DIMS), which involves continuous ionization of a static sample.4,5 This analytical technique has been used in untargeted ESI–MS profiling studies of plant, food, and environmental samples as well as in animal and human biofluid and tissue samples for the last two decades.6 The first reports on the use of DIMS for ESI–MS profiling of crude fungal extracts date back to 1996.7 Application of DIMS with nESI, coupled to a computational workflow for global metabolic profiling of various tissue extracts using ultrahigh mass accuracy and resolution Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometry, has been significantly developed and optimized in the last 5 years.8−10 In addition to operating in profiling mode for untargeted phenotyping of human plasma11 and urine,5 DIMS has been successfully applied to the quantification of serotonin-related compounds in urine samples12 and free fatty acids in human serum.13 The comparison studies of the DIMS and LC–MS approaches for global metabolic profiling of human serum14 and targeted analysis of heat shock proteins in cell line models15 have shown comparable classification, prediction, and quantification capabilities of both methods with a clear gain in analysis time when using DIMS.
The main limitations of any DIMS method include the strong matrix effect and ion suppression due to the fact that all ionizable components of a sample are infused at the same time and competing for charge. These limitations can be minimized by employing nanoelectrospray ionization conveniently available in chip-based ionization interfaces such as the Advion TriVersa NanoMate. The lower flow rates achieved with nanoelectrospray improve ionization efficiencies for all analytes in a mixture. This is explained by the formation of reduced size charged droplets compared to conventional electrospray, which leads to fewer droplet fusion events, less solvent evaporation prior to ion release into the gas phase, and thus a larger portion of analyte available for MS analysis.16 Despite advances in MS technology and instrument resolution, DIMS methods also suffer from an inability to distinguish structural isomers, which requires chromatographic separation and additional off-line MS/MS experiments. The separation of isomeric compounds cannot always be attained in general LC–MS profiling methods. Another limitation of DIMS methods is the simultaneous presence of multiple types of MS signals for each metabolite in the spectrum in addition to its molecular ion, such as adducts and in-source fragments. This complicates the processing and interpretation of the data. Nevertheless, this phenomenon can deliver useful structural and biochemical information for biomarker discovery by using statistical correlation approaches.17,18
The choice of mass analyzer can affect the quality of the data obtained by DIMS. With ultrahigh mass accuracy and high-resolution mass spectrometers such as Orbitrap and FT-ICR the masses obtained during the measurement are expected to be close to the theoretical mass of metabolites, which facilitates the prediction of ion elemental formulas. However, the increase in mass accuracy and resolution can be accompanied by the loss of sensitivity and scan speed and decrease in detector dynamic range, requiring specific data acquisition approaches to mitigate these effects such as the previously reported spectral stitching technique.9 The hybrid quadrupole-time-of-flight (Q-TOF) instrument offers high-resolution mode (up to 50 000) and flexible scan rates, and their costs are more accessible for many research laboratories compared with the costs of purchasing and maintaining ultrahigh mass accuracy and high-resolution mass spectrometers.
Despite the aforementioned limitations, DI-nESI-HRMS methods offer an appealing option for rapid MS sample screening and parallel semiquantitative analysis of large sample numbers and can be applied to large cohorts of biobanked samples. Although the targeted LC–MS/MS methods performed on triple quadrupole mass spectrometers are highly precise and accurate, they are usually focused on specific metabolic pathways, and the panels of analyzed metabolites are limited.19,20 Wider coverage of metabolite space requires consecutive implementation of several targeted LC–MS/MS assays, which does not meet the increasing demands of time and cost-efficiency in metabolic phenotyping applied to large clinical and epidemiological sample sets. In addition, it can be economically inefficient and environmentally unfriendly due to the increased solvent consumption. It is evident that a decision must be taken to balance the number of measured analytes and selectivity of an assay against throughput and sensitivity. Recently, we have reported the development and application of a direct infusion nanoelectrospray high-resolution mass spectrometry (DI-nESI-HRMS) method with time-of-flight detection for multiplexed parallel metabolic profiling of urine samples.21 As a proof of concept, this method was applied to a relatively small set of 101 urine samples and was validated for eight targeted metabolites showing to be precise and accurate according to the standard requirements.
We and others have shown that the DI-nESI-HRMS method using the NanoMate system, which requires minimal amounts of sample owing to the injectable volume being on the order of nanoliters, ensures a stable instrument performance and high sensitivity and is not compromised by contamination.13,11,21 In a single rapid analytical high-resolution sweep, the method provides wide metabolome coverage including amino acids, TCA cycle metabolites, fatty acids, acylcarnitines, gut microbial cometabolites, and other, mostly polar, metabolites. The quantification capability provided by the current method allows characterization of population phenotypes and selection of representative samples for further UPLC–MS (MS/MS) analysis. Moreover, DI-nESI-HRMS provides high-resolution accurate mass global profiles of samples that can be further explored for discovery and semiquantification of biomarkers, enabling interrogation of their association with metabolic phenotypes and clinical outcomes.
The proposed DI-nESI-HRMS method for multiplexed parallel untargeted and targeted semiquantitative metabolic analysis of the urine samples is not an alternative to a conventional UPLC–MS (MS/MS) analysis but a valuable complementary tool in the research portfolio of large-scale metabolic phenotyping laboratories.19,20,22 The main purpose of this work is to show the applicability of the optimized DI-nESI methodology with a Q-TOF mass analyzer to measure multiple compounds in parallel, in accordance with standard requirements of good precision, accuracy, stability, robustness, and reproducibility on a scale of thousands of samples that is able to deliver MWAS solutions. To validate the DI-nESI-HRMS method, we applied it to two relatively large population cohorts, The INTERnational study of MAcro/micronutrients and blood Pressure (INTERMAP)23 and Atherosclerosis Risk in Communities (ARIC),24 which address hypertension and cardiovascular risk factors, respectively. On the basis of the previous 1H NMR spectroscopic MWAS analysis of >10 000 24 h urine specimens from the INTERMAP study,3,25,26 we aimed to extend coverage of the blood pressure related metabolome and to quantify a panel of metabolites with varying biological functions. A subcohort of >2200 spot urine samples from the ARIC study was used for targeted metabolic analysis to measure specific urinary markers of oxidative capacity and assess their role as predictors of incident type 2 diabetes. Analysis was based on 47 selected metabolites for quantification in each study tailored to the research question, with 16 metabolites common for the two studies allowing comparison of the performance of the method across different populations (Table 1 and Table S-1).
Table 1. List of Metabolites for Quantification, Their Biochemical Function, and Linear Range (μg/mL) from Both INTERMAP and ARIC Studies.
| metabolite | biochemical function | WS6–WS1 (INTERMAP/ARIC) (μg/mL) | study (INTERMAP/ARIC) |
|---|---|---|---|
| hydroxycinnamic acid | marker of polyphenols consumption | 0.1–3.3 | INTERMAP |
| acetylcarnitine | fatty acid oxidation | 0.05–1.7 | both |
| arginine | urea cycle | 0.005–0.17 | ARIC |
| ascorbic acid | vitamin C | 0.1–3.3 | INTERMAP |
| benzoic acid | Phe, Tyr metabolism | 0.5–16.7 | both |
| caffeic acid | marker of polyphenols consumption | 0.03–0.8 | INTERMAP |
| carnitine | fatty acids metabolism | 0.05–1.7 | both |
| cholic acid | bile acid | 0.05–1.7 | INTERMAP |
| citric acid | TCA cycle | 0.3–12.5 | both |
| citrulline | urea cycle | 0.01–0.4 | ARIC |
| cotinine | marker of smoking | 0.01–0.4 | INTERMAP |
| creatine | cell’s energy shuttle | 0.5–16.7 | both |
| creatinine | cell’s energy shuttle | 1.6–50 | both |
| daidzein | marker of soya consumption | 0.01–0.4 | INTERMAP |
| deoxycholic acid | bile acid | 0.03–0.8 | INTERMAP |
| genistein | marker of soya consumption | 0.01–0.4 | INTERMAP |
| glutamic acid | urea cycle, glucose-Ala cycle | 0.2–6.7 | both |
| glycocholic acid | bile acid | 0.03–0.8 | INTERMAP |
| glycodeoxycholic acid | bile acid | 0.03–0.8 | INTERMAP |
| hippuric acid | marker of polyphenols consumption | 0.8–25/1.0–33.3 | both |
| homovanillic acid | metabolite of dopamine | 0.8–25 | INTERMAP |
| hydroxybenzoic acid | derivative of benzoic acid | 0.1–3.3 | ARIC |
| indoxyl sulfate | Trp metabolism | 0.4–12.5/0.3–8.3 | both |
| isovalerylglycine | BCAA metabolism | 0.05–1.7 | INTERMAP |
| 2-oxoglutaric acid | TCA cycle | 0.1–3.3/0.2–6.7 | both |
| ketoleucine | BCAA degradation | 0.03–0.8 | both |
| kynurenine | Trp metabolism | 0.3–8.3 | INTERMAP |
| leucine | BCAA | 0.1–3.3 | both |
| malic acid | TCA cycle | 0.005–0.17 | ARIC |
| methylsuccinic acid | fatty acid oxidation | 0.05–1.7 | ARIC |
| N-acetylneuraminic acid | amino sugar metabolism | 0.1–3.3/0.05–1.7 | both |
| nicotinamide | vitamin B3 | 0.05–1.7 | ARIC |
| nicotine | marker of smoking | 0.03–0.8 | INTERMAP |
| nicotinic acid | nicotinate and nicotinamide metabolism | 0.1–3.3/0.02–0.8 | both |
| N-α-acetyl-l-ornithine | renal function marker | 0.05–1.7 | ARIC |
| ornithine | urea cycle | 0.05–1.7 | ARIC |
| phenylacetic acid | Phe and Tyr metabolism | 0.05–1.7 | both |
| phenylacetylglutamine | Phe and Tyr metabolism | 0.4–12.5 | INTERMAP |
| phenylalanine | amino acid | 0.05–1.7 | ARIC |
| proline betaine | marker of citrus consumption | 0.3–8.3 | INTERMAP |
| propionylcarnitine | fatty acid oxidation | 0.03–0.8 | INTERMAP |
| saccharin | artificial sweetener | 0.01–0.4 | INTERMAP |
| succinic acid | TCA cycle | 0.4–12.5/0.3–8.3 | both |
| tryptophan | amino acid | 0.05–1.7 | ARIC |
| tyramine | Phe and Tyr metabolism | 0.3–8.3 | INTERMAP |
| tyrosine | amino acid | 0.05–1.7 | ARIC |
2. Materials and Methods
2.1. Materials and Preparation of Standard Solutions
A description of the solvents and reference standards (Table S-1) is provided in Supporting Information (SI). The labeled and nonlabeled standard stock solutions were prepared at a concentration of 1 mg/mL in methanol or methanol/water mixture if the standard was not soluble in methanol only. The multianalyte mixture of labeled internal standards and the multianalyte mixture of nonlabeled calibrators were prepared at different levels of concentration by mixing stock solutions in the total volume of 10 mL. In this way, the multianalyte mixture of the nonlabeled standards with different levels of concentration for each analyte was used as the most concentrated calibrator, Working Solution (WS1). The concentrations of the standards in the WS1 are shown as the upper limit of the linear range in Table 1 for the INTERMAP and ARIC studies, respectively. More working solutions (WS2–WS6) used for generation of calibration curves and spiked validation quality control (QC) samples were prepared from WS1 by serial dilution. The concentration of each labeled internal standard was fixed and within the expected linear range for each metabolite.
2.2. Sample Preparation
The dilution factor of urine samples was optimized for each study separately by assessing the effect of dilution on the intensities of endogenous metabolites and the added internal standards.
Various reference and QC materials were developed and used for method optimization and assessment of data quality during the experiments. Pooled Study Reference samples (SR) were prepared for both studies by pooling together 10 μL aliquots of all urine samples. The pooled SR samples were diluted by the same factor as urine samples from each study. The SR dilution series were prepared by diluting pooled SR by the factors of 1/10, 1/20, 1/50, and 1/100 and were used to ensure that the detected MS features positively correlate with sample dilution.
Urine Sample Preparation
Aliquots of 10 μL of thawed urine samples stratified by the four populations (USA, U.K., Japan, and PRC) and two separate clinic visits (first-F and repeated-R) from the INTERMAP study were pipetted in randomized order into deep-well plates and diluted by a factor of 50. Aliquots of 10 μL of each urinary sample were mixed to obtain eight pooled SR samples for each population and visit. The pooled SR samples were also diluted by a factor of 50. In ARIC study, the individual urine samples and pooled SR sample were prepared in the same way as for the INTERMAP study using a dilution factor of 20.
Calibration Series and QC Samples
Calibration series and QC samples were prepared by pipetting 50 μL of diluted pooled SR samples in a well-plate, adding 25 μL of the multianalyte mixture of labeled internal standards at fixed concentration and 25 μL of the corresponding calibrator solution (both prepared in methanol), and making up the total volume to 150 μL by adding methanol to maintain 1:2 water/methanol proportion. In this way, the urine samples were diluted in a well-plate by a factor of 150 (1/50 and 50 μL/150 μL) for the INTERMAP study and by a factor of 60 (1/20 and 50 μL/150 μL) for ARIC study, while the calibrators used for the calibration series and QC samples were diluted six times in a well-plate (25 μL/150 μL). All of these factors have been taken into account for the quantification of metabolites and calculations of the limits of quantification (LOQ). The description of the analyte-free matrix for external calibration is presented in the SI.
Each sample plate, in addition to the study samples, contained a calibration series, SR and dilution SR series, validation QC samples (two samples prepared at three different concentration levels), and blank solutions. The sample plates were sealed and subjected to 1 min ultrasonication, followed by 10 min of centrifugation at 1500g at 4 °C before MS analysis.
2.3. Validation Experiments
Linear ranges, inter- and intraday accuracy and precision, long-term stability, and method robustness were assessed for the DI-nESI-HRMS method using FDA guidelines27,28 for the validation of bioanalytical methods.
A standard addition method in pooled SR samples was used by spiking the multianalyte series of nonlabeled standards into each calibrator solution and validation QC sample. The mixture of labeled analogues, used as internal standards, was spiked into each sample at a known fixed concentration. The MS quantification was performed using the ratio of intensities of the metabolite to the intensity of its internal standard. The response for each QC sample was compared with the generated calibration curve to give concentration values.
Validation Study
The validation study was performed over three separate days. On the first day, the well-plate contained the calibration series consisting of six points plus zero point (spiked with the mixture of labeled internal standards only), six replicates of validation QC samples prepared at three different concentration levels for each metabolite (low, medium, and high), which were defined for each metabolite from the corresponding linear range (Table 1). Blanks (1:2 water/methanol) were injected after the calibration series and each QC series to test carry-over. On the second and third days, each plate contained a calibration series and one replicate of the QC series. The calibration series and QC samples in analyte-free matrix were prepared in the same way. The intra- and interday accuracy and precision were calculated as closeness of a measured value to a known value of spiked concentration (%) and coefficient of variance (CV%), respectively. Method LOQ was determined as 10 times the standard deviation of the y intercept divided by the slope of the calibration curves obtained in analyte-free matrix.
Stability Study
The stability study was carried out by preparing three additional validation and QC series from the INTERMAP study to assess the stability of the samples. One of them was measured immediately, while the second one was measured after three freeze–thaw cycles and the third one after long-term (3 months) storage at −80 °C. The CV% values of measurements were evaluated. Additionally, the untargeted analysis of the full-scan data was done to assess possible changes in global urine profiles.
Robustness of the Method
The robustness of the method was assessed using the data from the spiked validation QC samples, dilution SR series, and pooled SR samples measured during the analysis of 144 sample plates from INTERMAP study and 33 sample plates from ARIC study. The DI-nESI-HRMS method robustness was also assessed from the analysis of a synthetic test mixture (TM). The details of these measurements are described in the SI.
2.4. Nanoelectrospray MS System
Chip-based nanoelectrospray infusion analysis was performed using the TriVersa NanoMate system (Advion BioSciences, Ithaca, New York) coupled to a Waters Synapt G2-Si (Waters MS Technologies, U.K.) operating in high-resolution continuum mode in negative- and positive-ion modes with automatic polarity switching. The MS experimental conditions are detailed in the SI.
2.5. INTERMAP and ARIC Study Cohorts
To exemplify the application of the DI-nESI-HRMS method, it was applied for the analysis of two independent epidemiological studies. The INTERnational study of MAcro/micronutrients and blood Pressure (INTERMAP) is an international population-based cross-sectional study on relations of multiple dietary factors to hypertension among 4680 men and women ages 40 to 59 years from 17 diverse population samples in China, Japan, United Kingdom, and the United States.23,25 Data collected according to a common protocol include eight blood pressure measurements, four 24 h dietary recalls, and two timed 24 h urine collections per person (>10 000 urine specimens in total). Borate preservative was added to the urine specimen bottles prior to collection.23 Institutional ethics committee approval was obtained for each site, and all participants gave written informed consent.
Atherosclerosis Risk in Communities (ARIC) community-based prospective cohort addresses cardiovascular risk factors and aims to measure urinary markers of oxidative capacity and assess their role as predictors of incident type 2 diabetes. The participants, 15 792 predominantly black and white adults, aged 45–64 years at baseline, were from four U.S. communities and attended four scheduled visits during the period 1987–1998.24 Written informed consent was obtained from all participants, and the study protocol was approved by institutional review boards at each clinical site. In this work we analyzed a subcohort of >2200 spot urine specimens collected on the fourth visit.
2.6. Data Preprocessing
Basic data visualization and QC were achieved using the MassLynx 4.1 software (Waters Corporation, U.K.). For the analysis, the raw data were converted to the mzML format with ProteoWizard software,29 followed by processing using in-house scripts in Python 2.7.4, as described previously.21 These included averaging of the acquired scans, linear correction of the mass accuracy drift using the observed m/z values of internal standard compounds, interpolation of the data to a final resolution of 0.00056 m/z, peak picking using the Savitzky–Golay algorithm, and local linear baseline estimation.30 For the untargeted analysis, a data matrix containing all of the spectra in one ion mode interpolated on a common m/z scale was used. Calibration curves plotting, quantification, and corresponding calculations were performed in Microsoft Excel. The full-scan spectra were preprocessed and normalized in MATLAB R2014a (MathworksTM). The preprocessing consisted of removal of background signals by subtracting the spectrum of a blank sample from each sample profile and removal of the m/z features showing high variation in SR samples.
2.7. Data Analysis
The measured concentration of metabolites (μg/mL) in urine samples was normalized by creatinine concentration (mM, measured as well by DI-nESI-HRMS method in each sample) and used as μmol/mmol creatinine for further analyses. Differences in metabolite concentrations in USA and Japanese populations from INTERMAP study were investigated by linear regression analysis using the default linear regression modeling tools (lm) in R.31 The dependent variable (initially expressed in metabolite concentration normalized by creatinine, μmol/mmol creatinine) was log-transformed with country (USA or Japan), gender (discrete variables), age, and BMI (continuous variables) used as explanatory variables, without interactions (y ≈ population + gender + age + BMI).
Principal Component Analysis (PCA) and Orthogonal Projection to Latent Structures–Discriminant Analysis (OPLS-DA)32 were performed in SIMCA 14 (Umetrics, Sweden) to further investigate metabolic phenotypic differences in global urine profiles of two populations. The discriminant features were annotated using additional MS/MS experiments, statistical spectroscopy (STOCSY17), and online databases (HMDB33 and Metlin34).
3. Results and Discussion
3.1. DI-nESI-HRMS Method Optimization for Large-Scale Metabolic Profiling
The requirements for validation of regulatory studies and clinical diagnostic assays are normally very strict.27,35,36 In accordance with the FDA guidelines,27 QC samples are incorporated into a study to demonstrate that the analytical method is sufficiently accurate, precise, and sensitive enough to measure the actual concentration of a compound. The acceptable levels for accuracy and precision in these studies are set at 15% by FDA guidelines (except for the LLOQ level QC, where 20% is acceptable).27 However, the method must be “fit-for-purpose”, and the validation criteria should be appropriate for the intended use and meet the predefined needs of the study.37 Multiplexed studies, targeting tens or hundreds of compounds in a single analysis, present a particular challenge, especially in large population studies with a high degree of within- and between-individual variability of urinary metabolites. Different levels of assays have been proposed, each of them obeying a certain level of rigor of validation. For biomarker studies, FDA guidelines are slightly more “relaxed”, following the 20 and 30% rule for accuracy and precision of QC samples at high and LLOQ levels, respectively, which is more appropriate for metabolic profiling studies.28,37 In the present study we used these acceptance criteria taking into account not only the biological variability of the metabolites in urine samples from different populations but also the limitations of the DIMS method mentioned in the Introduction.
Prior to starting the method validation work, we carried out several prevalidation studies to assess the stability and purity of the labeled and nonlabeled standards (the details are described in the SI), selectivity of the method, dilution factor for the urine samples from each study, and matrix effect using analyte-free matrix and established the concentration ranges for each metabolite. To facilitate the view of the multistep process, the workflow followed in this work for DI-nESI-HRMS method optimization and validation is depicted in Figure S-1 in the Supporting Information.
Selectivity
Method selectivity refers to its ability to measure each analyte in the presence of the others in the matrix. This issue needs special control using the DIMS methods as the chromatographic separation step is omitted. We assessed the selectivity of DI–nESI–MS by measuring the response factor for labeled and nonlabeled compounds in neat methanol. The serially diluted mixtures of standard stock solutions prepared at the same concentration at four different levels were infused into mass spectrometer. The ratios of the intensities of nonlabeled standard to its labeled internal standard were calculated for each level of concentration. For those standards where the ratio value was different from 1, additional MS measurements using the mixtures with different number of the standards were carried out. The standards that interfered with the quantification of endogenous metabolites by having very close accurate mass or experiencing a strong matrix effect were excluded from further analysis. The remaining metabolites together with their biochemical function, internal standards, and corresponding m/z values are listed in Table 1 and Table S-1.
In addition, the signal purity of metabolites of interest was analyzed by off-line MS/MS analysis of the pooled urine sample and compared with the MS/MS spectra obtained for the authentic standards in neat solvent. The MS/MS spectra obtained for the standards of glutamic acid and phenylacetylglutamine in neat methanol as well as in a spiked and nonspiked urine sample from the INTERMAP study are shown in Figure S-3. For some low-level metabolites the MS/MS spectra in pooled urine sample showed the interference from other compounds with close accurate mass caused by resolution of the quadrupole and the absence of chromatographic separation.
Dilution Study
The assessment of the optimal dilution factor for urine samples is a key step in method optimization for each sample cohort. We characterized the dilution factors for the urine samples from both INTERMAP and ARIC studies by assessing the effect of dilution (1/10, 1/20, 1/50, and 1/100) on the intensities of endogenous metabolites and the added internal standards. In DIMS, sample dilution improves ionization efficiency and reduces the matrix suppression effect caused by the presence of inorganic salts. We found that the optimal dilution factor for the urine samples from the INTERMAP study was 1/50, whereas for the urine samples from ARIC study it was 1/20. The INTERMAP urine samples were preserved by adding boric acid during the sample collection.23 The presence of boric acid in the urine specimens from the INTERMAP study seemed to enhance the ionization of analytes in DI-nESI-HRMS analysis. We calculated the ratio of instrument responses obtained at 1/20 and 1/50 dilutions for selected metabolites and their internal standards (Figure S-4) in the urine samples from the INTERMAP study. The relative increase in intensities of the labeled compounds was 100% in more diluted urine sample (1/50), while the relative decrease in intensities of endogenous metabolites was minimal (<20%). The full-scan spectra of 1/50 diluted pooled urine samples of the USA-F and Japan-F populations (F-first visit) from the INTERMAP study and a pooled urine sample from the ARIC study are shown in Figure 1. The difference in sample preparation for two different sample cohorts underscores the importance of this optimization step and highlights DI-nESI-HRMS method versatility. For successful implementation, the assessment of the urine dilution factor for each study is a key step. The selected factor should also be appropriate for the analysis of metabolites tailored to the research question. Lower level metabolites will require less dilution than metabolites expected to be present at higher concentrations in urine samples.
Figure 1.

Full-scan spectra acquired in negative ion mode of 1/50 diluted pooled urine samples of the USA and Japan populations (first visit) from the INTERMAP study and 1/20 diluted pooled urine sample from the ARIC study.
Matrix Effects
Matrix effects consist of the suppression or enhancement of ionization of analytes by the presence of matrix components in biological samples and can be very significant in MS analysis, especially in DIMS, where there is no LC separation involved. To account for this effect, which is particularly accentuated in large-scale epidemiological studies due to the high sample variability, the method of standard additions can be used for metabolite quantification, as we previously demonstrated in the proof-of-concept study using DI-nESI-HRMS.21 The MS quantification was performed by the back-calculation of the ratio of each selected metabolite to its internal standard and the slope of the calibration curve obtained for that metabolite in the pooled urine sample. Here, to show the reliability of our method, we compared the method of standard additions using the pooled urine sample as a matrix with the method of external calibration in the analyte-free matrix.38 The details of this parallel assay are presented in the SI. To show the parallelism between the two matrices, standard addition and external calibration curves were plotted for some metabolites from the INTERMAP study (Figure S-5). The figures of merit of the QC samples quantification by both approaches are shown in Tables S-2 and S-3. However, because of the high biological variability expected in multipopulation studies, the use of an external calibration curve can result in under- or overestimation of metabolite concentration, in particular, at the extremes of the biological range.
Linear Ranges of Metabolites
The most challenging step in the development of any multiplexed assay of biofluids, in particular, urine, is to determine individual concentration levels for each of the targeted analytes. For the present DI-nESI-HRMS method development, we performed the initial estimation of expected concentrations for each metabolite from the values reported in the literature and compared the experimentally obtained response of metabolites in urine samples to the response obtained for the authentic standards in neat solvent. During the prevalidation stage the linear ranges for each metabolite were continuously refined on the basis of the linearity and analysis of QC samples until the results were acceptable for all analytes of interest. The concentrations of metabolites in total pooled urine samples, calculated from the standard addition curves, compared to the expected levels of concentration from the literature33 for the ARIC and INTERMAP studies are provided in Tables S-3 and S-4, respectively. In addition, the tables contain the LOQ values in nmol/mL estimated from the standard deviation of the y-intercept of the external calibration curves obtained in analyte-free matrix. These values are within or lower than the expected concentrations ranges, and thus our method is appropriate for the quantification of selected metabolites. The metabolite concentrations in mM quantified by DI-nESI-HRMS method using the standard addition curve for eight different pooled urine samples from the INTERMAP study and the pooled urine sample from the ARIC study are shown in Table S-5 in the SI.
3.2. DI-nESI-HRMS Method Validation
Accuracy and Precision
For each metabolite of interest, the linear range (LLOQ and ULOQ as the lower and upper extremes of the range shown in the Table 1), accuracy (%), and precision (CV%) were determined from the replicates of calibration curves and inter- and intraday analyses of the validation QC sample prepared at three different levels of concentration according to the FDA guidelines for bioanalytical method validation.27 The intra- and interday accuracy and precision values for the metabolites quantified in the validation QC samples from USA-F and Japan-F populations from the INTERMAP study shown in Table S-6 were acceptable according to the criteria adopted for this study. As observed from Table S-6, 8 of 37 quantified metabolites showed the values of error in accuracy and precision >30%. Some of them were metabolites expected to be present at low concentration level in urine such as ketoleucine or phenylacetic acid. It was surprising that three of the high concentration metabolites, namely, creatinine, creatine, and citric acid, were not as accurate and precise as expected according to the acceptance criteria of our study. We assume that this phenomenon could be due to their high concentration and likely saturation of the detector even at high sample dilution. In addition, citric acid was previously shown to form complexes with boric acid added as preservative to all INTERMAP urine samples.39 Their accurate analysis by DI-nESI-HRMS may require greater than 1/50 dilution of urine samples. During the analysis of the study samples, the in-study validation was carried out by analyzing the validation QC samples during the measurements of each sample plate to ensure that the assays continued to perform according to defined acceptable standards. As an example, the results of the in-study validation of the QC samples measured in sample plates from the ARIC study are presented in Table S-7. These results show that the optimized method gives accurate and reproducible results for the majority of selected metabolites. The sheer number of urine samples to be analyzed imposes a necessity to balance the number of analytes to quantify (with high degree of accuracy and precision) against the requirements of sample throughput.
Stability Study
The stability assay performed for standard stock solutions and biofluid samples is a key requirement for method development and validation. Freeze–thaw cycles may affect the stability of some targeted compounds in the course of the large-scale long-term studies. For the INTERMAP study, it has been previously shown that cryostorage and the use of boric acid preservation do not compromise the biochemical integrity of the urinary specimens.40−42
In the present study, three additional calibration and QC samples series were prepared to assess method performance and stability of the INTERMAP urine samples. One of the series was measured immediately after preparation, while the second one was measured following three freeze–thaw cycles and the third one was measured after the three months storage at −80 °C. The acceptance criteria for the stability study were set as for the validation study.
To demonstrate the effect of various storage protocols on the stability of metabolites, more detailed analysis was performed with the data from two different populations from the INTERMAP study, USA and Japan. The CV% values of the slopes of the standard addition curves and concentrations of the QC samples for selected metabolites measured as an example in Japan-F pooled urine samples in three stability series (Figure S-6) were <20% for the most part of metabolites. The values >20% were obtained for some metabolites measured in the QC samples prepared at low concentration level. The acquired full-scan data for stability validation series for USA and Japan populations were used for untargeted fingerprinting to further assess general sample integrity. The PCA scores plots obtained for pooled SR samples (1) and QC samples spiked at low (2) and medium (3) concentration levels in three stability validation series (fresh, green; three freeze–thaw cycles, blue; long-term storage, red) for the USA (U) and Japan (J) populations are shown in Figure 2. The separation of the global profiles observed in the plots is due to the biological difference between two populations (PC2 separating Japan and USA samples: Figure 2) and different levels of concentration (PC1 and PC3 in Figure 2 separating nonspiked and spiked urine samples for negative and positive ion mode, respectively). Almost perfect matching of the global profiles of the same type of sample (1, 2, and 3) for each population (U and J) confirmed the presence of minimal analytical variability and general sample stability with respect to the freeze–thaw cycles and long-term storage, which guarantees reliable and reproducible results using this protocol.
Figure 2.
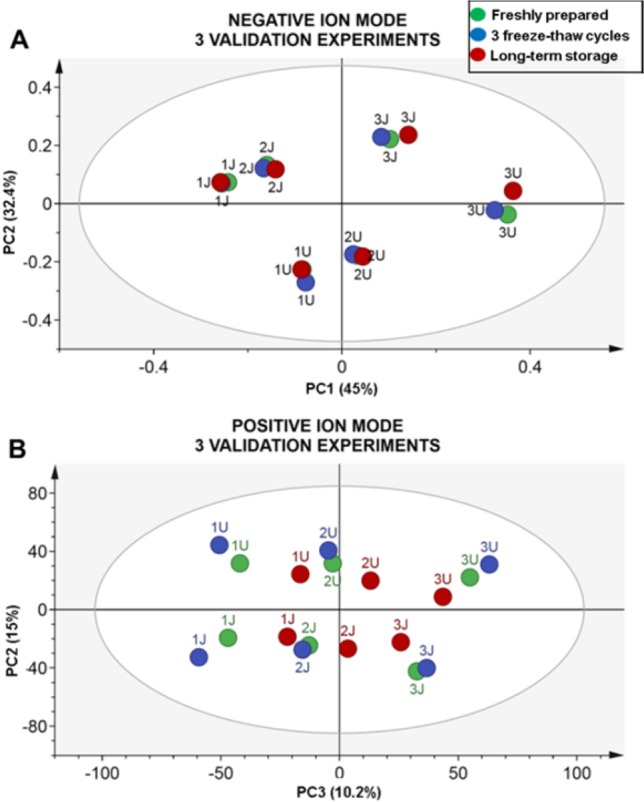
PCA scores plots obtained for USA-F (U) and Japan-F (J) nonspiked pooled urine samples (1), QC at low (2), and QC at medium (3) concentration level in three different validation series (freshly prepared, green; three freeze–thaw cycles, blue; long-term storage, red) in negative, R2X = 87.1%, Q2X = 75.6% (A), and positive, R2X = 85.7, Q2X = 76.0% (B) ion modes.
Method Robustness
The analysis of large-scale epidemiological sample cohorts may span over several months; therefore, the methods applied for these long-term metabolic studies should meet the requirements of robustness and repeatability.43
The DI–nESI–MS analysis of >10 000 INTERMAP samples was completed in 12 weeks in batches (sample plates) created for each population and clinic visit separately. To demonstrate that our method is robust for the long-term analysis, we used the data obtained for different types of SR and QC samples from randomly selected sample plates (14 and 10) from the USA and Japanese populations (first visit), respectively. The calibration series, dilution SR series, and validation QC samples from each selected sample plates were processed for targeted analysis for two populations. For the untargeted global profile analysis, only pooled SR samples from two populations were processed together to assess the analytical against biological variability.
The interbatch CV% values presented in Table S-8 obtained for each metabolite did not exceed 20% for the most part of quantified metabolites in validation QC samples. The largest CV% values were obtained for the most diluted SR samples 1/100 (Table S-8). However, for the majority of the metabolites the 30% cutoff level acceptable for biomarker assays was not exceeded. From the selected sample plates, the concentration values for six metabolites quantified in positive and negative ion modes in global QC samples are shown in Figure S-7. The dashed lines represent the mean concentration ±2 standard deviation values.
The untargeted analysis performed on the pooled SR samples from USA and Japan populations together allowed estimation of the biological and analytical variability. Using OPLS-DA fitted models to explore systematic differences in two populations, clear separation of the two populations was apparent, as demonstrated by the cross-validated scores plots in Figure S-8 obtained for the full-scan global profile analysis in negative (A) and positive (B) ion modes. The model characteristics describe models with high predictive values (Q2Y = 0.908 and 0.921, respectively), indicating that the biological differences are higher compared with analytical variability, which was <20%, as shown from the targeted analysis.
The precision of the targeted analysis was also assessed for the ARIC study using CV% values of the metabolites quantified in pooled SR samples and dilution SR series measured in all 33 sample plates (Table S-9). For the majority of metabolites in the pooled SR and 1/10 diluted SR samples the CV% values did not exceed 20%, with only 2 of 26 metabolites having CV% of 20–30%. The higher CV% values were observed, as expected, for the most diluted SR sample 1/50. Additionally, the analytical error and system stability was assessed using the synthetic test mixture (TM) analyzed using the same experimental protocol as urine samples. The details and results of this assessment are presented in the Supporting Information and in Figure S-8. In brief, using normalization by the total intensity, CV% values for all analytes from the mixture measured over 3 days and after instrument maintenance did not exceed 20%.
3.3. Metabolic Differences between Population Phenotypes: Application of DI-nESI-HRMS
The DI-nESI-HRMS method proposed herein provides a framework for high-throughput metabolic phenotyping of biological samples from large biobank sample collections.
After thorough optimization and validation of the method, we applied it for the analysis of >10 000 24 h collection urine samples from the INTERMAP study and >2200 spot urine samples from the ARIC study. With all of the data we obtained in a time window on the order of weeks, we are now able to explore metabolic phenotypes of different populations and obtain systems-level information reflecting genetic (inheritable) and environmental (e.g., diet-related) influences. As an example of DI-nESI-HRMS method application at the population level, we present the comparison of metabolic profiles of USA and Japanese population from the INTERMAP study. We used both semiquantitative and global profile data and applied univariate and multivariate statistical approaches to identify metabolic differences between the two populations.
Metabolite concentrations obtained from the targeted analyses of 2075 urine samples from USA (first visit; 1036 male and 1039 female) and 1119 samples from Japan (first visit; 560 male and 559 female) were normalized by creatinine measured by DI-nESI-HRMS in each sample, and log-transformed prior to the calculation of a linear regression model with concentration (as μmol/mmol creatinine) as the dependent variable and population (USA, Japan), gender, age, and BMI as the explanatory variables, which are also covariates of each other. The test comparing the levels of metabolites in two populations revealed significant differences for 32 out of 39 quantified metabolites (Table 2), p < 0.05. Metabolites that differed the most between the two population were ascorbic acid, citric acid, hippuric acid, vanillylmandelic acid, and p-cresol sulfate (higher in the USA population) and daidzein and (iso)valerylglycine (higher in the Japanese population). In the previous exploratory analysis of the INTERMAP samples using 1H NMR data3 it was shown that the urinary excretion patterns of East Asian and Western populations were different due to contrasting diets and diet-related major risk factors of hypertension, and the discriminant metabolites were predominantly of dietary origin. Here, we show higher levels of excretion of daidzein, a metabolite related to soy consumption, in the Japanese population. Ascorbic acid is a component of several medications, for example, mixtures with paracetamol used to treat fever and pain, the metabolites of which detected by DI-nESI-HRMS were higher in urine samples from the USA population. Analgesic use was previously reported to be higher in Western populations of the INTERMAP study.44 We also find that gut–microbial mammalian cometabolites, hippuric acid, p-cresol sulfate, vanillylmandelic acid, phenylacetylglutamine, and indoxyl sulfate were discriminant and higher in the USA population. This is consistent with previous findings,3,41 but also, analysis by mass spectrometry has revealed the detection and estimation of other phenolic compounds not previously seen by 1H NMR spectroscopy and thus facilitates deeper analysis of gut–microbial mammalian interactions in different populations.
Table 2. Metabolites Quantified by DI–nESI–MS Differing in USA (positive correlation) and Japanese (negative correlation) Populations and Their Association with Covariates Gender, Age, and BMIa.
| country |
gender |
BMI |
age |
|||||
|---|---|---|---|---|---|---|---|---|
| metabolite | β | p value | β | p value | β | p value | β | p value |
| hydroxycinnamic acid (isomers) | 0.004 | 5.05 × 10–1 | 0.048 | 1.22 × 10–18 | –0.002 | 6.64 × 10–5 | 0.005 | 4.42 × 10–19 |
| ascorbic acid | 0.564 | 4.85 × 10–94 | 0.337 | 2.78 × 10–49 | –0.007 | 1.17 × 10–3 | 0.013 | 1.05 × 10–9 |
| benzoic acid | 0.062 | 2.48 × 10–2 | 0.228 | 2.40 × 10–22 | –0.020 | 2.94 × 10–17 | 0.014 | 4.73 × 10–11 |
| caffeic acid | 0.019 | 1.26 × 10–20 | 0.024 | 8.69 × 10–43 | –0.002 | 9.36 × 10–25. | 0.001 | 2.22 × 10–8 |
| cholic acid | –0.026 | 2.91 × 10–11 | 0.010 | 2.97 × 10–3 | 0.0004 | 2.22 × 10–1 | 0.001 | 1.16 × 10–4 |
| citric acid | 0.246 | 4.91 × 10–29 | 0.521 | 1.17 × 10–156 | –0.009 | 2.70 × 10–7 | 0.006 | 1.35 × 10–3 |
| daidzein | –0.014 | 5.24 × 10–34 | 0.007 | 5.03 × 10–14 | –0.0003 | 3.42 × 10–3 | 0.000 | 1.22 × 10–3 |
| deoxycholic acid | 0.007 | 2.97 × 10–12 | 0.009 | 6.98 × 10–30 | –0.0004 | 4.91 × 10–6 | 0.000 | 3.51 × 10–8 |
| fumaric acid | 0.115 | 7.69 × 10–26 | 0.071 | 1.82 × 10–14 | –0.001 | 5.41 × 10–1 | 0.004 | 8.98 × 10–6 |
| genistein | –0.008 | 3.89 × 10–20 | 0.003 | 7.02 × 10–4 | –0.0001 | 1.34 × 10–1 | 0.000 | 1.15 × 10–1 |
| glutamic acid | 0.014 | 2.36 × 10–1 | 0.210 | 5.27 × 10–95 | –0.003 | 7.35 × 10–4 | 0.008 | 2.05 × 10–19 |
| glycocholic acid | –0.007 | 8.38 × 10–12 | 0.010 | 2.88 × 10–28 | –0.0003 | 4.41 × 10–4 | 0.000 | 4.29 × 10–1 |
| glycodeoxycholic acid | –0.004 | 1.18 × 10–5 | 0.007 | 6.71 × 10–25 | –0.0001 | 6.79 × 10–2 | 0.000 | 1.56 × 10–2 |
| hippuric acid | 0.643 | 1.48 × 10–84 | 0.306 | 6.08 × 10–29 | –0.020 | 2.25 × 10–13 | 0.013 | 5.79 × 10–7 |
| homovanillic acid | 0.013 | 4.17 × 10–1 | 0.235 | 9.74 × 10–65 | –0.014 | 1.54 × 10–25 | 0.011 | 2.75 × 10–17 |
| indoxyl sulfate | 0.056 | 8.83 × 10–5 | 0.173 | 1.26 × 10–44 | –0.003 | 4.65 × 10–3 | 0.004 | 9.62 × 10–4 |
| isovalerylglycine | –0.084 | 4.39 × 10–47 | 0.093 | 1.19 × 10–78 | –0.004 | 8.28 × 10–14 | 0.002 | 3.10 × 10–5 |
| 2-oxoglutaric acid | –0.206 | 1.70 × 10–33 | 0.342 | 4.97 × 10–116 | 0.002 | 2.31 × 10–1 | 0.005 | 1.69 × 10–4 |
| ketoleucine | 0.107 | 1.64 × 10–15 | 0.122 | 2.20 × 10–26 | –0.005 | 1.08 × 10–5 | 0.007 | 4.21 × 10–10 |
| leucine | 0.011 | 5.58 × 10–1 | 0.256 | 4.11 × 10–56 | –0.007 | 3.45 × 10–5 | 0.012 | 6.48 × 10–17 |
| N-acetylneuraminic acid | 0.058 | 6.00 × 10–11 | 0.157 | 1.29 × 10–92 | 0.001 | 1.79 × 10–1 | 0.006 | 2.97 × 10–17 |
| phenylacetylglutamine | 0.186 | 2.82 × 10–15 | 0.287 | 9.39 × 10–46 | –0.007 | 3.81 × 10–4 | 0.012 | 6.03 × 10–10 |
| saccharin | 0.084 | 6.52 × 10–4 | 0.086 | 3.37 × 10–5 | 0.006 | 3.47 × 10–3 | 0.001 | 6.55 × 10–1 |
| succinic acid | 0.155 | 7.44 × 10–28 | 0.235 | 2.85 × 10–82 | 0.001 | 4.09 × 10–1 | 0.007 | 2.81 × 10–10 |
| vanillylmandelic acid | 0.180 | 2.83 × 10–57 | 0.175 | 7.92 × 10–74 | –0.008 | 1.53 × 10–15 | 0.008 | 3.86 × 10–20 |
| p-cresol sulfate | 0.737 | 6.48 × 10–55 | 0.560 | 7.73 × 10–45 | –0.020 | 3.18 × 10–7 | 0.015 | 3.07 × 10–5 |
| phenylalanine | 0.011 | 1.48 × 10–4 | 0.033 | 1.04 × 10–41 | –0.001 | 4.15 × 10–3 | 0.001 | 2.96 × 10–7 |
| acetylcarnitine | 0.004 | 6.89 × 10–1 | 0.003 | 7.02 × 10–1 | 0.001 | 4.93 × 10–1 | –0.001 | 1.47 × 10–1 |
| carnitine | –0.008 | 6.01 × 10–1 | 0.019 | 1.55 × 10–1 | 0.002 | 9.40 × 10–2 | 0.001 | 6.00 × 10–1 |
| cotinine | –0.016 | 6.94 × 10–7 | –0.017 | 8.52 × 10–10 | –0.001 | 1.57 × 10–7 | 0.000 | 7.08 × 10–1 |
| creatine | –0.126 | 7.74 × 10–9 | 0.498 | 1.36 × 10–145 | –0.006 | 6.33 × 10–4 | 0.012 | 1.77 × 10–12 |
| creatinine | –0.142 | 2.15 × 10–40 | –0.156 | 2.63 × 10–65 | 0.008 | 6.29 × 10–19 | –0.008 | 5.47 × 10–24 |
| kynurenine | 0.073 | 7.82 × 10–7 | 0.122 | 7.58 × 10–22 | –0.003 | 6.69 × 10–3 | 0.007 | 2.19 × 10–10 |
| nicotine | 0.021 | 2.31 × 10–3 | –0.008 | 1.89 × 10–1 | –0.001 | 2.15 × 10–1 | 0.001 | 3.92 × 10–2 |
| nicotinic acid | –0.239 | 3.79 × 10–47 | 0.092 | 4.08 × 10–11 | –0.003 | 4.97 × 10–2 | 0.000 | 7.96 × 10–1 |
| phenylacetic acid | –0.002 | 3.03 × 10–1 | 0.006 | 3.52 × 10–6 | 0.000 | 4.00 × 10–2 | 0.000 | 8.74 × 10–2 |
| phenylethylamine | 0.125 | 8.68 × 10–9 | 0.110 | 2.31 × 10–9 | –0.008 | 4.85 × 10–6 | 0.009 | 4.87 × 10–7 |
| proline betaine | 0.201 | 7.99 × 10–9 | 0.148 | 5.12 × 10–7 | –0.014 | 1.95 × 10–6 | 0.015 | 1.16 × 10–7 |
| tyramine | –0.035 | 1.22 × 10–1 | 0.129 | 1.15 × 10–11 | –0.008 | 4.41 × 10–5 | 0.007 | 1.06 × 10–4 |
Correlation coefficients (β) and p values are listed for all metabolites. The correlation of metabolites with each variable is calculated by linear regression with other variables fixed.
The linear regression model also provided effect size estimates (β) and p values for the covariates that allow assessment of the differences in metabolite levels related to other factors such as gender, age, and BMI to be assessed (Table 2). We observed that some metabolites were significantly different between males and females in each of the two populations. Citric acid, creatine, and 2-oxoglutaric acid were the most discriminant metabolites and were excreted in higher concentration in females. Higher concentration of these metabolites in female urine has already been described.45 The comparison of several metabolite levels in two different populations stratified by gender is shown in the boxplots in Figure 3 and Figure S-10. The correlation of metabolite concentrations obtained by DI-nESI-HRMS was also assessed in relation to BMI (Table 2). Some of the measured metabolites have been previously shown to be associated with BMI and adiposity in an exploratory study of urinary metabolic signatures using 1H NMR spectroscopic data.46 In this study we found that citric acid, hippuric acid, ketoleucine, phenylacetylglutamine, p-cresol sulfate, and proline betaine were inversely correlated with BMI, and N-acetylneuraminic acid, creatinine, and acetylcarnitine were directly correlated with BMI, which underpins previous findings.
Figure 3.

Box plots showing difference in population (Japan and USA) and gender metabolite concentration levels obtained from DI-nESI-HRMS by the standard addition method.
To further explore the differences in metabolic phenotypes of USA and Japanese populations, we modeled the global MS metabolic profiles using multivariate approaches. Randomly selected 449 high-resolution metabolic profiles (225 male and 224 female) from the USA population and 451 high-resolution metabolic profiles (240 male and 211 female) from the Japanese population obtained in positive and negative ionization modes were preprocessed as described above and, after exclusion of the samples with high concentration of glucose (m/z 203.05 [M + Na]), were analyzed by PCA and OPLS-DA. The OPLS-DA cross-validated score plots of the DI-nESI-HRMS data (positive and negative ionization modes) shown in Figure 4 provide more evidence of clear discrimination between the USA and Japanese populations based on metabolic phenotype. Both models yielded high predictive values (Q2Y = 0.608 and 0.680, respectively), indicating a high degree of biological differences between the two populations. The loadings S-plots corresponding to both OPLS-DA models are illustrated in Figure S-11 and allow the observation and the assignment of the major features driving the separation between the two populations. In negative ion mode, these were mostly attributed to drug metabolites (such as acetaminophen and its glucuronide and sulfate and aspirin metabolites) excreted at higher concentration in USA population along with hippuric acid, citric acid, and p-cresol sulfate. Uric acid (m/z 167.02) and its fragment ion (m/z 124.01) were among the metabolites excreted in higher concentration in Japanese population. The excretion of uric acid can be related to the consumption of seafood. In positive ion mode, the putatively identified metabolites found at higher levels in USA urine samples, were isomers of 1- and 3-methylhistidine (m/z 170.10), creatinine, and proline betaine (H+ and Na+ adducts). Both 3-methylhistidine and creatinine are related to muscle turnover, whereas 1-methylhistidine is related to meat consumption and proline betaine is a biomarker of citrus fruit consumption. We applied statistical total correlation spectroscopy (STOCSY) to DI-nESI-HRMS data to assist in the assignment some of these discriminatory metabolites. An example of a spectra-like STOCSY plot illustrated in Figure 5 shows the correlation of the signal at m/z 170.10 detected in positive ion mode with the signals at m/z 141.07, m/z 126.10, and m/z 96.07 that are in-source fragments of 1- and 3-methylhistidine. The MS/MS spectrum for the ion at m/z 170.10 is shown in the Figure S-12 in the SI. The same approach was used to assign uric acid and its fragment in negative ion mode. In negative ionization mode, we also found an ion at m/z 182.00 to be present at higher concentration in the USA population. The STOCSY analysis revealed the correlation of this ion with a small signal at 105.96, which allowed the assignment of saccharin and its fragment ion. These preliminary results further highlight diet-related metabolites that drive metabolic differences between populations. The comparison of population phenotypes requires further data mining using a range of statistical approaches, metabolite structure elucidation, and the use of metabolic networks to translate metabolic spectroscopic data into biological knowledge.
Figure 4.

OPLS-DA cross-validated scores plots for the DI-nESI-HRMS data obtained in positive and negative ionization modes for the 449 and 451 urine samples from USA (blue) and Japanese (green) populations.
Figure 5.

STOCSY on the DI-nESI-HRMS full-scan data in positive ionization mode driven from the peak at m/z 170.10 (1- and 3-methylhistidine) highlighting correlations to the in-source fragments at (m/z 141.07, m/z 126.10, and m/z 96.07).
4. Conclusions
The current trend toward deep metabolic phenotyping of tens and hundreds of thousands of biobanked samples to gain an understanding of pathological conditions, physiological stimuli, or gene–environmental interactions on population-scale demands the urgent development of high-throughput analytical approaches and the creation of dedicated phenotyping centers.
In this work we demonstrated the use of a high-throughput direct infusion nanoelectrospray high-resolution mass spectrometry (DI-nESI-HRMS) method with the time-of-flight detection for parallel targeted analysis of multiple metabolites in urine samples for large population studies using two epidemiological sample cohorts, INTERMAP and ARIC, as exemplar data sets. We propose a workflow for optimization of this analytical platform for metabolic phenotyping on a scale of molecular epidemiology. The acceptance criteria for analytical quality were established taking into account the complexity of the samples, population variability, the nature of multiplexed assays, and the limitation of the experimental platform. For successful implementation, method optimization is a key step consisting of careful and detailed prevalidation assays to assess method selectivity, urine dilution factor, matrix effect, and established concentration ranges for each metabolite. The MS quantification was performed by back-calculation of the ratio of each metabolite to its internal standard and the slope of the standard addition calibration curve obtained in the pooled urine sample. The reliability of this approach was tested by comparing it with the method of external calibration curve obtained in an analyte-free matrix. The method has shown the intra- and interday accuracy and precision values for targeted metabolites in accordance with the adopted acceptance criteria. The analysis of sample stability under different measurement and storage conditions as well as robustness study using sample batches from different population and analysis time points from the two cohorts proved that the method is suitable for large-scale long-term epidemiological studies.
The main advantages accruing from this method are significantly decreased time of analysis (2 min for acquiring metabolic profiles in both positive and negative polarities), an easy experimental design due to instrumental simplicity and data processing requirements, and the possibility to avoid the problems related to LC column deterioration and LC system maintenance, which all increase the robustness of the analytical procedure and reduce associated costs. The profiling of 10 000 urine specimens in one polarity by reversed-phase UPLC–MS would span 100 days with 24 h continuous measurement cycle, whereas the same nonstop 24 h regime by DI-nESI-HRMS permitted analysis of these samples in two polarities in <30 days. The difference in costs of UPLC–MS profiling in only one polarity can be estimated to be 3 times higher than the costs of the DI-nESI-HRMS analysis in two polarities. The data presented here demonstrate that the application of DI-nESI-HRMS method is feasible for large-scale urinary metabolic phenotyping in molecular epidemiology with high degree of reproducibility and stability.
In conclusion, we propose the DI-nESI-HRMS method as a complementary and time-efficient tool in the research portfolio for molecular epidemiologists to define metabolic phenotypes and establish reference ranges in human populations. The use of DI-nESI-HRMS for large-scale epidemiological studies is an optimal and low-cost means of accelerating MS analysis, compared with conventional UPLC–MS methods.
Acknowledgments
The research was supported by grant R01 HL084228 from the National Heart, Lung, and Blood Institute, National Institutes of Health, by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Imperial College Healthcare NHS Trust and Imperial College London. We thank the MRC-NIHR National Phenome Centre, which is supported by the U.K. Medical Research Council (MRC) (in association with National Institute for Health Research (NIHR) England) Grant MC_PC_12025. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.6b01003.
Experimental section contains detailed description of the preparation of standard solutions, MS experimental conditions, and test mixture analysis. Table S-1. List of metabolites and internal standards. Tables S-2 and S-3. Comparison of standard addition and external calibration curves (INTERMAP and ARIC study, respectively). Table S-4. Concentration of metabolites measured in a pooled urine sample and LOQ value (INTERMAP study). Table S-5. Concentration of metabolites in mM calculated by standard addition method for eight pooled SR from the INTERMAP study. Table S-6. Parameters of method validation (INTERMAP) study. Table S-7. Parameters of in-study validation (ARIC study). Tables S-8 and S-9. Parameters of the method robustness (INTERMAP and ARIC study, respectively). Figure S-1. Flowchart for DI-nESI-HRMS method optimization. Figure S-2. 1H NMR spectra of labeled standards. Figure S-3. MS/MS spectra of metabolites. Figure S-4, histogram of metabolite and labeled standards intensities at different dilutions. Figure S-5. Calibration curves obtained in pooled urine sample by the method of standard additions and in analyte-free matrix. Figure S-6. CV% values for the QC samples in stability assay. Figure S-7. Concentration of metabolites measured in QC samples measured over 12 weeks in the INTERMAP study. Figure S-8. OPLS-DA scores plot from stability study. Figure S-9. Results of Test mixture analysis. Figure S-10. Box plot showing difference in metabolite levels in two different populations. Figure S-11. Loadings S-plots from OPLS-DA model comparing two populations. Figure S-12. MS/MS spectrum of the feature m/z 170.10 detected in urine samples in positive ion mode. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Tzoulaki I.; Ebbels T. M. D.; Valdes A.; Elliott P.; Ioannidis J. P. A. Design and Analysis of Metabolomics Studies in Epidemiologic Research: A Primer on -Omic Technologies. Am. J. Epidemiol. 2014, 180 (2), 129–139. 10.1093/aje/kwu143. [DOI] [PubMed] [Google Scholar]
- Bictash M.; Ebbels T. M.; Chan Q.; Loo R. L.; Yap I. K. S.; Brown I. J.; de Iorio M.; Daviglus M. L.; Holmes E.; Stamler J.; Nicholson J. K.; Elliott P. Opening up the ″Black Box″: Metabolic phenotyping and metabolome-wide association studies in epidemiology. Journal of Clinical Epidemiology 2010, 63 (9), 970–979. 10.1016/j.jclinepi.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E.; Loo R. L.; Stamler J.; Bictash M.; Yap I. K. S.; Chan Q.; Ebbels T.; De Iorio M.; Brown I. J.; Veselkov K. A.; Daviglus M. L.; Kesteloot H.; Ueshima H.; Zhao L. C.; Nicholson J. K.; Elliott P. Human metabolic phenotype diversity and its association with diet and blood pressure. Nature 2008, 453 (7193), 396–U50. 10.1038/nature06882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrer T.; Zamboni N. High-throughput discovery metabolomics. Curr. Opin. Biotechnol. 2015, 31, 73–78. 10.1016/j.copbio.2014.08.006. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Dominguez R.; Castilla-Quintero R.; Garcia-Barrera T.; Gomez-Ariza J. L. Development of a metabolomic approach based on urine samples and direct infusion mass spectrometry. Anal. Biochem. 2014, 465, 20–27. 10.1016/j.ab.2014.07.016. [DOI] [PubMed] [Google Scholar]
- Draper J.; Lloyd A. J.; Goodacre R.; Beckmann M. Flow infusion electrospray ionisation mass spectrometry for high throughput, non-targeted metabolite fingerprinting: a review. Metabolomics 2013, 9 (1), 4–29. 10.1007/s11306-012-0449-x. [DOI] [Google Scholar]
- Smedsgaard J.; Frisvad J. C. Using direct electrospray mass spectrometry in taxonomy and secondary metabolite profiling of crude fungal extracts. J. Microbiol. Methods 1996, 25 (1), 5–17. 10.1016/0167-7012(95)00073-9. [DOI] [Google Scholar]
- Kirwan J. A.; Broadhurst D. I.; Davidson R. L.; Viant M. R. Characterising and correcting batch variation in an automated direct infusion mass spectrometry (DIMS) metabolomics workflow. Anal. Bioanal. Chem. 2013, 405 (15), 5147–5157. 10.1007/s00216-013-6856-7. [DOI] [PubMed] [Google Scholar]
- Southam A. D.; Weber R. J.; Engel J.; Jones M. R.; Viant M. R. A complete workflow for high-resolution spectral-stitching nanoelectrospray direct-infusion mass-spectrometry-based metabolomics and lipidomics. Nat. Protoc. 2017, 12 (2), 255–273. 10.1038/nprot.2016.156. [DOI] [PubMed] [Google Scholar]
- Kirwan J. A.; Weber R. J.; Broadhurst D. I.; Viant M. R. Direct infusion mass spectrometry metabolomics dataset: a benchmark for data processing and quality control. Sci. Data 2014, 1, 140012. 10.1038/sdata.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boernsen K. O.; Gatzek S.; Imbert G. Controlled protein precipitation in combination with chip-based nanospray infusion mass spectrometry. An approach for metabolomics profiling of plasma. Anal. Chem. 2005, 77 (22), 7255–7264. 10.1021/ac0508604. [DOI] [PubMed] [Google Scholar]
- Moriarty M.; Lehane M.; O’Connell B.; Keeley H.; Furey A. Development of a nano-electrospray MSn method for the analysis of serotonin and related compounds in urine using a LTQ-orbitrap mass spectrometer. Talanta 2012, 90, 1–11. 10.1016/j.talanta.2011.11.085. [DOI] [PubMed] [Google Scholar]
- Zhang Y. P.; Qiu L.; Wang Y. M.; Qin X. Z.; Li Z. L. High-throughput and high-sensitivity quantitative analysis of serum unsaturated fatty acids by chip-based nanoelectrospray ionization-Fourier transform ion cyclotron resonance mass spectrometry: Early stage diagnostic biomarkers of pancreatic cancer. Analyst 2014, 139 (7), 1697–1706. 10.1039/c3an02130k. [DOI] [PubMed] [Google Scholar]
- Lin L.; Yu Q. A.; Yan X. M.; Hang W.; Zheng J. X.; Xing J. C.; Huang B. L. Direct infusion mass spectrometry or liquid chromatography mass spectrometry for human metabonomics? A serum metabonomic study of kidney cancer. Analyst 2010, 135 (11), 2970–2978. 10.1039/c0an00265h. [DOI] [PubMed] [Google Scholar]
- Xiang Y.; Koomen J. M. Evaluation of Direct Infusion-Multiple Reaction Monitoring Mass Spectrometry for Quantification of Heat Shock Proteins. Anal. Chem. 2012, 84 (4), 1981–1986. 10.1021/ac203011j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K. Q.; Page J. S.; Smith R. D. Charge competition and the linear dynamic range of detection in electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15 (10), 1416–1423. 10.1016/j.jasms.2004.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloarec O.; Dumas M. E.; Craig A.; Barton R. H.; Trygg J.; Hudson J.; Blancher C.; Gauguier D.; Lindon J. C.; Holmes E.; Nicholson J. Statistical total correlation spectroscopy: An exploratory approach for latent biomarker identification from metabolic H-1 NMR data sets. Anal. Chem. 2005, 77 (5), 1282–1289. 10.1021/ac048630x. [DOI] [PubMed] [Google Scholar]
- Posma J. M.; Garcia-Perez I.; De Iorio M.; Lindon J. C.; Elliott P.; Holmes E.; Ebbels T. M. D.; Nicholson J. K. Subset Optimization by Reference Matching (STORM): An Optimized Statistical Approach for Recovery of Metabolic Biomarker Structural Information from 1H NMR Spectra of Biofluids. Anal. Chem. 2012, 84 (24), 10694–10701. 10.1021/ac302360v. [DOI] [PubMed] [Google Scholar]
- Sarafian M. H.; Lewis M. R.; Pechlivanis A.; Ralphs S.; McPhail M. J. W.; Patel V. C.; Dumas M. E.; Holmes E.; Nicholson J. K. Bile Acid Profiling and Quantification in Biofluids Using Ultra-Performance Liquid Chromatography Tandem Mass Spectrometry. Anal. Chem. 2015, 87 (19), 9662–9670. 10.1021/acs.analchem.5b01556. [DOI] [PubMed] [Google Scholar]
- Wolfer A. M.; Gaudin M.; Taylor-Robinson S. D.; Holmes E.; Nicholson J. K. Development and Validation of a High-Throughput Ultrahigh-Performance Liquid Chromatography-Mass Spectrometry Approach for Screening of Oxylipins and Their Precursors. Anal. Chem. 2015, 87 (23), 11721–11731. 10.1021/acs.analchem.5b02794. [DOI] [PubMed] [Google Scholar]
- Chekmeneva E.; Correia G.; Denes J.; Gomez-Romero M.; Wijeyesekera A.; Perenyi D. R.; Koot Y.; Boomsma C.; Want E. J.; Dixon P. H.; Macklon N. S.; Chan Q.; Takats Z.; Nicholson J. K.; Holmes E. Development of nanoelectrospray high resolution isotope dilution mass spectrometry for targeted quantitative analysis of urinary metabolites: application to population profiling and clinical studies. Anal. Methods 2015, 7 (12), 5122–5133. 10.1039/C5AY00850F. [DOI] [Google Scholar]
- Gray N.; Lewis M. R.; Plumb R. S.; Wilson I. D.; Nicholson J. K. High-Throughput Microbore UPLC-MS Metabolic Phenotyping of Urine for Large-Scale Epidemiology Studies. J. Proteome Res. 2015, 14 (6), 2714–2721. 10.1021/acs.jproteome.5b00203. [DOI] [PubMed] [Google Scholar]
- Stamler J.; Elliott P.; Dennis B.; Dyer A. R.; Kesteloot H.; Liu K.; Ueshima H.; Zhou B. F. INTERMAP: background, aims, design, methods, and descriptive statistics (nondietary). J. Hum. Hypertens. 2003, 17 (9), 591–608. 10.1038/sj.jhh.1001603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams O. D. The Atherosclerosis Risk in Communities (Aric) Study - Design and Objectives. Am. J. Epidemiol. 1989, 129 (4), 687–702. 10.1093/oxfordjournals.aje.a115184. [DOI] [PubMed] [Google Scholar]
- Yap I. K. S.; Brown I. J.; Chan Q.; Wijeyesekera A.; Garcia-Perez I.; Bictash M.; Loo R. L.; Chadeau-Hyam M.; Ebbels T.; De Iorio M.; Maibaum E.; Zhao L. C.; Kesteloot H.; Daviglus M. L.; Stamler J.; Nicholson J. K.; Elliott P.; Holmes E. Metabolome-Wide Association Study Identifies Multiple Biomarkers that Discriminate North and South Chinese Populations at Differing Risks of Cardiovascular Disease INTERMAP Study. J. Proteome Res. 2010, 9 (12), 6647–6654. 10.1021/pr100798r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijeyesekera A.; Clarke P. A.; Bictash M.; Brown I. J.; Fidock M.; Ryckmans T.; Yap I. K. S.; Chan Q.; Stamler J.; Elliott P.; Holmes E.; Nicholson J. K. Quantitative UPLC-MS/MS analysis of the gut microbial co-metabolites phenylacetylglutamine, 4-cresyl sulphate and hippurate in human urine: INTERMAP Study. Anal. Methods 2012, 4 (1), 65–72. 10.1039/C1AY05427A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA Guidance for Industry, Bioanalytical Method Validation. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm368107.pdf.
- Dunn W. B.; Wilson I. D.; Nicholls A. W.; Broadhurst D. The importance of experimental design and QC samples in large-scale and MS-driven untargeted metabolomic studies of humans. Bioanalysis 2012, 4 (18), 2249–2264. 10.4155/bio.12.204. [DOI] [PubMed] [Google Scholar]
- Kessner D.; Chambers M.; Burke R.; Agus D.; Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 2008, 24 (21), 2534–2536. 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitzky A.; Golay M. J. E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36 (8), 1627–1639. 10.1021/ac60214a047. [DOI] [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2011. [Google Scholar]
- Bylesjo M.; Rantalainen M.; Cloarec O.; Nicholson J. K.; Holmes E.; Trygg J. OPLS discriminant analysis: combining the strengths of PLS-DA and SIMCA classification. J. Chemom. 2006, 20 (8–10), 341–351. 10.1002/cem.1006. [DOI] [Google Scholar]
- Bouatra S.; Aziat F.; Mandal R.; Guo A. C.; Wilson M. R.; Knox C.; Bjorndahl T. C.; Krishnamurthy R.; Saleem F.; Liu P.; Dame Z. T.; Poelzer J.; Huynh J.; Yallou F. S.; Psychogios N.; Dong E.; Bogumil R.; Roehring C.; Wishart D. S. The human urine metabolome. PLoS One 2013, 8 (9), e73076. 10.1371/journal.pone.0073076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. A.; O’Maille G.; Want E. J.; Qin C.; Trauger S. A.; Brandon T. R.; Custodio D. E.; Abagyan R.; Siuzdak G. METLIN - A metabolite mass spectral database. Ther. Drug Monit. 2005, 27 (6), 747–751. 10.1097/01.ftd.0000179845.53213.39. [DOI] [PubMed] [Google Scholar]
- Buick A. R.; Doig M. V.; Jeal S. C.; Land G. S.; Mcdowall R. D. Method Validation in the Bioanalytical Laboratory. J. Pharm. Biomed. Anal. 1990, 8 (8–12), 629–637. 10.1016/0731-7085(90)80093-5. [DOI] [PubMed] [Google Scholar]
- Viswanathan C. T.; Bansal S.; Booth B.; DeStefano A. J.; Rose M. J.; Sailstad J.; Shah V. P.; Skelly J. P.; Swann P. G.; Weiner R. Quantitative bioanalytical methods validation and implementation: Best practices for chromatographic and ligand binding assays. Pharm. Res. 2007, 24 (10), 1962–1973. 10.1007/s11095-007-9291-7. [DOI] [PubMed] [Google Scholar]
- Lee J. W.; Devanarayan V.; Barrett Y. C.; Weiner R.; Allinson J.; Fountain S.; Keller S.; Weinryb I.; Green M.; Duan L.; Rogers J. A.; Millham R.; O’Brien P. J.; Sailstad J.; Khan M.; Ray C.; Wagner J. A. Fit-for-purpose method development and validation for successful biomarker measurement. Pharm. Res. 2006, 23 (2), 312–328. 10.1007/s11095-005-9045-3. [DOI] [PubMed] [Google Scholar]
- Jones B. R.; Schultz G. A.; Eckstein J. A.; Ackermann B. L. Surrogate matrix and surrogate analyte approaches for definitive quantitation of endogenous biomolecules. Bioanalysis 2012, 4 (19), 2343–2356. 10.4155/bio.12.200. [DOI] [PubMed] [Google Scholar]
- Smith L. M.; Maher A. D.; Want E. J.; Elliott P.; Stamler J.; Hawkes G. E.; Holmes E.; Lindon J. C.; Nicholson J. K. Large-scale human metabolic phenotyping and molecular epidemiological studies via 1H NMR spectroscopy of urine: investigation of borate preservation. Anal. Chem. 2009, 81 (12), 4847–56. 10.1021/ac9004875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gika H. S.; Theodoridis G. A.; Wingate J. E.; Wilson I. D. Within-Day Reproducibility of an HPLC–MS-Based Method for Metabonomic Analysis: Application to Human Urine. J. Proteome Res. 2007, 6, 3291–3303. 10.1021/pr070183p. [DOI] [PubMed] [Google Scholar]
- Dumas M. E.; Maibaum E. C.; Teague C.; Ueshima H.; Zhou B. F.; Lindon J. C.; Nicholson J. K.; Stamler J.; Elliott P.; Chan Q.; Holmes E. Assessment of analytical reproducibility of 1H NMR spectroscopy based metabonomics for large-scale epidemiological research: the INTERMAP study. Anal. Chem. 2006, 78 (7), 2199–2208. 10.1021/ac0517085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton R. H.; Nicholson J. K.; Elliott P.; Holmes E. High-throughput 1H NMR-based metabolic analysis of human serum and urine for large-scale epidemiological studies: validation study. International Journal of Epidemiology 2008, 37, 31–40. 10.1093/ije/dym284. [DOI] [PubMed] [Google Scholar]
- Zelena E.; Dunn W. B.; Broadhurst D.; Francis-McIntyre S.; Carroll K. M.; Begley P.; O’Hagan S.; Knowles J. D.; Halsall A.; Wilson I. D.; Kell D. B. Development of a Robust and Repeatable UPLC-MS Method for the Long-Term Metabolomic Study of Human Serum. Anal. Chem. 2009, 81 (4), 1357–1364. 10.1021/ac8019366. [DOI] [PubMed] [Google Scholar]
- Loo R. L.; Chan Q.; Brown I. J.; Robertson C. E.; Stamler J.; Nicholson J. K.; Holmes E.; Elliott P. A comparison of self-reported analgesic use and detection of urinary ibuprofen and acetaminophen metabolites by means of metabonomics: the INTERMAP Study. Am. J. Epidemiol. 2012, 175 (4), 348–58. 10.1093/aje/kwr292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenot E. A.; Roux A.; Xu Y.; Ezan E.; Junot C. Analysis of the Human Adult Urinary Metabolome Variations with Age, Body Mass Index, and Gender by Implementing a Comprehensive Workflow for Univariate and OPLS Statistical Analyses. J. Proteome Res. 2015, 14 (8), 3322–3335. 10.1021/acs.jproteome.5b00354. [DOI] [PubMed] [Google Scholar]
- Elliott P.; Posma J. M.; Chan Q.; Garcia-Perez I.; Wijeyesekera A.; Bictash M.; Ebbels T. M.; Ueshima H.; Zhao L.; van Horn L.; Daviglus M.; Stamler J.; Holmes E.; Nicholson J. K. Urinary metabolic signatures of human adiposity. Sci. Transl. Med. 2015, 7 (285), 285ra62. 10.1126/scitranslmed.aaa5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.