

Abstract
Catalysis of H2 production and oxidation reactions is critical in renewable energy systems based around H2 as a clean fuel, but the present reliance on platinum-based catalysts is not sustainable. In nature, H2 is oxidized at minimal overpotential and high turnover frequencies at [NiFe] catalytic sites in hydrogenase enzymes. Although an outline mechanism has been established for the [NiFe] hydrogenases involving heterolytic cleavage of H2 followed by a first and then second transfer of a proton and electron away from the active site, details remain vague concerning how the proton transfers are facilitated by the protein environment close to the active site. Furthermore, although [NiFe] hydrogenases from different organisms or cellular environments share a common active site, they exhibit a broad range of catalytic characteristics indicating the importance of subtle changes in the surrounding protein in controlling their behavior. Here we review recent time-resolved infrared (IR) spectroscopic studies and IR spectroelectrochemical studies carried out in situ during electrocatalytic turnover. Additionally, we re-evaluate the significant body of IR spectroscopic data on hydrogenase active site states determined through more conventional solution studies, in order to highlight mechanistic steps that seem to apply generally across the [NiFe] hydrogenases, as well as steps which so far seem limited to specific groups of these enzymes. This analysis is intended to help focus attention on the key open questions where further work is needed to assess important aspects of proton and electron transfer in the mechanism of [NiFe] hydrogenases.
Keywords: hydrogenase mechanism, hydrogenase electrocatalysis, infrared spectroscopy, Raman spectroscopy, NRVS, proton-coupled electron transfer
1. Introduction
Hydrogenases are ancient enzymes that evolved to allow primitive organisms to extract energy from the H2-rich primordial environment. The ability to oxidize or produce H2 is prevalent in the microbial world and has emerged through three convergent pathways that have yielded enzymes with [Fe], [FeFe], and [NiFe] catalytic centers.1 The [NiFe] hydrogenases tend to be more robust and have attracted significant interest from chemists because their turnover frequencies, reaching >1000 s–1 for H2 oxidation, rival that of Pt active sites.1−3 Achieving a detailed structure–functional understanding of these enzymes therefore offers promise for informing the design of non-noble-metal H2-splitting catalysts.4 Here we address mechanistic questions relating to the catalytic cycle of [NiFe] hydrogenases, focusing on insight obtained from recent vibrational spectroscopic studies into proton- and electron-transfer steps.
The [NiFe] family of hydrogenases share a core composition of a “large” subunit comprising the [NiFe] bimetallic active site together with a smaller electron-transfer subunit containing iron sulfur clusters which facilitate long-range transfer of electrons released during H2 oxidation or required during H+ reduction.1 This pair of catalytic and electron-transfer subunits recur in a modular fashion, coupled with other functional subunits, in hydrogenases with diverse physiological roles ranging from energy conservation to cellular redox balancing and disposal of excess reducing equivalents.5,6 The bimetallic [NiFe] catalytic site is coordinated to the protein via two terminal cysteine ligands to Ni and two cysteines that bridge the Ni and Fe, and the Fe is additionally coordinated by CO and two CN– ligands which keep the Fe site low spin (Figure 1A). Amino acids surrounding, but not directly coordinated to, the active site are also important in proton-transfer steps in the mechanism of [NiFe] hydrogenases and additional amino acids which are highly conserved across different [NiFe] hydrogenases are shown in Figure 1B.
Figure 1.
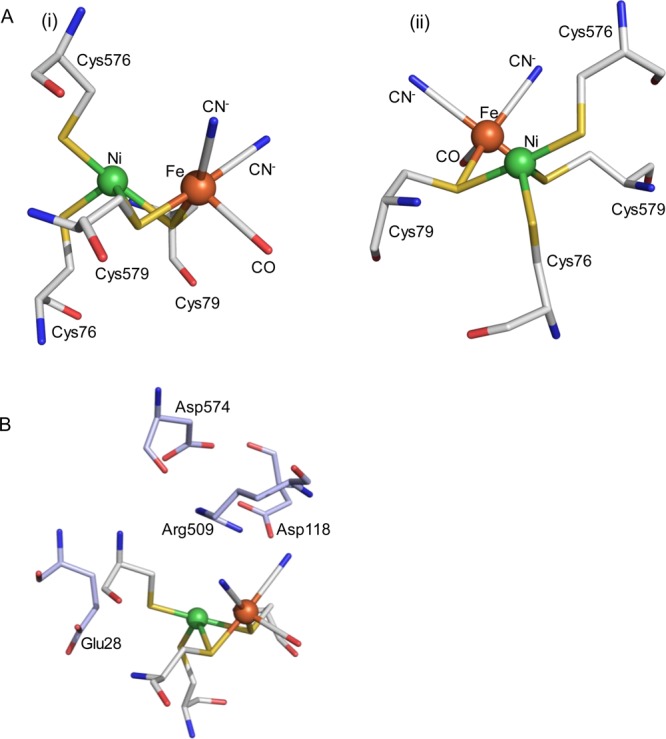
Highly conserved bimetallic active site of [NiFe] hydrogenases. Numbering refers to the amino acid sequence for E. coli Hyd-1, and we use this numbering throughout. Panel (A) shows two different orientations of the same structure to highlight the coordination geometry around the metals. Panel (B) presents the same structure, with the position of additional highly conserved amino acid residues shown. The active site structure is representative, but taken from crystallographic data for E. coli Hyd-1, PDB code 3USE, and prepared using Pymol.7
Mechanistic studies have focused on a small subset of this broad family of enzymes, and it has largely been assumed that a single mechanism should apply to all [NiFe] hydrogenases. The fact that hydrogenases operate on the smallest possible substrate molecule, H2, has so far prevented identification of a substrate-bound Michaelis–Menten complex, although there have been suggestions from computational and experimental studies regarding the nature of a transient H2 complex that precedes heterolytic cleavage of H2.8−10 The challenges are compounded by the difficulty in identifying the location of H atoms crystallographically within a large protein molecule.11
The deceptively simple oxidation of H2 to release two protons and two electrons necessarily involves coordinated proton and electron-transfer processes, and our understanding of these steps in [NiFe] hydrogenases remains limited so far. Electron transfer clearly requires, as a first step, outer-sphere electron tunnelling from the active site to the proximal iron sulfur cluster. Several long-range proton-transfer pathways involving both amino acids and ordered water molecules have been discussed.12−18,11 There are several well-conserved amino acid residues close to the active site of [NiFe] hydrogenases, and therefore, multiple possibilities exist for the initial steps in proton transfer during catalysis, as we discuss below.
Vibrational spectroscopy, and in particular infrared (IR) spectroscopy, has played an important role in characterizing the active site states of hydrogenases, and indeed provided the first identification of the diatomic ligands detected in an early crystal structure of a [NiFe] hydrogenase from Desulfovibrio gigas as the biologically unusual CO and CN– groups.19−22 Subsequently, a number of [NiFe] hydrogenases have been characterized through solution IR studies that focus on shifts in the vibrational bands arising from the active site CO and CN– ligands according to changes in electron density at the active site.1,23 The position of the CO stretching band, νCO, is particularly diagnostic because an increase in electron density at the active site increases back-donation to π* orbitals of the ligand, weakening the CO bond and shifting the νCO band to lower wavenumber (relative energy). Solution IR spectroelectrochemical studies were thus of particular importance in developing an understanding of the different redox states of [NiFe] hydrogenases.1,23,24 These are typically carried out in a transmission cell incorporating a gold mini-grid working electrode for electron transfer to hydrogenase solutions, mediated via a system of soluble small-molecule redox molecules.1,23,25Table 1 lists νCO positions for the main states observed for [NiFe] hydrogenases that have been characterized most thoroughly by IR spectroscopy, and we return to a more detailed analysis of this data below.
Table 1. Infrared peak positions (in cm–1) of the intrinsic active site νCO band for a range of [NiFe] hydrogenases in different redox or coordination states.a.
| hydrogenase (group) | Ni-Ab | Ni-B | Ni-SUb | Ni-SIrb | Nia-SI | Nia-C | Nia-LI | Nia-LII | Nia-LIII | Nia-RI | Nia-RII | Nia-RIII |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D. vulgaris Miyazaki F (1)26−29 | 1956 | 1955 | 1946c | 1922 | 1943 | 1961 | - | 1911 | 1890 | 1948 | 1932 | 1919 |
| D. gigas (1)30 | 1947 | 1946 | 1950 | 1914 | 1934 | 1952 | - | - | - | 1940 | 1923 | - |
| D. fructosovorans (1)31 | 1947 | 1946 | 1950 | 1913 | 1933 | 1951 | - | - | - | 1938 | 1922 | - |
| A. vinosum MBH (1)32 | 1945 | 1943 | 1948 | 1910 | 1931 | 1951 | 1898 | - | - | 1936 | 1921 | 1913 |
| T. roseopersicina MBH (1)33 | - | 1944 | - | - | 1930 | 1951 | 1899 | - | - | - | 1921 | 1915d |
| R. eutrophaMBH (1)34 | - | 1948 | 1943 | 1910 | 1936 | 1957 | 1899e | - | - | 1948 | 1926 | 1919 |
| E. coli Hyd-1 (1)35,36 | - | 1943 | - | - | 1927 | 1949 | 1898f | 1877f | 1867f | - | 1922 | 1914 |
| A. aeolicus MBH (1)37−39 | - | 1939 | - | - | 1927 | 1949 | 1862 | 1876 | 1900 | - | 1910 | - |
| R. eutrophaRH (2)40 | - | 1951 | 1957 | 1938 | 1942 | 1961 | - | - | - | 1949 | 1934 | 1918 |
| P. furiosus SH1 (3)41,42 | - | 1960 | - | 1931 | 1950 | 1967 | 1917 | 1922 | - | 1954 | 1940 | 1931 |
| R. eutropha SH (3)43,44 | - | 1957 | - | 1946 | - | 1961 | - | - | - | 1958 | 1922 | 1913 |
| Synechocystis SH (3)45 | - | 1957 | - | - | 1947 | 1968 | - | - | - | 1955 | - | - |
The precise νCO peak positions are dependent upon the measurement conditions so there are small variations throughout the literature.
The Ni-A state is an inactive state at the Ni-B redox level that is slow to undergo reductive activation; the Ni-SU and Ni-SIr states are additional states at the Nia-SI redox level that are not thought to be involved in catalysis but are intermediates in activation of the oxidized Ni-A and Ni-B states, respectively.1
The Ni-SU state of D. vulgaris Miyazaki F was initially assigned as 1946 cm–126 and is generally cited with this value,1,3 although it has also been assigned as 1958 cm–1.28,46
We have reassigned this state as a Nia-R state based upon its appearance upon extended reductive treatment.
In a later report on R. eutropha MBH,47 a single Nia-L state with νCO at ∼1910 cm–1 was observed upon illumination at cryogenic temperature; the origin of this discrepancy is unclear.
We have reassigned our previously reported Nia-L states of E. coli Hyd-1 in order of decreasing νCO band position, all other Nia-L states are reported as in the primary literature.
The ease of following the relatively intense CO and CN stretching vibrational bands means that IR studies on hydrogenases have focused almost entirely on the active site. They have been complemented by electron paramagnetic resonance (EPR) spectroscopy studies on frozen hydrogenase samples which reveal not only the paramagnetic NiIII and NiI active site states but also the redox properties of the iron sulfur relay clusters.24,43,48−52 The outline mechanism for H2 oxidation (or the reverse sequence for proton reduction) shown in Scheme 1 has emerged from combined analysis of these studies together with DFT studies that target the active site region.
Scheme 1. Skeletal Mechanism for H2 Oxidation and Production at the Active Site of [NiFe] Hydrogenases.
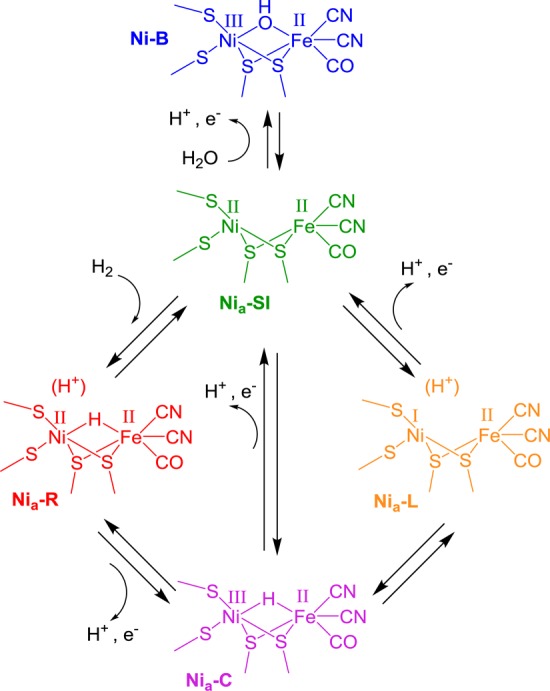
The colors used here for specific states of the active site are utilized throughout this Perspective. Catalytically active states are labeled “Nia-X”, where X = SI, C, R, or L. The oxidized, inactive, Ni-B state is included in the scheme as it is observed in spectroscopic data presented elsewhere in this Perspective.
[NiFe] hydrogenases tend to be isolated in a mixture of oxidized, inactive states at the NiIII FeII level, as well as states at the NiII FeII level.3 The Ni-B (blue) and Nia-SI (green) states shown in Scheme 1 are important states at these redox levels, although other oxidized forms are observed in some hydrogenases.1,44 Nia-SI is directly relevant to catalysis and is the state that undergoes initial interaction with H2 (hence the subscript “a” for “active”); however, Ni-B requires reductive activation by electrons or H2 and is reformed reversibly when the enzyme becomes oxidized. Heterolytic cleavage of H2 yields the diamagnetic NiII FeII state, Nia-R (red), with a bridging hydride between the metals and a proton residing on, or close to, the active site. Transfer of the proton and an electron away from the active site gives Nia-C (purple). Further transfer of a second proton and electron away from the active site recovers Nia-SI, either directly or via a NiI intermediate. This NiI intermediate is termed Nia-L because in earlier studies it was observed during illumination of Nia-C (hence L, for light), usually only at low temperatures.24 Nia-C and Nia-L are at the same redox level and are simply tautomers in which electrons from the hydride in Nia-C are transferred to the Ni to give the formally NiI species in Nia-L with the proton residing on a nearby basic residue. Simulation of IR and advanced EPR data has indicated the presence of a metal–metal bond in Nia-L,53 accounting for high electron density on Fe in this state as evidenced by low νCO band positions (Table 1). There is no particular reason why the acceptor site for the first proton (in Nia-R) and the second proton (in Nia-L) need be the same.54 In Scheme 1, the proton acceptor sites are not specified, nor is the redox state of the iron–sulfur electron acceptor cluster proximal to the active site, and these points are discussed in more detail below.
Questions of the proton-transfer pathways in hydrogenases are highly relevant to the development of functional biomimetic catalysts. For example, Dubois and co-workers have demonstrated significant improvements in the activity of nickel complexes for H2 oxidation or proton reduction when an amine-based proton relay system is included.55 Developments in the second coordination sphere of biomimetic systems have been described recently.56
Much of the spectroscopic work on hydrogenases has focused on identification of the different redox and coordination states exhibited by the active site of [NiFe] hydrogenases, but more recently, the focus has shifted to examining transitions between states and the relevance of these transitions to catalytic turnover. The approach known as protein film electrochemistry permits efficient catalytic turnover to be controlled as a function of electrode potential in a range of solution conditions,57−59 and although this technique itself does not provide structural information, it is useful when combined with IR spectroscopy to allow spectra to be collected during steady state turnover of hydrogenase (an approach we have termed protein film infrared electrochemistry, PFIRE).35 More transient states have also been probed in time-resolved infrared spectroscopic techniques in which transitions between states of a hydrogenase have been triggered by the release of caged electrons or by photolysis of light-sensitive states of the enzyme.41,42,60 Attention has also focused on the role of conserved amino acids in the region around the active site in controlling proton transfer,12,54,61−63 and the correlation between the redox state of the proximal cluster and that of the active site.36,60 These studies have been aided by the availability of site-directed variants of [NiFe] hydrogenases with particular amino acid exchanges in the region of the active site and the proximal iron sulfur cluster.12,54,61,63 However, one limitation to recent studies of [NiFe] hydrogenases has been that each vibrational spectroscopy approach has only been applied to one or two different hydrogenases, and it remains to be seen whether the findings from each approach represent aspects of a common [NiFe] hydrogenase mechanism or whether certain details are specific to individual hydrogenases.
Classification of [NiFe] Hydrogenases
Although this Perspective focuses on mechanistic aspects of H2 oxidation in [NiFe] hydrogenases, some discussion of the biological classification of [NiFe] hydrogenases is necessary in order to appreciate the structural and functional variations between different enzymes of this type, and in turn the possible influence of the protein environment on mechanistic steps in H2 oxidation at the active site. Classification of hydrogenases is mainly based on phylogenetic analysis of their amino acid sequences and is surveyed briefly here, but has been discussed in detail and tabulated elsewhere.5,6
Many of the well-studied [NiFe] hydrogenases belong to the group of membrane-bound H2-uptake hydrogenases known as Group 1. X-ray crystallographic structures are available for a number of members of this Group,11,63−71 and several have been characterized extensively using vibrational spectroscopic methods (Table 1, and discussed in more detail below). The Group I hydrogenases incorporate a large subunit, housing the [NiFe] active site, a small subunit comprising a chain of three electron-relay iron sulfur clusters, and may also comprise one or more membrane anchor subunits. Some of these enzymes are likely to exist as multimers of the large and small subunit pair. The typical large/small subunit arrangement is shown in Figure 2A, represented by Escherichia coli hydrogenase 1 (Hyd-1). Subclassifications within Group 1 distinguish the well-studied “ancestral” or “prototypical” [NiFe] hydrogenases, which are typically sensitive to O2, from the “O2-tolerant” [NiFe] hydrogenases. The former link H2 oxidation to a range of inorganic electron acceptors such as sulfate or nitrate, whereas “O2-tolerant” [NiFe] hydrogenases are often involved in aerobic respiration. Certain structural characteristics appear to be associated with O2-tolerant hydrogenases. Crystallographically characterized enzymes of this type, including the membrane bound hydrogenase (MBH) from Ralstonia eutropha,65,72 Hyd-1 from E. coli,69,73 and MBH from Hydrogenovibrio marinus(64) which have an unusual high potential [4Fe-3S] electron relay iron sulfur cluster in their small subunit in the position proximal to the active site, coordinated by 6 Cys residues (Figure 2B).74 The MBH from Aquifex aeolicus also has a high potential proximal cluster capable of transferring two electrons to/from the active site.49 This contrasts with the standard [4Fe-4S] proximal cluster coordinated by 4 Cys residues (Figure 2C) that is typical of the O2-sensitive [NiFe] hydrogenases. The biological function of other subgroups within Group 1 is less well-established. It has been suggested that the so-called “actinobacterial type” [NiFe] hydrogenases, some of which can scavenge H2 at levels found in the lower atmosphere, may be appropriately included in Group 1 rather than segregated in Group 5.6 These enzymes comprise a 3Cys1Asp coordinated proximal [4Fe-4S] cluster (Figure 2D).75
Figure 2.

(A) Arrangement of the large and small subunit in E. coli Hyd-1, a member of Group 1 [NiFe] hydrogenases, PDB code 3USE. Variability in structure and coordination of the proximal cluster in [NiFe] hydrogenases: (B) E. coli Hyd-1 oxidized [4Fe-3S] cluster with 6Cys coordination (PDB code 3USC); (C) regular [4Fe-4S] cluster with all-cysteine coordination in the periplasmic hydrogenase from Desulfovibrio gigas (PDB code 1YQ9); and (D) [4Fe-4S] cluster with Asp-3Cys coordination in the actinobacterial-type hydrogenase from Ralstonia eutropha (PDB code 5AA5). Prepared using Pymol.7
A related set of [NiFeSe] hydrogenases exist, in which a terminal cysteine residue coordinated to Ni at the active site is replaced by selenocysteine. The active site chemistry of the [NiFeSe] hydrogenases is less well understood. The selenium atom of the selenocysteine residue is more polarizable and has a lower pKa than the cysteine equivalent, and these enzymes are found to be extremely active.1,76,77 Although these hydrogenases do not exhibit EPR-active oxidized states (at the Ni-B redox level, Scheme 1), H2 oxidation activity is inhibited by O2,76 and the enzymes undergo oxidative anaerobic inactivation78 in protein film electrochemistry measurements. There is IR spectroscopic evidence for the Nia-C and Nia-R states of the active site,79,80 and Nia-L states have been observed by EPR spectroscopy upon illumination at cryogenic temperatures.80,81
Hydrogenases of Groups 2–4 have been studied less extensively. The Group 2 [NiFe] hydrogenases are cytosolic and include the sensing hydrogenases, of which the regulatory hydrogenase (RH) from R. eutropha has been most studied.3,40,52,82−87R. eutropha RH has been shown to have a particularly low potential proximal cluster, below ca. −0.5 V,52 and this may be significant in its behavior as this means the proximal cluster potential lies more negative than the potential of the H+/H2 couple potential at neutral pH. The Group 3 enzymes are also cytosolic and comprise a dimeric [NiFe] hydrogenase unit coupled via an extended iron sulfur electron-transfer chain to an enzyme moiety equipped for reducing biological cofactors. These include nicotinamide adenine dinucleotide, NAD+, nicotinamide adenine dinucleotide phosphate, NADP+, or “cofactor 420”, also known as F420 (8-hydroxy-5-deazaflavin).6 The Group 3 enzymes are known as “bidirectional” because they function reversibly in vivo (although note that most [NiFe] hydrogenases function reversibly in vitro, oxidizing H2, and reducing H+ in the presence of appropriate acceptors or donors). Only one crystal structure exists for Group 3, that of the F420-reducing hydrogenase from Methanothermobacter marburgensis,18Figure 3, although it is far from representative of the Group as a whole because there is considerable variation in the number and type of subunits and the redox cofactors present in different enzymes of this Group. The Group 3 hydrogenases also vary in their tolerance to O2.3,5,6,43,44 Limited spectroscopic data exist for the Group 3 hydrogenases, but those for which some data exist include NAD+-reducing soluble hydrogenases HoxHYFUI2 from R. eutropha,43,44,51 the HoxHY module of HoxEFUYH from the photosynthetic bacterium Allochromatium vinosum,88 and the bidirectional [NiFe] hydrogenase from Synechocystis.45 The NADP+-reducing soluble hydrogenase (SH) 1 from Pyrococcus furiosus, a hyperthermophilic archaeon, has been studied more extensively using time-resolved IR methods.41,42,61
Figure 3.
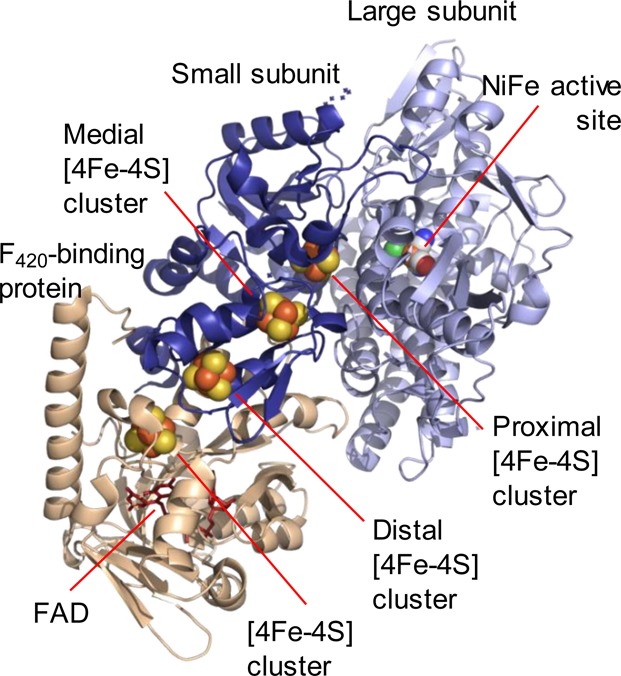
Representation of the X-ray crystallographic structure of the F420-reducing hydrogenase from Methanothermobacter marburgensis, the only structurally characterized member of Group 3 [NiFe] hydrogenases. Prepared using Pymol,7 PDB code 4OMF. The active site and iron sulfur clusters are shown as spheres in elemental colors, and the flavin adenine dinucleotide (FAD) cofactor is shown in red sticks. Considerable variation in subunit composition, structure, and physiological function exists in the Group 3 enzymes.
The IR data shown in Table 1 represents examples of [NiFe] hydrogenases from Groups 1, 2, and 3.
2. Experimental Evidence for States Involved in the [NiFe] Hydrogenase Catalytic Cycle
In this section, we introduce evidence for each step in the [NiFe] hydrogenase catalytic cycle (as outlined in Scheme 1) viewed in the direction of H2 oxidation, and we discuss recent developments in the context of recent vibrational spectroscopic data and reanalysis of earlier data.
There is relatively little evidence for the structure of the EPR-silent NiIIFeII Nia-SI state,89 and it is generally depicted with a vacant coordination site at the bridging position between Ni and Fe. The Nia-SI state is in acid–base equilibrium with an inactive state known as Ni-SIr, which is deprotonated relative to Nia-SI.1,26 The pKa value of this equilibrium is 7.8 ± 0.1 in the [NiFe] hydrogenase from Desulfovibrio vulgaris Miyazaki F (MF).26 Computational studies have suggested that one of the terminal cysteine ligands to the active site may be protonated in Nia-SI,90,91 although models in which these ligands are deprotonated have also been considered.92 The Nia-SI state reacts with H2, which is cleaved heterolytically and reduces the active site to the Nia-R state (also EPR silent),93 leaving a bridging hydride ligand at the active site. The initial acceptor for the proton released upon H2 cleavage is still a matter of debate, as is the precise location of H2 binding and the nature of a (probably transient) Michaelis–Menten complex between Nia-SI and H2.
The most common representation of the Nia-R state is that shown in Figure 4A, in which a terminal cysteine (C) thiolate coordinated to NiII acted as the initial proton acceptor (C546 in D. vulgaris MF notation, C576 in E. coli Hyd 1 notation). Experimental evidence for this representation of Nia-R and the presence of a bridging hydride ligand was first provided by a 0.89 Å resolution crystal structure of the [NiFe] hydrogenase from D. vulgaris MF (electron density map shown in Figure 4B),11 where a shortened Ni–H distance relative to Fe–H showed tighter binding of the hydride to Ni at the active site. The presence of a bridging hydride was confirmed by nuclear resonance vibrational spectroscopy (NRVS)94,95 where a vibration at 675 cm–1 was shown to be sensitive to H/D isotope exchange and assigned to a Ni–H–Fe wag vibrational mode by comparison with DFT calculations and spectra of model compounds (Figure 4C). Protonation of a terminal thiolate is supported by earlier DFT calculations on the EPR silent states of the active site; thiolate protonation was required to model the Nia-SI state, and both thiolate protonation and a bridging hydride ligand were necessary to accurately reproduce the IR peak positions of the Nia-RI state of D. vulgaris MF [NiFe] hydrogenase.91 In agreement with the crystallographic data of Ogata et al.,11 calculations have shown that protonation of the terminal cysteine thiolate, C576 (E. coli notation), gives a state that is 14–51 kJ mol–1 lower in energy than protonation at any other active site coordinated cysteine.96 A key feature of the Nia-R crystal structure is asymmetric binding of the bridging hydride, which is more closely associated with Ni. A common feature of bimetallic hydride-containing mimetic compounds is Fe-centered reactivity (for a comprehensive review, see Schilter et al.),4 but a recent structural and electronic analogue of the Nia-R state has been reported which contains a Ni-bound hydride.97
Figure 4.
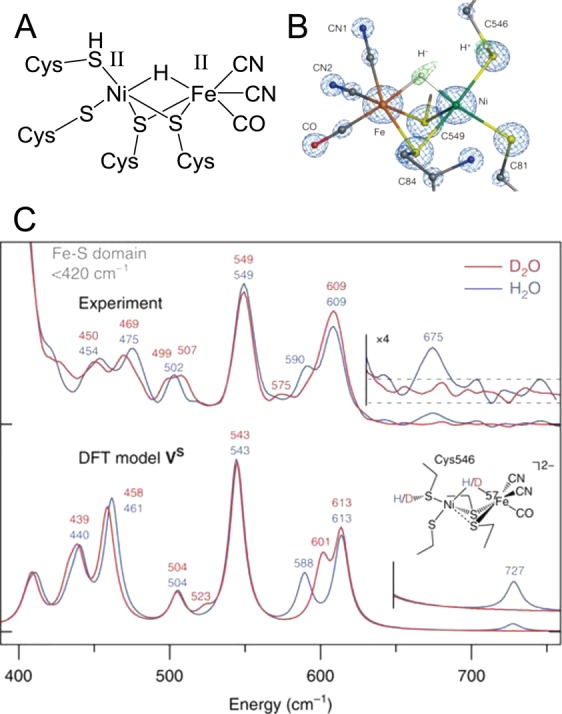
X-ray crystallographic and nuclear resonance vibrational spectroscopic (NRVS) evidence for a bridging hydride in the Nia-R state of [NiFe] hydrogenase from D. vulgaris MF. (A) The most common representation of the Nia-R state. (B) Electron density map showing the density associated with crystallographically assigned H atoms in green mesh. (C) Experimental and DFT calculated NRVS spectra at the Nia-R redox level, showing a H/D isotope-dependent band assigned to the Ni-H–Fe wag vibration of a bridging hydride (multiplied by a factor of 4 in the inset). Panel (B) reproduced with permission from ref (11). Copyright 2015 Nature Publishing Group. Panel (C) adapted with permission from ref (94). Copyright 2015 Nature Publishing Group.
On the basis of the representation of the Nia-R state shown in Figure 4A, the mechanism of the transition between Nia-SI and Nia-R is generally suggested to involve initial side-on binding of H2 to Ni in a Kubas-complex type arrangement,98,99 with a cysteine thiolate sulfur acting as a base and accepting the initial proton during H2 splitting (Figure 5A). Reversible protonation of a Ni-coordinated terminal thiolate has been demonstrated in a bimetallic mimetic compound (Figure 5B) that is also an active proton reduction catalyst.100 Treatment of a dichloromethane solution of [1] with HBF4 results in quantitative formation of the salt [1H][BF4], which can then be deprotonated by addition of water. However, biomimetic catalysts are generally poor at H2 oxidation and require strong acids or bases to produce or oxidize H2, respectively.100−102
Figure 5.
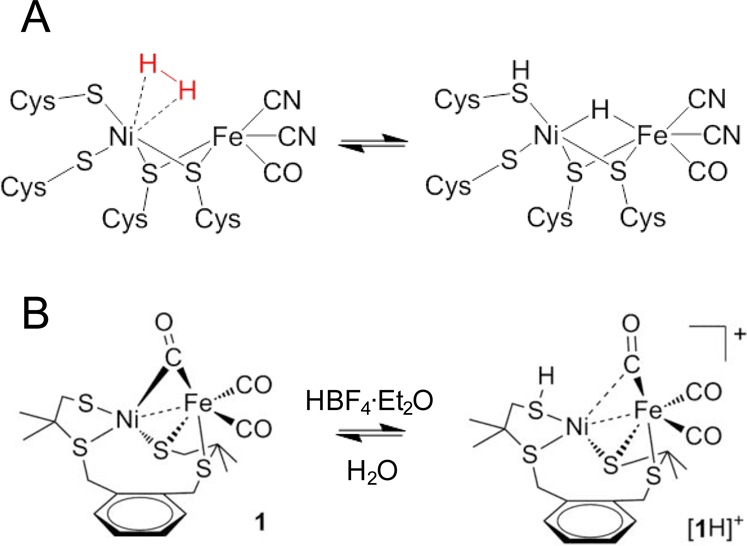
(A) H2 activation via side-on binding in a Kubas-type complex, with a terminal cysteine thiolate sulfur acting as the initial proton acceptor during Nia-R formation. (B) Reversible protonation of a Ni-coordinated terminal thiolate ligand has been demonstrated in a bimetallic [NiFe] hydrogenase mimetic compound. (B) Reproduced with permission from ref (100). Copyright 2012 American Chemical Society.
An alternative mechanism for the Nia-SI to Nia-R transition was proposed by Armstrong and co-workers, who demonstrated that a strictly conserved arginine residue (R509 in E. coli Hyd-1 notation) was important for H2 oxidation activity in E. coli Hyd-1.54 The guanidinium side chain of this arginine residue suspends a nitrogen atom 4.5 Å above the bridging coordination position of the Ni-Fe active site in the wild type enzyme (Figure 6A). Substitution of the arginine residue for lysine generated a variant, R509K, with 100-fold lower H2 oxidation activity than wild type Hyd-1 (Figure 6B) despite the structure of the inner coordination sphere of the active site being virtually unchanged (Figure 6C). This led to the suggestion of a frustrated Lewis pair (FLP)-type mechanism for H2 splitting in which the guanidinium side chain is transiently deprotonated and the resulting guanidine provides the strong base required for H2 cleavage (Figure 7A). This mechanism has obvious similarities to FeFe hydrogenases, in which an azadithiolate bridging ligand positions a basic nitrogen atom above the distal Fe site of the active site H-cluster (Figure 7B),103 and also to enzyme-inspired mimetic Ni pincer complexes, which contain pendant amine groups acting as the initial proton acceptor during H2 oxidation (Figure 7C).104,56
Figure 6.
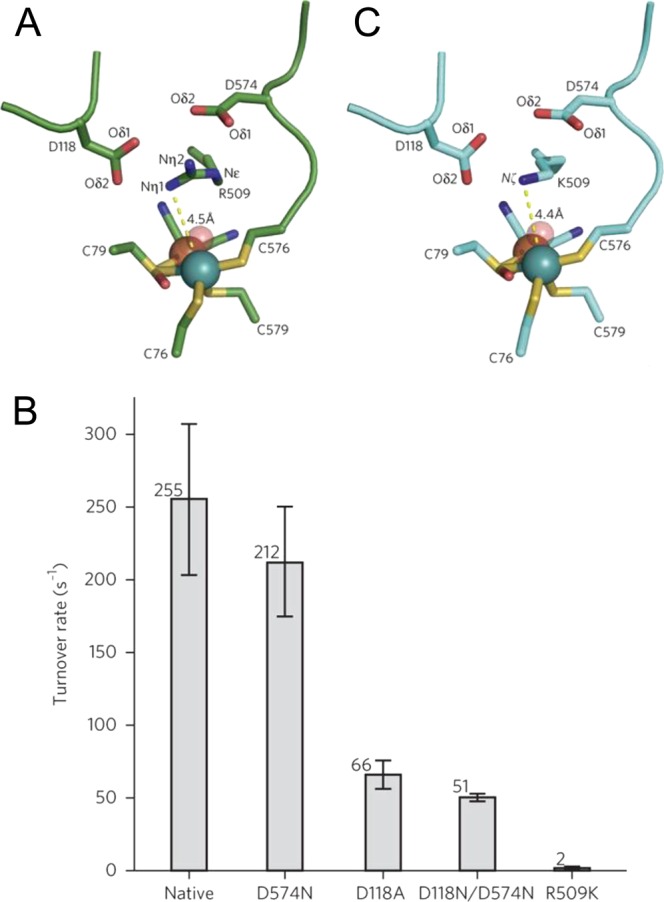
Genetic variants have demonstrated the importance of a conserved arginine (R) residue for H2 activation by E. coli Hyd-1. (A) X-ray crystal structure of wild type Hyd-1, with a guanidinium group of arginine (R) 509 positioned above the active site. (B) Activity assays for wild type and variant Hyd-1, showing the effect of different mutations in the active site canopy. The aspartic acid (D) 118 and 574 variants are discussed later. (C) X-ray crystal structure of the R509K variant. Adapted with permission from ref (54). Copyright 2015 Nature Publishing Group.
Figure 7.
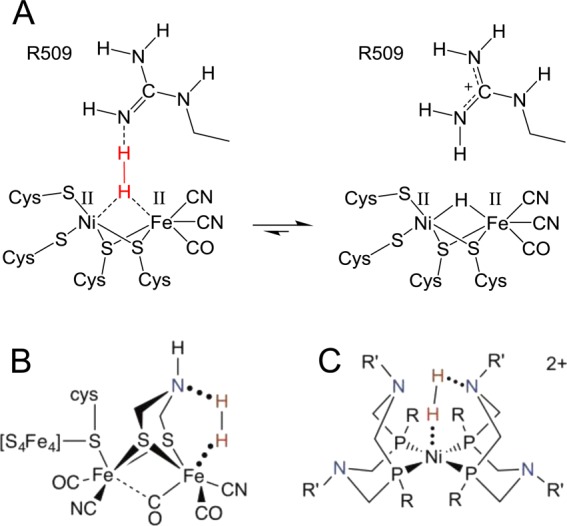
(A) Proposed mode of H2 activation based on a frustrated Lewis pair mechanism involving transient deprotonation of the guanidinium side chain of a conserved arginine residue. Analogous mechanisms have been reported for H2 activation by [FeFe] hydrogenases (B) and enzyme-inspired Ni pincer complexes (C), both of which contain a pendant amine that acts as a base. (B) and (C) reproduced with permission from ref (55). Copyright 2013 Elsevier B.V.
The representations of the Nia-R state suggested by these mechanisms for H2 activation are oversimplified. IR studies on a range of [NiFe] hydrogenases show that there are (at least) three different (sub)states at the Nia-R redox level (labeled Nia-RI, Nia-RII, and Nia-RIII according to literature convention, see Table 1 and references therein). In the case of E. coli Hyd-1, two Nia-R states are observed in the reduced enzyme and are nearly equally populated.35 IR spectra of the crystal samples used to determine the structure of the Nia-R state of D. vulgaris MF [NiFe] hydrogenase were predominantly in the state Nia-RI (see Table 1), but the crystals also contained up to 18% of a second Nia-R state, Nia-RII.11 Since all of the Nia-R states in these studies were generated by reaction with H2, it seems reasonable to assume that they are all equally likely to be functionally relevant, and therefore, inclusion of a single picture of the Nia-R state in the catalytic cycle may be an oversimplification (although the presence of “unproductive” Nia-R states, not along the reaction coordinate but in rapid equilibrium remains a possibility, see Discussion below).
The Nia-R and Nia-C states have long been assumed to be catalytic intermediates general to all [NiFe] hydrogenases. The catalytic relevance of these states is supported by recent vibrational spectroscopic studies.35,41 Vincent and co-workers used protein film IR electrochemistry (PFIRE, an in situ application of IR spectroscopy to protein film electrochemistry) to demonstrate the presence of Nia-SI, Nia-R, and Nia-C during electrocatalytic turnover.35Figure 8A,B show IR spectra collected from the same sample of E. coli Hyd-1 under both nonturnover (Ar, Figure 8A) and steady-state turnover (H2, Figure 8B) conditions. In the absence of H2, a significant population of both Nia-SI and the oxidized, inactive Ni-B state (blue) are present at −0.074 V versus the standard hydrogen electrode (SHE), whereas only the Ni-B state is observed at +0.356 V. In the presence of H2 the distribution of active site states reflects the steady-state populations of catalytically active intermediates. Two Nia-R states (red) are observed as the majority species at −0.074 V, and the population of the Nia-SI state (green) has diminished suggesting rapid reaction with H2. The most striking differences occur at +0.356 V, however, as the Nia-SI, Nia-R, Nia-C and Nia-L (orange) states are all present under H2 when they are completely absent at this potential under nonturnover conditions in an Ar atmosphere. The accompanying current–time traces are shown in Figure 8C. The stable, constant current observed at −0.074 V indicates efficient mass transport to the electrode-immobilized hydrogenase, whereas the monotonic decay in current at +0.356 V is due to the well-established slow oxidative inactivation which leads to conversion of a proportion of the enzyme to the Ni-B state. The presence of a species during steady-state turnover is not enough on its own to confirm involvement in catalysis, however. Dyer and co-workers used time-resolved IR spectroscopy to provide the first direct evidence that the Nia-SI, Nia-C, and Nia-R states can interconvert on time scales faster than the turnover frequency of the hydrogenase being studied (P. furiosus SH1 has a modest turnover frequency of 62 s–1 for H+ reduction under the experimental conditions in Figure 8D,41 although higher turnover frequencies are observed at elevated temperatures105,106) and thus are sufficiently kinetically competent to be catalytic intermediates.41 A rapid photoinduced reductive potential jump of ca. 50 mV was applied to P. furiosus SH1, using the cofactor NADH as a “caged electron” source. Figure 8D shows the absorption transients from one such experiment, demonstrating the conversion of Nia-C (purple) to Nia-R (red) from roughly 0.5 ms onward. An additional intermediate between Nia-R and Nia-C may be implied by the slight delay in Nia-R formation relative to Nia-C decay and is accounted for in the kinetic scheme used to model the transient absorption spectra (inset, Figure 8D).
Figure 8.

Infrared spectroscopic measurements recorded under catalytically relevant conditions. (A) Nonturnover (under an Ar atmosphere) and (B) steady-state electrocatalytic turnover (under a H2 atmosphere) spectra of E. coli Hyd-1 recorded using the PFIRE technique, revealing states that are formed in response to electrocatalytic H2 oxidation. The spectra in (A) demonstrate that the redox state of the active site can also be controlled electrochemically, whereas in (B) the active site states are controlled both by the electrode potential and by H2 oxidation. (C) Steady-state current recorded simultaneously during collection of the spectra shown in (A) and (B). (D) Transient absorption measurements of P. furiosus SH1 following photoinduced reductive treatment, showing interconversion of Nia-C (purple), Nia-L (orange), Nia-SI (green), and Nia-R (red) states at time scales shorter than the turnover frequency (62 s–1). The inset in (D) shows the kinetic scheme used to fit the transient absorption spectra (black lines). Panels (A), (B), and (C) adapted with permission from ref (35). Copyright 2015 Wiley-VCH. Panel (D) adapted with permission from ref (41). Copyright 2015 American Chemical Society.
In terms of the outline catalytic states shown in Scheme 1, the regulatory hydrogenase from R. eutropha was, initially, a puzzling case. Despite the fact that solution assays on this enzyme demonstrated catalytic H2 oxidation (albeit with a very low turnover frequency), the Nia-R state(s) were not observed upon treatment with H2,82,83,87 or electrochemical reduction in solution.86 This lead to suggestion of a distinct two-state catalytic cycle for this enzyme involving only Nia-SI and Nia-C.1,3 Recently, however, PFIRE spectra have revealed the presence of at least one Nia-R state during catalytic H2 oxidation,40 suggesting that a common set of redox levels is conserved across the [NiFe] hydrogenases from quite different Groups.
For a long time , the catalytic cycle of [NiFe] hydrogenases were thought to contain only three states, Nia-SI, Nia-C, and Nia-R, at least from an experimentalist’s perspective, as these were the only states that had been observed under “normal”, “physiologically relevant” conditions.1 Additional state(s) had been observed at the Nia-C redox level, the Nia-L states, but were thought to be an artifact due to the unusual conditions of illumination at cryogenic temperature seemingly required for their formation.24 The tacit assumption behind these assertions was that the second proton-coupled electron-transfer step in the H2 oxidation mechanism, the transition from Nia-C to Nia-SI, must be concerted and that no other intermediates were involved (Scheme 1). From a theoretical viewpoint, however, additional intermediates during the transition from Nia-C to Nia-SI have been considered for quite some time. Nia-L, or a structurally similar NiI state formed upon deprotonation of a NiIII species containing a bridging hydride, has been implicated in computational studies of the catalytic cycle by the groups of Siegbahn, Hall, and Ryde8,107,96 and was suggested as an intermediate by Lindahl108 while discussing the role of metal–metal bonds in [NiFe] hydrogenases. Sequential proton- and electron-transfer steps have been established for small-molecule Ni-based H2 production catalysts.55 Recently Brazzolotto et al. reported a bimetallic [NiFe] biomimetic compound that produces H2 at a rate of 250 s–1 via a catalytic pathway that contains structural analogues of both Nia-L and Nia-R states.97
Experimentally, growing evidence supports the involvement of Nia-L as an intermediate. Hirota and co-workers demonstrated that it is possible to produce significant quantities of Nia-SI during photolysis of the Nia-C state at low temperature, but only from samples in which the proximal iron–sulfur cluster is oxidized and therefore can accept electrons from the active site (Figure 9A).60Figure 9B shows prephotolysis spectra and light-minus-dark difference spectra following photolysis for samples of the O2-sensitive [NiFe] hydrogenase from D. vulgaris MF under a H2 atmosphere. Positive bands relate to species that are accumulated as a result of photolysis, at the expense of species with negative bands. At all temperatures studied, the “normal” photoconversion of Nia-C to Nia-L is observed, with the stability (lifetime) of the photoproduct increasing at lower temperatures. In contrast, Figure 9C shows a similar set of pre- and postphotolysis spectra, this time under a N2 atmosphere. Again, photoconversion of Nia-C to Nia-L is observed, but now a significant photoinduced increase in the population of Nia-SI is observed. The difference between these two sets of photolysis spectra lies in the redox state of the proximal cluster. Under a N2 atmosphere, the proximal cluster is oxidized in a significant proportion of the D. vulgaris MF hydrogenase sample in the Nia-C state, as determined by EPR spectroscopy,60 and can therefore accept an electron from the photogenerated NiI active site. Under a H2 atmosphere, the proximal cluster is reduced and unable to accept an electron from Nia-L. These results suggest a pathway exists between Nia-L and Nia-SI, gated by the oxidation state of the proximal cluster (Figure 9A).
Figure 9.

Pathway for interconversion between Nia-L and Nia-SI, demonstrated by photolysis measurements. (A) Scheme showing Nia-SI formation from Nia-L, only in the presence of an oxidized proximal cluster. (B) Photolysis spectra of the [NiFe] hydrogenase from D. vulgaris MF under a H2 atmosphere show “normal” formation of Nia-L. (C) Photolysis spectra of [NiFe] hydrogenase from D. vulgaris MF recorded under a N2 atmosphere show formation of both Nia-L and Nia-SI, under conditions where the proximal cluster is at least partially oxidized. Panels (B) and (C) adapted with permission from ref (60). Copyright 2014 Wiley-VCH.
A related effect has been observed by Murphy et al.,36 who demonstrated a pH-dependent equilibrium between the Nia-C and Nia-L states of E. coli Hyd-1. The reversible conversion of Nia-C to Nia-L in E. coli Hyd-1 proceeds readily in the dark at ambient temperature, and the Nia-C and Nia-L species share the same potential dependence over the pH range studied. The proximal clusters of Group 1 O2-tolerant hydrogenases have unusually high reduction potentials, more positive than the Nia-L/Nia-SI potential at all pH values. The proximal cluster will therefore predominantly be in the reduced state, consistent with the ease of observation of Nia-L in these hydrogenases (see Figure 9A). Vincent and co-workers have demonstrated that Nia-L is formed reversibly in response to steady-state H2 oxidation by E. coli Hyd-1 (Figure 8B).35 A low level of Nia-SI production from Nia-C is also evident in earlier low-temperature photoinduced IR spectra of R. eutropha MBH.47
Transient, reversible formation of Nia-SI from Nia-L on subturnover frequency time scales has been demonstrated during light-triggered measurements at room temperature by Dyer109 and co-workers.41,42 Essentially instantaneous formation of two Nia-L states was observed upon photolysis, within 100 ns of illumination (i.e., faster than the experimental time resolution), and the relaxation of both these states followed approximately equal decay kinetics. Kinetic modeling of transient absorption spectra of the Nia-C, Nia-SI, and both Nia-L states (similar to those shown in Figure 8D) revealed that the Nia-L species are sequential, on-pathway intermediates during the transition between Nia-C and Nia-SI. The relative intensities of the two Nia-L states were found to be pH dependent below pH 7.5, with only one Nia-L species observed at pH 6.1. Dyer and co-workers interpreted this observation as evidence that a proton acceptor site becomes unavailable as the pH is lowered, blocking proton transfer away from the active site.42 Hirota and co-workers observed pH-dependence of Nia-L formation from Nia-C, with two Nia-L states, differing in νCO by 20 cm–1, observed at high pH.110
3. Discussion
Implications of Multiple Nia-L States on the [NiFe] Hydrogenase Mechanism
Significant variation is found in the absolute νCO band positions of all states of the [NiFe] active site between different hydrogenases (Table 1). However, in all [NiFe] hydrogenases studied to date, the Nia-C state has the highest νCO band of the catalytically active states. For any given hydrogenase, there is good agreement between the νCO band positions of all the active states relative to νCO of Nia-C, and so Nia-C can be used as an internal standard (see Supporting Information Figure S1). This concept is demonstrated particularly well for the Nia-SI state in Figure 10A, which plots the relative νCO band positions of the Nia-SI and Nia-L states for the hydrogenases listed in Table 1. The νCO band of the Nia-SI state is on average 19.7 cm–1 lower than that of Nia-C, and all the relative positions lie within 1.5 standard deviations of this value (the shaded regions in Figure 10 represent ±2 standard deviations of the mean). Treating the Nia-L states in the same way results in three groupings of relative νCO energies (not all Nia-L states have been observed by IR spectroscopy in all the hydrogenases listed in Table 1 so the precise assignment to “I”, “II”, or “III” is less well-defined than for the Nia-R states, which are simply labeled in order of decreasing νCO band position). The relative Nia-L positions highlight differences between different types of hydrogenases. Group 1 O2-sensitive hydrogenases tend to have a single higher wavenumber Nia-L as the dominant species (the lower νCO species in D. vulgaris MF [NiFe] hydrogenase is only evident as a minor component in photolysis spectra recorded at high pH),110 whereas the Group 1 O2-tolerant E. coli Hyd-1 and A. aeolicus Hase 1 display additional Nia-L species with νCO positions that are ≥20 cm–1 lower. In addition, the Nia-L state(s) of O2-sensitive Group 1 hydrogenases have only been observed during photolysis at low temperatures, whereas for O2-tolerant hydrogenases Nia-L is observed under a variety of more ambient conditions. These observations possibly reflect a more weakly bound hydride in the Nia-C state of O2-tolerant Group 1 hydrogenases, as demonstrated for A. aeolicus Hase 1.38
Figure 10.

Relative νCO positions of catalytically active states of [NiFe] hydrogenases show remarkably similar wavenumber positions with respect to the Nia-C state. (A) Relative positions of the Nia-SI and Nia-L states, showing that the Nia-L states fall into three groupings of νCO. (B) Relative positions of the Nia-R states demonstrates three νCO groupings. Calculated using the data in Table 1. Shaded regions represent ±2 standard deviations of the mean relative νCO position for each state (νCO values for R. eutropha SH have been omitted from these calculations as they are less well-characterized but are plotted for comparison; open squares/triangles). Included for comparison are results from DFT calculations of the Nia-SI,91 Nia-L,53 and Nia-R91 states (open diamonds).
Limited data are available for Group 3 hydrogenases. In the only reports to date on Nia-L for these hydrogenases, for P. furiosus SH1,41,42 only higher wavenumber Nia-L states have been reported. The Nia-L states of P. furiosus SH1 are a noteworthy case as transient IR spectra reveal that at least two Nia-L states are catalytically relevant, despite the fact that a stable Nia-L population is not normally observed under dark conditions (although a small population of Nia-L is observed under laboratory illumination at pH ≥ 8.5).41,42 Observation of a stable Nia-L state under “physiologically relevant” conditions is therefore not a prerequisite for involvement of Nia-L in the catalytic cycle. This could have implications for the catalytic relevance of Nia-L in O2-sensitive Group 1 hydrogenases, and the Group 2 RH from R. eutropha, where Nia-L species have so far only been observed following photolysis of Nia-C.1,40,82
The pH dependence of Nia-L formation36,110 confirms earlier suggestions that the difference between Nia-L states is the location of the proton released from the hydride bridge in Nia-C. The change in νCO upon Nia-L formation depends upon how far removed the proton becomes from the active site, with higher wavenumber states likely to contain a proton more closely associated with the active site. Deprotonation of the active site (complete removal of the proton from the primary coordination sphere) would increase electron density at the active site and lead to a substantially lower νCO.91,100,53 Theoretical calculations of the Nia-L state, included for comparison as orange open diamonds in Figure 8A, indicate a lowering of the νCO band position by ∼30 cm–1 between models with a protonated terminal cysteine thiolate and a completely deprotonated active site.53 Although neither of these models precisely reproduce the experimental Nia-L νCO positions relative to the calculated νCO band of Nia-C, the difference between the protonated cysteine thiolate and deprotonated active site models is very similar to the difference in νCO between the highest and lowest wavenumber Nia-L groups. A similar change in νCO (−45–50 cm–1) following protonation of a terminal thiolate ligand has been observed in the biomimetic cluster shown in Figure 5.100 These observations suggest that the three groupings of Nia-L states in Figure 10A (in order of decreasing νCO position) could correspond to protonation of the primary coordination sphere of the active site, protonation of an acceptor within hydrogen-bonding distance of the active site and complete deprotonation of the active site. Dyer and co-workers have demonstrated that changes in νCO position as a result of either a change in redox state of the proximal iron sulfur cluster or protonation changes at nearby amino acid residues are much smaller (< ∼5 cm–1).42 The catalytic relevance of the Nia-L substates is not yet clear, and it is possible that one or more of these substates correspond to off-pathway species.
The location of possible protonation sites is still under investigation, although a number of proton-transfer pathways have been postulated on the basis of site-directed mutagenesis, theoretical modeling, and crystallographic data. Raman spectroscopic evidence on R. eutropha MBH47 and theoretical studies53,96 suggest that a terminal cysteine thiolate is the initial proton acceptor in the Nia-C to Nia-L transition: a recent computational study by Ryde has shown that, of the four cysteine thiolates coordinated to the active site, protonation of C576 (in Hyd-1 notation) leads to the lowest energy structure.96 A conserved glutamate residue (E28 in Hyd-1 notation) located close to the Ni-bound terminal cysteine thiolates is suggested to be important for proton transfer beyond the primary coordination sphere of the active site on the basis of mutagenesis studies,12 and is the active-site terminus of a number of putative proton-transfer pathways.11−14,30 Time-resolved photolysis measurements by Dyer and co-workers have established this residue as a proton acceptor site during the Nia-L to Nia-SI transition in P. furiosus SH1,61 consistent with their earlier study which showed that proton transfer is mediated by an amino acid residue with pKa ∼ 7.42
Taken together, these results support mechanism(s) in which some or all of the Nia-L states represent sequential proton movement away from the active site ahead of electron transfer to the proximal cluster during the transition from Nia-C to Nia-SI. This is shown in Scheme 2, which considers possible proton-transfer events in the vicinity of the [NiFe] active site. Proton migration to a terminal cysteine thiolate results in two distinct Nia-L states (Scheme 2a,b) that are in rapid equilibrium and differ in the degree of hydrogen bonding to the carboxylate group of E28; these are the proposed structures of the two Nia-L species observed in P. furiosus SH1, with closely spaced νCO (Figure 10A).42,61 The transition to Nia-SI can then proceed either through concerted elementary proton and electron transfer (Scheme 2b to Nia-SI) or through sequential chemical (C) and electrochemical/electron-transfer (E) steps (in electrochemistry notation)111 via one (or more) Nia-L states with a deprotonated primary coordination sphere (Scheme 2c,d) in a C(C)E mechanism.
Scheme 2. Possible Sequential Involvement of Nia-L States in the Transition between Nia-C and Nia-SI.

With reference to the groupings of Nia-L states shown in Figure 10A: (a) and (b) are proposed structures of the high wavenumber Nia-L species, such as those observed in P. furiosus SH1, in which a proton is retained in the primary coordination sphere of the [NiFe] active site; (c) and (d) are possible structures of the two lower wavenumber Nia-L species, with a deprotonated primary coordination sphere.
Because of the large H/D kinetic isotope effect (>40)41 and pH dependence of the Nia-L to Nia-S transition,42 Dyer and co-workers favor concerted transfer of an electron and a proton (Scheme 2b to Nia-SI). They calculate a ΔG value of −1 kJ/mol for the concerted process on the basis of the relative concentrations of Nia-L and Nia-SI during a defined time window following photolysis of Nia-C, and they make the assumption that ΔG > 0 for oxidation of (b) without coupled deprotonation.61 While entirely consistent with available data for P. furiosus SH1, this model is not necessarily consistent with the work of Hirota and co-workers on D. vulgaris MF hydrogenase,110 who showed that the relative populations of protonated (b) and deprotonated (c or d) Nia-L states are pH-dependent with a calculated ΔG value of −1.2 ± 0.9 kJ/mol for their interconversion. Neither is it consistent with the observation that Nia-L species observed in Group 1 O2-tolerant hydrogenases, even in the dark and at ambient temperature, tend to be biased toward lower wavenumber species and are likely to have a deprotonated active site (vide infra, Figure 10A). The energy landscape at the Nia-C/L redox level is therefore presumably relatively flat, at least in terms of the energy difference between the range of available protonation sites, as shown in previous DFT studies that included a NiI intermediate.9,10 The distance over which the proton can transfer in the Nia-L state(s) during catalysis is likely to depend upon the rate of electron transfer from the active site to the proximal cluster at the midpoint potential of the Nia-SI/Nia-L redox couple. In the case of the O2-tolerant E. coli Hyd-1 both the proximal and medial iron sulfur clusters have potentials significantly more positive than the Nia-SI/Nia-L couple and so are reduced, and therefore fully occupied, when the active site is at the Nia-C/Nia-L redox level (although there is no direct experimental evidence at present reporting on the redox behavior of the iron sulfur relay during turnover). This would retard the electron transfer required for conversion of Nia-L to Nia-SI, which could then become rate-limiting. Under these circumstances, it is possible that the proton associated with Nia-L could travel further along the proton-transfer pathway than the glutamate acceptor. This idea of multiple proton acceptor sites for Nia-L states of E. coli Hyd-1 is consistent with the data reported by Murphy et al.,36 where the pH-dependence of the relative populations of Nia-C and Nia-L does not fit a simple single proton equilibrium for the Nia-C to Nia-L transition. Figure 11A shows a pH titration equivalent to that reported by Murphy et al.,36 but now focusing on the pH-dependent populations of Nia-C and the two low νCO Nia-L states of E. coli Hyd-1 separately. It is clear that, in this case, the two Nia-L species correspond either to unique proton acceptor sites with quite different pKa values, or a single proton acceptor site with multiple pH-dependent conformations, the origin of which is unclear at present. Low wavenumber νCO Nia-L states have also been observed in the O2-tolerant Hase 1 from A. aeolicus (Table 1 and Figure 10A). Molecular dynamics simulations of proton transport in A. aeolicus Hase 1 by Sacquin-Mora and co-workers112 have revealed that the proton-accepting glutamate residue E13 (Glu13, circled in Figure 11B, and equivalent to E28 in E. coli Hyd-1) is conformationally flexible and takes up two orientations; 30% of the residues have the carboxylate side chain oriented toward the active site (Figure 11B, purple), with 70% oriented toward the iron sulfur clusters and two glutamate residues, E118 and E58, in the small subunit (Figure 11B, orange). In contrast, the equivalent residue in D. vulgaris MF [NiFe] hydrogenase was more conformationally rigid in the simulations. Sacquin-Mora and co-workers postulate that the conformational flexibility of glutamate might increase the rate of proton transfer to and from the active site.112 It is interesting to note that an increased rate of proton transfer via conformational flexibility of a glutamate residue has been observed in similar molecular dynamics simulations of the FeFe hydrogenase from Clostridium pasterianum.113 It is possible that the two low νCO Nia-L states observed in E. coli Hyd-1 and A. aeolicus Hase 1 correspond to protons located on the E13-equivalent glutamate residue (A. aeolicus numbering, E28 in E. coli Hyd-1) in both the conformations shown in Figure 11B, although this will require further investigation. While this description of the low wavenumber νCO Nia-L states in O2-tolerant hydrogenases is appealing, it is important to keep in mind the caveat that these states could be off-pathway species in fast equilibrium with catalytically active states. At present, the only Nia-L states109 conclusively demonstrated as on-pathway intermediates are those described by Dyer and co-workers for P. furiosus SH1.41,42
Figure 11.
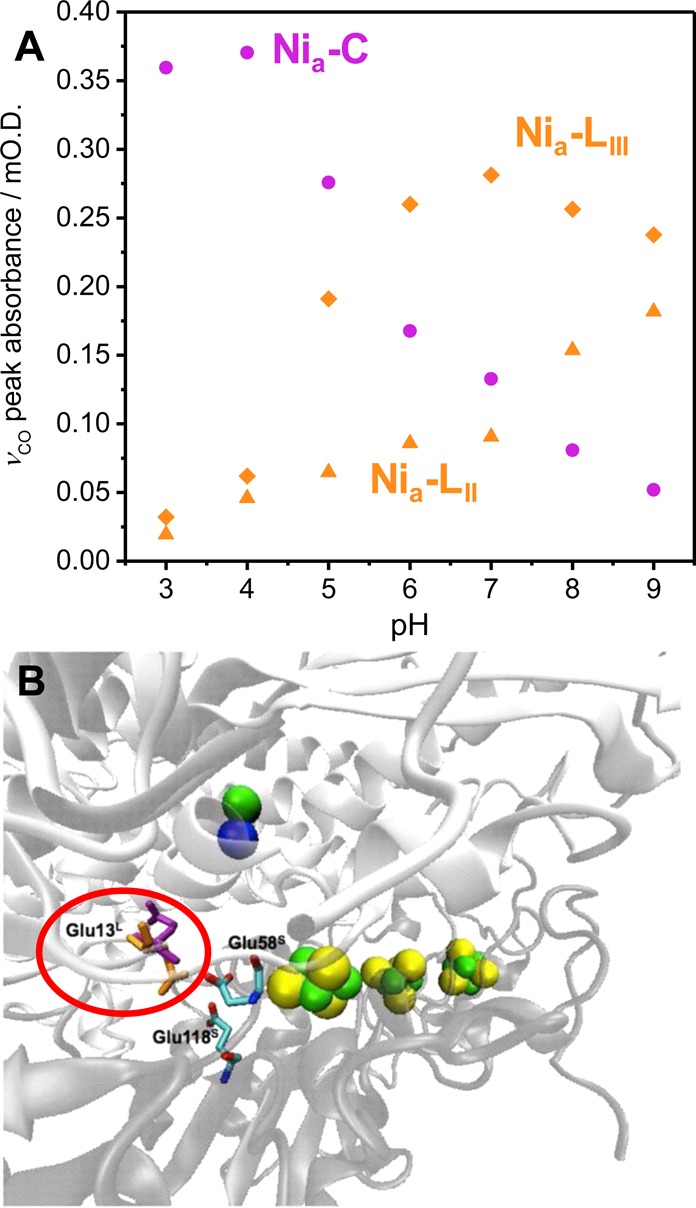
(A) pH dependence of the relative populations of Nia-C (purple circles), Nia-LII (orange triangles), and Nia-LIII (orange diamonds) states as determined from the respective νCO band intensities in a series of PFIRE measurements similar to those previously reported.36 The Nia-L states of E. coli Hyd-1 do not share the same pH dependence, implying multiple protonation sites, or multiple orientations of the same protonation site. (B) Conformational flexibility of a conserved glutamate (Glu13 in A. aeolicus Hase 1 numbering, Glu28 in E. coli Hyd-1 numbering, circled in red) has been suggested on the basis of molecular dynamics simulations in Group 1 O2-tolerant hydrogenases; the two conformations are shown in orange and purple. Panel (B) reproduced with permission from ref (112). Copyright 2014 American Chemical Society.
Implications of Multiple Nia-R States on the [NiFe] Hydrogenase Mechanism
Figure 10B shows that the Nia-R states can also be split into three distinct groupings of νCO positions relative to the νCO of Nia-C, which correspond to the Nia-RI, Nia-RII, and Nia-RIII states. (In calculating the averages and standard deviations used in Figure 10B, we have omitted the νCO positions of R. eutropha SH; these are less well-characterized and display obvious discrepancies with the other hydrogenases, although we have plotted the data for comparison.) As is the case for the Nia-L states, only one Nia-R substate is widely discussed in the literature. The highest wavenumber state, Nia-RI, tends to be the target of theoretical modeling of experimental data,91 is the predominant state in crystals from which Ogata and co-workers have reported a high resolution X-ray structure,11 and is the state implicated in computational studies of the [NiFe] hydrogenase mechanism.8−10 Therefore, this is the representation most commonly used for “the Nia-R state” (Figure 4A) even for hydrogenases where the Nia-RI state is only a minority species (R. eutropha MBH, for example) or has not been observed at all (A. aeolicus Hase 1, for example), and despite the fact that the crystal samples (D. vulgaris MF) used for structural characterization also contained up to 18% of the Nia-RII state. Thus, there are clearly open questions regarding the relationship between the Nia-R substates. Also included in Figure 10B (open diamonds) are relative νCO values from DFT calculations of the Nia-R state both with cysteine thiolate protonation and a bridging hydride (as shown in Figure 4A) and as a deprotonated model in which only the bridging hydride is retained.91 The DFT calculations accurately reproduce both the experimental νCO positions relative to Nia-C and the difference in νCO between the Nia-RI and Nia-RIII states. Therefore, it is possible that the three Nia-R states represent successive proton-transfer steps away from the active site following initial heterolytic cleavage of H2.
As was the case with the Nia-L states the highest νCO Nia-R state, Nia-RI, tends to dominate in spectra of the Group 1 O2-sensitive hydrogenases, whereas either Nia-RII or Nia-RIII are the majority species for Group 1 O2-tolerant hydrogenases. In the case of E. coli Hyd-1, the Nia-RI state has not been observed during turnover and nonturnover studies carried out over a wide pH range.35,36 If our interpretation of the Nia-RII and Nia-RIII states as having a proton somewhat removed from the primary coordination sphere of the active site is correct, then the lack of a Nia-RI species for E. coli Hyd-1 is consistent with the mechanism proposed by Armstrong and co-workers54 (Figure 7A): heterolytic cleavage of H2 via a frustrated Lewis pair (FLP) mechanism, with the guanidinium group of an arginine residue in the active site canopy as the initial proton acceptor, results in a bridging hydride between Ni and Fe and a deprotonated active site. It should be noted, however, that this observation is not universally true for Group 1 O2-tolerant hydrogenases as a Nia-RI state is observed in R. eutropha MBH. If a FLP mechanism is occurring in E. coli Hyd-1, it might be expected that only one Nia-R state should be observed, rather than two. One possible explanation for this apparent discrepancy is that one of either Nia-RII or Nia-RIII might be closely related to the Michaelis–Menten complex between H2 and the [NiFe] active site, although further structural, kinetic, spectroscopic, and theoretical work would be needed to test this hypothesis. It is also possible that alternative proton-transfer routes exist and this would be consistent with the observation that a low-level of activity (1% native activity) is still observed in the R509K variant of E. coli Hyd-1 (Figure 6B,C), and that 20% native activity remains after exchange of one or both of the highly conserved aspartic acid residues in the active site canopy (D118 and D574) with asparagine (Figure 6B). A proton-transfer pathway has been proposed that terminates at an aspartic acid residue connected via a salt bridge to R509.17 The mutagenesis studies suggest that these aspartic acid residues are not essential for proton transfer, and Armstrong and co-workers note several crystallographically ordered water molecules in the vicinity of R509 that could mediate proton transfer.54Figure 12 shows the position of these water molecules in wild-type Hyd-1; similar conserved water structures are evident in membrane bound hydrogenases from Hydrogenovibrio marinus,64R. eutropha,65 and D. vulgaris MF.11 An additional water molecule is present in the R509K variant (Figure 12, blue sphere), and it is possible that proton transfer mediated by this water molecule is responsible for the residual activity of the R509K variant.
Figure 12.
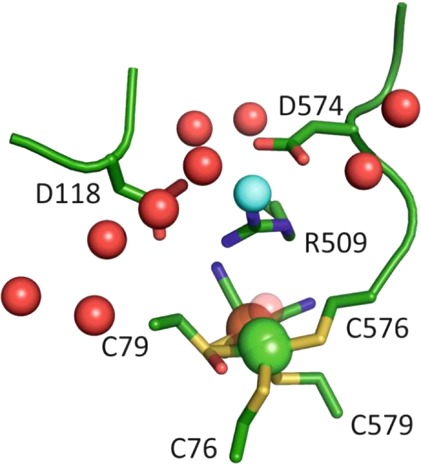
Active-site region of [NiFe] hydrogenases contain ordered crystallographic water molecules, demonstrated here for E. coli Hyd-1.54 The water molecules denoted in red spheres were found to be highly conserved in a range of Hyd-1 variants, whereas an additional water (blue sphere) was present in the R509K variant reported by Armstrong and co-workers. A stereo view of this figure is presented in the Supporting Information, Figure S2. Adapted with permission from ref (54). Copyright 2015 Nature Publishing Group.
The range of Nia-R states observed for different [NiFe] hydrogenases remains largely unexplained, although here we have suggested that the different Nia-R states, as with the Nia-L states, could represent steps involved in proton transfer during catalysis. The fact that amino acid mutations of basic residues close to the active site do not completely suppress activity suggests that multiple pathways, or a range of possible initial acceptor sites, exist for proton transfer. Understanding is still lacking on an initial H2 complex. Spectroscopic study of the base-variants around the active site will be informative in assessing the roles of these residues during H2 activation.
4. Summary and Outlook
Recent spectroscopic studies have confirmed the involvement of Nia-SI, Nia-R, and Nia-C in the [NiFe] hydrogenase catalytic cycle, and these studies also reinforce the likely importance of a range of NiI species, Nia-L, as on-pathway intermediates between Nia-C and Nia-SI. Two mechanisms have been proposed for initial splitting of H2 during the transition from Nia-SI to Nia-R, with either a Ni-bound terminal thiolate or a highly conserved arginine residue acting as the primary proton acceptor. The initial proton acceptor during the transition from Nia-C to Nia-L is thought to be the terminal cysteine thiolate, with a conserved glutamate residue demonstrated to be important for proton transfer beyond the active site. The postulation of a single acceptor site for each of these proton-transfer steps is appealing in its simplicity but belies the fact that multiple Nia-L and Nia-R states are observed. Proton-transfer pathways have been postulated that terminate close to both the conserved arginine and the terminal cysteine thiolates, and so it is possible (likely, even) that the two protons produced during H2 oxidation leave the active site region by different routes.
By considering data from across the range of [NiFe] hydrogenases that have been studied using vibrational spectroscopy, we have identified broad trends in the relative energies of νCO vibrations, and we suggest that these can be interpreted in terms of protonation behavior in the vicinity of the active site. In particular, we note that the relative energies of the νCO bands of the Nia-L and Nia-R states each fall into groupings that are consistent with theoretical calculations for protonation and deprotonation of the primary coordination sphere of Ni at the [NiFe] active site. We therefore propose that the collection of Nia-L and Nia-R states could represent rapid equilibration of the protons produced during the transitions from Nia-SI to Nia-R and from Nia-C to Nia-L to multiple acceptor sites along the first stages in proton-transfer pathway(s) leading to the enzyme surface.
In the case of states at the Nia-C/Nia-L redox level, interconversion between distinct Nia-L states is observed, in both time-resolved and static IR measurements, supporting the involvement of at least two Nia-L species during the transition from Nia-C to Nia-SI. The distance over which the proton can travel, and therefore, which Nia-L states are involved for any given hydrogenase, depends upon the relative rates of proton- and electron-transfer away from the active site. The rate of electron transfer will depend upon the ability of the proximal iron sulfur cluster to accept an electron, which requires the cluster to be (at least transiently) at the oxidized redox level. Several different proximal clusters are found in [NiFe] hydrogenases with reduction potentials that vary over a wide range: from very low potentials (<−0.5 V, as in R. eutropha RH), through O2-sensitive Group 1 hydrogenases (where the proximal cluster potential is close to the potential of the Nia-C(L)/Nia-SI couple), to the O2-tolerant Group 1 hydrogenases, which have very high potential proximal clusters (>0 V in E. coli Hyd-1). The rate of this electron-transfer step is likely to vary widely between different hydrogenases.
Open questions about the [NiFe] hydrogenase mechanism concern proton and electron-transfer steps as well as the initial mode of activation of H2. Different mechanistic pathways may be present in different groups of hydrogenases, and perhaps even within groups, so a common mechanism may not apply to all [NiFe] hydrogenases. Instead, it is possible that the [NiFe] active site provides a mechanistically flexible framework that can support H2-cycling via a range of pathways depending upon factors such as the potentials of clusters in the iron sulfur relay, solution pH, and the exact location of proton acceptor sites. Genetic variants are becoming available that are designed to affect specific steps in the [NiFe] catalytic cycle. The novel transient and turnover IR techniques discussed in this Perspective should be particularly informative when applied to these variants to elucidate the mechanistic significance of specific amino acid residues around the active site in hydrogenases from different Groups and different organisms within these Groups.
Acknowledgments
The work of K.A.V. and P.A.A. was supported by the European Research Council (EnergyBioCatalysis-ERC-2010-StG-258600), Engineering and Physical Sciences Research Council IB Catalyst award EP/N013514/1, and Biotechnology and Biological Sciences Research Council (BB/L009722/1 and BB/N006321/1). R.H. was supported by Ministerio de Ciencia y Tecnología, Universidad de Costa Rica, and Lincoln College, Oxford.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.6b03182.
Illustrative use of Nia-C as an internal standard, stereo view showing the position of crystallographically ordered water molecules in the vicinity of the [NiFe] hydrogenase active site (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Lubitz W.; Ogata H.; Rüdiger O.; Reijerse E. Chem. Rev. 2014, 114, 4081–4148. 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- Vincent K. A.; Parkin A.; Armstrong F. A. Chem. Rev. 2007, 107, 4366–4413. 10.1021/cr050191u. [DOI] [PubMed] [Google Scholar]
- Shafaat H. S.; Rüdiger O.; Ogata H.; Lubitz W. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 986–1002. 10.1016/j.bbabio.2013.01.015. [DOI] [PubMed] [Google Scholar]
- Schilter D.; Camara J. M.; Huynh M. T.; Hammes-Schiffer S.; Rauchfuss T. B. Chem. Rev. 2016, 116, 8693–8749. 10.1021/acs.chemrev.6b00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignais P. M.; Billoud B. Chem. Rev. 2007, 107, 4206–4272. 10.1021/cr050196r. [DOI] [PubMed] [Google Scholar]
- Greening C.; Biswas A.; Carere C. R.; Jackson C. J.; Taylor M. C.; Stott M. B.; Cook G. M.; Morales S. E. ISME J. 2016, 10, 761–777. 10.1038/ismej.2015.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, version 1.8.0.7; Schrödinger, LLC: New York, 2016.
- Pardo A.; De Lacey A. L.; Fernández V. M.; Fan H.-J.; Fan Y.; Hall M. B. JBIC, J. Biol. Inorg. Chem. 2006, 11, 286–306. 10.1007/s00775-005-0076-3. [DOI] [PubMed] [Google Scholar]
- Siegbahn P. E. M.; Tye J. W.; Hall M. B. Chem. Rev. 2007, 107, 4414–4435. 10.1021/cr050185y. [DOI] [PubMed] [Google Scholar]
- Lill S. O. N.; Siegbahn P. E. M. Biochemistry 2009, 48, 1056–1066. 10.1021/bi801218n. [DOI] [PubMed] [Google Scholar]
- Ogata H.; Nishikawa K.; Lubitz W. Nature 2015, 520, 571–574. 10.1038/nature14110. [DOI] [PubMed] [Google Scholar]
- Dementin S.; Burlat B.; De Lacey A. L.; Pardo A.; Adryanczyk-Perrier G.; Guigliarelli B.; Fernandez V. M.; Rousset M. J. Biol. Chem. 2004, 279, 10508–10513. 10.1074/jbc.M312716200. [DOI] [PubMed] [Google Scholar]
- Fontecilla-Camps J. C.; Volbeda A.; Cavazza C.; Nicolet Y. Chem. Rev. 2007, 107, 4273–4303. 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]
- Teixeira V. H.; Soares C. M.; Baptista A. M. Proteins: Struct., Funct., Genet. 2008, 70, 1010–1022. 10.1002/prot.21588. [DOI] [PubMed] [Google Scholar]
- Galván I. F.; Volbeda A.; Fontecilla-Camps J. C.; Field M. J. Proteins: Struct., Funct., Genet. 2008, 73, 195–203. 10.1002/prot.22045. [DOI] [PubMed] [Google Scholar]
- Sumner I.; Voth G. A. J. Phys. Chem. B 2012, 116, 2917–2926. 10.1021/jp208512y. [DOI] [PubMed] [Google Scholar]
- Szőri-Dorogházi E.; Maróti G.; Szőri M.; Nyilasi A.; Rákhely G.; Kovács K. L. PLoS One 2012, 7, e34666. 10.1371/journal.pone.0034666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitt S.; Ma K.; Warkentin E.; Moll J.; Pierik A. J.; Shima S.; Ermler U. J. Mol. Biol. 2014, 426, 2813–2826. 10.1016/j.jmb.2014.05.024. [DOI] [PubMed] [Google Scholar]
- Bagley K. A.; Van Garderen C. J.; Chen M.; Woodruff W. H.; Duin E. C.; Albracht S. P. J. Biochemistry 1994, 33, 9229–9236. 10.1021/bi00197a026. [DOI] [PubMed] [Google Scholar]
- Bagley K. A.; Duin E. C.; Roseboom W.; Albracht S. P. J.; Woodruff W. H. Biochemistry 1995, 34, 5527–5535. 10.1021/bi00016a026. [DOI] [PubMed] [Google Scholar]
- de Lacey A. L.; Hatchikian E. C.; Volbeda A.; Frey M.; Fontecilla-Camps J. C.; Fernandez V. M. J. Am. Chem. Soc. 1997, 119, 7181–7189. 10.1021/ja963802w. [DOI] [Google Scholar]
- Happe R. P.; Roseboom W.; Pierik A. J.; Albracht S. P. J.; Bagley K. A. Nature 1997, 385, 126–126. 10.1038/385126a0. [DOI] [PubMed] [Google Scholar]
- De Lacey A. L.; Fernández V. M.; Rousset M.; Cammack R. Chem. Rev. 2007, 107, 4304–4330. 10.1021/cr0501947. [DOI] [PubMed] [Google Scholar]
- Pandelia M.-E.; Ogata H.; Lubitz W. ChemPhysChem 2010, 11, 1127–1140. 10.1002/cphc.200900950. [DOI] [PubMed] [Google Scholar]
- Gutiérrez-Sanz O.; Rüdiger O.; De Lacey A. L. Methods Mol. Biol. 2014, 1122, 95–106. 10.1007/978-1-62703-794-5_7. [DOI] [PubMed] [Google Scholar]
- Fichtner C.; Laurich C.; Bothe E.; Lubitz W. Biochemistry 2006, 45, 9706–9716. 10.1021/bi0602462. [DOI] [PubMed] [Google Scholar]
- Kellers P.; Pandelia M.-E.; Currell L. J.; Görner H.; Lubitz W. Phys. Chem. Chem. Phys. 2009, 11, 8680–8683. 10.1039/b913635e. [DOI] [PubMed] [Google Scholar]
- Pandelia M.-E.; Ogata H.; Currell L. J.; Flores M.; Lubitz W. JBIC, J. Biol. Inorg. Chem. 2009, 14, 1227–1241. 10.1007/s00775-009-0566-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandelia M.-E.; Ogata H.; Currell L. J.; Flores M.; Lubitz W. Biochim. Biophys. Acta, Bioenerg. 2010, 1797, 304–313. 10.1016/j.bbabio.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Volbeda A.; Garcin E.; Piras C.; de Lacey A. L.; Fernandez V. M.; Hatchikian E. C.; Frey M.; Fontecilla-Camps J. C. J. Am. Chem. Soc. 1996, 118, 12989–12996. 10.1021/ja962270g. [DOI] [Google Scholar]
- De Lacey A. L.; Stadler C.; Fernandez V. M.; Hatchikian C. E.; Fan H.-J.; Li S.; Hall M. B. JBIC, J. Biol. Inorg. Chem. 2002, 7, 318–326. 10.1007/s00775-001-0301-7. [DOI] [PubMed] [Google Scholar]
- Bleijlevens B.; van Broekhuizen F. A.; De Lacey A. L.; Roseboom W.; Fernandez V. M.; Albracht S. P. J. JBIC, J. Biol. Inorg. Chem. 2004, 9, 743–752. 10.1007/s00775-004-0570-z. [DOI] [PubMed] [Google Scholar]
- Greene B. L.; Joseph C. A.; Maroney M. J.; Dyer R. B. J. Am. Chem. Soc. 2012, 134, 11108–11111. 10.1021/ja3042367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saggu M.; Zebger I.; Ludwig M.; Lenz O.; Friedrich B.; Hildebrandt P.; Lendzian F. J. Biol. Chem. 2009, 284, 16264–16276. 10.1074/jbc.M805690200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo R.; Ash P. A.; Healy A. J.; Vincent K. A. Angew. Chem., Int. Ed. 2015, 54, 7110–7113. 10.1002/anie.201502338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy B. J.; Hidalgo R.; Roessler M. M.; Evans R. M.; Ash P. A.; Myers W. K.; Vincent K. A.; Armstrong F. A. J. Am. Chem. Soc. 2015, 137, 8484–8489. 10.1021/jacs.5b03182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandelia M.-E.; Fourmond V.; Tron-Infossi P.; Lojou E.; Bertrand P.; Léger C.; Giudici-Orticoni M.-T.; Lubitz W. J. Am. Chem. Soc. 2010, 132, 6991–7004. 10.1021/ja910838d. [DOI] [PubMed] [Google Scholar]
- Pandelia M.-E.; Infossi P.; Stein M.; Giudici-Orticoni M.-T.; Lubitz W. Chem. Commun. 2012, 48, 823–825. 10.1039/C1CC16109A. [DOI] [PubMed] [Google Scholar]
- Pandelia M.-E.; Infossi P.; Giudici-Orticoni M. T.; Lubitz W. Biochemistry 2010, 49, 8873–8881. 10.1021/bi1006546. [DOI] [PubMed] [Google Scholar]
- Ash P. A.; Liu J.; Coutard N.; Heidary N.; Horch M.; Gudim I.; Simler T.; Zebger I.; Lenz O.; Vincent K. A. J. Phys. Chem. B 2015, 119, 13807–13815. 10.1021/acs.jpcb.5b04164. [DOI] [PubMed] [Google Scholar]
- Greene B. L.; Wu C.-H.; McTernan P. M.; Adams M. W. W.; Dyer R. B. J. Am. Chem. Soc. 2015, 137, 4558–4566. 10.1021/jacs.5b01791. [DOI] [PubMed] [Google Scholar]
- Greene B. L.; Wu C.-H.; Vansuch G. E.; Adams M. W. W.; Dyer R. B. Biochemistry 2016, 55, 1813–1825. 10.1021/acs.biochem.5b01348. [DOI] [PubMed] [Google Scholar]
- Horch M.; Lauterbach L.; Saggu M.; Hildebrandt P.; Lendzian F.; Bittl R.; Lenz O.; Zebger I. Angew. Chem., Int. Ed. 2010, 49, 8026–8029. 10.1002/anie.201002197. [DOI] [PubMed] [Google Scholar]
- Horch M.; Lauterbach L.; Mroginski M. A.; Hildebrandt P.; Lenz O.; Zebger I. J. Am. Chem. Soc. 2015, 137, 2555–2564. 10.1021/ja511154y. [DOI] [PubMed] [Google Scholar]
- Germer F.; Zebger I.; Saggu M.; Lendzian F.; Schulz R.; Appel J. J. Biol. Chem. 2009, 284, 36462–36472. 10.1074/jbc.M109.028795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millo D.; Pandelia M.-E.; Utesch T.; Wisitruangsakul N.; Mroginski M. A.; Lubitz W.; Hildebrandt P.; Zebger I. J. Phys. Chem. B 2009, 113, 15344–15351. 10.1021/jp906575r. [DOI] [PubMed] [Google Scholar]
- Siebert E.; Horch M.; Rippers Y.; Fritsch J.; Frielingsdorf S.; Lenz O.; Velazquez Escobar F.; Siebert F.; Paasche L.; Kuhlmann U.; Lendzian F.; Mroginski M.-A.; Zebger I.; Hildebrandt P. Angew. Chem., Int. Ed. 2013, 52, 5162–5165. 10.1002/anie.201209732. [DOI] [PubMed] [Google Scholar]
- Lubitz W.; Reijerse E.; van Gastel M. Chem. Rev. 2007, 107, 4331–4365. 10.1021/cr050186q. [DOI] [PubMed] [Google Scholar]
- Pandelia M.-E.; Nitschke W.; Infossi P.; Giudici-Orticoni M.-T.; Bill E.; Lubitz W. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6097–6102. 10.1073/pnas.1100610108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler M. M.; Evans R. M.; Davies R. A.; Harmer J.; Armstrong F. A. J. Am. Chem. Soc. 2012, 134, 15581–15594. 10.1021/ja307117y. [DOI] [PubMed] [Google Scholar]
- Karstens K.; Wahlefeld S.; Horch M.; Grunzel M.; Lauterbach L.; Lendzian F.; Zebger I.; Lenz O. Biochemistry 2015, 54, 389–403. 10.1021/bi501347u. [DOI] [PubMed] [Google Scholar]
- Roncaroli F.; Bill E.; Friedrich B.; Lenz O.; Lubitz W.; Pandelia M.-E. Chem. Sci. 2015, 6, 4495–4507. 10.1039/C5SC01560J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa M.; Pandelia M.-E.; Lubitz W.; van Gastel M.; Neese F. J. Am. Chem. Soc. 2013, 135, 3915–3925. 10.1021/ja3115899. [DOI] [PubMed] [Google Scholar]
- Evans R. M.; Brooke E. J.; Wehlin S. A. M.; Nomerotskaia E.; Sargent F.; Carr S. B.; Phillips S. E. V.; Armstrong F. A. Nat. Chem. Biol. 2016, 12, 46–50. 10.1038/nchembio.1976. [DOI] [PubMed] [Google Scholar]
- Shaw W. J.; Helm M. L.; DuBois D. L. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 1123–1139. 10.1016/j.bbabio.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Priyadarshani N.; Dutta A.; Ginovska B.; Buchko G. W.; O’Hagan M.; Raugei S.; Shaw W. J. ACS Catal. 2016, 6, 6037–6049. 10.1021/acscatal.6b01433. [DOI] [Google Scholar]
- Léger C.; Bertrand P. Chem. Rev. 2008, 108, 2379–2438. 10.1021/cr0680742. [DOI] [PubMed] [Google Scholar]
- Armstrong F. A.; Evans R. M.; Hexter S. V.; Murphy B. J.; Roessler M. M.; Wulff P. Acc. Chem. Res. 2016, 49, 884–892. 10.1021/acs.accounts.6b00027. [DOI] [PubMed] [Google Scholar]
- Flanagan L. A.; Parkin A. Biochem. Soc. Trans. 2016, 44, 315–328. 10.1042/BST20150201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai H.; Nishikawa K.; Suzuki M.; Higuchi Y.; Hirota S. Angew. Chem., Int. Ed. 2014, 53, 13817–13820. 10.1002/anie.201408552. [DOI] [PubMed] [Google Scholar]
- Greene B. L.; Vansuch G. E.; Wu C.-H.; Adams M. W. W.; Dyer R. B. J. Am. Chem. Soc. 2016, 138, 13013–13021. 10.1021/jacs.6b07789. [DOI] [PubMed] [Google Scholar]
- Greco C.; Fourmond V.; Baffert C.; Wang P.; Dementin S.; Bertrand P.; Bruschi M.; Blumberger J.; de Gioia L.; Léger C. Energy Environ. Sci. 2014, 7, 3543–3573. 10.1039/C4EE01848F. [DOI] [Google Scholar]
- Abou-Hamdan A.; Ceccaldi P.; Lebrette H.; Gutiérrez-Sanz O.; Richaud P.; Cournac L.; Guigliarelli B.; De Lacey A. L.; Léger C.; Volbeda A.; Burlat B.; Dementin S. J. Biol. Chem. 2015, 290, 8550–8558. 10.1074/jbc.M114.630491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shomura Y.; Yoon K.-S.; Nishihara H.; Higuchi Y. Nature 2011, 479, 253–256. 10.1038/nature10504. [DOI] [PubMed] [Google Scholar]
- Frielingsdorf S.; Fritsch J.; Schmidt A.; Hammer M.; Löwenstein J.; Siebert E.; Pelmenschikov V.; Jaenicke T.; Kalms J.; Rippers Y.; Lendzian F.; Zebger I.; Teutloff C.; Kaupp M.; Bittl R.; Hildebrandt P.; Friedrich B.; Lenz O.; Scheerer P. Nat. Chem. Biol. 2014, 10, 378–385. 10.1038/nchembio.1500. [DOI] [PubMed] [Google Scholar]
- Volbeda A.; Charon M.-H.; Piras C.; Hatchikian E. C.; Frey M.; Fontecilla-Camps J. C. Nature 1995, 373, 580–587. 10.1038/373580a0. [DOI] [PubMed] [Google Scholar]
- Ogata H.; Hirota S.; Nakahara A.; Komori H.; Shibata N.; Kato T.; Kano K.; Higuchi Y. Structure 2005, 13, 1635–1642. 10.1016/j.str.2005.07.018. [DOI] [PubMed] [Google Scholar]
- Ogata H.; Kellers P.; Lubitz W. J. Mol. Biol. 2010, 402, 428–444. 10.1016/j.jmb.2010.07.041. [DOI] [PubMed] [Google Scholar]
- Volbeda A.; Darnault C.; Parkin A.; Sargent F.; Armstrong F. A.; Fontecilla-Camps J. C. Structure 2013, 21, 184–190. 10.1016/j.str.2012.11.010. [DOI] [PubMed] [Google Scholar]
- Bowman L.; Flanagan L.; Fyfe P. K.; Parkin A.; Hunter W. N.; Sargent F. Biochem. J. 2014, 458, 449–458. 10.1042/BJ20131520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volbeda A.; Martin L.; Barbier E.; Gutiérrez-Sanz O.; De Lacey A. L.; Liebgott P.-P.; Dementin S.; Rousset M.; Fontecilla-Camps J. C. JBIC, J. Biol. Inorg. Chem. 2015, 20, 11–22. 10.1007/s00775-014-1203-9. [DOI] [PubMed] [Google Scholar]
- Fritsch J.; Scheerer P.; Frielingsdorf S.; Kroschinsky S.; Friedrich B.; Lenz O.; Spahn C. M. T. Nature 2011, 479, 249–252. 10.1038/nature10505. [DOI] [PubMed] [Google Scholar]
- Volbeda A.; Amara P.; Darnault C.; Mouesca J.-M.; Parkin A.; Roessler M. M.; Armstrong F. A.; Fontecilla-Camps J. C. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 5305–5310. 10.1073/pnas.1119806109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukey M. J.; Roessler M. M.; Parkin A.; Evans R. M.; Davies R. A.; Lenz O.; Friedrich B.; Sargent F.; Armstrong F. A. J. Am. Chem. Soc. 2011, 133, 16881–16892. 10.1021/ja205393w. [DOI] [PubMed] [Google Scholar]
- Schäfer C.; Bommer M.; Hennig S. E.; Jeoung J.-H.; Dobbek H.; Lenz O. Structure 2016, 24, 285–292. 10.1016/j.str.2015.11.010. [DOI] [PubMed] [Google Scholar]
- Parkin A.; Goldet G.; Cavazza C.; Fontecilla-Camps J. C.; Armstrong F. A. J. Am. Chem. Soc. 2008, 130, 13410–13416. 10.1021/ja803657d. [DOI] [PubMed] [Google Scholar]
- Wombwell C.; Caputo C. A.; Reisner E. Acc. Chem. Res. 2015, 48, 2858–2865. 10.1021/acs.accounts.5b00326. [DOI] [PubMed] [Google Scholar]
- Ceccaldi P.; Marques M. C.; Fourmond V.; Pereira I. C.; Léger C. Chem. Commun. 2015, 51, 14223–14226. 10.1039/C5CC05930E. [DOI] [PubMed] [Google Scholar]
- De Lacey A. L.; Gutiérrez-Sánchez C.; Fernández V. M.; Pacheco I.; Pereira I. A. C. JBIC, J. Biol. Inorg. Chem. 2008, 13, 1315–1320. 10.1007/s00775-008-0412-5. [DOI] [PubMed] [Google Scholar]
- Riethausen J.; Rüdiger O.; Gärtner W.; Lubitz W.; Shafaat H. S. ChemBioChem 2013, 14, 1714–1719. 10.1002/cbic.201300120. [DOI] [PubMed] [Google Scholar]
- Medina M.; Claude Hatchikian E.; Cammack R. Biochim. Biophys. Acta, Bioenerg. 1996, 1275, 227–236. 10.1016/0005-2728(96)00007-2. [DOI] [Google Scholar]
- Pierik A. J.; Schmelz M.; Lenz O.; Friedrich B.; Albracht S. P. FEBS Lett. 1998, 438, 231–235. 10.1016/S0014-5793(98)01306-4. [DOI] [PubMed] [Google Scholar]
- Bernhard M.; Buhrke T.; Bleijlevens B.; De Lacey A. L.; Fernandez V. M.; Albracht S. P. J.; Friedrich B. J. Biol. Chem. 2001, 276, 15592–15597. 10.1074/jbc.M009802200. [DOI] [PubMed] [Google Scholar]
- Brecht M.; van Gastel M.; Buhrke T.; Friedrich B.; Lubitz W. J. Am. Chem. Soc. 2003, 125, 13075–13083. 10.1021/ja036624x. [DOI] [PubMed] [Google Scholar]
- Buhrke T.; Lenz O.; Krauss N.; Friedrich B. J. Biol. Chem. 2005, 280, 23791–23796. 10.1074/jbc.M503260200. [DOI] [PubMed] [Google Scholar]
- Healy A. J.; Ash P. A.; Lenz O.; Vincent K. A. Phys. Chem. Chem. Phys. 2013, 15, 7055–7059. 10.1039/c3cp00119a. [DOI] [PubMed] [Google Scholar]
- Horch M.; Schoknecht J.; Mroginski M. A.; Lenz O.; Hildebrandt P.; Zebger I. J. Am. Chem. Soc. 2014, 136, 9870–9873. 10.1021/ja505119q. [DOI] [PubMed] [Google Scholar]
- Long M.; Liu J.; Chen Z.; Bleijlevens B.; Roseboom W.; Albracht S. P. J. JBIC, J. Biol. Inorg. Chem. 2006, 12, 62–78. 10.1007/s00775-006-0162-1. [DOI] [PubMed] [Google Scholar]
- Ogata H.; Lubitz W.; Higuchi Y. J. Biochem. 2016, 160, 251–258. 10.1093/jb/mvw048. [DOI] [PubMed] [Google Scholar]
- Jayapal P.; Sundararajan M.; Hillier I. H.; Burton N. A. Phys. Chem. Chem. Phys. 2008, 10, 4249–4257. 10.1039/b804035d. [DOI] [PubMed] [Google Scholar]
- Krämer T.; Kampa M.; Lubitz W.; van Gastel M.; Neese F. ChemBioChem 2013, 14, 1898–1905. 10.1002/cbic.201300104. [DOI] [PubMed] [Google Scholar]
- De Gioia L.; Fantucci P.; Guigliarelli B.; Bertrand P. Inorg. Chem. 1999, 38, 2658–2662. 10.1021/ic9811446. [DOI] [Google Scholar]
- George S. J.; Kurkin S.; Thorneley R. N. F.; Albracht S. P. J. Biochemistry 2004, 43, 6808–6819. 10.1021/bi049853k. [DOI] [PubMed] [Google Scholar]
- Ogata H.; Krämer T.; Wang H.; Schilter D.; Pelmenschikov V.; van Gastel M.; Neese F.; Rauchfuss T. B.; Gee L. B.; Scott A. D.; Yoda Y.; Tanaka Y.; Lubitz W.; Cramer S. P. Nat. Commun. 2015, 6, 7890. 10.1038/ncomms8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Yoda Y.; Ogata H.; Tanaka Y.; Lubitz W. J. Synchrotron Radiat. 2015, 22, 1334–1344. 10.1107/S1600577515017816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong G.; Ryde U. JBIC, J. Biol. Inorg. Chem. 2016, 21, 383–394. 10.1007/s00775-016-1348-9. [DOI] [PubMed] [Google Scholar]
- Brazzolotto D.; Gennari M.; Queyriaux N.; Simmons T. R.; Pécaut J.; Demeshko S.; Meyer F.; Orio M.; Artero V.; Duboc C. Nat. Chem. 2016, 8, 1054–1060. 10.1038/nchem.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubas G. J.; Ryan R. R.; Swanson B. I.; Vergamini P. J.; Wasserman H. J. J. Am. Chem. Soc. 1984, 106, 451–452. 10.1021/ja00314a049. [DOI] [Google Scholar]
- Kubas G. J.Metal-dihydrogen and σ-bond complexes; Kluwer Academic: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Weber K.; Krämer T.; Shafaat H. S.; Weyhermüller T.; Bill E.; van Gastel M.; Neese F.; Lubitz W. J. Am. Chem. Soc. 2012, 134, 20745–20755. 10.1021/ja309563p. [DOI] [PubMed] [Google Scholar]
- Ogo S.; Ichikawa K.; Kishima T.; Matsumoto T.; Nakai H.; Kusaka K.; Ohhara T. Science 2013, 339, 682–684. 10.1126/science.1231345. [DOI] [PubMed] [Google Scholar]
- Chambers G. M.; Mitra J.; Rauchfuss T. B.; Stein M. Inorg. Chem. 2014, 53, 4243–4249. 10.1021/ic500389p. [DOI] [PubMed] [Google Scholar]
- Berggren G.; Adamska A.; Lambertz C.; Simmons T. R.; Esselborn J.; Atta M.; Gambarelli S.; Mouesca J.-M.; Reijerse E.; Lubitz W.; Happe T.; Artero V.; Fontecave M. Nature 2013, 499, 66–69. 10.1038/nature12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A.; DuBois D. L.; Roberts J. A. S.; Shaw W. J. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 16286–16291. 10.1073/pnas.1416381111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant F. O.; Adams M. W. J. Biol. Chem. 1989, 264, 5070–5079. [PubMed] [Google Scholar]
- Ma K.; Weiss R.; Adams M. W. W. J. Bacteriol. 2000, 182, 1864–1871. 10.1128/JB.182.7.1864-1871.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu S.; Thomson L. M.; Hall M. B. J. Am. Chem. Soc. 1999, 121, 4000–4007. 10.1021/ja983469r. [DOI] [Google Scholar]
- Lindahl P. A. J. Inorg. Biochem. 2012, 106, 172–178. 10.1016/j.jinorgbio.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer and co-workers have relabeled the Nia-L states they observe in P. furiosus SH1 as “Nia-I” to reflect their role as active intermediates. However, we prefer the reclassification as Nia-L to reflect a role in catalysis while maintaining a link with existing literature on the Ni-L states.
- Tai H.; Nishikawa K.; Inoue S.; Higuchi Y.; Hirota S. J. Phys. Chem. B 2015, 119, 13668–13674. 10.1021/acs.jpcb.5b03075. [DOI] [PubMed] [Google Scholar]
- Bard A.; Faulkner L. R.. Electrochemical Methods: Fundamentals and Applications, 2nd ed..; John Wiley & Sons, Inc.: Hoboken, NJ, 2001. [Google Scholar]
- Oteri F.; Baaden M.; Lojou E.; Sacquin-Mora S. J. Phys. Chem. B 2014, 118, 13800–13811. 10.1021/jp5089965. [DOI] [PubMed] [Google Scholar]
- Long H.; King P. W.; Chang C. H. J. Phys. Chem. B 2014, 118, 890–900. 10.1021/jp408621r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.