

Abstract
The use of a combination of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and β-pinene permits the removal of 2-naphthylmethyl (Nap) ether protecting groups on highly sensitive substrates. The reaction tolerates both acid and base sensitive protecting groups, and products are afforded in 68–96% yield. The utility of the method is demonstrated by the removal of the Nap protecting groups on highly sensitive 2,6-dideoxy-sugar disaccharides.
Like most other forms of chemical synthesis, carbohydrate chemistry relies heavily on protecting groups. In addition to masking functional groups from reacting, protecting groups often play a role of both controlling selectivity in traditional glycosylation reactions,1,2 and modulating the reactivity of coupling partners through either “arming” or “disarming” effects.3 As a consequence, when planning the synthesis of an oligosaccharide, it is often necessary to carefully consider the nature of the protecting groups on every monosaccharide. Typically, protecting groups in carbohydrate chemistry can be divided into two groups; “permanent” protecting groups, such as benzyl ethers, which are removed at the end of the synthesis, and “temporary” protecting groups, which are removed during the course of the synthesis to unmask nucleophilic alcohols for further coupling.4 Among the temporary protecting groups that have found increased usage over the course of the past decade, the 2-naphthylmethyl (Nap) protecting group has emerged as a particularly valuable addition to the chemist’s arsenal.5 Two factors give rise to the utility of this protecting group. First, it does not significantly attenuate carbohydrate reactivity (it is not a disarmed protecting group). Furthermore, it can be readily removed under a variety of conditions that are orthogonal to benzyl ethers, including deprotection under oxidative6−8 and acid-mediated conditions.9−11
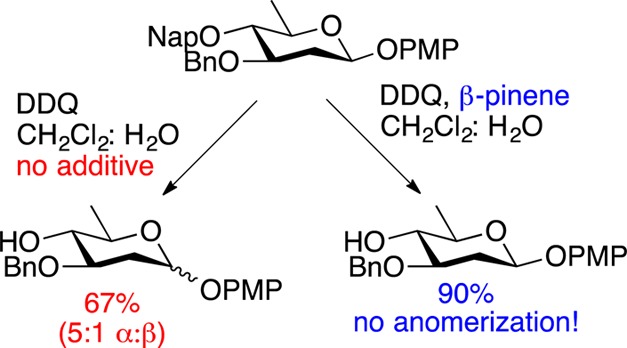
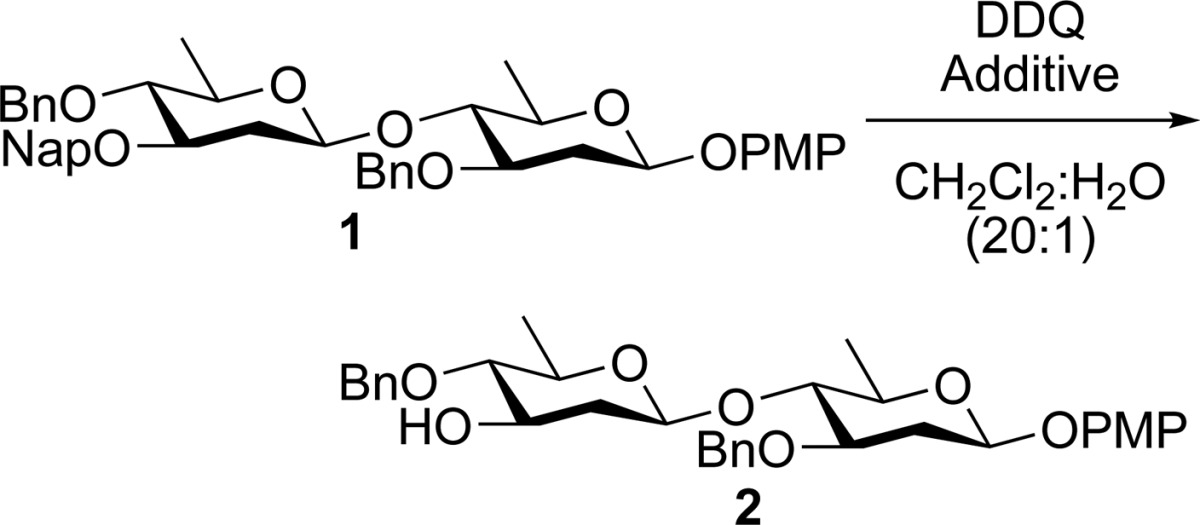
Given the versatility of the Nap protecting group, we sought to examine its applicability as a temporary protecting group in our ongoing studies in 2-deoxy-sugar oligosaccharide synthesis.12,13 Owing to the highly labile nature of deoxy-sugars, we had to limit our studies to reactions that did not require the use of strong acid. To this end, we studied the reaction of disaccharide 1 with DDQ in the presence of CH2Cl2 and water under conditions described by Danishefsky and co-workers.14 In the absence of any additives, DDQ removed the Nap ether to afford the deprotected disaccharide 2 in moderate yield (56%, Table 1, entry 1). Reasoning that the acidic 2,3-dichloro-5,6-dicyanohydroquinone byproduct of the DDQ deprotection was causing product decomposition,15 we next looked at the use of additives in the reaction. The use of the non-nucleophilic base tri-tert-butyl pyrimidine (TTBP) did not improve the yield of the reaction (Table 1, entry 2), presumably due to the low pKa of the conjugate base of TTBP.16 The more basic hindered amines, such as 2,6-di-tert-butyl-4-methylpyridine (DTBMP) resulted in a slight increase in the yield of the desired product (Table 1, entry 3). Surprisingly, 2,6-di-tert-butylpyridine (DTBP) resulted in greatly increased yields of the desired product (Table 1 entry 4), despite the fact that this compound is slightly less basic than DTBMP.17
Table 1. Screen of Additives on DDQ-Mediated Nap Ether Removala.
| entry | additive | additive equiv. | yield |
|---|---|---|---|
| 1 | 54% | ||
| 2 | TTBP | 1 | 60% |
| 3 | DTBMP | 3.4 | 65% |
| 4 | DTBP | 3.4 | 86% |
| 5 | β-pinene | 3.4 | 96% |
PMP = 4-methoxyphenyl.
While the use of DTBP afforded the desired product in good yield, we sought to further improve the yield and mildness of the reaction while avoiding the use of expensive additives. To this end, we turned our attention to the use of nonbasic proton scavengers. Recently, Shen and co-workers described the use of the strained alkene β-pinene as an alternative acid scavenger in glycosylations where amine bases were problematic.18,19 Reasoning that we could take advantage of this proton scavenger in our studies, we examined the outcome of the reaction when β-pinene was used in place of an amine base. This approach proved to be highly effective, and treating 1 with a combination of DDQ and β-pinene led to the formation of the desired product 2 in 96% yield (Table 1, entry 5).20
Having established the optimal conditions for Nap ether removal, we turned our attention to examining the scope of the reaction, and its compatibility with various common sugar protecting groups (Table 2). The synthesis of these substrates were carried out under standard conditions as outlined in Scheme 1. Benzylidene acetals are tolerated under the reaction conditions (Table 2, entries 1 and 2). Similarly, Nap ethers on ethyl or phenyl thioglycosides of both fucose and rhamnose can be readily removed (Table 2, entries 5–7). The reaction tolerates deoxy-sugars possessing sensitive β-linked anomeric 4-methoxyphenyl (PMP) protecting groups (Table 2, entry 8). Omitting β-pinene from this latter reaction led to both reduced yield and anomerization of the PMP group to give predominantly α-linked product (Table 2, entry 8). This latter result highlights the importance of the use of scavangers in the deprotection of sensitive substrates. With less sensitive substrates similar yields were obtained both in the presence and absence of β-pinene (Table 2, entries 1, 3, 4, 6, and 7). Finally, the reaction conditions also tolerated highly sensitive glycal structures (Table 2, entries 9 and 10), although removal of an allylic Nap group proceeded in lower yield than the corresponding homoallylic Nap ether. Notably, in the absence of β-pinene we were only able to obtain the product in low (18%) yield along with significant quantity of a byproduct. The NMR spectra of this product appeared to contain a mixture of hemiacetal and aldehyde. In order to elucidate the identity of this material, it was acetylated to afford the 2,3-anhydro sugar 32 in 60% yield over two steps (Scheme 2). This latter result clearly demonstrates the advantage of the use of β-pinene in the reaction.
Table 2. Substrate Scope.
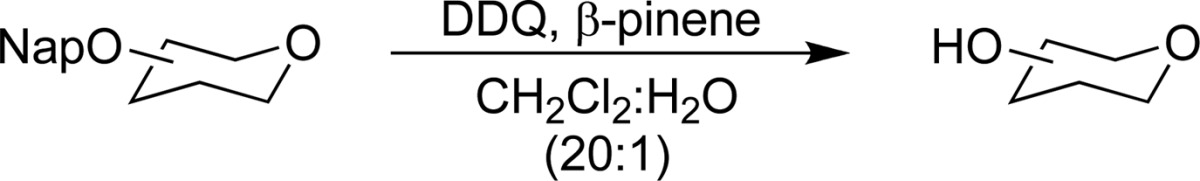
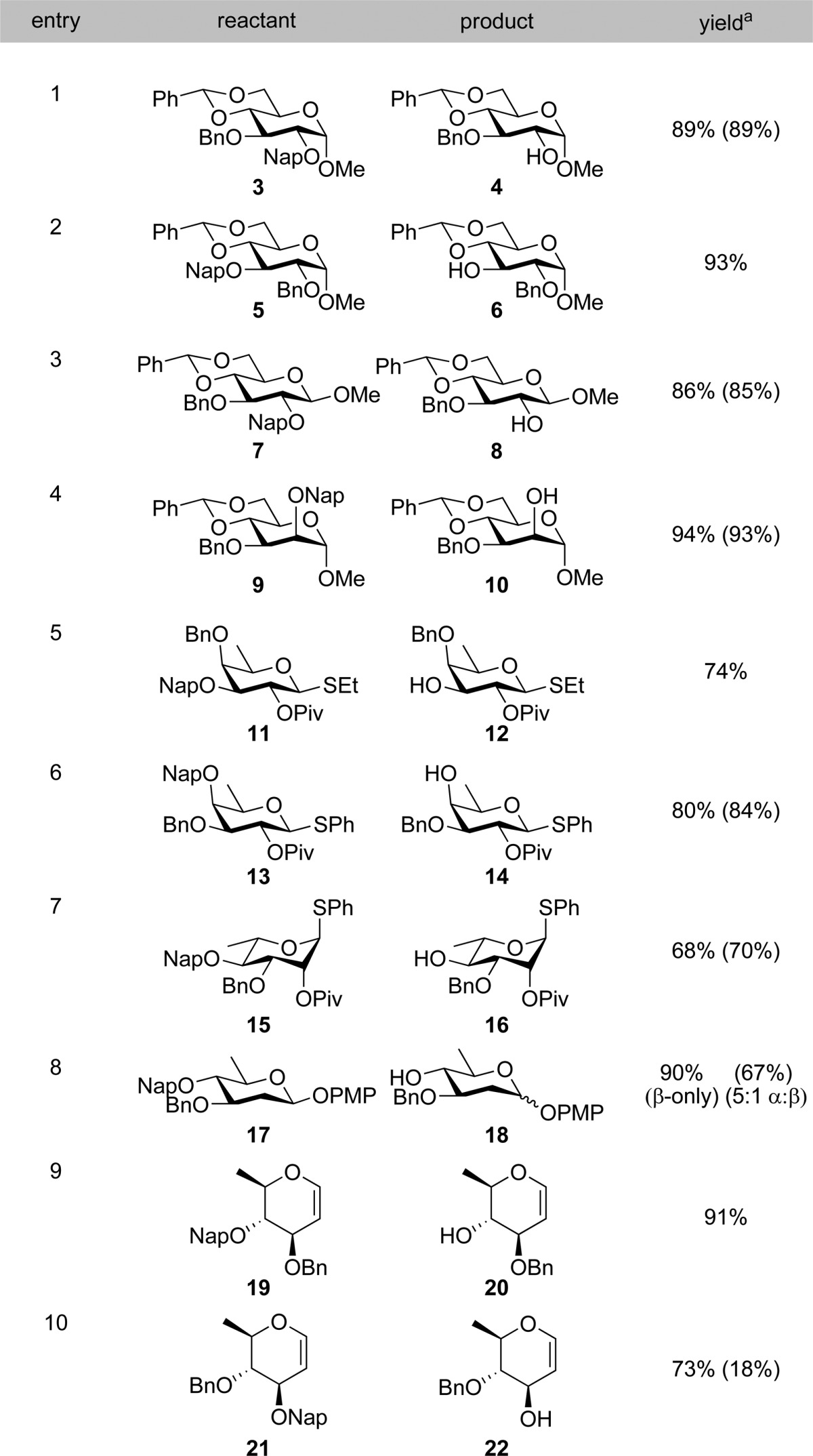
Yield in the parentheses is without β-pinene.
Scheme 1. Synthesis of Substrates Used in this Study.

Scheme 2. Identification of Byproduct from the Reaction of 17 without β-Pinene.

In conclusion, we have demonstrated that a combination of DDQ and β-pinene permits the removal of Nap ethers under particularly mild conditions. The reaction tolerates a number of highly sensitive substrates, and does not suffer from problems of in situ anomerization of the substrates observed when DDQ is used alone. Given the mildness of these conditions, and the inexpensive nature of β-pinene, we anticipate that these conditions will prove useful to chemists who wish to use Nap ethers as protecting groups in all areas of synthetic chemistry.
Experimental Section
General Considerations
All reactions were performed under inert argon atmosphere, unless otherwise noted. Flash column chromatography was performed on 230–400 mesh silica gel. Analytical and preparative thin layer chromatography was carried out on silica gel 60 F-254 plates. Products were visualized using UV or by staining with 5% aqueous sulfuric acid or ceric ammonium molybdate. NMR spectra were recorded on a NMR spectrometer at 500 MHz for 1H NMR and 125 MHz for 13C NMR. Chemical shifts are reported in ppm relative to TMS (for 1H NMR in CDCl3) or CDCl3 (for 13C NMR in CDCl3). For 1H NMR spectra, data are reported as follows: δ shift, multiplicity (s = singlet, m = multiplet, t = triplet, d = doublet, dd = doublet of doublets, td = triplet of doublets, q = quartet, brs = broad singlet), coupling constants are reported in Hz. Low-resolution mass spectra (LRMS) were recorded using a ESI-MS with an additional APCI source. High-resolution mass spectra (HRMS) were obtained on Fourier Transform Ion Cyclotron Resonance Mass Spectrometer (FT-ICR-MS) with direct analysis in real time (DART) ionization source. Optical rotations were measured at 589 nm in a 5 cm cell at 24 °C. Solvents for reactions were dried immediately prior to use. All other chemicals were purchased at the highest possible quality and used as received. Compounds 4,216,218,2210,2315,2420,2523,2627,27 and 30(25) were prepared according to literature methods
Procedure A: General Procedure Nap Ether Removal with β-Pinene
To a solution of 1 (0.59 mmol) and β-pinene (2.0 mmol) in DCM (58 mL) and H2O (2.9 mL), was added DDQ (1.1 mmol). The reaction was stirred at room temperature under argon for 4 h and monitored by TLC. The reaction mixture was then diluted with DCM (50 mL), and the organic layer was washed 3 times with 50 mL of 2 M NaOH. The pooled organic layers were washed with 100 mL of sat. NaHCO3, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (30% ethyl acetate in hexanes) to afford 2 (323 mg, 96%) as a white powder.
Procedure B: General Procedure Nap Ether Removal without β-Pinene
To a solution of 1 (0.59 mmol) in DCM (59 mL) and H2O (2.9 mL), was added DDQ (1.1 mmol). The reaction was stirred at room temperature under argon for 4 h and monitored by TLC. The reaction mixture was then diluted with DCM (75 mL), and the organic layer was washed 3 times with 50 mL of 2 M NaOH. The pooled organic layers were washed with 50 mL of sat. NaHCO3, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (30% ethyl acetate in hexanes) to afford 2 (183 mg, 54%) as a white powder.
p-Methoxylphenyl 4-(4′-O-Benzyl-2′,6′-dideoxy-β-d-arabino-hexopyranosyl)-3-O-benzyl-2,6-dideoxy-β-d-arabino-hexopyranose (2)
mp: 151–152 °C; [α] D24 = −30.0 (c 1.05, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.41–7.26 (m, 10H), 6.97–6.91 (m, 2H), 6.84–6.78 (m, 2H), 4.95 (dd, J = 9.9, 2.0 Hz, 1H), 4.82–4.64 (m, 5H), 3.76 (s, 3H), 3.71–3.56 (m, 1H), 3.50–3.40 (m, 1H), 3.39–3.25 (m, 2H), 2.99 (t, J = 8.9 Hz, 1H), 2.45 (ddd, J = 11.4, 6.3, 1.7 Hz, 1H), 2.26 (ddd, J = 13.7, 6.8, 1.7 Hz, 1H), 2.08 (d, J = 3.2 Hz, 1H), 1.88 (td, J = 12.1, 10.1 Hz, 1H), 1.60 (td, J = 12.0, 9.8 Hz, 1H), 1.35 (t, J = 6.5 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 155.0, 151.2, 138.7, 138.2, 128.7, 128.4, 128.1, 127.9, 127.6, 127.5, 117.9, 114.5, 100.5, 98.5, 85.8, 82.5, 77.6, 75.2, 71.9, 71.6, 71.3, 71.0, 55.6, 39.3, 37.0, 18.5, 18.4; LRMS (ESI) m/z: [M+Na]+ Calcd for C33H40O8Na 587.26; Found 587.45; HRMS (DART) m/z: [M+NH4]+ Calcd for C33H44NO8 582.3061; Found 582.3058.
Methyl 3-O-Benzyl-4,6-O-benzylidine-α-d-glucopyranoside (4)
The title compound was prepared according to procedure A using 3 (250 mg, 0.48 mmol) and β-pinene (0.24 mL, 11.6 mmol) dissolved in DCM (48 mL) and H2O (22.4 mL), the solution was then treated with DDQ (220 mg, 0.97 mmol) for 4 h. The title compound was purified by flash column chromatography on silica gel (20% ethyl acetate in hexanes) to afford compound 4 (159 mg, 89%) as a white solid. The spectroscopic data is in good agreement with previously reported data.211H NMR (500 MHz, CDCl3) δ 7.52–7.46 (m, 2H), 7.43–7.27 (m, 8H), 5.58 (s, 1H), 4.97 (d, J = 11.6 Hz, 1H), 4.82 (d, J = 3.8 Hz, 1H), 4.79 (d, J = 11.6 Hz, 1H), 4.30 (dd, J = 10.0, 4.6 Hz, 1H), 3.89–3.80 (m, 2H), 3.80–3.70 (m, 2H), 3.65 (t, J = 9.2 Hz, 1H), 3.46 (s, 3H), 2.29 (d, J = 7.4 Hz, 1H).
Methyl 3-O-Benzyl-4,6-O-benzylidine-α-d-glucopyranoside (4)
The title compound was prepared according to procedure B using 3 (250 mg, 0.48 mmol) in DCM (48 mL) and H2O (2.4 mL), DDQ (220 mg, 0.97 mmol) was then added. The title compound was purified by flash column chromatography on silica gel (20% ethyl acetate in hexanes) to afford compound 4 (158 mg, 89%). The spectroscopic data is in good agreement with previously reported data.21
Methyl 2-O-Benzyl-4,6-O-benzylidine-α-d-glucopyranoside (6)
The title compound was prepared according to procedure A using 5 (234 mg, 0.45 mmol) and β-pinene (0.24 mL, 1.5 mmol) dissolved in DCM (46 mL) and H2O (2.3 mL), the solution was then treated with DDQ (206 mg, 0.9 mmol) for 4 h. The title compound was purified by flash column chromatography on silica gel (20% ethyl acetate in hexanes) to afford compound 6 (155 mg, 93%) as a white solid. The spectroscopic data is in good agreement with previously reported data.211H NMR (500 MHz, CDCl3) δ 7.52–7.44 (m, 2H), 7.42–7.28 (m, 8H), 5.52 (s, 1H), 4.79 (d, J = 12.2 Hz, 1H), 4.71 (d, J = 12.2 Hz, 1H), 4.61 (d, J = 3.6 Hz, 1H), 4.26 (dd, J = 10.2, 4.8 Hz, 1H), 4.15 (td, J = 9.3, 2.2 Hz, 1H), 3.81 (td, J = 10.0, 4.8 Hz, 1H), 3.70 (t, J = 10.3 Hz, 1H), 3.53–3.43 (m, 2H), 3.38 (s, 3H), 2.55 (d, J = 2.2 Hz, 1H).
Methyl 3-O-Benzyl-4,6-O-benzylidine-β-d-glucopyranoside (8)
The title compound was prepared according to procedure A using 7 (100 mg, 0.19 mmol) and β-pinene (0.10 mL, 0.64 mmol) dissolved in DCM (19 mL) and H2O (0.95 mL), the solution was then treated with DDQ (86 mg, 0.38 mmol) for 4 h. The title compound was purified by flash column chromatography on silica gel (40% ethyl acetate in hexanes) to afford compound 8 (63 mg, 86%) as a white solid. The spectroscopic data is in good agreement with previously reported data.221H NMR (500 MHz, CDCl3) δ 7.52–7.46 (m, 2H), 7.44–7.26 (m, 8H), 5.58 (s, 1H), 4.98 (d, J = 11.6 Hz, 1H), 4.79 (d, J = 11.6 Hz, 1H), 4.40–4.31 (m, 2H), 3.81 (t, J = 10.3 Hz, 1H), 3.75–3.63 (m, 2H), 3.58 (s, 3H), 3.57–3.52 (m, 1H), 3.46 (ddd, J = 10.1, 8.8, 5.0 Hz, 1H), 2.43 (d, J = 2.2 Hz, 1H).
Methyl 3-O-Benzyl-4,6-O-benzylidine-β-d-glucopyranoside (8)
The title compound was prepared according to procedure B using 7 (100 mg, 0.19 mmol) dissolved in DCM (19 mL) and H2O (0.95 mL), the solution was then treated with DDQ (86 mg, 0.38 mmol) for 3 h. The title compound was purified by flash column chromatography on silica gel (40% ethyl acetate in hexanes) to afford compound 20 (62 mg, 85%) as a white solid. The spectroscopic data is in good agreement with previously reported data.221H NMR (500 MHz, CDCl3) δ 7.52–7.46 (m, 2H), 7.44–7.26 (m, 8H), 5.58 (s, 1H), 4.98 (d, J = 11.6 Hz, 1H), 4.79 (d, J = 11.6 Hz, 1H), 4.40–4.31 (m, 2H), 3.81 (t, J = 10.3 Hz, 1H), 3.75–3.63 (m, 2H), 3.58 (s, 3H), 3.57–3.52 (m, 1H), 3.46 (ddd, J = 10.1, 8.8, 5.0 Hz, 1H), 2.43 (d, J = 2.2 Hz, 1H).
Methyl 3-O-Benzyl-4,6-O-benzylidine-α-d-mannopyranoside (10)
The title compound was prepared according to procedure A using 9 (100 mg, 0.19 mmol) and β-pinene (0.10 mL, 0.64 mmol) dissolved in DCM (19 mL) and H2O (0.95 mL), the solution was then treated with DDQ (86 mg, 0.38 mmol) for 4 h. The title compound was purified by flash column chromatography on silica gel (40% ethyl acetate in hexanes) to afford compound 10 (69 mg, 94%) as colorless oil. The spectroscopic data is in good agreement with previously reported data.221H NMR (500 MHz, CDCl3) δ 7.52–7.46 (m, 2H), 7.42–7.25 (m, 8H), 5.61 (s, 1H), 4.84 (d, J = 11.8 Hz, 1H), 4.76–4.67 (m, 2H), 4.27 (dd, J = 9.7, 4.3 Hz, 1H), 4.10 (t, J = 9.3 Hz, 1H), 4.04–3.99 (m, 1H), 3.93–3.76 (m, 3H), 3.36 (s, 3H), 2.80 (d, J = 1.5 Hz, 1H).
Methyl 3-O-Benzyl-4,6-O-benzylidine-α-d-mannopyranoside (10)
The title compound was prepared according to procedure B using 9 (100 mg, 0.19 mmol) dissolved in DCM (19 mL) and H2O (0.95 mL), the solution was then treated with DDQ (86 mg, 0.38 mmol) for 4 h. The title compound was purified by flash column chromatography on silica gel (40% ethyl acetate in hexanes) to afford compound 10 (62 mg, 85%) as colorless oil. The spectroscopic data is in good agreement with previously reported data.231H NMR (500 MHz, CDCl3) δ 7.52–7.46 (m, 2H), 7.42–7.25 (m, 8H), 5.61 (s, 1H), 4.84 (d, J = 11.8 Hz, 1H), 4.76–4.67 (m, 2H), 4.27 (dd, J = 9.7, 4.3 Hz, 1H), 4.10 (t, J = 9.3 Hz, 1H), 4.04–3.99 (m, 1H), 3.93–3.76 (m, 3H), 3.36 (s, 3H), 2.80 (d, J = 1.5 Hz, 1H).
Ethyl 6-Deoxy-4-O-benzyl-2-O-pivaloyl-1-thio-β-d-galactopyranoside (12)
The title compound was prepared according to procedure A using 11 (462.8 mg, 0.885 mmol) and β-pinene (0.47 mL, 3.03 mmol) dissolved in DCM (89 mL) and H2O (4.4 mL), the solution was then treated with DDQ (400 mg, 1.77 mmol) for 5 h. The title compound was purified by flash column chromatography on silica gel (5% ethyl acetate in toluene) to afford compound 12 (251.4 mg, 74%) as a white solid. mp: 144–145 °C; [α] D24 = −8.4 (c = 1.14, CH2Cl2); 1H NMR (500 MHz, CDCl3): δ 7.37–7.31 (m, 5H), 5.02 (t, J = 10.0 Hz, 1H), 4.83 (d, J = 11.7 Hz, 1H), 4.69 (d, J = 11.7 Hz, 1H), 4.37 (d, J = 10.0 Hz, 1H), 3.67–3.63 (m, 3H), 2.77–2.72 (m, 1H), 2.69–2.62 (m, 1H), 2.25 (d, J = 9.5 Hz, 1 H), 1.34 (d, J = 6.5 Hz, 3H), 1.25 (t, J = 7.5 Hz, 3H), 1.23 (s, 9H); 13C NMR (125 MHz, CDCl3): δ 178.5, 138.0, 128.6, 128.0, 127.9, 82.9, 80.1, 76.1, 75.0, 74.4, 71.1, 38.9, 27.1, 23.6, 17.3, 14.9; LRMS (ESI) m/z: [M+Na]+ Calcd for C20H30O5SNa 405.17; Found 405.29; HRMS (DART) m/z: [M+NH4]+ Calcd for C20H34NO5S 400.2152; Found 400.2144.
Phenyl 6-Deoxy-3-O-benzyl-2-O-pivaloyl-1-thio-β-d-galactopyranoside (14)
The title compound was prepared according to procedure A using 13 (100 mg, 0.18 mmol) and β-pinene (0.09 mL, 0.60 mmol) dissolved in DCM (17 mL) and H2O (1.9 mL), the solution was then treated with DDQ (79.5 mg, 0.35 mmol) for 2 h. The title compound was purified by flash column chromatography on silica gel (5% to 10% ethyl acetate in toluene) to afford compound 14 (60.7 mg, 80%) as an amorphous solid. [α] D24 = +5.5 (c = 0.95, CH2Cl2); 1H NMR (500 MHz, CDCl3): δ 7.50–7.48 (m, 2H), 7.34–7.26 (m, 8H), 5.23 (t, J = 9.8 Hz, 1H), 4.66–4.58 (m, 3H), 3.84 (t, J = 2.5 Hz, 1H), 3.62–3.57 (m, 2H), 2.38 (s, 1H), 1.40 (d, J = 6.5 Hz, 3H), 1.24 (s, 9H); 13C NMR (125 MHz, CDCl3): δ 177.0, 137.4, 133.9, 132.2, 128.9, 128.7, 128.2, 127.8, 127.7, 86.9, 80.4, 74.5, 71.9, 69.2, 68.6, 38.9, 27.3, 16.9; LRMS (ESI) m/z: [M+Na]+ Calcd for C24H30O5SNa 453.17; Found 453.36; HRMS (DART) m/z: [M+NH4]+ Calcd for C24H34NO5S 448.2152; Found 448.2155.
Phenyl 6-Deoxy-3-O-benzyl-2-O-pivaloyl-1-thio-β-d-galactopyranoside (14)
The title compound was prepared according to procedure B using 13 (116.7 mg, 0.20 mmol) dissolved in DCM (20 mL) and H2O (2.2 mL), the solution was then treated with DDQ (96.5 mg, 0.43 mmol) for 5 h. The title compound was purified by flash column chromatography on silica gel (5% to 10% ethyl acetate in toluene) to afford compound 14 (73.8 mg, 84%) as an amorphous solid. The spectroscopic data is in good agreement with data as shown above. 1H NMR (500 MHz, CDCl3): δ 7.50–7.48 (m, 2H), 7.34–7.26 (m, 8H), 5.23 (t, J = 9.5 Hz, 1H), 4.66–4.58 (m, 3H), 3.84 (t, J = 2.5 Hz, 1H), 3.62–3.57 (m, 2H), 2.35 (s, 1H), 1.40 (d, J = 6.5 Hz, 3H), 1.24 (s, 9H).
Phenyl 6-Deoxy-3-O-benzyl-2-O-pivaloyl-1-thio-α-l-rhamnopyranose (16)
The title compound was prepared according to procedure A using 15(24) (60.5 mg, 0.10 mmol) and β-pinene (0.06 mL, 0.36 mmol) dissolved in DCM (10.6 mL) and H2O (0.53 mL), the solution was then treated with DDQ (48 mg, 0.212 mmol) for 4 h. The title compound was purified by flash column chromatography on silica gel (5% ethyl acetate in toluene) to afford compound 16 (30.9 mg, 68%) as a white solid. The spectroscopic data is in good agreement with previously reported data.241H NMR (500 MHz, CDCl3): δ 7.49–7.47 (m, 2H), 7.38–7.26 (m, 8H), 5.60 (s, 1H), 5.40 (s, 1H), 4.72 (d, J = 10.9 Hz, 1H), 4.42 (d, J = 10.9 Hz, 1H), 4.23–4.20 (m, 1H), 3.72 (dd, J = 2.4, 9.5 Hz, 1H), 3.62 (t, J = 9.2 Hz, 1H), 2.33 (s, 1H), 1.35 (d, J = 6.1 Hz, 3H), 1.21 (s, 9H).
Phenyl 6-Deoxy-3-O-benzyl-2-O-pivaloyl-1-thio-α-l-rhamnopyranose (16)
The title compound was prepared according to procedure B using 15(24) (115.5 mg, 0.20 mmol) dissolved in DCM (20 mL) and H2O (2.2 mL), the solution was then treated with DDQ (93.3 mg, 0.41 mmol) for 4 h. The title compound was purified by flash column chromatography on silica gel (5% ethyl acetate in toluene) to afford compound 16 (61.3 mg, 70%) as a white solid. The spectroscopic data is in good agreement with previously reported data.241H NMR (500 MHz, CDCl3): δ 7.49–7.47 (m, 2H), 7.37–7.26 (m, 8H), 5.60 (s, 1H), 5.40 (s, 1H), 4.72 (d, J = 11.1 Hz, 1H), 4.42 (d, J = 11.1 Hz, 1H), 4.23–4.20 (m, 1H), 3.72 (dd, J = 2.4, 9.5 Hz, 1H), 3.62 (t, J = 9.2 Hz, 1H), 2.33 (s, 1H), 1.35 (d, J = 6.0 Hz, 3H), 1.21 (s, 9H).
β-p-Methoxylphenyl 3-O-Benzyl-2,6-dideoxy-d-arabino-hexopyranose (18)
The title compound was prepared according to procedure A using 17 (1.5 g, 3.6 mmol) and β-pinene (1.9 mL, 12.3 mmol) dissolved in DCM (360 mL) and H2O (18 mL), the solution was then treated with DDQ (1.6 g, 7.2 mmol) for 3 h. The title compound was purified by flash column chromatography on silica gel (100% dichloromethane then switched to 10% ethyl acetate in hexanes) to afford compound 18 (1.0 g, 90%) as a white solid. mp: 68–69 °C; [α] D24 = −64.3 (c = 0.97, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.42–7.29 (m, 5H), 6.98–6.93 (m, 2H), 6.85–6.77 (m, 2H), 4.98 (dd, J = 9.8, 2.1 Hz, 1H), 4.72 (d, J = 11.6 Hz, 1H), 4.51 (d, J = 11.6 Hz, 1H), 3.76 (s, 3H), 3.52–3.37 (m, 2H), 3.34–3.26 (m, 1H), 2.54–2.47 (m, 2H), 1.83 (td, J = 12.0, 9.9 Hz, 1H), 1.38 (d, J = 6.1 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 155.2, 151.3, 138.0, 128.8, 128.1, 127.9, 118.0, 117.9, 98.6, 78.8, 75.6, 71.9, 71.1, 55.8, 36.0, 18.1; LRMS (ESI) m/z: [M+Na]+ Calcd for C20H24O5Na 367.15; Found 367.16; HRMS (DART) m/z: [M+NH4]+ Calcd for C20H28NO5 362.1962; Found 362.1947.
α-p-Methoxylphenyl-3-O-benzyl-2,6-dideoxy-d-arabino-hexopyranose (18a)
The title compound was prepared according to procedure B using 17 (825 mg, 1.9 mmol) dissolved in DCM (20 mL) and H2O (2 mL), DDQ (882 mg, 3.1 mmol) was then added. The reaction was stirred at room temperature under argon for 2 h and monitored by TLC. The reaction mixture was diluted with DCM (25 mL), and the organic layer was washed 3 times with 2 M NaOH. The pooled organic layers were washed with H2O, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (20% ethyl acetate in hexanes) to afford 18 (438 mg, 67% 5:1 α:β) as a white powder. mp: 84–85 °C; [α] D24 = +98.75 (c 1.28, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.42–7.27 (m, 5H), 6.98 (d, J = 12.2 Hz, 2H), 6.83 (d, J = 12.2 Hz, 2H), 5.50 (d, J = 3.7, 1.4 Hz, 1H), 4.74 (d, J = 11.5 Hz, 1H), 4.57 (d, J = 11.5 Hz, 1H), 3.95 (ddd, J = 11.4, 8.9, 4.8 Hz, 1H), 3.83 (dq, J = 9.5, 6.2 Hz, 1H), 3.77 (s, 3H), 3.31 (td, J = 9.2, 2.1 Hz, 1H), 2.48 (dd, J = 13.0, 4.9, 1.5 Hz, 1H), 2.43 (d, J = 2.1 Hz, 1H), 1.77 (ddd, J = 12.9, 11.4, 3.6 Hz, 1H), 1.26 (d, J = 6.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 154.9, 150.8, 138.4, 128.7, 128.0, 127.9, 117.9, 114.7, 96.8, 77.0, 76.2, 71.4, 68.4, 55.8, 35.1, 18.0; LRMS (ESI) m/z: [M+NH4]+ Calcd for C20H28NO5 (M+NH4) 362.41; Found 362.18; HRMS (DART) m/z: [M+NH4]+ Calcd for C20H28NO5 362.1962; Found 362.1971.
3-O-Benzyl-6-deoxy-d-glucal (20)
The title compound was prepared according to procedure A using 19 (325 mg, 0.9 mmol) and β-pinene (0.8 mL, 3.6 mmol) dissolved in DCM (90 mL) and H2O (4.5 mL), the solution was then treated with DDQ (410 mg, 1.8 mmol) for 4 h. The title compound was purified by flash column chromatography on silica gel (10% ethyl acetate in toluene) to afford compound 20 (180 mg, 91%) as colorless oil. The spectroscopic data is in good agreement with previously reported data.251H NMR (500 MHz, CDCl3) δ 7.36 (d, J = 4.4 Hz, 4H), 7.35–7.26 (m, 1H), 6.35 (dd, J = 6.2, 1.5 Hz, 1H), 4.85 (dd, J = 6.1, 2.2 Hz, 1H), 4.71 (d, J = 11.7 Hz, 1H), 4.55 (d, J = 11.8 Hz, 1H), 4.05 (dt, J = 7.1, 1.9 Hz, 1H), 3.90 (dq, J = 9.2, 6.3 Hz, 1H), 3.62 (ddd, J = 9.3, 7.0, 3.6 Hz, 1H), 2.19 (d, J = 3.6 Hz, 1H), 1.39 (d, J = 6.4 Hz, 3H).
4-O-Benzyl-6-deoxy-d-glucal (22)
The title compound was prepared according to procedure A using 21 (250 mg, 0.69 mmol) and β-pinene (0.37 mL, 2.3 mmol) dissolved in DCM (58 mL) and H2O (2.9 mL), the solution was then treated with DDQ (322 mg, 1.3 mmol) for 2 h. The title compound was purified by flash column chromatography on silica gel (30% ethyl acetate in hexane) to afford compound 22 (111 mg, 73%) as a pink-white solid. mp: 107–108 °C; [α] D24 = +10.9 (c 1.01, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.39–7.30 (m, 5H), 6.33–6.32 (dd, J = 6.0, 1.4 Hz, 1H), 4.82 (d, J = 11.6 Hz, 1H), 4.80 (d, J = 11.6 Hz, 1H), 4.70 (dd, J = 6.0, 2.3 Hz, 1H), 4.37–4.33 (m, 1H), 3.91 (dq, J = 9.6, 6.4 Hz, 1H), 3.28 (dd, J = 9.6, 6.9 Hz, 1H), 1.67 (d, J = 5.6 Hz, 1H), 1.41 (d, J = 6.4 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 144.7, 138.3, 128.6, 127.9, 103.1, 82.5, 74.3, 74.1, 70.0, 17.6; HRMS (DART) m/z: [M–H]− Calcd for C13H15O3 219.1027; Found 219.1029.
Acetyl 4-O-Benzyl-6-deoxy-d-erythro-hex-2-enopyranoside (32)
The title compound was prepared according to procedure B using 21 (250 mg, 0.69 mmol) dissolved in DCM (58 mL) and H2O (2.9 mL), the solution was then treated with DDQ (322 mg, 1.3 mmol) for 2 h. The title compound was purified by flash column chromatography on silica gel (30% ethyl acetate in hexane) to afford a white solid. This solid was dissolved in pyridine (1.2 mL), and treated with 4-dimethylaminopyridine (2 mg, 0.01 mmol) followed by dropwise addition of acetic anhydride (0.05 mL, 0.58 mmol). After stirring for 3 h, the reaction mixture was concentrated in vacuo and azeotroped with toluene (2 × 25 mL). The title compound was purified by flash column chromatography on silica gel (20% ethyl acetate in hexane) to afford compound 32 (75 mg, 60% over 2 steps 10:1 α:β) as yellow oil. [α] D24 = +79.9 (c 1.07, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.40–7.27 (m, 5H), 6.21 (s, 1H), 6.18 (d, J = 9.7 Hz, 1H), 5.78 (ddd, J = 10.3, 2.9, 1.9 Hz, 1H), 4.69 (d, J = 11.5 Hz, 1H), 4.57 (d, J = 11.5 Hz, 1H), 3.90 (dq, J = 9.0, 6.2 Hz, 1H), 3.73 (dq, J = 9.0, 1.7 Hz, 1H), 2.08 (s, 3H), 1.32 (d, J = 6.2 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 170.3, 137.9, 132.1, 128.6, 128.1, 128.0, 124.9, 88.9, 75.8, 71.5, 68.4, 21.4, 18.3; LRMS (ESI) m/z: [M+Na]+ Calcd for C15H18NaO4 285.29; Found 285.09; HRMS (DART) m/z: [M - OAc]+ Calcd for C13H15O2 203.1067; Found 203.1085.
Preparation of Substrates
Methyl-2-O-(2-naphthylmethyl)-3-O-benzyl-4,6-O-benzylidene-α-d-glucopyranoside (3)
Sodium hydride (60% in mineral oil, 80 mg, 1.9 mmol) and tetrabutylammonium iodide (48 mg, 0.13 mmol) were suspended in DMF (10 mL). The suspension was cooled to 0 °C and treated with a solution of 4(21) (250 mg, 0.67 mmol) in DMF (3 mL), while additional DMF (3 mL) was simultaneously added. After the mixture was stirred for 10 min at 0 °C, 2-(bromomethyl)-naphthalene (287 mg, 1.30 mmol) in 4 mL DMF was added dropwise to the mixture and the reaction was then allowed to warm to room temperature. After being stirred for 2 h, the reaction mixture was quenched with saturated aqueous NH4Cl and extracted three times with diethyl ether. The pooled organic layers were washed with water and brine, and then dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (20% ethyl acetate in hexanes) to afford compound 3 (336 mg, 98%) as a white solid. The spectroscopic data is in good agreement with previously reported data.281H NMR (500 MHz, CDCl3) δ 7.86–7.74 (m, 4H), 7.54–7.44 (m, 5H), 7.44–7.27 (m, 8H), 5.55 (s, 1H), 5.02 (d, J = 12.3 Hz, 1H), 4.94 (d, J = 11.2 Hz, 1H), 4.87 (d, J = 11.8 Hz, 2H), 4.59 (d, J = 3.7 Hz, 1H), 4.25 (dd, J = 10.2, 4.9 Hz, 1H), 4.08 (t, J = 9.3 Hz, 1H), 3.83 (td, J = 10.0, 4.8 Hz, 1H), 3.69 (t, J = 10.3 Hz, 1H), 3.64–3.56 (m, 2H), 3.41 (s, 3H).
Methyl-2-O-benzyl-3-O-(2-naphthylmethyl)-4,6-O-benzylidene-α-d-glucopyranoside (5)
Sodium hydride (60% in mineral oil, 160 mg, 3.9 mmol) and tetrabutylammonium iodide (96 mg, 0.26 mmol) were suspended in DMF (6 mL). The suspension was cooled to 0 °C and treated with a solution of 6(21) (0.5 g, 1.3 mmol) in DMF (3 mL), while additional DMF (3 mL) was simultaneously added. After the mixture was stirred for 10 min at 0 °C, 2-(bromomethyl)naphthalene (574 mg, 2.6 mmol) in 3 mL DMF was added dropwise to the mixture and the reaction was then allowed to warm to room temperature. After being stirred for 2 h, the reaction mixture was quenched with saturated aqueous NH4Cl and extracted three times with diethyl ether. The pooled organic layers were washed with water and brine, and then dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (10% ethyl acetate in hexanes) to afford compound 5 (656 mg, 98%) as a white solid. The spectroscopic data is in good agreement with previously reported data.281H NMR (500 MHz, CDCl3) δ 7.84–7.74 (m, 3H), 7.73–7.67 (m, 1H), 7.54–7.26 (m, 13H), 5.57 (s, 1H), 5.07 (d, J = 11.6 Hz, 1H), 5.00 (d, J = 11.6 Hz, 1H), 4.88 (d, J = 12.1 Hz, 1H), 4.73 (d, J = 12.2 Hz, 1H), 4.61 (d, J = 3.7 Hz, 1H), 4.27 (dd, J = 10.2, 4.8 Hz, 1H), 4.10 (t, J = 9.3 Hz, 1H), 3.84 (td, J = 9.9, 4.8 Hz, 1H), 3.72 (t, J = 10.3 Hz, 1H), 3.64 (t, J = 9.4 Hz, 1H), 3.59 (dd, J = 9.3, 3.8 Hz, 1H), 3.41 (s, 3H).
Ethyl-6-deoxy-3,4-O-isopropylidene-2-O-pivaloyl-1-thio-β-d-galactoside (24)
Synthesis of compound 24 was performed using procedures adapted from Chan et al.29 A solution of 23(26) (3.43 g, 13 mmol) in pyridine (21 mL) was cooled to 0 °C and treated dropwise with pivaloyl chloride (8.5 mL, 69 mmol). The reaction was then allowed to warm to room temperature and stirred overnight. The mixture was then diluted with ethyl acetate and washed with dilute aqueous HCl solution, saturated NaHCO3 solution, and brine. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (25% ethyl acetate in hexanes) to afford compound 24 (4.46 g, 97%) as a white solid. mp: 54–55 °C; [α] D24 = +12.9 (c = 1.4, CH2Cl2); 1H NMR (500 MHz, CDCl3): δ 5.00 (dd, J = 10.2, 7.3 Hz, 1H), 4.32 (d, J = 10.2 Hz, 1H), 4.13 (dd, J = 7.2, 5.4 Hz, 1H), 4.06 (dd, J = 5.3, 2.0 Hz, 1H), 3.87 (td, J = 6.5, 1.9 Hz, 1H), 2.76–2.69 (m, 1H), 2.68–2.62 (m, 1H), 1.57 (s, 3H), 1.42 (d, J = 6.5 Hz, 3H), 1.36 (s, 3H), 1.24 (d, J = 2.5 Hz, 3H), 1.23 (s, 9H); 13C NMR (125 MHz, CDCl3): δ 177.1, 110.1, 82.3, 76.8, 76.5, 72.7, 71.0, 38.7, 27.9, 26.5, 26.4, 23.6, 16.9, 14.8; LRMS (ESI) m/z: [M+Na]+ Calcd for C16H28O5SNa 355.16; Found 355.27; HRMS (DART) m/z: [M+NH4]+ Calcd for C16H32NO5S 350.1996; Found 350.2002.
Ethyl-6-deoxy-2-O-pivaloyl-1-thio-β-d-galactopyranoside (25)
Synthesis of compound 25 was performed using procedures adapted from van Boom et al.26 A solution of 24 (2.79 g, 8.4 mmol) in acetic acid (41 mL) and water (10 mL) was refluxed at 110 °C for 24 h. The reaction mixture was cooled to room temperature. Upon cooling, the reaction was quenched with saturated NaHCO3 solution and extracted 3 times with CH2Cl2. The pooled organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (30% ethyl acetate in hexanes) to afford compound 25 (1.56 g, 64%) as an amorphous solid. [α] D24 = −23.7 (c = 1.02, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 4.91 (t, J = 9.6 Hz, 1H), 4.41 (d, J = 10.0 Hz, 1H), 3.78 (ddd, J = 6.0, 3.5, 1.1 Hz, 1H), 3.72–3.63 (m, 2H), 2.96 (d, J = 7.5 Hz, 1H), 2.79–2.61 (m, 2H), 2.25 (d, J = 5.9 Hz, 1H), 1.37 (d, J = 6.5 Hz, 3H), 1.27 (t, J = 7.4 Hz, 3H), 1.25 (s, 9H); 13C NMR (125 MHz, CDCl3): δ 179.0, 82.9, 74.6, 74.2, 72.1, 71.4, 38.9, 27.1, 23.9, 16.6, 15.0; LRMS (ESI) m/z: [M+Na]+ Calcd for C13H24O5SNa 315.12; Found 314.82; HRMS (DART) m/z: [M+NH4]+ Calcd for C13H28NO5S 310.1683; Found 310.1693.
Ethyl-6-deoxy-3-O-(2-naphthylmethyl)-2-O-pivaloyl-1-thio-β-d-galactopyranoside (26)
Synthesis of compound 26 was performed using procedures adapted from Takahashi et al.25 Compound 25 (1 g, 3.4 mmol) and Bu2SnO (0.93 g, 3.7 mmol) were suspended in toluene (33 mL) at room temperature. The reaction mixture was stirred at reflux at 120 °C under Dean–Stark conditions for 3 h, then cooled to room temperature. Tetrabutylammonium iodide (0.25 g, 0.68 mmol) and 2-(bromomethyl)naphthylene (1.1 g, 5.1 mmol) were added to the reaction mixture at room temperature. The reaction was returned to reflux, stirred for 3 h, allowed to cool to room temperature, and concentrated in vacuo. The crude residue was purified by flash column chromatography on silica gel (20–30% ethyl acetate in hexanes) to afford compound 26 (680 mg, 46%) as an amorphous solid. [α] D24 = −20.8 (c = 1.06, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.85–7.82 (m, 3H), 7.74 (s, 1H), 7.51–7.41 (m, 3H), 5.29 (t, J = 9.7 Hz, 1H), 4.80–4.79 (m, 2H), 4.33 (d, J = 10.0 Hz, 1H), 3.85 (s, 1H), 3.62 (dd, J = 9.4, 3.3 Hz, 1H), 3.58 (d, J = 6.3 Hz, 1H), 2.79–2.64 (m, 2H), 2.43 (s, 1H), 1.36 (d, J = 6.5 Hz, 3H), 1.26–1.21 (m, 12H); 13C NMR (125 MHz, CDCl3): δ 177.2, 134.9, 133.3, 133.2, 128.6, 128.0, 127.9, 126.6, 126.5, 126.3, 125.6, 83.2, 80.3, 74.6, 72.1, 69.4, 68.5, 38.9, 27.4, 23.4, 16.8, 14.9; LRMS (ESI) m/z: [M+Na]+ Calcd for C24H32O5SNa 455.19; Found 455.64; HRMS (DART) m/z: [M+NH4]+ Calcd for C24H36NO5S 450.2309; Found 450.2302.
Ethyl-6-deoxy-4-O-benzyl-3-O-(2-naphthylmethyl)-2-O-pivaloyl-1-thio-β-d-galactopyranoside (11)
A solution of 26 (540 mg, 1.2 mmol) and sodium hydride (60% in mineral oil, 190 mg, 4.7 mmol) in DMF (8 mL) was cooled to 0 °C and treated dropwise with benzyl bromide (0.29 mL, 2.4 mmol); the reaction was then allowed to warm to room temperature for 1 h. The reaction mixture was quenched with saturated aqueous NH4Cl solution. The reaction was diluted with Et2O and H2O, and extracted 3 times with Et2O. The pooled organic layers were washed two times with H2O and brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (10–15% ethyl acetate in hexanes) to afford compound 11 (462.8 mg, 74%) as a white powder. mp: 97–98 °C; [α] D24 = −4.0 (c = 0.97, CH2Cl2); 1H NMR (500 MHz, CDCl3): δ 7.85–7.81 (m, 3H), 7.76 (s, 1H), 7.50–7.48 (m, 2H), 7.44–7.42 (m, 1H), 7.34–7.24 (m, 5H), 5.49 (t, J = 6.5 Hz, 1H), 5.00 (d, J = 12.0 Hz, 1H), 4.79 (s, 2H), 4.68 (d, J = 11.5 Hz, 1H), 4.35 (d, J = 10.0 Hz, 1H), 3.67 (d, J = 2.7 Hz, 1H), 3.65 (s, 1H), 3.54 (q, J = 6.4 Hz, 1H), 2.79–2.72 (m, 1H), 2.71–2.64 (m, 1H), 1.25–1.22 (m, 15H); 13C NMR (125 MHz, CDCl3): δ 177.1, 138.7, 135.6, 133.4, 133.1, 128.4, 128.3, 128.2, 128.0, 127.9, 127.6, 126.4, 126.2, 126.1, 125.6, 83.5, 82.5, 76.1, 75.1, 74.5, 72.8, 69.3, 38.9, 27.4, 23.4, 17.4, 14.9; LRMS (ESI) m/z: [M+Na]+ Calcd for C31H38SO5Na 545.23; Found 545.35; HRMS (DART) m/z: [M+NH4]+ Calcd for C31H42NSO5 540.2778; Found 540.2783.
Phenyl-6-deoxy-2-O-pivaloyl-1-thio-β-d-galactopyranoside (28)
Synthesis of compound 28 was performed using procedures adapted from van Boom et al.26 A solution of 27(27) (1.92 g, 5.0 mmol) in acetic acid (24 mL) and water (6 mL) was refluxed at 110 °C for 2 h. The reaction mixture was then allowed to cool to room temperature. Upon cooling, the solution was diluted with CH2Cl2. The mixture was washed with saturated NaHCO3 solution and brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (50% ethyl acetate in hexanes) to afford compound 28 (1.64 g, 96%) as a white powder. mp: 149–150 °C; [α] D24 = −11.0 (c = 1.22, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.53–7.45 (m, 2H), 7.36–7.28 (m, 3H), 4.88 (t, J = 9.6 Hz, 1H), 4.64 (d, J = 10.1 Hz, 1H), 3.77 (ddd, J = 6.1, 3.4, 1.0 Hz, 1H), 3.74–3.64 (m, 2H), 2.97 (d, J = 7.5 Hz, 1H), 2.15 (d, J = 6.1 Hz, 1H), 1.39 (d, J = 6.5 Hz, 3H), 1.28 (s, 9H); 13C NMR (125 MHz, CDCl3): δ 179.0, 133.2, 132.5, 129.1, 128.1, 86.2, 74.6, 74.5, 72.1, 71.4, 39.1, 27.3, 16.8; LRMS (ESI) m/z: [M+Na]+ Calcd for C17H24O5SNa 363.12; Found 363.27; HRMS (DART) m/z: [M+NH4]+ Calcd for C17H28NO5S 358.1683; Found 358.1682.
Phenyl-6-deoxy-3-O-benzyl-2-O-pivaloyl-1-thio-β-d-galactopyranoside (14)
Synthesis of 14 was performed using procedures adapted from Takahashi et al.25 Compound 28 (250 mg, 0.74 mmol) and Bu2SnO (200 mg, 0.81 mmol) were suspended in toluene (7 mL) at room temperature. The reaction mixture was stirred at reflux at 130 °C under Dean–Stark conditions for 3 h, then cooled to room temperature. Tetrabutylammonium iodide (54.3 mg, 0.15 mmol) and benzyl bromide (0.13 mL, 1.1 mmol) were added to the reaction mixture at room temperature. The reaction was returned to reflux, stirred for 3 h, the reaction mixture was allowed to cool to room temperature, and then was concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel (30% ethyl acetate in hexanes) to afford 14 (287.5 mg, 91%) as an amorphous solid. [α] D24 = +5.5 (c = 0.95, CH2Cl2); 1H NMR (500 MHz, CDCl3): δ 7.50–7.48 (m, 2H), 7.34–7.26 (m, 8H), 5.23 (t, J = 9.8 Hz, 1H), 4.66–4.58 (m, 3H), 3.84 (t, J = 2.5 Hz, 1H), 3.62–3.57 (m, 2H), 2.38 (s, 1H), 1.40 (d, J = 6.5 Hz, 3H), 1.24 (s, 9H); 13C NMR (125 MHz, CDCl3): δ 177.0, 137.4, 133.9, 132.2, 128.9, 128.7, 128.2, 127.8, 127.7, 86.9, 80.4, 74.5, 71.9, 69.2, 68.6, 38.9, 27.3, 16.9; LRMS (ESI) m/z: [M+Na]+ Calcd for C24H30O5SNa 453.17; Found 453.36; HRMS (DART) m/z: [M+NH4]+ Calcd for C24H34NO5S 448.2152; Found 448.2155.
Phenyl-6-deoxy-4-O-(2-naphthylmethyl)-3-O-benzyl-2-O-pivaloyl-1-thio-β-d-galactopyranoside (13)
A solution of 14 (1.6 g, 3.7 mmol) and sodium hydride (60% in mineral oil, 0.46 g, 11 mmol) in DMF (8.6 mL) was cooled to 0 °C and treated with 2-(bromomethyl)naphthylene (1.6 g, 7.5 mmol), and then the reaction was allowed to warm to room temperature. After being stirred overnight, the reaction mixture was quenched using saturated NH4Cl aqueous solution. The reaction was diluted with Et2O and H2O and extracted 3 times with Et2O. The pooled organic layer was washed two times with H2O then washed with brine. The pooled organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (100% toluene) to afford compound 13 (1.97 g, 91%) as a white powder. mp: 86–87 °C; [α] D24 = +4.4 (c = 1.29, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.82–7.75 (m, 3H), 7.67 (s, 1H), 7.50–7.45 (m, 5H), 7.35–7.30 (m, 5H), 7.26–7.17 (m, 3H), 5.51 (t, J = 9.8 Hz, 1H), 5.11 (d, J = 11.9 Hz, 1H), 4.82 (d, J = 11.9 Hz, 1H), 4.69–4.59 (m, 3H), 3.68 (d, J = 2.8 Hz, 1H), 3.64 (dd, J = 9.6, 2.7 Hz, 1H), 3.56 (q, J = 6.9 Hz, 1H), 1.27 (d, J = 6.3 Hz, 3H), 1.24 (s, 9H); 13C NMR (125 MHz, CDCl3): δ 176.8, 137.9, 135.9, 134.3, 133.1, 133.0, 131.8, 131.5, 128.7, 128.5, 127.9, 127.8, 127.7, 127.6, 127.4, 126.9, 126.5, 125.9, 125.8, 87.1, 82.6, 75.4, 74.9, 74.3, 72.7, 69.3, 38.8, 27.2, 17.4; LRMS (ESI) m/z: [M+Na]+ Calcd for C35H38O5SNa 593.23; Found 593.36; HRMS (DART) m/z: [M+NH4]+ Calcd for C35H42NO5S 588.2778; Found 588.2785.
3-O-Benzyl-4-O-(2-naphthylmethyl)-6-deoxy-d-glucal (19)
Sodium hydride (60% by weight, 1 g, 27 mmol) and tetrabutylammonium iodide (664 mg, 1.8 mmol) were suspended in DMF (38 mL). The suspension was cooled to 0 °C and treated with a solution of 20(25) (2 g, 9.0 mmol) in DMF (15 mL), while additional DMF (15 mL) was simultaneously added. After the mixture was stirred for 10 min at 0 °C, 2-(bromomethyl)naphthalene (3.9 g, 18 mmol) in 10 mL DMF was added dropwise to the mixture and the reaction was then allowed to warm to room temperature. After being stirred for 2 h, the reaction mixture was quenched with saturated aqueous NH4Cl and extracted three times with diethyl ether. The pooled organic layers were washed with water and brine, and then dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (2% ethyl acetate in hexanes) to afford compound 19 (2.9 g, 90%) as a white powder. mp: 60–61 °C; [α] D24 = −40.1 (c = 1.32, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.87–7.78 (m, 3H), 7.75 (s, 1H), 7.47 (ht, J = 5.3, 2.5 Hz, 3H), 7.34 (dt, J = 6.9, 2.1 Hz, 3H), 7.32–7.22 (m, 2H), 6.37 (d, J = 6.0 Hz, 1H), 5.03 (dd, J = 11.9, 4.1 Hz, 1H), 4.91–4.83 (m, 2H), 4.67 (dt, J = 12.3, 2.9 Hz, 1H), 4.58 (dd, J = 11.6, 1.9 Hz, 1H), 4.26 (dd, J = 6.9, 2.9, 2.3 Hz, 1H), 4.04–3.94 (m, 1H), 3.54 (ddd, J = 8.7, 6.4, 1.9 Hz, 1H), 1.40 (dd, J = 6.4, 1.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 144.9, 138.5, 135.8, 133.4, 133.1, 128.5, 128.3, 128.0, 127.9, 127.8, 127.7, 126.8, 126.2, 126.1, 126.0, 100.2, 79.5, 76.6, 74.2, 74.1, 70.6, 17.7; LRMS (ESI) m/z: [M+NH4]+ Calcd for C24H28NO3 378.45; Found 378.02; HRMS (DART) m/z: [M+NH4]+ Calcd for C24H28NO3 378.2064; Found 378.2076.
3-O-(2-Naphthylmethyl)-4-O-benzyl-6-deoxy-d-glucal (21)
Sodium hydride (60% in mineral oil, 738 mg, 18 mmol) and tetrabutylammonium iodide (679 mg, 1.8 mmol) were suspended in DMF (42 mL). The suspension was cooled to 0 °C and treated with a solution of 29(25) (2.5 g, 9.2 mmol) in DMF (10 mL), while additional DMF (10 mL) was simultaneously added. After the mixture was stirred for 10 min at 0 °C, benzyl bromide (2.1 mL, 18 mmol) was added dropwise to the mixture and the reaction was then allowed to warm to room temperature. After being stirred for 3 h, the reaction mixture was quenched with saturated aqueous NH4Cl and extracted three times with diethyl ether. The pooled organic layers were washed with water and brine, and then dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (2% ethyl acetate in hexanes) and afforded compound 21 (2.8 g, 87%) as an amorphous solid: [α] D24 = −54.4 (c = 1.14, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.90–7.67 (m, 4H), 7.57–7.37 (m, 3H), 7.37–7.17 (m, 5H), 6.37 (d, J = 6.1, 1.4 Hz, 1H), 4.98–4.83 (m, 2H), 4.82 (d, J = 12.0 Hz, 1H), 4.76–4.69 (m, 2H), 4.27 (dd, J = 6.6, 1.4 Hz, 1H), 3.96 (dq, J = 9.1, 6.5 Hz, 1H), 3.52 (dd, J = 9.0, 6.5 Hz, 1H), 1.39 (d, J = 6.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 144.9, 138.3, 136.0, 133.0,, 128.4, 128.2, 128.1, 128.0, 127.9, 127.8, 126.5, 126.5, 126.2, 126.0, 125.9, 100.3, 79.7, 76.6, 74.1, 70.8, 17.6;; LRMS (ESI) m/z: [M+Na]+ Calcd for C24H24O3Na 383.16; Found 383.18; HRMS (DART) m/z: [M+NH4]+ Calcd for C24H28NO3 378.2064; Found 378.2059.
3-O-Benzyl-4-O-(2-naphthylmethyl)-2,6-dideoxy-d-arabino-hexopyranose (30)
Synthesis of compound 30 was performed using procedures adapted from Blumenstein et al.30 To a solution of 19 (1.5 g, 5.2 mmol) in THF (20 mL), Ph3·HBr (361 mg, 1.0 mmol) was added to the reaction and allowed to stir at room temperature. After 10 min, H2O (0.47 mL, 26 mmol) was added to the reaction. The reaction was stirred for 4 h. The reaction was quenched with saturated, aqueous NaHCO3, diluted with ethyl acetate and water, and extracted 3 times with ethyl acetate. The pooled organic layer was washed with H2O and brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (25% ethyl acetate in hexanes) to afford compound 30 (1.8 g, 94%) as a white powder. mp: 150–151 °C; [α] D24 = +14.8 (c = 1.05, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.86–7.74 (m, 4H), 7.51–7.42 (m, 3H), 7.39–7.25 (m, 5H), 5.35 (d, J = 3.7 Hz, 1H), 5.11 (d, J = 11.2 Hz, 1H), 4.82 (d, J = 10.8 Hz, 1H), 4.73–4.63 (m, 2H), 4.09–3.97 (m, 2H), 3.20 (dd, J = 19.2, 0.9 Hz, 1H), 2.38–2.30 (m, 2H), 1.70 (ddd, J = 13.2, 11.2, 3.6, 2.2 Hz, 1H), 1.30 (d, J = 6.3 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 138.7, 136.1, 133.39, 133.0, 128.6, 128.5, 128.2, 128.1, 128.0, 127.8, 127.77, 127.76, 127.7, 126.8, 126.7, 126.2, 126.1, 126.0, 125.9, 93.9, 92.1, 84.4, 83.5, 79.1, 76.9, 75.3, 75.28, 71.9, 71.6, 71.6, 67.5, 53.5, 38.5, 35.9, 18.4, 18.38; LRMS (ESI) m/z: [M+Na]+ Calcd for C24H26O4Na 401.17; Found 401.19; HRMS (DART) m/z: [M+NH4]+ Calcd for C24H30NO4 396.2169; Found 396.2157.
3-O-(2-Naphthylmethyl)-4-O-benzyl-2,6-dideoxy-d-arabino-hexopyranose (31)
Synthesis of compound 31 was performed using procedures adapted from Blumenstein et al.30 To a solution of 21 (890 mg, 2.4 mmol) in THF (10 mL), Ph3·HBr (168 mg, 0.49 mmol) was added to the reaction and allowed to stir at room temperature. After 10 min, H2O (0.21 mL, 12 mmol) was added to the reaction. The reaction was stirred for 6 h. The reaction was quenched with saturated, aqueous NaHCO3, diluted with ethyl acetate and water, and extracted 3 times with ethyl acetate. The pooled organic layers were washed with H2O and brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (25% ethyl acetate in hexanes) to afford compound 31 (852 mg, 93%) as a white powder. mp: 97–98 °C; [α] D24 = +15.0 (c = 1.15, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.85–7.73 (m, 4H), 7.49–7.42 (m, 3H), 7.37–7.26 (m, 5H), 5.35 (s, 1H), 4.99 (d, J = 11.0 Hz, 1H), 4.88–4.74 (m, 2H), 4.70 (d, J = 11.0 Hz, 1H), 4.16–3.95 (m, 2H), 3.17 (t, J = 9.1 Hz, 1H), 2.35 (dd, J = 24.0, 7.9 Hz, 1H), 1.77–1.67 (m, 1H), 1.29 (d, J = 6.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 138.7, 136.3, 133.5, 133.1, 128.6, 128.5, 128.4, 128.2, 128.1, 128.07, 128.03, 128.0, 127.8, 127.7, 126.6, 126.4, 126.3, 126.2, 126.1, 125.9, 93.9, 92.2, 84.5, 83.6, 79.0, 77.4, 77.0, 75.4, 75.3, 72.0, 71.7, 67.6, 38.6, 36.0, 18.4, 18.3; LRMS (ESI) m/z: [M+Na]+ Calcd for C24H26O4Na 401.17; Found 401.18; HRMS (DART) m/z: [M+NH4]+ Calcd for C24H30NO4 396.2169; Found 396.2155.
β-p-Methoxylphenyl 3-O-benzyl-4-O-(2-naphthylmethyl)-2,6-dideoxy-D-arabino-hexopyranose (17)
A solution of donor 30 (142 mg, 0.375 mmol) and 2,4,6-tritert-butylpyrimidine (TTBP, 74 mg, 0.375 mmol) in 3 mL THF was cooled to −78 °C and treated dropwise with potassium hexamethyldisilazane (1 M in THF, 0.375 mL, 0.375 mmol). After 15 min, a solution of p-toluenesulfonic anhydride (122 mg, 0.375 mmol) in 2 mL THF was added slowly to the reaction. The solution was maintained at −78 °C for 30 min. Meanwhile in a separate flask, 4-methoxyphenol (31 mg, 0.250 mmol) was dissolved in 2 mL THF, cooled to −78 °C, and treated with potassium hexamethyldisilazane (1 M in THF, 0.25 mL, 0.25 mmol). After 30 min, this solution was transferred dropwise by syringe to the primary reaction vessel. The reaction mixture was then allowed to gradually warm to room temperature over the course of 4 h. The reaction was quenched with 0.3 mL of saturated, aqueous NH4Cl, diluted with water, and extracted 3 times with diethyl ether. The pooled organic layers were washed with H2O and brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (10% ethyl acetate in hexanes) to afford 17 as a single β-anomer (84 mg, 54%) as a white powder. mp: 93–94 °C; [α] D24 = −31.2 (c = 1.12, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.86–7.75 (m, 4H), 7.52–7.43 (m, 3H), 7.41–7.26 (m, 5H), 6.98–6.92 (m, 2H), 6.84–6.78 (m, 2H), 5.13 (d, J = 11.1 Hz, 1H), 4.97 (dd, J = 9.9, 2.1 Hz, 1H), 4.85 (d, J = 11.1 Hz, 1H), 4.75 (d, J = 11.7 Hz, 1H), 4.66 (d, J = 11.7 Hz, 1H), 3.76 (s, 3H), 3.76–3.69 (m, 1H), 3.54–3.45 (m, 1H), 3.28 (t, J = 8.9 Hz, 1H), 2.53 (dd, J = 12.5, 5.0 Hz, 1H), 1.90 (dd, J = 12.0, 9.8 Hz, 1H), 1.39 (d, J = 6.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 155.1, 151.4, 138.4, 135.9, 133.4, 133.1, 128.6, 128.3, 128.1, 127.9, 127.8, 126.9, 126.22, 126.2, 118.0, 114.6, 98.5, 83.6, 79.2, 75.5, 71.7, 55.8, 37.0, 18.5; LRMS (ESI) m/z: [M+Na]+ Calcd for C31H32O5Na 507.21; Found 507.26; HRMS (DART) m/z: [M+NH4]+ Calcd for C31H36NO5 502.2588; Found 502.2592.
β-p-Methoxylphenyl 4-(3′-O-Naphthylmethyl-4′-O-benzyl-2′,6′-dideoxy-β-d-arabino-hexopyranoyl)-3-O-benzyl-2,6-dideoxy-β-d-arabino-hexopyranose (1)
A solution of donor 31 (138 mg, 0.36 mmol) and 2,4,6-tritert-butylpyrimidine (TTBP, 89 mg, 0.36 mmol) in 5 mL THF was cooled to −78 °C and treated dropwise with potassium hexamethyldisilazane (1 M in THF, 0.36 mL, 0.36 mmol). After 15 min, a solution of p-toluenesulfonic anhydride (117 mg, 0.36 mmol) in 2 mL THF was added slowly to the reaction. The solution was maintained at −78 °C for 30 min. Meanwhile in a separate flask, the acceptor 18 (84 mg, 0.24 mmol) was dissolved in 2 mL THF, cooled to −78 °C, and treated with potassium hexamethyldisilazane (1 M in THF, 0.24 mL, 0.24 mmol). After 30 min, this solution was transferred dropwise by syringe to the primary reaction vessel. The reaction mixture was then allowed to gradually warm to room temperature over the course of 4.5 h. The reaction was quenched with 0.3 mL of saturated, aqueous NH4Cl, diluted with water, and extracted 3 times with DCM. The pooled organic layers were washed with H2O, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (15% ethyl acetate in hexanes) to afford 1 as a single β-anomer (127 mg, 75%) as a white powder. mp: 115–116 °C; [α] D24 = −33.4 (c = 1.28, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.86–7.72 (m, 4H), 7.49–7.43 (m, 3H), 7.40–7.22 (m, 10H), 6.94 (d, J = 9.4 Hz, 2H), 6.80 (d, J = 9.7 Hz, 2H), 5.00–4.91 (m, 2H), 4.83–4.63 (m, 6H), 3.76 (s, 3H), 3.69–3.59 (m, 2H), 3.49–3.39 (m, 1H), 3.38–3.25 (m, 2H), 3.15 (t, J = 9.0 Hz, 1H), 2.45 (ddd, J = 12.6, 5.2, 2.1 Hz, 1H), 2.39 (ddd, J = 21.7, 8.3, 4.0 Hz, 1H), 1.86 (td, J = 12.2, 10.0 Hz, 1H), 1.63 (td, J = 12.1, 9.9 Hz, 1H), 1.34 (d, J = 8.9 Hz, 3H), 1.30 (d, J = 6.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 155.2, 151.3, 138.7, 138.6, 136.0, 128.5, 128.4, 128.3, 128.2, 128.0, 127.9, 127.8, 127.7, 127.6, 126.5, 126.3, 126.0, 125.9, 118.1, 114.6, 100.3, 98.5, 83.9, 82.4, 79.3, 77.8, 75.4, 72.0, 71.8, 71.4, 55.8, 37.6, 37.1, 18.6, 18.4; LRMS (ESI) m/z: [M+Na]+ Calcd for C44H48O8Na 727.32; Found 727.45; HRMS (ESI) m/z: [M+NH4]+ Calcd for C44H52NO8 722.3687; Found 722.3695.
Methyl-2-O-(2-naphthylmethyl)-3-O-benzyl-4,6-O-benzylidine-β-d-glucopyranoside (7)
To a solution of 8(22) (1 g, 2.6 mmol) in DMF (12 mL) at 0 °C, sodium hydride (60% in mineral oil, 322 mg, 8 mmol) was added. After the mixture was stirred for 10 min at 0 °C, 2-(bromomethyl)naphthalene (1.1 g, 5.2 mmol) in 6 mL DMF was added dropwise to the mixture and the reaction was then allowed to warm to room temperature. After being stirred for 16 h, the reaction mixture was quenched with saturated aqueous NH4Cl and extracted three times with diethyl ether. The pooled organic layers were washed with water and brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (15% ethyl acetate in hexanes) to afford compound 7 (900 mg, 70%) as a white solid. mp: 154–155 °C; [α] D24 = −15.5 (c 1.07, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.85–7.75 (m, 4H), 7.52–7.44 (m, 5H), 7.44–7.27 (s, 8H), 5.58 (s, 1H), 5.03 (d, J = 11.2 Hz, 1H), 4.93 (t, J = 6.0, 5.3 Hz, 2H), 4.82 (d, J = 11.4 Hz, 1H), 4.47 (d, J = 7.6 Hz, 1H), 4.38 (dd, J = 10.5, 5.0 Hz, 1H), 3.79 (q, J = 9.5, 8.6 Hz, 2H), 3.70 (t, J = 9.3 Hz, 1H), 3.61 (s, 3H), 3.51 (t, J = 8.2 Hz, 1H), 3.44 (td, J = 9.7, 5.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 138.6, 137.5, 136.0, 133.4, 133.2, 129.1, 128.4, 128.4, 128.2, 128.1, 127.8, 127.7, 126.8, 126.3, 126.1, 126.1, 125.9, 105.4, 101.3, 82.3, 81.7, 80.9, 75.4, 75.3, 68.9, 66.1, 57.6; LRMS (ESI) m/z: [M+Na]+ Calcd for C32H32NaO6 535.59; Found 535.36; HRMS (DART) m/z: [M+H]+ Calcd for C32H33O6 513.2272; Found 513.2281.
Methyl-2-O-(2-naphthylmethyl)-3-O-benzyl-4,6-O-benzylidine-β-d-mannopyranoside (9)
Sodium hydride (60% in mineral oil, 480 mg, 12 mmol) and tetrabutylammonium iodide (295 mg, 0.8 mmol) were suspended in DMF (18 mL). The suspension was cooled to 0 °C and treated with a solution of 10(23) (1.5 g, 4.0 mmol) in DMF (9 mL), while additional DMF (9 mL) was simultaneously added. After the mixture was stirred for 10 min at 0 °C, 2-(bromomethyl)naphthalene (1.7 g, 8 mmol) in 9 mL DMF was added dropwise to the mixture and the reaction was then allowed to warm to room temperature. After being stirred for 3 h, the reaction mixture was quenched with saturated aqueous NH4Cl and extracted three times with diethyl ether. The pooled organic layers were washed with water and brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (15% ethyl acetate in hexanes) to afford compound 9 (1.8 g, 87%) as colorless foam. [α] D24 = −14.6 (c 1.16, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.87–7.76 (m, 4H), 7.57–7.45 (m, 5H), 7.42–7.25 (m, 8H), 5.66 (d, J = 2.7 Hz, 1H), 4.94 (q, J = 12.4, 2.6 Hz, 2H), 4.83 (d, J = 12.2, 2.9 Hz, 1H), 4.72 (s, 1H), 4.65 (d, J = 12.3, 2.7 Hz, 1H), 4.33–4.23 (m, 2H), 3.99–3.85 (m, 3H), 3.79 (td, J = 9.8, 4.5 Hz, 1H), 3.31 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 138.8, 137.8, 135.6, 133.3, 133.2, 128.9, 128.4, 128.3, 128.2, 128.0, 127.8, 127.6, 127.6, 127.1, 126.3, 126.2, 126.1, 126.0, 101.6, 100.6, 79.3, 76.6, 76.3, 73.8, 73.3, 69.0, 64.2, 54.9; LRMS (ESI) m/z: [M+Na]+ Calcd for C32H32NaO6 535.59; Found 535.36; HRMS (DART) m/z: [M–OCH3]+ Calcd for C31H29O5 481.2010; Found 481.2021.
Acknowledgments
We thank the National Science Foundation (NSF 1566233) and National Institutes of Health (5R01GM115779) for generous financial support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.7b00065.
NMR spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Codée J. D. C.; Ali A.; Overkleeft H. S.; van der Marel G. A. C. R. Chim. 2011, 14, 178–193. 10.1016/j.crci.2010.05.010. [DOI] [Google Scholar]
- Guo J.; Ye X.-S. Molecules 2010, 15, 7235–7265. 10.3390/molecules15107235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser-Reid B.; López J. C. Top. Curr. Chem. 2010, 301, 1–29. 10.1007/128_2010_105. [DOI] [PubMed] [Google Scholar]
- Seeberger P. H. Chem. Soc. Rev. 2008, 37, 19–28. 10.1039/B511197H. [DOI] [PubMed] [Google Scholar]
- Gaunt M. J.; Yu J.; Spencer J. B. J. Org. Chem. 1998, 63, 4172–4173. 10.1021/jo980823v. [DOI] [Google Scholar]
- Xia J.; Abbas S. A.; Locke R. D.; Piskorz C. F.; Alderfer J. L.; Matta K. L. Tetrahedron Lett. 2000, 41, 169–173. 10.1016/S0040-4039(99)02046-8. [DOI] [Google Scholar]
- Cattaneo V.; Oldrini D.; Corrado A.; Berti F.; Adamo R. Org. Chem. Front. 2016, 3, 753–758. 10.1039/C6QO00144K. [DOI] [Google Scholar]
- Csuk R.; Dörr P. Tetrahedron 1994, 50, 9983–9988. 10.1016/S0040-4020(01)89613-7. [DOI] [Google Scholar]
- Li Y.; Roy B.; Liu X. Chem. Commun. 2011, 47, 8952–8954. 10.1039/c1cc13264d. [DOI] [PubMed] [Google Scholar]
- Li Y.; Liu X. Chem. Commun. 2014, 50, 3155–3158. 10.1039/c3cc49205b. [DOI] [PubMed] [Google Scholar]
- Volbeda A. G.; Kistemaker H. A. V.; Overkleeft H. S.; van der Marel G.; Filippov D. V.; Codée J. D. C. J. Org. Chem. 2015, 80, 8796–8806. 10.1021/acs.joc.5b01030. [DOI] [PubMed] [Google Scholar]
- Issa J. P.; Bennett C. S. J. Am. Chem. Soc. 2014, 136, 5740–5744. 10.1021/ja500410c. [DOI] [PubMed] [Google Scholar]
- Nogueira J.; Bylsma M.; Bright D. K.; Bennett C. S. Angew. Chem., Int. Ed. 2016, 55, 10088–10092. 10.1002/anie.201605091. [DOI] [PubMed] [Google Scholar]
- Kim H. M.; Kim I. J.; Danishefsky S. J. J. Am. Chem. Soc. 2001, 123, 35–48. 10.1021/ja0022730. [DOI] [PubMed] [Google Scholar]
- Horita K.; Yoshioka T.; Tanaka T.; Oikawa Y.; Yonemitsu O. Tetrahedron 1986, 42, 3021–3028. 10.1016/S0040-4020(01)90593-9. [DOI] [Google Scholar]
- Crich D.; Smith M.; Yao Q.; Picione J. Synthesis 2001, 2001, 0323–0326. 10.1055/s-2001-10798. [DOI] [Google Scholar]
- Deutsch E.; Cheung N. K. V. J. Org. Chem. 1973, 38, 1123–1126. 10.1021/jo00946a013. [DOI] [Google Scholar]
- Gu X.; Chen L.; Wang X.; Liu X.; You Q.; Xi W.; Gao L.; Chen G.; Chen Y.-L.; Xiong B.; Shen J. J. Org. Chem. 2014, 79, 1100–1110. 10.1021/jo402551x. [DOI] [PubMed] [Google Scholar]
- Chen G.; Yin Q.; Yin J.; Gu X.; Liu X.; You Q.; Chen Y.-L.; Xiong B.; Shen J. Org. Biomol. Chem. 2014, 12, 9781–9785. 10.1039/C4OB01807A. [DOI] [PubMed] [Google Scholar]
- During the course of the reaction, a white precipitate was formed, which was tentatively identified by ESI-MS as the adduct between the 2,3-dichloro-5,6-dicyanohydroquinone and β-pinene.
- Khanbabaee K.; Lötzerich K.; Borges M.; Grosser M. J. Prakt. Chem. 1999, 341, 159–166. 10.1002/(SICI)1521-3897(199902)341:2<159::AID-PRAC159>3.0.CO;2-6. [DOI] [Google Scholar]
- van der Ven J.; Wijkmans J. C.H.M.; Kamerling J. P.; Vliegenthart J. F. G. Carbohydr. Res. 1994, 253, 121–139. 10.1016/0008-6215(94)80060-X. [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Honda T. J. Am. Chem. Soc. 1994, 116, 5647–5656. 10.1021/ja00092a014. [DOI] [Google Scholar]
- Lee Y. J.; Fulse D. B.; Kim K. S. Carbohydr. Res. 2008, 343, 1574–1584. 10.1016/j.carres.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Tanaka H.; Yamaguchi S.; Yoshizawa A.; Takagi M.; Shin-ya K.; Takahashi T. Chem. - Asian J. 2010, 5, 1407–1424. 10.1002/asia.200900640. [DOI] [PubMed] [Google Scholar]
- Zegelaar-Jaarsveld K.; van der Plas S. C.; van der Marel G. A.; van Boom J. H. J. Carbohydr. Chem. 1996, 15, 591–610. 10.1080/07328309608005677. [DOI] [Google Scholar]
- Stick R. V.; Tilbrook M. G.; Williams S. J. Aust. J. Chem. 1999, 52, 895–904. 10.1071/CH99031. [DOI] [Google Scholar]
- Wang C.-C.; Lee J.-C.; Luo S.-Y.; Kulkarni S. S.; Huang Y.-W.; Lee C.-C.; Chang K.-L.; Hung S.-C. Nature 2007, 446, 896–899. 10.1038/nature05730. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Chan T.-H. J. Org. Chem. 1998, 63, 6035–6038. 10.1021/jo980294v. [DOI] [PubMed] [Google Scholar]
- Chen G.; Franck R. W.; Yang G.; Blumenstein M. Can. J. Chem. 2002, 80, 894–899. 10.1139/v02-095. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.