

Summary
The interface between the membrane (MS) and cytoplasmic (C) rings of the bacterial flagellar motor couples torque generation to rotation within the membrane. The structure of the C-terminal helices of the integral membrane protein FliF (FliFC) bound to the N-terminus of the switch complex protein FliG (FliGN) reveals that FliGN folds around FliFC to produce a topology that closely resembles both the middle and C-terminal domains of FliG. The interface is consistent with solution-state NMR, SAXS, in vivo interaction studies and cellular motility assays. Co-folding with FliFC induces substantial conformational changes in FliGN, and suggests that FliF and FliG have the same stoichiometry within the rotor. Modeling the FliFC:FliGN complex into cryoEM rotor density updates the architecture of the middle and upper switch complex and shows how domain shuffling of a conserved interaction module anchors the cytoplasmic rotor to the membrane.
Keywords: switch complex, flagellar motor, domain shuffling
eTOC Blurb
FliF and FliG comprise the MS-ring and upper C-ring of the bacterial flagellar motor. Lynch et al. use X-ray crystallography, SAXS, NMR, and in vivo studies to reveal how FliF:FliG fold into a single domain, whose toplogy is found elsewhere in FliG, and generate an updated model of the upper flagellar rotor.
Introduction
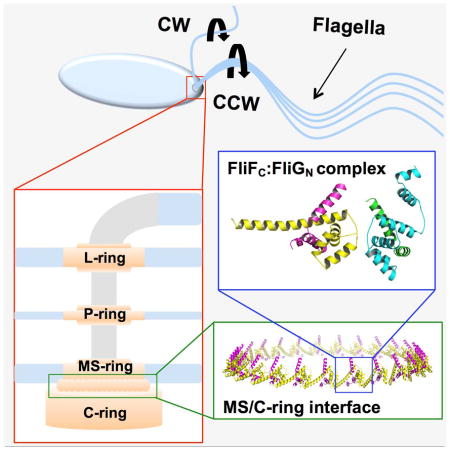
The bacterial flagellar motor is the principle organelle that enables motile bacteria to move within their environment. The motor can be divided into three major components: (1) the filament, which serves as the propeller of the motor, (2) the hook, a flexible adaptor between the filament and cell body, and (3) the basal body, which generates torque and induces flagellar rotation switching. The basal body (Figure 1) is comprised of a series of transmembrane rings that enclose a central rod. Powered by the proton or sodium gradient that spans the inner membrane, torque is generated by interactions between the membrane-embedded ion channels and the switch complex located in the cytosolic space (Berg, 2003; Minamino et al., 2008, 2015; Sowa, 2008; Chen et al., 2011) The switch complex forms the C-ring and is composed of the proteins FliG, FliM, and FliN (or FliY) (Figure 1). The switch complex rotates either counterclockwise (CCW) or clockwise (CW) and in doing so dictates whether the cell swims smoothly or tumbles in solution. Interaction of the switch complex with the phosphorylated form of the response regulator CheY causes switching of rotation direction (Berg, 2003; Sowa, 2008).
Figure 1.

Schematic diagram of the bacterial flagellar motor.
A prerequisite for motor operation and assembly during the early stages of flagellar biosynthesis is the correct positioning of the switch complex relative to the MS-ring (Macnab, 2003; Grünenfelder et al., 2003; Chevance et al., 2008; Minamino et al., 2008, 2015). Located in the inner membrane and composed of approximately 25 copies of a single transmembrane protein, FliF, the MS-ring is the first circular structure to form during flagellar assembly (Figure 1) (Macnab, 2003; Chevance et al., 2008; Sowa, 2008). The MS-ring adheres to the switch complex through interactions mediated between the C-terminal tail of FliF (FliFC) and the N-terminal region of FliG (FliGN) (Francis et al., 1992, Thomas et al., 2001; Brown et al., 2002; Grünenfelder et al., 2003). Electron microscopy, site-directed alanine-scanning mutagenesis, and fusion/deletion studies have revealed that these two proteins directly interact with one another in a 1:1 stoichiometric ratio (Thomas et al., 1999, 2001, 2006; Ogawa et al., 2015). The structure of FliG has been determined for the full-length protein (Lee et al., 2010), and the individual domains (Brown et al., 2002, Minamino et al. 2011), alone and in complex with domains of FliM (Paul et al., 2011; Vartanian et al., 2012; Lam et al., 2013; Sircar et al., 2015). FliF is an integral membrane protein with extensive periplasmic domains homologous to the injectosome SctJ/D proteins and the sporulation factors SpoIIIAG/H (Bergeron, 2016). FliG and FliF orthologs are found in essentially all flagellated (and some non-flagellated) bacteria and thus this interaction can be considered a universal contact made in bacterial flagella (Levenson et al., 2012; Bergeron, 2016).
To characterize the interaction between FliFC and FliGN, Levenson et al. employed tryptophan fluorescence and 1H-15N TROSY-HSQC nuclear magnetic resonance spectroscopy to map the binding surface of T. maritima FliFC on FliGN (Levenson et al., 2012). The Kd between FliGN and FliFC was measured in the low nanomolar range (< 40 nM) and FliFC binding caused chemical shift perturbations of backbone resonances across the entirety of FliGN. As a whole, the data suggested that the FliGN domain orders as a result of FliFC binding – an effect consistent with multi-angle light scattering experiments that demonstrated the dissociation of FliG homodimers to form mixed FliFC:FliG heterodimers upon FliFC addition (Levenson et al., 2012). A strong interaction between FliFC and FliGN agrees well with previous observations made when purifying intact flagellar motors from Salmonella cells. Intact basal bodies readily lose the FliM and FliN regions of the C-ring; however, very low pH is required to induce dissociation of the FliF:FliG interface, owing to the unique strength of the interaction (Francis et al., 1994). More recently, a similar interaction between FliF and FliG was reconstituted with a soluble full-length form of FliF from a marine Vibro species (Ogawa et al., 2015).
Herein, we report the crystal structure of a complex between FliF495–532 (FliFC) and FliG1–98 (FliGN). All three domains of full-length FliG have a strikingly similar topology when FliGN is bound to FliFC. Indeed, FliGN folds around the FliFC helices and produces a structure similar to that of the FliG middle domain (FliGM) and the FliG C-terminal domain (FliGC) (Brown et al, 2002; Lee et al., 2010). Solution NMR, small-angle X-ray scattering (SAXS), and biochemical data are fully consistent with the structure, and help differentiate between two alternative complexes found in the crystal lattice. The FliFC:FliGN structure was then used to model the MS/C-ring interface in the context of cryo-EM density for whole Salmonella rotors (Thomas et al., 2006). An extended C-ring structure was generated taking into account previously placed FliGMC:FliMM units (Sircar et al., 2015). Targeted cross-linking and motility assays of structure-guided variant proteins reveal the functional importance of the FliFC:FliGN interaction
Results
Solution-state properties of an engineered FliFC:FliGN fusion protein
FliFC:FliGN {FliFC(495–532):FliGN(1–98) for crystallography and FliFC(495–532):FliGN(1–102) for NMR} were produced as chimeric fusion proteins to ensure a 1:1 component ratio (SI Figure 1). The constructs were engineered such that FliG and FliF are linked together via a sequence containing a His8 tag bracketed by two TEV protease sites (or one TEV protease site between His8 and FliGN for the NMR studies). 1H-15N TROSY-HSQC NMR spectroscopy was employed to evaluate the resulting complex before and after TEV proteolysis (Figure 2B). Additionally, the cleaved complex was compared to that of 15N labeled FliGN incubated with a synthetic FliFC peptide corresponding to the identical sequence encoded by the fusion construct (Figure 2A) (Levenson et al., 2012) The 1H-15N TROSY-HSQC spectra of the FliFC:FliGN fusion construct after TEV proteolysis (Figure 2A, red) is nearly identical to that of 15N labeled FliGN incubated with stoichiometric amounts of the corresponding FliFC peptide (Figure 2A, black). Thus, the cleaved fusion protein has a conformation similar to that of FliGN mixed with FliFC. Similarly, there are little differences in the measured 1H-15N resonances before (black) and after (red) TEV proteolysis (Figure 2B) indicating that the intact and cleaved proteins are remarkably similar in organization. Resonances of isotopically labeled TEV-proteolyzed FliF495–532:FliG1–102 complex were assigned using standard 3D NMR techniques, yielding assignments for 93% of residues (SI Figure 2A). Cα chemical shift deviations from random coil values showed both FliF495–532 and FliG1–102 within the complex to be well-folded, with alpha helical structure (SI Figure 2B). Overall, the data indicate the FliFC:FliGN complex is the same whether produced as a fusion protein or as separate components and that the presence of a linker does not prevent complex assembly.
Figure 2.

NMR Characterization of FliF495–532:FliG1–102 complex. (A) Black: TROSY-HSQC data of 15N-labeled FliG1–102 bound to unlabeled FliF495–532 peptide. Red: TROSY-HSQC data of 15N-labeled FliF495–532:FliG1–102 fusion protein after TEV proteolysis. (B) Black: TROSY-HSQC data of 15N-labeled FliF495–532:FliG1–102 fusion protein before TEV proteolysis Red: TROSY-HSQC data of 15N-labeled FliF495–532:FliG1–102 fusion protein after TEV proteolysis. See also SI Figures 1, 2 and 6.
Solution-state structure of the FliFC:FliGN complex by SAXS
SAXS was used to evaluate the FliFC:FliGN solution-state structure. For these experiments, size-exclusion chromatography (SEC) was coupled to the SAXS measurements to separate different oligomeric or conformational states and limit contamination of soluble aggregates. The SEC chromatogram of the purified FliFC:FliGN complex (SI Figure 3A) reveals a well-behaved protein complex. SAXS was measured for the area enclosed by the black dotted lines on the SEC chromatogram shown in SI Figure 3A. Kratky analysis of the scattering data (SI Figure 3B) converges towards zero at high q values, characteristic of a globular protein complex (Lipfert et al., 2007). Generation of a low-resolution de novo molecular envelope of the protein complex results in a globular envelope with an extended region (SI Figure 3B). Interestingly, there appears to be poor agreement between the envelope and the structure of FliGN from a full-length FliG structure determined from Aquifex aeolicus (SI Figure 3D; Lee et al., 2010). Either the binding of FliFC to FliGN produces a substantial conformational change in FliGN or the FliFC peptide is large enough to account for the unfilled area within the envelope. It is likely that FliF binding alters FliG, as the interaction converts FliG from a homodimer (FliG:FliG) to heterodimer (FliG:FliF) (Levenson et al., 2012). For more information regarding SAXS data acquisition and envelope generation, see SI Methods.
Crystal Structure of FliFC:FliGN complex
Crystals of the FliFC:FliGN complex were grown by vapor diffusion at pH 7.5 (see Methods). Attempts to determine the structure of FliFC:FliGN by molecular replacement (MR) failed, therefore a selenomethionine (seMet) variant of the FliFC:FliGN complex was crystallized (see Methods). The seMet FliFC:FliGN crystals diffracted to 2.6 Å resolution and yielded an initial SAD phased map with a figure of merit of 0.341. A de novo model was built and refined into the map with excellent final agreement statistics (Table 1). The unit cell consisted of two heterodimers per asymmetric unit, with one dimer having an extended α-helix towards the C-terminal portion of the FliG peptide, and the other having three segmented helices (Figure 3A). Although the two conformations of the last α-helix in FliGN may indicate flexibility of this helix, the dimer with the extended α-helix (Figure 3) agrees better with the SAXS envelopes and is thus presumed to represent the dominant solution-state structure (SI Figure 4C).
Table 1.
Data collection and Refinement Statistics
| Wild Type FliFC:FliGN complex | Selenomethionine FliFC:FliGN complex | |
|---|---|---|
| Wavelength (Å) | 0.97700 | 0.97921 |
| Synchrotron | CHESSa | APSb |
| Beam-line | A1 | 24-ID-E |
| Space group | P21 | P21 |
| a, b, c (Å) | 49.18, 59.33, 51.72 | 48.81, 59.24, 51.76 |
| α, β, γ (°) | 90.00, 115.59, 90.00 | 90.00, 115.76, 90.00 |
| Resolution (Å) | 50.0 - 2.10 (2.15 - 2.10) | 50.0 - 2.60 (2.66 - 2.60) |
| †Rmerge (%) | 8.2 | 12.2 |
| †Rp.i.m. (%) | 5.5 (21.8) | 5.3 (15.8) |
| †Rmeas (%) | 9.9 (35.5) | 13.3 (38.8) |
| ▪I/σ(I)▪ | 13.4 (3.2) | 14.8 (7.0) |
| Completeness (%) | 99.5 (99.6) | 99.2 (99.5) |
| Multiplicity | 3.1 (2.4) | 6.0 (6.0) |
| Anomalous completeness (%) | 98.1 | |
| Mosaicity | 0.43 – 0.81 | 0.63 – 2.55 |
| Total Reflections | 47,958 | 46, 599 |
| Phaser FOM | 0.341 | |
| Refinement | ||
| Resolution (Å) | 44.35 - 2.10 (2.18–2.10) | |
| No. of unique reflections | 15,618 (1559) | |
| Reflections used for Rfree | 1556 (161) | |
| Rwork/Rfree (%) | 16.9 (17.4)/21.7 (23.7) | |
| Clash Score | 4.70 | |
| No. of non-hydrogen atoms | 2263 | |
| Protein | 2090 (260 residues) | |
| Water | 173 | |
| B factors (Å2) | ||
| Wilson | 18.1 | |
| Average B-factors | 24.3 | |
| Protein | 23.7 | |
| Water | 31.8 | |
| R.M.S. deviations | ||
| Bond lengths (Å) | 0.007 | |
| Bond angles (°) | 0.9 | |
| Ramachandran outliers (%) | 0.4 | |
| Rotamer outliers (%) | 1.3 | |
Statistics for the highest-resolution shell are shown in parenthesis,
Ri = ΣΣi|Ii − <Ii>|/ΣΣi Ii
Cornell High Energy Synchrotron Source - Cornell University
Advanced Photon Source - Argonne National Lab
Figure 3.

Crystal Structure of FliFC:FliGN. The two unique heterodimers per asymmetric unit oriented as in the crystal (A) or with the same perspective (B) (FliFC - violet and green, FliGN - yellow and cyan). The two different morphologies of the terminal FliGN helix are circled. See also SI Figures 3, 5, 6, and 7.
The FliFC terminal peptide forms two α-helices connected by a short extended linker (FCα1 residues 497–514, and FCα2 517–529, Figure 3B). FliGN is an all α-helical structure, similar to the all α-helical T. maritima FliGM and FliGC domains (Brown et al., 2002; Paul et al., 2011; Sircar et al., 2015), as well as the full-length FliG from A. aeolicus (Lee et al., 2010). FliGN consists of four helices, denoted GNα1–4 in Figure 3B (residues 6–19, 21–30, 33–45, and 51–87, respectively) where GNα1–3 form an ARM-like domain that is also present in FliGM and FliGC (Brown et al., 2002; Lee et al., 2010). Together with FCα2, GNα1–3 comprise a 4-membered right-handed super-helix that has been well-documented as a structural motif in other FliG structures (Brown et al., 2002; Lee et al., 2010; Paul et al., 2011) and is also found in the N-terminal cytoplasmic domain of the Mg2+ transporter MgtE (Hattori et al., 2007). We will refer to these FliG super-helical domains as ARM-like motifs, following previous designations ((Lee et al., 2010; Vartanian et al., 2012), although we note that these domains differ substantially from traditional ARM motifs in helix crossing angles and packing interactions (Andrade et al., 2001). FCα1 and FCα2 assume a hook-like structure that latches into the V-shaped cavity formed from the GNα1–3 super helix and GNα4. In the unbound FliGN of A. aeolicus (Lee et al., 2010), GNα1–3 forms a similar super helix but GNα4 is instead composed of two helices connected by a linker. Although similar in secondary structure, the unbound FliGN domain GNα4 from A. aeolicus does not superimpose as well with FliGN in complex with FliFC (SI Figure 4A). In the absence of FliFC this region may be highly flexible, which would explain the difficulty in crystallizing unbound FliGN.
FliFC and FliGN associate to form a shared hydrophobic core. Interacting regions of FliFC and FliGN mapped by NMR involve the burial of conserved hydrophobic residues on both FliFC and FliGN (Levenson et al., 2012). Similarly, the electrostatic potential at the molecular surface of FliGN reveals a hydrophobic, non-charged FliF binding region. The FliFC:FliGN complex buries ~1,700 Å2 of hydrophobic binding surface on the FliFC peptide, and is stabilized at the periphery by electrostatic interactions (SI Figure 5). A nearly invariant tryptophan residue essential for the FliFC:FliGN interaction (T. maritima FliF W527) (Thomas et al., 2001; Levenson et al., 2012) is tightly sequestered within a hydrophobic pocket formed from GNα1, GNα4, and FCα2 (SI Figure 5E).
From the extensive hydrophobic interactions it appears as though FliGN folds around FliFC. Surprisingly, the resulting topology matches that of the FliGM and FliGC domains (Figure 4A and 4B). The structure of the FliFC:FliGN complex superimposes well with the tertiary structure of FliGM and FliGC from T. maritima (Figure 4; left - FliGM PDB: 3SOH, right - FliGC PDB: 1LKV), producing RMS values of 1.053 Å (40 residues) and 1.227 Å (63 residues), respectively (Paul et al., 2011; Brown et al., 2002). Superposition of the FliFC:FliGN complex with the middle and C-terminal domains from A. aeolicus FliGFL results in a similar outcome (SI Figure 4) (Lee et al., 2010).
Figure 4.

Domain repeats within FliG. (A) Superimposition of T. maritima FliFC:FliGN complex (FliFC - violet, FliGN - yellow) and Thermotoga maritima (left) FliGM (grey, PDB 3SOH, RMS 1.053 Å, 40 residues) and (right) FliGC (grey, PDB 1LKV, RMS 1.227 Å, 63 residues). The black circle signifies the absence of a corresponding ARMC domain. (B) Comparison of secondary structure motifs and primary sequence alignment of the superimposed regions. See also SI Figure 4.
Mapping of the FliFC:FliGN interface by paramagnetic resonance enhancement
To further investigate the solution-state structure of the FliFC:FliGN complex, paramagnetic relaxation enhancement (PRE) experiments were carried out in which 1H-15N TROSY-HSQC NMR spectra were collected on a FliF495–532 (E508C):FliG1–102 (E. coli FliF numbering) mutant that was conjugated to a MTSL nitroxide spin-label. Proximity to the conjugated MTSL causes neighboring 15N backbone nuclei to experience increased resonance peak broadening. Mapping of the maximally broadened residues in FliGN from the spin label on FCα1 produces a binding interface that is in excellent agreement with the crystal structure (SI Figure 6 and SI Table 2). Notably, a small portion of the C-terminal tail of GNα4 also experiences a significant amount of resonance peak broadening with peak intensities 27–65% compared to that observed in the protein after the MTSL label had been reduced to a spectrally inactive form by addition of ascorbic acid, despite these residues being relatively far removed from the spin-label in the crystal structure. Perhaps the suspected flexibility of this helix brings it into proximity of the label for some conformational states of the complex.
Modeling of MS/C-Ring Interface of the Bacterial Flagellar Motor
The FliFC:FliGN complex was then docked into 3D cryo-EM reconstructions of the intact CW locked Salmonella motors (Thomas et al., 2006). It was assumed that the FliFC peptide descends perpendicular to the inner membrane, despite the 32-residue gap present between FCα1 and the second transmembrane helix (TM2) region of FliFNM. Symmetric rings were generated that contained either 24, 25, or 26 copies of FliFC:FliGN, consistent with previously determined FliF:FliG stoichiometry (Thomas et al., 2006). When FliFC:FliGN rings with 24 or 26 subunits were docked into the C25 cryo-EM density, there were either clashes from overlapping secondary structural motifs (26 subunits), or unaccounted areas of electron density (24 subunits). Rings modeled with C25 symmetry had minimal steric clashes within the ring, minimized areas of unassigned density and produced a correlation coefficient of 0.83 to 30 Å resolution (Figure 5A–C). Thus, the dimensions of the FliGN:FliGC complex agree well with the MS/C-ring symmetry. In modeling the MS/C-ring interface, FCα1 was directed upwards towards the inner-membrane, an orientation that conferred biological relevance, as well as allowed for systematic packing of all 25 subunits within minimal steric clashes (Figure 5A–C). In agreement with the model, recent bioinformatics-based modeling of the upper periplasmic region of FliFNM indicates 25 FliF subunits per MS-ring (Bergeron, 2016).
Figure 5.

Modeling of the FliFC:FliGN ring with C25 symmetry. (A) Fully assembled (FliFC:FliGN)n ring (n = 25 copies) showing the side (left) and top (right) view. (B) (FliFC:FiGN)25 ring and (FliMM:FliGMC)34 ring, (red -FliMM, blue -FliGMC) superimposed with the EM density of the Salmonella rotor (Thomas et al., 2006) Ring density simulated at a 30 Å from the structures gave correlation coefficients with the EM density as follows: (FliFC:FliGN) 0.83, and (FliMM:FliGMC) 0.79.
In an effort to extend the modeling further, we carried out the same fitting technique with a FliGMC:FliMM complex structure previously generated via x-ray crystallography and pulse-dipolar ESR (PDB ID 4QRM, Sircar et al., 2015). In total, 34 copies of a single FliGMC:FliMM complex were fit into the rotor density, producing a correlation coefficient of 0.79 (Figure 5C). Notably there remains a disparity between the FliGMC and FliGN stoichiometry by 9 copies. This symmetry mismatch is well recognized (Thomas et al., 1999, 2001; Manson, 2007; Berg 2003; Sowa et al., 2008), and could result from unoccupied positions of FliGMC in the C-ring (Sircar et al., 2015), non-equivalence in the FliG:FliM contacts (Paul et al., 2011; Sakar et al., 2011) or as a result of adaptive remodeling of both FliM and FliN (Fukuoka et al., 1994; Lele et al., 2012; Delalez et al., 2014). Nonetheless, the current model suggests that 34 copies of FiGN could not fit within the upper portion of the EM density.
Functional analysis of the FliFC:FliGN complex
To validate the structure of FliFC:FliGN, site-directed mutants were evaluated by in vivo motility assays. All experiments were performed in E. coli with homologous E. coli proteins, where FliF and FliG share 29% and 36% sequence similarity with the T. maritima proteins, respectively, and conserve nearly all of the key residues involved in the complex interface (SI Figure 7) (Levenson et al., 2012; Thomas et al., 2001). Additionally, residue substitutions at the FliF-FliG interface were tested in protein interaction studies and crosslinking of engineered cysteine mutants were further employed to probe the functional FliF:FliG interface.
Hydrophobic to aspartate mutations FliG were made and cell migration rates were measured relative to wild type (Figure 6A,B). Residue substitutions along the FliF:FliG interface observed in the crystal structure proved most detrimental to motility. Specifically, motility defects were strongest for changes that are in close proximity to FC α 2, especially at positions Leu13, Leu29, Leu37, Ile17 (E. coli numbering), which is consistent with the interactions between FliFC and FliG found in the crystal structure.
Figure 6.

In vivo E. coli motility and interaction assays. (A) Two views of the FliGN domain, showing motility defects that result upon replacement of the indicated hydrophobic residues with aspartate. Motility phenotypes were tested by expressing mutant FliG proteins in the fliG null strain. Yellow-green, rate less than 100% but greater than 70% of wild-type; yellow, rate between 20% and 70% of wild type; orange, motile but at less than 20% of wild type; red, immotile. (B) A table summarizing the relative rates, values are the mean ± s.d. for 3 determinations. (C) Interaction between FliFC (residues 505–552) and FliGN (residues 1–87) observed in the bacterial adenylate cyclase two-hybrid system. Positive interactions drive expression of β-galactosidase. Negative controls and a leucine-zipper positive control are shown. (D) Effects of hydrophobic-to-aspartate replacements in FliGN on the FliF-FliG interaction. Coloring is based on β-galactosidase activities: Green, activity indistinguishable from wild type; yellow-green, activity less than 100% but greater than 70% of wild type; yellow, activity between 20% and 70% of wild type; red, activity decreased to the level of negative controls. FliFC shown light purple in all figures. See also SI Figure 8.
Analysis with the bacterial adenylate cyclase two-hybrid (BACTH) method provided independent support for a strong FliF:FliG interaction involving the interfaces observed in the crystal structure (Figure 6D) (Miller et al., 1972; Karimova et al., 2001; Battesti et al., 2012). BACTH experiments used hybrid constructs containing the FliFC (E. coli residues 505–552) and FliGN (E. coli residues 1–87). In experiments with wild-type FliFC and FliGN, strong color development was observed on MacConkey plates, and measurements of β-galactosidase activity afforded values comparable to the leucine-zipper positive control (Figure 6C). Hydrophobic-to-aspartate replacements of FliG on the predicted interface weakened the interaction in a pattern similar to the motility phenotypes. Residue substitutions made to FliF provided similar validation of the expected contact sites (SI Figure 8). These results suggest that the interface involving FCα2 may be chiefly responsible for the strength of the interaction. Consistent with this, a hydrophobic-to-aspartate replacement at FliF residue 542 in FCα2 eliminated both motility and the two-hybrid interaction. A mutation at position Val538 in FCα1 caused a significant reduction in migration rate, while not diminishing the FliF:FliG interaction. In liquid culture, this mutant exhibited relatively weak motility and a substantial fraction of immotile cells, indicating a delay or partial defect in flagellar assembly. Thus, the V538D replacement alters the FliF:FliG relationship while not greatly weakening the interaction.
To further probe the FliF:FliG interaction in vivo, cross-linking of engineered cysteine mutants were employed to map the binding site interface between FliF and FliG in E. coli. Targeted disulfide crosslinking experiments indicate that the FliF:FliG relationship in E. coli is similar to that observed in the structure of the T. maritima proteins (Figure 7C). Fifty-five double-cysteine mutants were made (Figure 7A; SI Table 3). In total, seven relatively strong crosslinks were observed, all involving positions that are in proximity in the crystal structure (Cβ-Cβ distances 6–10.5 Å). All of the cysteine double mutants that displayed strong crosslinking retained significant function (45% – 85% of wild-type) in soft-agar. Crosslinking experiments yielded one surprising result; Cys543 in FliF showed only weak crosslinking though predicted to be close to several of the FliGN cysteine replacements. The strongest crosslink of Cys543 (still weak in absolute terms) was to Cys29 in FliG, which according to the structure is the closest (to 543) of the 11 FliG replacements examined. Residue 543, normally Ile, resides in FCα1 and is buried at the FliG:FliF interface. We hypothesized that packing in this region might be so stable as to prevent deprotonation or necessary movements of the buried cysteine side chains for crosslinking. In an attempt to loosen the interface and increase the reactivity of Cys543, the adjacent Arg544 was replaced with alanine; however, crosslinking was not significantly increased. Crosslinking was then carried out in the presence of urea (Figure 7B). The crosslink to FliG at Cys29 was slightly enhanced in the presence of 3 M urea, while crosslinking to a more distant position (Cys44) remained negligible.
Figure 7.

In vivo FliG-FliF crosslinking. (A) Products of iodine-induced disulfide crosslinking were detected using anti-FliG immunoblots. The seven cysteine pairs that gave strong crosslinking are shown together with single cysteine controls. Crosslinking was weak or undetectable for forty-eight other cysteine pairs tested (SI Table 3). In addition to the indicated cys-mutant FliF, a comparable amount of wild-type FliF (expressed from the chromosome) was present in the crosslinking experiments; thus, crosslinking yields should underestimate what would occur if all of the FliF carried the Cys replacement. (B) A low-yield crosslink between FliG position 543 and FliG position 29 that is made somewhat stronger (by ~50%) in the presence of 3 M urea. The more-distant position Cys44 did not crosslink. (C) Mapping the crosslinks onto the FliG-FliF structure. Thicker red lines indicate the crosslinks observed under standard conditions, and the thin red line indicates the weaker, urea-enhanced crosslink involving the buried position 543. See also SI Figure 8.
Overall, results from the in vivo experiments fit well with the crystallographic structure of the FliFC:FliGN complex, and indicate that the FliF:FliG interfaces are essentially identical in E. coli and T. maritima. As expected, the FliF:FliG interaction is critical for function. Mutant phenotypes and the non-reactivity of Cys543 highlight the importance of the interactions involving FCα2 and indicate that packing at this interface may be unusually stable.
Discussion
The MS/C-ring interface is an essential contact site of the flagellar motor. Not only does it serve as a checkpoint during flagellar morphogenesis, but it positions FliG with respect to the inner membrane such that it can (1) bind to the MS-ring to propagate rotation, (2) interact with the stator complexes to confer torque generation to the switch complex, and (3) position FliG such that it can bind to FliM to anchor CW/CCW switching and rotor dynamics (Berg, 2003; Sowa, 2008). Although fusion-deletion and mutagenesis studies have established that FliF and FliG interact through their N and C terminal domains, respectively, a molecular level view of this interaction remained elusive. The 2.1 Å resolution crystal structure of FliFC:FliGN defines the interface between the MS and C-rings within the flagellar motor. Furthermore, by reconciling the FliFC:FliGN crystal structure with NMR, SAXS, in vivo assays and modeling to cryo-EM reconstructions, we validate the structure as biologically relevant and further expand current understanding of protein organization at the MS/C-ring interface.
Fusion of the FliFC peptide to FliGN was an effective strategy for producing the complex. Comparing TROSY-HSQC 1H-15N NMR spectroscopy of the fused protein to the complex formed by its components indicates that the fusion folds as the native complex (Figure 2). Given the intimate contact formed by the two separate proteins, it is not surprising that they appear to fold as one unit. Notably, the ability of the cleaved and non-cleaved moieties to form essentially the same structure places spatial restrictions on the location of the FliFC binding site. Fortunately, these restrictions were satisfied in the native FliFC:FliGN complex (Figure 3), and found to be in agreement with the FliFC:FliGN interface mapped from the PRE studies (SI Figure 6 and SI Table 2).
FliFC:FliGN crystallizes in two conformations (Figure 3A). It was important to consider both when modeling the MS/C-ring interface. The FliGN core that houses the FliFC binding site remains invariant, owing to the strength of the FliF:FliG interaction. However, the C-terminal tail of FliGN (GNα4) assumes either an extended α-helix, or three segmented helices (Figure 3B). Although the crystal lattice may influence these conformations, the solution-state complex favors the extended helix (SI Figure 3B–C). Nonetheless, the more compact conformation may play a role in motor operation, as it confers a higher degree of flexibility thus could serve as a flexible hinge to absorb stress during rotation reversals. Modeling of the FliFC:FliGN structure into the ring density (Figure 5A–B) directs FCα1 toward the inner membrane of TM2 of FliF. The 32 unaccounted residues between FliFC FCα2 and TM2 of FliFNM could similarly serve as a flexible hinge.
Importantly, FliGN requires FliFC to fold into a stable FliGM/FliGC-like domain, and thus the stoichiometry of FliF to FliG is most likely to be 1:1. Such a 1:1 complex has important implications for the overall structure of the rotor, as FliG must interact with FliM, of which there are ~34 copies. Therefore, either not every FliM interacts with one FliG in the same way, or the protein stoichiometry in the MS and C-rings are not correct. The latter seems unlikely, as 25 copies of the FliF:FliG complex fit well around the upper switch, and it is difficult to accommodate more subunits without significant steric clashes. Further, the current finding that the MS/C-ring interface has C25 symmetry is in agreement with recent homology modeling of the periplasmic region of FliF (Bergeron, 2016).
FliG is divided into three distinct domains, where each domain has evolved to carry out a specific function within the flagellar switch (Lloyd et al., 1996; Berg, 2003; Sowa, 2008). FliGN contains the binding site for FliFC and anchors the C-ring to the inner-membrane, FliGM interacts with FliGC and FliMM to propagate CCW/CW switching, and FliGC contains conserved charged residues to interact with membrane-bound stators in torque generation (Lloyd et al., 1997; Berg, 2003; Sowa, 2008). The structure of FliG determined from A. aeolicus agrees with structures of the M and C terminal domains (Brown et al, 2002; Paul et al., 2002). A notable feature of unbound FliGN identified in the full-length structure was the structural similarity between AqFliGN helices N1–4, and AqFliGC terminal helices 3–6 (Lee et al., 2010). A similar observation was made previously from the crystal structure of FliGMC from T. maritima (PDB 1LKV) regarding the ARM motifs present in FliGM and FliGC (ARMM/C) (Brown et al., 2002). Interestingly, the FliFC:FliGN complex shows how the structural replication within FliGC is even more extensive than in FliGMC (Figure 4; SI Figure 4). When FliF is bound to FliG, all three domains adopt similar conformations such that FliF:FliG aligns well with the ARMM motif and helixMC/NM in FliGM, as well as with helices 1–6 in FliGC. The striking similarity of the FliF:FliG fold with that of FliGM and FliGC suggests domain shuffling may relate all three of the FliG domains (Di Roberto et al., 2014). Either the N-terminal helix of FliGN was transferred to the C-terminus of FliF, or the FliGN:FliFC unit was fused and propagated to generate FliGM and FliGC. The genes for FliF and FliG are often adjacent, in the same orientation and expressed from the same promoter. Thus, in many instances the coding sequences for the two helices that complete the FliGN superhelix are tightly coupled to the FliG gene. Furthermore, in Chlamydia the C-terminal region of FliF has been replaced with an entire FliGM-like domain (Bergeron, 2016). Thus, the interface between the FliF TM2 and the C-ring is always a FliGM-like domain, whether it is composed of a single polypeptide or two that mate FliFC to FliGN. Interestingly, upon comparing the sequences of the FliFC:FliGN complex to FliGM, the region where FliFC terminates and FliGN begins coincides with the FliMM-binding EHPQR motif of FliGM (Paul et al., 2011; Vartanian et al., 2012) (Figure 4B). If FliGM and FliFC:FliGN share a common origin, severing a continuous FliGN domain into two at what would be the FliM interaction site on the analogous FliGM may have been advantageous to prevent incorrect binding of FliGN to FliMM during MS/C-ring assembly.
The contact residues of the FliF:FliG structure mediate protein interactions in cells and are critical for generating functional flagella. Substitution of key hydrophobic residues to aspartic acid along the FliF:FliG interface of both FliG (Figure 6A) and FliF (SI Figure 8) disrupt cell motility and protein-interactions. Although FCα1 buries ~570 Å2 of surface area, the functional specificity and strength of the FliF:FliG interaction can largely be attributed to FCα2. Disulfide crosslinking data provides direct biochemical evidence in support of these findings. One surprising observation was the relatively small extent of crosslink formation between buried FliF Cys543 and any potential disulfide partner (Figure 7B, SI Table 4). This result likely reflects the uniquely strong interaction between FCα2 and FliG that prevents movement of the cysteine residues or access to oxidants. These results provide evidence for the conjecture that FliFC and FliGN essentially fold as one domain to provide a robust anchor for the C-ring to the membrane. It is not surprising then that the FliF:FliG interaction is much stronger than that of the FliG:FliM or FliM:FliN and can persist longer at low pH without dissociation (Francis et al, 1994).
Overall, by combining data from x-ray crystallography, SAXS, NMR, modeling to cryo-EM structures and in vivo functional studies, we have defined the FliF:FliG interaction at the MS/C-ring interface. The structure has then been elaborated into an updated model for the upper switch. Importantly, the structure suggests that the FliF:FliG stoichiometry is likely the same, and that domain shuffling appears to have driven the association of these key building blocks of the flagellar motor. We propose a model where a domain is duplicated and then genetically split so that a folding unit can associate two otherwise non-interacting proteins with high affinity. It is notable that protein engineering has used a similar strategy of split protein complementation to effect interactions in cells through the development of tools such as split GFP, ubiquitin and luciferase proteins. (Xing et al., 2016)
Experimental Procedures
i. Protein Expression and Purification
Native proteins were expressed in BL21 (DE3) E. coli cells, grown at 37°C in LB media containing 50 μg-mL−1 kanamycin, and induced with 1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) at 25°C after reaching an OD600 of ~0.6. Selenomethionine variants were expressed in B834 methionine auxotrophic E. coli cells at 37°C in minimal media containing 100 μg-mL−1 ampicillin hydrochloride and 50 mg-L−1 +/− L-selenomethionine.
All proteins were purified according to the following procedure: cell pellets were re-suspended in lysis buffer (25 mM HEPES pH 7.5, 500 mM NaCl, 5 mM imidazole), lysed via sonication, and the lysate was then centrifuged at 20,000 rpm for 45 minutes at 4°C. The supernatant was passed through a Ni-NTA column, washed with 50 mL wash buffer (25 mM HEPES pH 7.5, 500 mM NaCl, 25 mM imidazole), and eluted with elution buffer (25 mM HEPES pH 7.5, 500 mM NaCl, 200 mM imidazole). The linker between FliGN and FliFC was removed by overnight incubation with TEV protease at 30°C. TEV was removed via centrifugation and the FliFC:FliGN complex was dialyzed extensively against 50 mM sodium phosphate pH 6.5. For the preparation of selenomethionine FliFC:FliGN, all buffers were supplemented with 10 mM DTT. Successful selenomethionine incorporation was confirmed via whole-protein positive ion electrospray LC-MS/MS.
Crystallization, Data Collection, and Phasing
The FliFC:FliGN complex was subjected to sparse course matrix screening in 600 nL drops at a protein concentration of 65 mg/mL. Crystals grew in 100 mM HEPES pH 7.5, 25% (w/v) PEG 3000, 200 mM NaCl at 25°C after ~7 days. Selenomethionine-incorporated crystals grew from 33 mg/mL protein, 100 mM imidazole pH 7.2, 130 mM NaCl, 30% (w/v) PEG 8000 in ~14 days.
Briefly, data sets were integrated and scaled in HKL2000 and SCALPACK (See Table 1). Phasing of the anomalous data set was done with Phenix HYSS and the model built with Autobuild (Phenix) programs (Adams et al., 2010). Manual adjustments to model were made with COOT and subsequent refinements were carried out with Phenix Refine. The final FliFC:FliGN model has an Rfree of 21.74 % and a Rwork of 16.92 % with excellent stereochemistry.
ii. Ring Simulation and Fitting into cryo-EM Maps
Low-resolution cryo-EM maps of whole rotor density containing the C, MS, L, and P rings were provided by Dennis Thomas (Thomas et al., 2006). To construct a model of the upper C-ring/MS-ring interface, a single FliFC:FliGN complex was aligned to the whole rotor density such that the coordinate systems of the crystal structure PDB file coincided with that of the rotor MRC map file. The center of mass was calculated for the placed complex, and the structure was duplicated n number of times and rotated 360/n degrees around an axis chosen perpendicular to the membrane. The ring was simulated to a radius of 15 nm, a distance corresponding to the radius of the EM map. Three rings of n = 24, 25 and 26 Cn fold symmetry were generated and evaluated with respect to the EM density. By inspection, only rings of C25 fold symmetry produced a clash-free model that has minimal gaps in density when comparing. The C25 upper C/MS-ring model was fit into the EM map using the Fit to Map program in Chimera. When simulating the EM map to a resolution of 30 Å, the C25 fold FliFC:FliGN ring provided good agreement with the whole rotor density and produced a correlation coefficient of 0.83. The above procedure was repeated with a FliGMC:FliMM model (PDB 4QRM), to give a mid rotor subunit copy number of 34 and a correlation coefficient of 0.79.
iii. Generation, purification, and NMR analysis of isotopically enriched FliFC:FliGN complex
The fusion FliFC(495–532):FliGN(1–102) construct was generated by traditional molecular cloning methods. The FliFC portion was amplified from a plasmid obtained from the JCSG and cloned into a plasmid containing FliGN(1–102) (Levenson et al., 2012). All NMR data was collected with a construct similar to SI Figure 1, however, the construct only included one TEV protease site between the His8 tag and the FliGN domain. Isotopically-labeled proteins were grown in M9 minimal media supplemented with 15NH4Cl, 13C glucose, and/or D2O (when necessary). Rosetta (DE3) E. coli cells were used for expression of all constructs. Cells were grown at 37 °C, and upon reaching an OD600 of ~0.6, were induced with a final concentration of 1 mM IPTG and expressed for approximately 6 hours. Purification was identical as described previously. After SEC, the protein samples were dialyzed into 50 mM sodium phosphate, 100 mM NaCl, pH 6.5 for NMR analysis. After dialysis, all samples were concentrated to their final concentration, at which point 0.02% (w/v) NaN3 was added. NMR spectra were collected on a Bruker Avance 800 MHz spectrometer equipped with a cryoprobe.
E. coli strains and media for interaction and motility studies
Escherichia coli strains and plasmids are listed in SI Table 1. All strains were derivatives of the wild type strain RP437. Chromosomal in-frame deletions or point mutations were made by using the lambda red method (Datsenko et al, 2000). TB medium contained (per liter) 10 g tryptone and 5 g NaCl, LB medium contained the same plus 5 g yeast extract. Soft-agar motility plates used TB media and Bacto-agar at 2.6 g-liter−1. Ampicillin was used at 100 μg-ml in liquid media, 100 μg-ml−1 in selective plates, and 50 μg-/ml−1 in soft-agar motility plates. Chloramphenicol was used at 50 μg-ml−1 in liquid media and selective plates and at 25 μg-ml−1 in motility plates. IPTG and sodium salicylate were prepared as aqueous 0.1 M and 10 mM stocks and used at the concentrations indicated in the figures.
iv. Motility assay
All of the experiments examining motility defects were carried out in the respective null backgrounds (DB225, EKS10, see SI Table 1). Soft-agar plates were spotted with 3 μl of overnight cultures. Once migration began, colony size was measured at regular intervals and plots of diameter vs. time fitted to a line to determine rates. Rates are reported relative to wild-type controls measured in the same experiment. Effects of Asp replacements on motility were measured in strains expressing the mutant FliG from a plasmid (derivatives of pKP619), induced with 100 μM IPTG. Effects of Asp replacements in FliF were measured in strains expressing FliF from the plasmid pEK16, induced with 0.3μM salicylate, and additionally expressing wild-type FliG from plasmid pKP619 (induced as above), in a ΔfliF strain. The additional, plasmid-expressed FliG was found to be necessary for optimal function. Effects on motility of cysteine replacements in FliG and FliF were measured in a ΔfliFfliG double-deletion strain, with both proteins expressed from plasmids.
v. In vivo and crosslinking mutagenesis
Site-directed mutations were made using the QuikChange method (Stratagene). DNA sequencing and oligonucleotide synthesis were carried out by core facilities at the University of Utah.
vi. Double-cysteine cross-linking
Disulfide crosslinking between FliF and FliG was studied in strains expressing cysteine-containing FliG variants from the chromosome and cysteine-containing FliF variants from plasmid pEK16. For these experiments, wild type protein (FliF) was present along with the Cys-mutant FliF. Levels of the two were roughly equal. The plasmid was induced with 0.2 μM salicylate, which gave FliF levels sufficient to compete with the wild-type (cysteine-less) FliF expressed from the chromosome but not so high as to impair motility. (Motility impairment occurred with induction by 1 μM or higher.) Cells were cultured overnight at 37° C and diluted 100-fold into TB containing antibiotic and 0.2 μM sodium salicylate then grown at 32° C for 5–6 h (to OD600 0.7 – 0.8). OD600 was measured to adjust the cell density, then equal numbers of cells were pelleted and re-suspended with XL buffer (20mM Na-phosphate pH7.4, 150mM NaCl). For cross linking with iodine, 0.1ml of cell mixed with 4 μl of 25 mM iodine for 3 min at room temperature, then sulfhydryl groups were blocked by addition of 2 μl 0.5 M NEM and incubation at room temperature for 3 min. Samples were mixed with an equal volume of 2X non-reducing loading buffer and heated at 95 °C for 10 min before loading on SDS-PAGE gels. Some experiments included urea during the cross-linking step, at the concentrations indicated in the figures, to test the effects of partially destabilizing the proteins.
vii. SDS page and immunoblotting
Proteins were resolved in 7.5% SDS-PAGE gels and transferred onto nitrocellulose using a Transblot turbo apparatus (BioRad). Rabbit polyclonal antibody against FliG was used at 1:1,000 dilution in a solution containing phosphate-buffered saline pH 7.4, 0.1% gelatin, and 0.01% sodium azide. Immunoblots were visualized and analyzed using the LiCor Odyssey infrared-imaging system.
viii. Two-hybrid interaction assay
Bacterial adenylate cyclase two-hybrid (BACTH) measurements of the FliF:FliG interaction used vectors provided in a kit from Euromedex. Procedures followed previously published procedures (Miller et al., 1972; Battesti et al., 2012) with minor modifications. Cells from a single colony were grown overnight at 32° C in LB containing ampicillin and 0.5 mM IPTG, then pelleted and re-suspended in PBS buffer. 0.04 ml of cells were mixed with 0.96 ml Z buffer (0.06 M Na2HPO4, 0.04 M NaH2PO4, 0.01 M KCl, 0.001 M MgSO4, pH 7.0, 0.08% SDS, 0.22% (v/v) BME). Cells were permeabilized by addition 100 μl chloroform and vortexed for 10 sec, followed by incubation at 30° C for 5 min. Reaction was started by adding 0.2 ml ONPG solution (4mg-ml−1). After measured times of reaction at 30° C, reaction was stopped by addition of 0.5 ml 1 M NaHCO3. Product was quantified by measurement of OD420, with a correction due to cell scattering. Activity in Miller Units was computed according to the formula: {1000 × [(OD420 − 1.75 × OD550)]/(time of reaction × volume of culture used × OD600)}; where OD420 and OD550 are read from the reaction mixture and OD600 reflects cell density in the washed cell suspension.
Highlights.
FliFC:FliGN fold together to produce a topology that repeats throughout FliG
FliFC:FliGN interact through hydrophobic contacts critical for motor function
FliF:FliG 1:1 stoichiometry produces an MS/C-ring interface with ~C25-fold symmetry
Acknowledgments
Research reported in this publication was supported by the NIGMS of the NIH under Award Number T32GM008500 (MJL, BRC) and R01GM59544 (FWD). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank NECAT (NIH grant P41 GM103403) and CHESS/MacCHESS (NSF: DMR-1332208; NIH: P41 GM-103485) for access to data collection facilities.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Accession Numbers
FliFC:FliGN complex PDB ID: 5TDY
FliFC:FliGN NMR assignments BMRB ID: 26908
Author Contributions
Conceptualization: B.R.C., F.W.D. D.F.B.; Methodology, R.L., M.J.L.; Investigation: M.J.L., R.L., E.K., and R.S.; Writing – Original Draft: M.J.L. and B.R.C. (In vivo data/discussion – E.K. and D.F.B., NMR data/discussion – R.L. and F.W.D.); Writing – Review & Editing: M.J.L., R.L., B.R.C., F.W.D., D.F.B., and R.S.; Funding Acquisition, Resources, and Supervision: B.R.C., F.W.D., and D.F.B.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade MA, Petosa C, O’Donoghue SI, Muller CW, Bork P. Comparison of ARM and HEAT protein repeats. Structure. 2001;309:1–18. doi: 10.1006/jmbi.2001.4624. [DOI] [PubMed] [Google Scholar]
- Battesti A, Bouveret E. The bacterial two-hybrid system based on adenylate cyclase reconstitution in Escherichia coli. Methods. 2012;5894:325–334. doi: 10.1016/j.ymeth.2012.07.018. [DOI] [PubMed] [Google Scholar]
- Berg HC. The Rotary Motion of the Bacterial Flagella. Annu Rev Biochem. 2003;72:19–54. doi: 10.1146/annurev.biochem.72.121801.161737. [DOI] [PubMed] [Google Scholar]
- Bergeron JR. Structural modeling of the flagellum MS ring protein FliF reveals similarities to the type III secretion system and sporulation complex. PeerJ. 2016;4:e1718. doi: 10.7717/peerj.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PN, Hill CP, Blair DF. Crystal structure of the middle and C-terminal domains of the flagellar rotor protein FliG. The EMBO Journal. 2002;21:3225–3234. doi: 10.1093/emboj/cdf332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Beeby M, Murphy GE, Leadbetter JR, Hendrixson DR, Briegel A, Jensen GJ. Structural diversity of bacterial flagellar motors. The EMBO Journal. 2011;30:2972–2981. doi: 10.1038/emboj.2011.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevance FFV, Hughes KT. Coordinating assembly of a bacterial macromolecular machine. Nature Reviews Microbiology. 2008;6:455–465. doi: 10.1038/nrmicro1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delalez NJ, Berry RM, Armitage JP. Stoichiometry and Turnover of the Bacterial Flagellar Switch Protein FliN. mBio. 2014;5:e01216. doi: 10.1128/mBio.01216-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Roberto RB, Peisajovich SG. The role of domain shuffling in the evolution of signaling networks. Journal of Experimental Zoology Part B, Molecular and Developmental Evolution. 2014;322:65–72. doi: 10.1002/jez.b.22551. [DOI] [PubMed] [Google Scholar]
- Francis NR, Irikura VM, Yamaguchi S, DeRosier DJ, Macnab RM. Localization of the Salmonella typhimurium flagellar switch protein FliG to the cytoplasmic M-ring face of the basal body. Proc Natl Acad Sci USA. 1992;89:6304–6308. doi: 10.1073/pnas.89.14.6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis NR, Sosinsky GE, Thomas D, DeRosier DJ. Isolation, characterization and structure of bacterial flagellar motors containing the switch complex. Journal of Molecular Biology. 1994;235:1261–70. doi: 10.1006/jmbi.1994.1079. [DOI] [PubMed] [Google Scholar]
- Fukuoka H, Inoue Y, Terasawa S, Takahashi H, Ishijima A. Exchange of rotor components in functioning bacterial flagellar motor. Biochemical and Biophysical Research Comm. 2010;394:130–135. doi: 10.1016/j.bbrc.2010.02.129. [DOI] [PubMed] [Google Scholar]
- Grünenfelder B, Gehrig S, Jenal U. Role of the cytoplasmic C terminus of the FliF motor protein in flagellar assembly and rotation. Journal of Bacteriology. 2003;185:1624–1633. doi: 10.1128/JB.185.5.1624-1633.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori M, Tanaka Y, Fukai S, Ishitani R, Nureki O. Crystal structure of the MgtE Mg2+ transporter. Nature. 2007;448:1072–1076. doi: 10.1038/nature06093. [DOI] [PubMed] [Google Scholar]
- Karimova G, Ullmann A, Ladant D. Protein-protein interaction between Bacillus stearothermophilus tyrosyl-tRNA synthetase subdomains revealed by a bacterial two-hybrid system. J Mol Microbial Biotechnol. 2001;3:73–82. [PubMed] [Google Scholar]
- Lam KH, et al. Structural basis of FliG-FliM interaction in Helicobacter pylori. Molecular Microbiology. 2013;88:798–812. doi: 10.1111/mmi.12222. [DOI] [PubMed] [Google Scholar]
- Lee LK, Ginsburg MA, Crovace C, Donohoe M, Stock D. Structure of the torque ring of the flagellar motor and the molecular basis for rotational switching. Nature. 2010;466:996–1000. doi: 10.1038/nature09300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lele PP, Branch RW, Nathan VSJ, Berg HC. Mechanism for adaptive remodeling of the bacterial flagellar switch. Proc Natl Acad Sci USA. 2012;109:20018–20022. doi: 10.1073/pnas.1212327109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson R, Zhou H, Dahlquist FW. Structural insights into the interaction between the bacterial flagellar motor proteins FliF and FliG. Biochemistry. 2012;51:5052–5060. doi: 10.1021/bi3004582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipfert J, Doniach S. Small-Angle X-Ray Scattering from RNA, Proteins, and Protein Complexes. Annual Review of Biophysics and Biomolecular Structure. 2007;36:307–327. doi: 10.1146/annurev.biophys.36.040306.132655. [DOI] [PubMed] [Google Scholar]
- Lloyd SA, Blair DF. Charged residues of the rotor protein FliG essential for torque generation in the flagellar motor of Escherichia coli. Journal of Molecular Biology. 1997;266:733–744. doi: 10.1006/jmbi.1996.0836. [DOI] [PubMed] [Google Scholar]
- Lloyd SA, Tang H, Wang X, Billings S, Blair DF. Torque generation in the flagellar motor of Escherichia coli: evidence of a direct role for FliG but not for FliM or FliN. Journal of Bacteriology. 1996;178:223–231. doi: 10.1128/jb.178.1.223-231.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macnab RM. How Bacteria Assemble Flagella. Annual Review of Microbiology. 2003;57:77–100. doi: 10.1146/annurev.micro.57.030502.090832. [DOI] [PubMed] [Google Scholar]
- Manson MD. How 34 pegs fit into 26 + 8 holes in the flagellar motor. Journal of Bacteriology. 2007;189:291–293. doi: 10.1128/JB.01556-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH. Experiments in molecular genetics: Assay of β-Galactosidase. Cold Spring Harbor Laboratory press; Cold Spring Harbor NY: 1972. pp. 352–355. [Google Scholar]
- Minamino T, Imada K. The bacterial flagellar motor and its structural diversity. Trends in Microbiology. 2015;23:267–274. doi: 10.1016/j.tim.2014.12.011. [DOI] [PubMed] [Google Scholar]
- Minamino T, Imada K, Kinoshita M, Nakamura S, Morimoto YV, Namba K. Structural Insight into the Rotational Switching Mechanism of the Bacterial Flagellar Motor. PLoS Biology. 2011;9:e1000616. doi: 10.1371/journal.pbio.1000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamino T, Imada K, Namba K. Molecular motors of the bacterial flagella. Current Opinion in Structural Biology. 2008;18:693–701. doi: 10.1016/j.sbi.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Ogawa R, Abe-Yoshizumi R, Kishi T, Homma M, Kojima S. Interaction of the C-terminal tail of FliF with FliG from the Na+-driven flagellar motor of Vibrio alginolyticus. Journal of Bacteriology. 2015;197:63–72. doi: 10.1128/JB.02271-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul K, Gonzalez-Bonet G, Bilwes AM, Crane BR, Blair D. Architecture of the flagellar rotor. The EMBO Journal. 2011;30:2962–2971. doi: 10.1038/emboj.2011.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar MK, Paul K, Blair DF. Subunit organization and reversal-associated movements in the flagellar switch of Escherichia coli. Journal of Biological Chemistry. 2010;285:675–684. doi: 10.1074/jbc.M109.068676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sircar R, Borbat PP, Lynch MJ, Bhatnagar J, Beyersdorf MS, Halkides CJ, Crane BR. Assembly States of FliM and FliG within the Flagellar Switch Complex. Journal of Molecular Biology. 2015;427:867–886. doi: 10.1016/j.jmb.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa Y, Berry RM. Bacterial flagellar motor. Quarterly Reviews of Biophysics. 2008;41:103–132. doi: 10.1017/S0033583508004691. [DOI] [PubMed] [Google Scholar]
- Thomas DR, Francis NR, Xu C, DeRosier DJ. The three-dimensional structure of the flagellar rotor from a clockwise-locked mutant of Salmonella enterica serovar Typhimurium. Journal of Bacteriology. 2006;188:7039–48. doi: 10.1128/JB.00552-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DR, Morgan DG, DeRosier DJ. Rotational symmetry of the C ring and a mechanism for the flagellar rotary motor. Proc Natl Acad Sci USA. 1999;96:10134–10139. doi: 10.1073/pnas.96.18.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Morgan DG, DeRosier DJ. Structures of bacterial flagellar motors from two FliF-FliG gene fusion mutants. Journal of Bacteriology. 2001;183:6404–6412. doi: 10.1128/JB.183.21.6404-6412.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian AS, Paz A, Fortgang EA, Abramson J, Dahlquist FW. Structure of flagellar motor proteins in complex allows for insights into motor structure and switching. Journal of Biological Chemistry. 2012;287:35779–35783. doi: 10.1074/jbc.C112.378380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing S, Wallmeroth N, Berendzen KW, Grefen C. Techniques for the Analysis of Protein-Protein Interactions in Vivo. Plant Physiology. 2016;171:727–758. doi: 10.1104/pp.16.00470. [DOI] [PMC free article] [PubMed] [Google Scholar]