

Abstract
The NO synthases (NOS) catalyze a two-step oxidation of L-Arginine (Arg) to generate NO. In the first step, O2 activation involves one electron being provided to the heme by an enzyme-bound 6R-tetrahydro-L-biopterin cofactor (H4B), which then forms a H4B radical that must be reduced back to H4B in order for NOS to continue catalysis. Although an NADPH-derived electron is used to reduce the H4B radical, how this occurs is unknown. We hypothesized that the NOS flavoprotein domain might reduce the H4B radical by utilizing the NOS heme porphyrin as a conduit to deliver the electron. This model predicts that factors influencing NOS heme reduction should also influence the extent and rate of H4B radical reduction in kind. To test this, we utilized single catalytic turnover and stop-freeze methods, along with electron paramagnetic resonance spectroscopy, to measure the rate and extent of reduction of the 5-methyl-H4B radical formed in neuronal NOS (nNOS) as a consequence of its catalyzing Arg hydroxylation. We used several nNOS variants that supported either a slower or faster than normal rate of ferric heme reduction. We found that the rates and extents of nNOS heme reduction correlated well with the rates and extents of 5-methyl-H4B radical reduction among the various nNOS enzymes. This supports a model where the heme porphyrin transfers an electron from the NOS flavoprotein to the H4B radical formed during catalysis, revealing that the heme plays a dual role in catalyzing O2 activation or electron transfer at distinct points in the reaction cycle.
Keywords: nNOS, Fast reaction kinetics, heme reduction, Electron transfer, Biopterin, 5-Methbiopterin, Freeze quench, EPR
Graphical Abstract
An H4B radical forms in NOS during NO synthesis and must be reduced to continue. Using NOS variants, we found the rates and extents of their heme reduction correlate with their H4B radical reduction. Thus, the NOS heme (red) may act like a wire to pass an electron from the FMN (gold) to the H4B radical (green) during catalysis.
INTRODUCTION
Nitric Oxide Synthases (NOSs3, EC 1.14.13.39) catalyze a stepwise oxidation of L-Arginine (Arg) to form nitric oxide (NO) and L-citrulline, with N-hydroxy-L-Arg (NOHA) forming as a bound intermediate [1–3] (Fig. 1). Catalysis takes place within a heme-containing oxygenase domain that is attached to a flavoprotein domain by an intervening hinge and adjacent calmodulin (CaM) binding motif. The flavoprotein domain contains FMN and FAD, binds NADPH, and provides electrons to the heme for dioxygen activation.
Fig. 1. Reaction catalyzed by NOS Enzymes.

The reaction involves an initial hydroxylation of Arg, leading to the formation of Nω-hydroxy-L-arginine (NOHA), followed by oxidation of NOHA to form L-citrulline and NO. The reaction consumes 1.5 mol of NADPH, and 2 mol of dioxygen per mol of L-citrulline and NO formed [51].
Although NOS activates dioxygen like in the cytochromes P450 [4], some aspects of the process are unique to NOS enzymes. For example, the electron transfer (ET) from the NOS FMN domain to the heme is CaM-dependent, and CaM must be in its Ca2+-replete state to bind to the NOS [1;2]. This links NOS heme reduction and NO synthesis to Ca2+ signaling cascades [1;2]. The NOS enzymes are also unique in their utilizing 6R-tetrahydro-L-biopterin (H4B) as a cofactor. The H4B remains bound in NOS through multiple catalytic turnovers [5–9], and functions to reduce the heme ferrous-dioxy intermediate (FeIIIO2−) that forms during catalysis in the NOS oxygenase domain [10–14]. This one-electron transfer from H4B forms a bound H4B radical, and is critical because it enables NOS to form the heme-oxy species that reacts with Arg, resulting in its hydroxylation to NOHA [1–3] (Fig. 2).
Fig. 2. Electron transfer steps and the possible uncoupling events that occur during Arg hydroxylation to NOHA by NOS.

The solid boxes indicate detected NOS heme species that form in the reaction, and contain the substrate species and H4B redox species (red) that are bound within the enzyme at each step. The dashed blue arrows indicate three uncoupling events that compete kinetically with the productive steps: Decay of the heme-dioxy species to form superoxide and ferric heme, NOHA dissociation from the enzyme, and oxidation of the H4B radical to inactive H2B. See text for details.
After the H4B radical forms within NOS it must be reduced back to H4B in order for the enzyme to continue catalysis [1–3] (Fig. 2). However, the H4B radical can also undergo a non-enzymatic, time-dependent oxidation to dihydrobiopterin (H2B) [10;13;14] (Fig. 2), which cannot redox cycle in NOS, and therefore places a time constraint on the H4B radical reduction step. In a previous study, we found that the H4B radical that forms in neuronal NOS (nNOS) during Arg hydroxylation is reduced back to H4B by an electron provided by the attached flavoprotein domain, in a CaM-dependent process [15]. This finding satisfied earlier NADPH stoichiometry studies [16], and revealed that H4B radical reduction in NOS is similar to its ferric heme reduction because it has the same flavoprotein electron source and dependence on bound CaM.
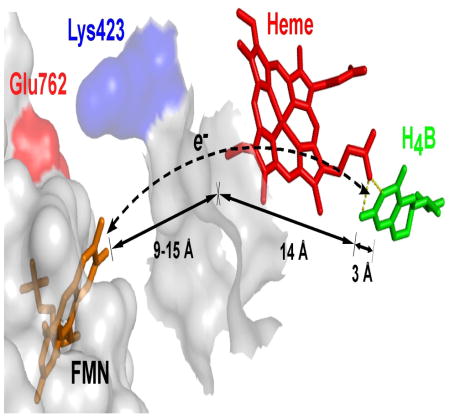
Related work suggested a structural barrier may exist for H4B radical reduction in NOS enzymes. Specifically, model studies [15–19] of docked FMN domain-oxygenase domain complexes indicated that while the FMN cofactor could approach the bound heme within an edge-to-edge distance of ≤ 18 Å, which is amenable for rapid ET, the closest approach of the FMN to bound H4B would be 26 to 32 Å, a distance that is not amenable for ET [20]. This led us to propose that the FMN domain of NOS might utilize a through-heme ET pathway to achieve reduction of the H4B radical [15]. It follows that if a through-heme ET pathway is operative (Fig. 3), then the structural and electronic features that control NOS ferric heme reduction should also control the rate and extent of H4B radical reduction.
Fig. 3. Proposed through-heme pathway for the reduction of H4B radical in NOS.

The cartoon shows a portion of the FMN domain (semi-transparent white with an orange-bound FMN) docked onto a portion of the NOSoxy domain (semi-transparent white). The bound heme (red) in NOSoxy has an edge positioned near the proposed docking site for the FMN domain, and docking is possibly facilitated by electrostatic interaction of surface residues Glu762 and Lys423. The bound H4B (green) in NOSoxy is at least 17 Å away from the surface. A through-heme electron transfer pathway from FMN to H4B (dashed line) is indicated, along with the relevant distances marked by arrows. Adapted from Ref. [15].
To test this hypothesis, we utilized a collection of nNOS and CaM variants that support rates of ferric heme reduction that are slower or faster than in wild-type nNOS, and then studied and compared their rates and extents of 5-methyl-H4B radical reduction following catalysis of Arg hydroxylation in a single turnover setting. Our findings provide experimental evidence in support of a through-heme ET pathway for H4B radical reduction during NOS catalysis.
RESULTS
To test if facets that regulate nNOS heme reduction also regulate H4B radical reduction, we utilized combinations of nNOS or CaM mutants that we expected either to have heme reduction blocked or to have heme reduction rates that are slower or faster than in native CaM-bound nNOS. We measured the heme reduction rates of the various nNOS proteins under anaerobic conditions by following the build up of their ferrous heme-CO complex after mixing each ferric enzyme with excess NADPH, as done previously [19;21–24]. Table 1 lists the various nNOS and CaM protein combinations that we used and their measured rates of heme reduction. As expected, CaM-free nNOS had no measureable heme reduction while CaM-bound nNOS had a normal heme reduction rate [19;21;23;24]. The heme reduction in CaM-bound nNOS was entirely blocked by incorporating charge reversal substitutions (Glu to Arg) at three residues (E762, E816, E819) that are located on the surface of the FMN domain and are known to be important for supporting heme reduction in nNOS [22;24]. Heme reduction was slowed in CaM-bound nNOS by using two different established strategies: Incorporating a charge reversal mutation at Lys423, which is located on the surface of the nNOSoxy domain and is thought to help dock the FMN domain during its ET to the heme [19]; or by using the CaM Glu47Ala mutant, which is known to support a slower heme reduction in nNOS compared to native CaM [25]. We increased the rate of heme reduction above that in wild-type nNOS by simultaneously incorporating three changes that are each known to support a faster heme reduction: A charge-neutralizing point mutation of a nNOSoxy domain surface residue (Lys452Asn) [19], a two-residue deletion (Δ2) in the linker element that connects the FMN and FAD domains in NOS enzymes [23], and a charge reversal point mutation in the C-terminal tail of the reductase domain (Arg1400Glu) [26]. We found that all of the nNOS proteins that showed measurable heme reduction rates also catalyzed NO synthesis from Arg (data not shown). These findings indicate that the proteins and circumstances described here would be useful for investigating connections between heme reduction and H4B radical reduction in nNOS.
Table 1. Rate of NO synthesis, heme reduction and radical disappearance in nNOS proteins.
Rates of NO synthesis were measured at 25 °C as described under “Experimental Procedures”. Values (min−1) represent the mean ± S.D. of three independent measurements with two preparations of each enzyme. WT, wild-type. Heme reduction rates were measured at 10 °C, See text for details.
| Enzyme | Conditions | NO Synthesis Rate (min−1) | Heme Reduction Rate (s−1) | Radical Disappearance Rate (s−1) |
|---|---|---|---|---|
| WT nNOS | Without CaM | 0 | 0 | <0.1 |
| WT nNOS | + WT CaM | 38 ± 3 | 5.9 ± 0.3 | 1.2 |
| WT nNOS | + E47A CaM | 32 ± 1 | 2.6 ± 0.5 | 0.64 |
| K423E nNOS | + WT CaM | 0.14 ± 0.05 | 0.48 ± 0.1 | <0.1 |
| Δ2-R1400E_K452N nNOS | + WT CaM | 11 ± 1 | 8.1 ± 0.5 | 2.1 |
| E762R/E816R/E819R nNOS | + WT CaM | 0 | 0 | <0.1 |
We next utilized a freeze-quench technique to follow the build up and disappearance of the 5MeH4B radical during single-turnover Arg hydroxylation reactions catalyzed by the various nNOS proteins noted above. We specifically used 5MeH4B in place of H4B in these experiments because it has a faster radical formation rate (approximately 51 s−1) and a slower rate of oxidative decay (approximately 0.2 s−1) in nNOS compared to the native H4B [13], and thus provides a wider time window to observe catalytic reduction of the bound pterin radical following Arg hydroxylation. The 5MeH4B radical also has a characteristic hyperfine structure that helps to distinguish its EPR signal from the signals arising from the flavin or superoxide radical species that also may form in nNOS during the reaction [27–29]. Otherwise, it is established that 5MeH4B closely mimics H4B in where it binds in NOS [29;30] and in its ability to support a normal rate of NO synthesis [12;31;32].
The single turnover reactions were initiated by rapidly mixing an O2 saturated buffer with anaerobic solutions of each NADPH pre-reduced, CaM-bound or CaM-free ferrous nNOS protein that contained saturating concentrations of Arg and 5MeH4B. Under this condition, O2 binding to the ferrous heme is rapid compared to all subsequent reaction steps, including the ET from 5MeH4B to the ferrous heme-dioxy species, as noted in our previous single-turnover reaction studies of nNOS [14;29;33] (Fig, 2). Following mixing, the reaction solutions were aged for different times within the instrument before undergoing rapid freezing, and the frozen samples had their 5MeH4B free radical signal quantified by EPR.
Fig. 4A illustrates the build up and disappearance of the 5MeH4B radical signal in each nNOS protein during the single turnover Arg hydroxylation reactions described above. The lines of best fit were derived according to an A to B to C kinetic model [13;15;33], and the fitted rates of 5MeH4B radical disappearance for each nNOS protein are listed in Table 1. All of the nNOS proteins showed a peak build up of 5MeH4B radical signal within 500 ms after the reactions were initiated, consistent with previous results [15] (Fig. 2). Subsequently, we observed different rates of 5MeH4B radical disappearance among the proteins. There were slow rates of radical disappearance in nNOS proteins that had either none or very slow heme reduction, specifically in CaM-free nNOS, and the CaM-bound forms of the E762R/E816R/E819R nNOS and the K423E nNOS. In comparison, the radical disappearance rates were faster in nNOS proteins that had faster heme reduction rates. A plot of the data in Fig. 4B shows that a correlation exists between the rates of heme reduction and the rates of 5MeH4B radical disappearance across the group of nNOS proteins (r2 = 0.97).
Fig. 4. Kinetics of 5MeH4B formation and disappearance and the relationship to the heme reduction rate.

(A) The 5MeH4B radical content in the various nNOS enzymes versus time during their Arg hydroxylation reactions that were run at 10 °C under conditions as indicated in the inset box. Double Mutant nNOS is the Δ2-R1400E_K452N nNOS and Trip Mut nNOS is the E762R/E816R/E819R nNOS. The solid lines are the best fit derived using a kinetic equation of A → B → C. Data are the mean and S.D. of three individual experiments each. (B) The rates of 5MeH4B radical disappearance are plotted against the corresponding rates of heme reduction for the group of nNOS proteins, with the colored point identifications as noted in panel A. The dashed line of best fit and the correlation coefficient are shown.
In NOS enzymes, the H4B (or 5MeH4B) radicals that form during catalysis can disappear by two pathways: A reductive pathway whereby the NOS flavoprotein domain provides an electron to reduce the radical and regenerate the tetrahydro species (5MeH4B or H4B), or an oxidative pathway that is non-enzymatic and generates the dihydro species (5MeH2B or H2B), which cannot redox cycle within NOS enzymes (Fig. 2). The relative rates of these two processes will determine the amount of reduced versus oxidized cofactor that is left in a NOS at the end of its single turnover Arg hydroxylation reaction. We therefore measured the final oxidation state of the bound H4B cofactor in each nNOS protein after letting their Arg hydroxylation reactions go to completion. In this circumstance, we utilized the native H4B cofactor because there is no advantage to using 5MeH4B in its place, as there was in the kinetic studies above. The percentage of oxidized (H2B) or reduced (H4B) cofactor that was present in each enzyme is listed in Table 2. The data show that CaM-free nNOS primarily contained H2B (approximately 80% of total) at the end of its Arg hydroxylation reaction, while the CaM-bound nNOS primarily contained H4B (approximately 80% of total), confirming our previous results that showed reduction of the H4B radical in nNOS requires that CaM be bound [15]. For the other nNOS proteins, the percentages of H2B and H4B at the end of their reactions also varied in a reciprocal manner, from approximately 20% to 96%. A plot of the percentages versus the heme reduction rates (Fig. 5) shows that the mechanism of H4B radical disappearance shifted from the oxidative pathway to the reductive pathway as the heme reduction rate increased, in an approximate linear relationship (r2 = 0.98).
Table 2. Fate of the H4B radical following its formation in the Arg hydroxylation reaction.
The percentage of oxidized (H2B) or reduced (H4B) cofactor that was present in each NOS enzyme at the end of their Arg hydroxylation reaction was determined. Values represent the mean ± S.D. of at least three independent measurements of each enzyme. WT, wild-type.
| Enzyme | Conditions | % of H4B | % of H2B |
|---|---|---|---|
| WT nNOS | Without CaM | 15 ± 2 | 82 ± 4 |
| WT nNOS | +WT CaM | 65 ± 9 | 32 ± 6 |
| WT nNOS | +E47A CaM | 40 ± 6 | 55 ± 9 |
| K423E nNOS | + WT CaM | 20 ± 5 | 76 ± 2 |
| Δ2-R1400E_K452N nNOS | + WT CaM | 97 ± 6 | 5 ± 2 |
| E762R/E816R/E819R nNOS | + WT CaM | 18 ± 8 | 78 ± 8 |
Fig. 5. The influence of the nNOS heme reduction rate on H4B radical processing after Arg hydroxylation.

The measured percentages of H2B and H4B that were present in each nNOS protein at the end of its Arg hydroxylation reaction are plotted as a function of the corresponding rates of heme reduction. The colored point identifications are as listed in panel Fig. 4, panel A. The lines of best fit and the correlation coefficients are shown.
DISCUSSION
NOS enzymes form a H4B (or 5MeH4B) radical during their Arg hydroxylation reaction that must be transformed back to the fully-reduced cofactor before NO synthesis can continue. Prior work indicated that the electron used to reduce the 5MeH4B radical comes from the NOS flavoprotein domain [15], and protein structural models that bring the NOS FMN and oxygenase domains in contact revealed that the FMN-to-5MeH4B cofactor distance was too great for direct ET to take place between them. This led us to propose that a through-heme ET pathway (FMN to heme to radical) might operate to reduce the 5MeH4B radical [15], but this was never tested. Here, we used a group of nNOS and CaM variants to test the model, reasoning that if a through-heme ET was operative, then the extent and speed of 5MeH4B radical reduction in a given nNOS enzyme should be similarly impacted by protein structural changes that impact its heme reduction. The results showed that the rates of 5MeH4B radical reduction, and the extents of H4B radical reduction, both correlate with the rates and extents of heme reduction across the group.
For interpreting these results, it is important to note that we altered heme reduction in our nNOS proteins in different ways, by incorporating a variety of site-directed mutations into nNOS or into CaM. Although all these mutations ultimately impact ET from the FMN domain to the nNOS heme, they do so by different mechanisms that alter functions of different regions in nNOS. Briefly, the charge reversal mutations that we incorporated on the FMN domain surface (triple mutant) or on the oxygenase domain surface (K423E) are each thought to antagonize charge pairing interactions that likely form within the interface of the transient FMN-NOSoxy domain complex to help direct ET from FMN to heme [19;24;34]. Accordingly, all these charge reversals inhibited heme reduction in nNOS. Incorporating the E47A mutation in CaM also slowed nNOS heme reduction but by a mechanism that is independent of alteration within nNOS itself. The E47A CaM mutation was designed to partly eliminate a salt bridge interaction that forms between the NOS FMN domain and bound CaM [35], which diminishes nNOS heme reduction [25] presumably by influencing conformational behaviors that promote ET from the FMN to the heme. In contrast, the charge reversal mutation that we incorporated into the C-terminal regulatory element of nNOS (R1400E) increases the nNOS heme reduction rate [26] because in this case the charge reversal antagonizes a charge pairing interaction between Arg1400 and bound NADPH that is needed in order for the C-terminal regulatory element to fulfill its repressive function on FMN domain conformational behaviors [26]. Likewise, the two-residue deletion that we made in the FAD-FMN domain linker element supports an increased heme reduction rate in nNOS [23]. Notably, 4 of the 6 protein changes that alter the nNOS heme reduction rate are located either in the nNOS flavoprotein domain, which is physically separated from the oxygenase domain where the 5MeH4B and heme is bound, or are located in a different protein altogether (i.e., E47A CaM). This means that their observed impacts on the rate of 5MeH4B radical reduction, and on the extent of H4B radical reduction, cannot be due to their having direct effects on the bound heme or on the bound 5MeH4B or H4B radicals, and this concept in turn helps to simplify and strengthen our interpretation of the results.
In general, slowing down H4B radical reduction in NOS has several consequences for catalysis. First, as demonstrated here, it will result in a greater proportion of the bound H4B radical becoming oxidized to H2B during turnover, which means that a greater proportion of the NOS enzyme would have to dissociate its H2B cofactor and bind a H4B molecule from solution in its place to continue catalysis. The time required for this process depends on the relative solution concentrations of H4B and H2B, which do vary in biological settings as a consequence of oxidative stress [36;37], and together with an inherently slower H4B radical reduction rate, would increase the probability of competing unproductive processes occurring in the NOS enzyme (see Fig. 2). Specifically, these include the dissociation of the NOS ferrous heme-O2 intermediate to generate ferric enzyme and superoxide, which occurs in nNOS at rates ranging from 0.1 to 20 s−1 depending on conditions [38;39], and the dissociation of the bound NOHA reaction intermediate from NOS [16;40–42]. Both events would diminish NO synthesis and make the NOS enzyme become more uncoupled with regard to its ratio of NADPH oxidized per NO formed. Interestingly, the mammalian NOS enzyme with the slowest heme reduction rate is endothelial NOS [43], and it is known to be uncoupled and highly dependent on the cellular H4B to H2B concentration ratio [44]. It would be interesting to examine if these behaviors are due to endothelial NOS having an inherently slower H4B radical reduction rate and thus poor H4B radical redox cycling, as we saw occur in our nNOS variants that have slower than normal heme reduction rates. This topic can now be addressed in future studies.
In summary, we found that a link exists between the rate and extent of ferric heme reduction and the rate and extent of 5MeH4B and H4B radical reduction in nNOS, suggesting that that the processes are regulated by the same mechanisms. When one takes into account the different geometric constraints for ET from FMN to the NOS heme versus to its bound H4B radical, our results can best be explained by a through-heme ET pathway for H4B radical reduction, which because of their structural similarity is likely to operate in all three mammalian NOS. Mechanistically, it is remarkable to consider that the heme porphyrin ring in NOS enzymes may enable ET to its iron for O2 binding, or enable ET to the H4B radical, depending on the redox state of the bound H4B cofactor, which in turn depends on where the enzyme is in the catalytic cycle. This circumstance raises interesting new questions that can now be addressed.
EXPERIMENTAL PROCEDURES
Chemicals
H4B, H2B, and 5-methyl-H4B (5MeH4B) were obtained from Dr. Schirck’s laboratory (Jona, Switzerland). A 5MeH4B stock solution was prepared fresh in 40 mm EPPS, pH 7.6 in the presence of either ascorbic Acid or DTT. All other chemicals were obtained either from Sigma or Fisher Scientific International, Inc.
General Methods and Materials
Absorption spectra and steady-state kinetic data were obtained using a Shimadzu UV-2401PC spectrophotometer. All plots and some additional curve-fitting were done using Origin® 8.0 (OriginLab, Northampton, MA). All experiments were repeated two or more times with at least two independently prepared batches of proteins to ensure consistent reproducibility of the results. Data were analyzed and are expressed as mean ± S.D.
Protein Purification
Rat nNOS proteins were overexpressed in Escherichia coli and purified in the absence of H4B as described previously [45;46].The ferrous heme-CO adduct absorbing at 444 nm was used to measure hemeprotein content with an extinction coefficient of ε444 = 74 mM−1 cm−1 (A444 – A500) [47]. The individual nNOSoxy and flavoprotein domains were overexpressed and purified as described previously [46;48]. The concentrated proteins were stored at −80 °C in a buffer containing 40 mM EPPS, pH 7.6, containing 10% glycerol and 0.15 M NaCl.
Sample Preparation for the Stop-Freeze Experiments
Reduction of ferric enzymes to ferrous was done as follows, with a slight modification of the previously reported methods [33;46]. Both wild-type and mutant nNOS proteins (with either bound wild-type CaM / mutant CaM / CaM-free) were reduced by careful titration using sodium dithionite in a gastight cuvette under anaerobic conditions. Briefly, a buffered solution containing ~120 μM nNOS protein, 10 mM Arg, 1 mM 5MeH4B (and an additional 200 μM CaM, and 600 μM Ca2+ for studies involving CaM bound enzymes) was made anaerobic by several cycles of vacuum and flushing with deoxygenated N2, and then was titrated using deoxygenated and N2 purged sodium dithionite solution. Heme reduction was monitored by the appearance of a 550-nm absorbance peak and the disappearance of the 650-nm absorbance peak. The samples were periodically scanned in the UV-visible spectrophotometer to assure that the ferrous heme did not oxidize to ferric heme prior to transfer to the rapid quench instrument.
Rapid-freeze Kinetic Experiments
Ferrous nNOS samples prepared as described above [33] were transferred with an anaerobic syringe to a rapid quench instrument (RQF-63, TGK Scientific, Bradford on Avon, UK) maintained at 10 °C, and the samples were rapid-mixed with an O2-saturated buffer (40 mM EPPS, 10% glycerol, 150 mM NaCl, pH 7.6) to initiate the reaction (this resulted in a post-mix O2 concentration in the reaction of <0.8 mM). The reaction mixture was then aged for various times in the instrument followed by rapid injection onto the wall of a Stainless Steel funnel attached to an EPR tube, all bathed in a liquid N2/isopentane freezing solution as previously reported [15;33]. The frozen samples were carefully packed into the EPR tubes, surrounded by the liquid N2/isopentane freezing solution, and then stored in liquid N2 until analysis. All experiments were repeated three times with at least two independently prepared batches of proteins to ensure consistent reproducibility of the results.
Electron paramagnetic resonance (EPR) analysis
EPR spectra of each frozen reaction sample was recorded in a Bruker EMX instrument equipped with an Hewlett-Packard 5352B microwave power controller. Temperature was maintained constant at 150 K using continuous-flow of Pre-cooled nitrogen gas and a temperature controller. Data acquisition for all samples were averaged out of 10 scans and the spectra were recorded at 150 K using a microwave power of 2 milliwatts or 20 microwatts, a frequency of 9.5 GHz, a modulation amplitude of 1 G, and a modulation frequency of 100 kHz. The EPR spectrum of the 5MeH4B radical was obtained by mixing 5MeH4B-bound ferrous nNOSoxy with O2-saturated buffer and then rapid freezing after 400 ms of reaction [14]. The EPR spectrum of the flavin semiquinone radical (FSQ) was obtained for the nNOS flavoprotein domain by adding a slight molar excess of NADPH in air-saturated buffer and then waiting for the flavins to air oxidize to get an air-stable FMN semiquinone radical [28]. The 5MeH4B and FSQ radicals saturate at different microwave power, with P1/2, the power at half-saturation values, being 38mW and 65 microwatts, for the 5MeH4B and FSQ radicals respectively [15]. At 2 mW the FSQ radical is largely saturated and no longer responsive to power, whereas the 5MeH4B radical is not saturated at 2 mW. The EPR intensities contributed by the 5MeH4B and FSQ radical in each nNOS protein reaction sample were calculated using the following equations:
where DI(2 mW, t) and DI(20 μ, t) are the double integrations of EPR spectra of the same sample measured at 2 milliwatts and 20 microwatts power at time t, respectively. I5MeH4B, t and IFSQ, t are only the two variables representing the intensities of 5MeH4B and FSQ radicals at 2 milliwatts. The coefficients of 0.1132 and 0.4490 were determined from power saturation measurements for 5MeH4B radical and FSQ radical, indicating the fold intensity dropped from 2 milliwatts to 20 microwatts. Solving the above 2 equations gives the following equations to calculate the I5MeH4B and IFSQ.
The EPR data for 5MeH4B radical formation and disappearance was analyzed as described in detail previously [15]. Data were fitted using the sequential kinetic reaction model, A goes to B goes to C [33], where A is the beginning 5MeH4B species, B is the radical species, and C is the sum of the radical species that has become reduced (i.e., 5MeH4B), or oxidized (5MeH2B), to obtain rates of 5MeH4B radical formation and disappearance, and the radical concentration versus time.
NO Synthesis Assay
The initial rate of NO synthesis for wild type or mutant nNOS enzymes was measured at 25 °C using the oxyhemoglobin assay for NO [23;49]. The nNOS proteins (0.1–0.2 μM) were added to a cuvette containing 40 mM EPPS, pH 7.6, containing 2 μM CaM, 0.6 mM CaCl2, 0.3 mM DTT, 5 mM Arg, 4 μM each of FAD, FMN and H4B, 100 units/ml catalase, and 10 μM oxyhemoglobin to give a final volume of 0.4 ml. The reaction was started by adding NADPH to give 0.2 mM. The NO-mediated conversion of oxyhemoglobin to methemoglobin was monitored over time as an absorbance increase at 401 nm and quantified using an extinction coefficient of 38 mM−1cm−1.
Heme Reduction Measurements
The kinetics of ferric heme reduction in the nNOS enzymes was analyzed at 10 °C as described previously [19;21–24] using a stopped-flow apparatus and diode array detector (TGK Scientific KinetAsyst SF-61DX2) equipped for anaerobic analysis. Heme reduction was monitored by formation of the ferrous-CO complex and the kinetics determined by fitting traces of absorbance change versus time at 444 nm. A protein solution containing 10 μM nNOS, 100 mM EPPS, pH 7.6, 100 mM NaCl, 10 μM H4B, 2 mM L-Arg, 0.5 mM DTT, 100 μM CaM (WT or mutant depending on the experiment), 5 mM CaCl2, and 1 mM EDTA, at least half CO saturated, was mixed with an anaerobic, CO-saturated solution containing 100 mM EPPS, pH 7.6, 100 mM NaCl, and 100 μM NADPH. Heme reduction was followed by the formation of the ferrous-CO complex with maximum around 444 nm.
Redox State of Bound Biopterin
The oxidation state of the bound biopterin in wild-type and mutant nNOS proteins (with wild-type CaM/mutant CaM /CaM-free) following the single catalytic turnover Arg hydroxylation reactions was determined using established methods with some modifications [13;50]. Approximately 20 μM of each H4B-free nNOS protein was incubated with 25 μM H4B and 1 mM Arg at 10 C for ~2 hours. A portion of the sample also received 100 μM CaM and 500 μM CaCl2. The samples were made anaerobic and then reduced to ferrous by titrating with a near stoichiometric amount of dithionite solution. To initiate the single turnover reaction, 200 μL of each anaerobic ferrous nNOS protein sample was mixed at room temperature with 50 μL of O2-saturated buffer (40 mm EPPS, pH 7.5) containing 10 μL of iodine solution (0.9% (w/v) iodine in H2O). After mixing thoroughly and allowing for the completion of the single turnover reaction, 100 μL aliquots were transferred either into a vial containing 10 μL of 1M NaOH and 10 μL alkaline iodine solution (0.9% (w/v) iodine and 1.8% (w/v) KI in 0.1M NaOH) or into a vial containing 10 μL of 1M HCl and 10 μL acidic iodine solution (0.9% (w/v) iodine and 1.8% (w/v) KI in 0.1M HCl). These samples were incubated in the dark for at least 90 min. The sample in alkaline solution was then neutralized by adding 10 μL of 1M HCl, and all of the remaining iodine in all samples was quenched by adding 10 μL of freshly prepared 4% (w/v) ascorbic acid solution. The precipitated protein was removed from each sample by centrifuging at 10,000 X g for 10 min. A 25-μL aliquot from each sample was then injected onto a 4.6 × 250-mm Partisil 5μ ODS-3 column (Microsolv) that was equilibrated with 15 mM potassium phosphate buffer, pH 3.0. A methanol gradient was applied to a maximum of 15% B until 35 min; then within 5 min, the gradient was returned to 0% B and the column was washed with 100% 15mM Potassium Phosphate buffer, pH 3.0. A fluorescence detector was used to detect biopterin products with excitation wavelength set at 350 nm and emission wavelength set at 440 nm. Total biopterin (H4B + H2B) was determined from the area of the peak eluting at 26.5 min that had undergone acidic workup, while oxidized biopterin (H2B) was determined from the area of this same peak from the sample that had undergone basic workup. The amount of pterin remaining as H4B after the reaction was calculated by subtracting the amount of biopterin present in the alkaline solution from the amount of biopterin present in the acidic solution. Any dilution difference between the acidic and basic condition during the work-up was corrected for in the calculation.
Acknowledgments
We thank Deborah Durra from Stuehr lab for technical assistance.
Funding- This work was supported by National Institutes of Health Grants [GM051491] and [HL076491] (to D.J.S.).
Footnotes
Abbreviations used are: NOS, nitric-oxide synthase; NOSoxy, NOS oxygenase domain; NOSred, NOS reductase domain; NOHA, Nω-hydroxyl-L-arginine; H4B, (6R)-5,6,7,8-tetrahydro-L-biopterin; 5MeH4B, (6R)-5-methyl-6,7,8-trihydro-L-biopterin; EPPS, 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid; NADPH, nicotinamide adenine dinucleotide phosphate, reduced form; FSQ, flavin semiquinone; EPR, electron paramagnetic resonance; CaM, calmodulin; ET, electron transfer; nNOS, neuronal NOS; Cpd I, compound I; Cpd II, compound II.
Conflict of interest- The authors declare that they have no conflicts of interest with the contents of this article.
Author Contributions- S.R. designed the study, performed the experiments, analyzed data and prepared the manuscript; M.M.H. carried out the experiments and analyzed data and prepared the manuscript; D.J.S. designed the study, analyzed data and prepared the manuscript. All authors performed critical reading of the manuscript prior to submission and approved the final version of the paper.
References
- 1.Daff S. NO synthase: structures and mechanisms. Nitric Oxide. 2010;23:1–11. doi: 10.1016/j.niox.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Gorren ACF, Mayer B. Nitric-oxide synthase: A cytochrome P450 family foster child. Biochimica et Biophysica Acta-General Subjects. 2007;1770:432–445. doi: 10.1016/j.bbagen.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 3.Wei CC, Crane BR, Stuehr DJ. Tetrahydrobiopterin radical enzymology. Chem Rev. 2003;103:2365–2383. doi: 10.1021/cr0204350. [DOI] [PubMed] [Google Scholar]
- 4.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 5.Giovanelli J, Campos KL, Kaufman S. Tetrahydrobiopterin, A Cofactor for Rat Cerebellar Nitric-Oxide Synthase, Does Not Function As A Reactant in the Oxygenation of Arginine. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:7091–7095. doi: 10.1073/pnas.88.16.7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorren ACF, List BM, Schrammel A, Pitters E, Hemmens B, Werner ER, Schmidt K, Mayer B. Tetrahydrobiopterin-free neuronal nitric oxide synthase: Evidence for two identical highly anticooperative pteridine binding sites. Biochemistry. 1996;35:16735–16745. doi: 10.1021/bi961931j. [DOI] [PubMed] [Google Scholar]
- 7.List BM, Klosch B, Volker C, Gorren ACF, Sessa WC, Werner ER, Kukovetz WR, Schmidt K, Mayer B. Characterization of bovine endothelial nitric oxide synthase as a homodimer with down-regulated uncoupled NADPH oxidase activity: Tetrahydrobiopterin binding kinetics and role of haem in dimerization. Biochemical Journal. 1997;323:159–165. doi: 10.1042/bj3230159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reif A, Frohlich LG, Kotsonis P, Frey A, Bommel HM, Wink DA, Pfleiderer W, Schmidt HHHW. Tetrahydrobiopterin inhibits monomerization and is consumed during catalysis in neuronal NO synthase. Journal of Biological Chemistry. 1999;274:24921–24929. doi: 10.1074/jbc.274.35.24921. [DOI] [PubMed] [Google Scholar]
- 9.Witteveen CFB, Giovanelli J, Kaufman S. Reactivity of tetrahydrobiopterin bound to nitric-oxide synthase. Journal of Biological Chemistry. 1999;274:29755–29762. doi: 10.1074/jbc.274.42.29755. [DOI] [PubMed] [Google Scholar]
- 10.Berka V, Yeh HC, Gao D, Kiran F, Tsai AL. Redox function of tetrahydrobiopterin and effect of L-arginine on oxygen binding in endothelial nitric oxide synthase. Biochemistry. 2004;43:13137–13148. doi: 10.1021/bi049026j. [DOI] [PubMed] [Google Scholar]
- 11.Hurshman AR, Krebs C, Edmondson DE, Marletta MA. Ability of tetrahydrobiopterin analogues to support catalysis by inducible nitric oxide synthase: Formation of a pterin radical is required for enzyme activity. Biochemistry. 2003;42:13287–13303. doi: 10.1021/bi035491p. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt PP, Lange R, Gorren ACF, Werner ER, Mayer B, Andersson KK. Formation of a protonated trihydrohiopterin radical cation in the first reaction cycle of neuronal and endothelial nitric oxide synthase detected by electron paramagnetic: Resonance spectroscopy. Journal of Biological Inorganic Chemistry. 2001;6:151–158. doi: 10.1007/s007750000185. [DOI] [PubMed] [Google Scholar]
- 13.Wei CC, Wang ZQ, Hemann C, Hille R, Stuehr DJ. A tetrahydrobiopterin radical forms and then becomes reduced during Nomega-hydroxyarginine oxidation by nitric-oxide synthase. J Biol Chem. 2003;278:46668–46673. doi: 10.1074/jbc.M307682200. [DOI] [PubMed] [Google Scholar]
- 14.Wei CC, Wang ZQ, Durra D, Hemann C, Hille R, Garcin ED, Getzoff ED, Stuehr DJ. The three nitric-oxide synthases differ in their kinetics of tetrahydrobiopterin radical formation, heme-dioxy reduction, and arginine hydroxylation. J Biol Chem. 2005;280:8929–8935. doi: 10.1074/jbc.M409737200. [DOI] [PubMed] [Google Scholar]
- 15.Wei CC, Wang ZQ, Tejero J, Yang YP, Hemann C, Hille R, Stuehr DJ. Catalytic Reduction of a Tetrahydrobiopterin Radical within Nitric-oxide Synthase. J Biol Chem. 2008;283:11734–11742. doi: 10.1074/jbc.M709250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stuehr DJ, Kwon NS, Nathan CF, Griffith OW, Feldman PL, Wiseman J. N omega-hydroxy-L-arginine is an intermediate in the biosynthesis of nitric oxide from L-arginine. J Biol Chem. 1991;266:6259–6263. [PubMed] [Google Scholar]
- 17.Campbell MG, Smith BC, Potter CS, Carragher B, Marletta MA. Molecular architecture of mammalian nitric oxide synthases. Proc Natl Acad Sci U S A. 2014;111:E3614–E3623. doi: 10.1073/pnas.1413763111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng C, Chen L, Li W, Elmore BO, Fan W, Sun X. Dissecting regulation mechanism of the FMN to heme interdomain electron transfer in nitric oxide synthases. J Inorg Biochem. 2014;130:130–140. doi: 10.1016/j.jinorgbio.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tejero J, Hannibal L, Mustovich A, Stuehr DJ. Surface charges and regulation of FMN to heme electron transfer in nitric-oxide synthase. J Biol Chem. 2010;285:27232–27240. doi: 10.1074/jbc.M110.138842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moser CC, Page CC, Dutton PL. Darwin at the molecular scale: selection and variance in electron tunnelling proteins including cytochrome c oxidase. Philosophical Transactions of the Royal Society B-Biological Sciences. 2006;361:1295–1305. doi: 10.1098/rstb.2006.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adak S, Aulak KS, Stuehr DJ. Chimeras of nitric-oxide synthase types I and III establish fundamental correlates between heme reduction, heme-NO complex formation, and catalytic activity. J Biol Chem. 2001;276:23246–23252. doi: 10.1074/jbc.M102509200. [DOI] [PubMed] [Google Scholar]
- 22.Haque MM, Fadlalla M, Wang ZQ, Ray SS, Panda K, Stuehr DJ. Neutralizing a surface charge on the FMN subdomain increases the activity of neuronal nitric-oxide synthase by enhancing the oxygen reactivity of the enzyme heme-nitric oxide complex. J Biol Chem. 2009;284:19237–19247. doi: 10.1074/jbc.M109.013144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haque MM, Fadlalla MA, Aulak KS, Ghosh A, Durra D, Stuehr DJ. Control of electron transfer and catalysis in neuronal nitric-oxide synthase (nNOS) by a hinge connecting its FMN and FAD-NADPH domains. J Biol Chem. 2012;287:30105–30116. doi: 10.1074/jbc.M112.339697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Panda K, Haque MM, Garcin-Hosfield ED, Durra D, Getzoff ED, Stuehr DJ. Surface charge interactions of the FMN module govern catalysis by nitric-oxide synthase. J Biol Chem. 2006;281:36819–36827. doi: 10.1074/jbc.M606129200. [DOI] [PubMed] [Google Scholar]
- 25.Tejero J, Haque MM, Durra D, Stuehr DJ. A bridging interaction allows calmodulin to activate NO synthase through a bi-modal mechanism. J Biol Chem. 2010;285:25941–25949. doi: 10.1074/jbc.M110.126797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tiso M, Konas DW, Panda K, Garcin ED, Sharma M, Getzoff ED, Stuehr DJ. C-terminal tail residue Arg1400 enables NADPH to regulate electron transfer in neuronal nitric-oxide synthase. J Biol Chem. 2005;280:39208–39219. doi: 10.1074/jbc.M507775200. [DOI] [PubMed] [Google Scholar]
- 27.Berka V, Wu G, Yeh HC, Palmer G, Tsai AL. Three different oxygen-induced radical species in endothelial nitric-oxide synthase oxygenase domain under regulation by L-arginine and tetrahydrobiopterin. J Biol Chem. 2004;279:32243–32251. doi: 10.1074/jbc.M404044200. [DOI] [PubMed] [Google Scholar]
- 28.Galli C, MacArthur R, AbuSoud HM, Clark P, Stuehr DJ, Brudvig GW. EPR spectroscopic characterization of neuronal NO synthase. Biochemistry. 1996;35:2804–2810. doi: 10.1021/bi9520444. [DOI] [PubMed] [Google Scholar]
- 29.Wei CC, Wang ZQ, Arvai AS, Hemann C, Hille R, Getzoff ED, Stuehr DJ. Structure of tetrahydrobiopterin tunes its electron transfer to the heme-dioxy intermediate in nitric oxide synthase. Biochemistry. 2003;42:1969–1977. doi: 10.1021/bi026898h. [DOI] [PubMed] [Google Scholar]
- 30.Crane BR, Arvai AS, Ghosh S, Getzoff ED, Stuehr DJ, Tainer JA. Structures of the N(omega)-hydroxy-L-arginine complex of inducible nitric oxide synthase oxygenase dimer with active and inactive pterins. Biochemistry. 2000;39:4608–4621. doi: 10.1021/bi992409a. [DOI] [PubMed] [Google Scholar]
- 31.Gorren AC, Bec N, Schrammel A, Werner ER, Lange R, Mayer B. Low-temperature optical absorption spectra suggest a redox role for tetrahydrobiopterin in both steps of nitric oxide synthase catalysis. Biochemistry. 2000;39:11763–11770. doi: 10.1021/bi0007775. [DOI] [PubMed] [Google Scholar]
- 32.Gorren ACF, Kungl AJ, Schmidt K, Werner ER, Mayer B. Electrochemistry of pterin cofactors and inhibitors of nitric oxide synthase. Nitric Oxide-Biology and Chemistry. 2001;5:176–186. doi: 10.1006/niox.2001.0332. [DOI] [PubMed] [Google Scholar]
- 33.Wei CC, Wang ZQ, Wang Q, Meade AL, Hemann C, Hille R, Stuehr DJ. Rapid kinetic studies link tetrahydrobiopterin radical formation to heme-dioxy reduction and arginine hydroxylation in inducible nitric-oxide synthase. J Biol Chem. 2001;276:315–319. doi: 10.1074/jbc.M008441200. [DOI] [PubMed] [Google Scholar]
- 34.Shimanuki T, Sato H, Daff S, Sagami I, Shimizu T. Crucial role of Lys(423) in the electron transfer of neuronal nitric-oxide synthase. J Biol Chem. 1999;274:26956–26961. doi: 10.1074/jbc.274.38.26956. [DOI] [PubMed] [Google Scholar]
- 35.Xia C, Misra I, Iyanagi T, Kim JJ. Regulation of interdomain interactions by calmodulin in inducible nitric-oxide synthase. J Biol Chem. 2009;284:30708–30717. doi: 10.1074/jbc.M109.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bendall JK, Douglas G, McNeill E, Channon KM, Crabtree MJ. Tetrahydrobiopterin in cardiovascular health and disease. Antioxid Redox Signal. 2014;20:3040–3077. doi: 10.1089/ars.2013.5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crabtree MJ, Channon KM. Synthesis and recycling of tetrahydrobiopterin in endothelial function and vascular disease. Nitric Oxide. 2011;25:81–88. doi: 10.1016/j.niox.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ost TW, Daff S. Thermodynamic and kinetic analysis of the nitrosyl, carbonyl, and dioxy heme complexes of neuronal nitric-oxide synthase. The roles of substrate and tetrahydrobiopterin in oxygen activation. J Biol Chem. 2005;280:965–973. doi: 10.1074/jbc.M411191200. [DOI] [PubMed] [Google Scholar]
- 39.Stuehr D, Pou S, Rosen GM. Oxygen reduction by nitric-oxide synthases. J Biol Chem. 2001;276:14533–14536. doi: 10.1074/jbc.R100011200. [DOI] [PubMed] [Google Scholar]
- 40.Chenais B, Yapo A, Lepoivre M, Tenu JP. N omega-hydroxy-L-arginine, a reactional intermediate in nitric oxide biosynthesis, induces cytostasis in human and murine tumor cells. Biochem Biophys Res Commun. 1993;196:1558–1565. doi: 10.1006/bbrc.1993.2429. [DOI] [PubMed] [Google Scholar]
- 41.Hecker M, Boese M, Schini-Kerth VB, Mulsch A, Busse R. Characterization of the stable L-arginine-derived relaxing factor released from cytokine-stimulated vascular smooth muscle cells as an NG-hydroxyl-L-arginine-nitric oxide adduct. Proc Natl Acad Sci U S A. 1995;92:4671–4675. doi: 10.1073/pnas.92.10.4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wigand R, Meyer J, Busse R, Hecker M. Increased serum NG-hydroxy-L-arginine in patients with rheumatoid arthritis and systemic lupus erythematosus as an index of an increased nitric oxide synthase activity. Ann Rheum Dis. 1997;56:330–332. doi: 10.1136/ard.56.5.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abu-Soud HM, Ichimori K, Presta A, Stuehr DJ. Electron transfer, oxygen binding, and nitric oxide feedback inhibition in endothelial nitric-oxide synthase. J Biol Chem. 2000;275:17349–17357. doi: 10.1074/jbc.M000050200. [DOI] [PubMed] [Google Scholar]
- 44.Bendall JK, Douglas G, McNeill E, Channon KM, Crabtree MJ. Tetrahydrobiopterin in cardiovascular health and disease. Antioxid Redox Signal. 2014;20:3040–3077. doi: 10.1089/ars.2013.5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adak S, Crooks C, Wang Q, Crane BR, Tainer JA, Getzoff ED, Stuehr DJ. Tryptophan 409 controls the activity of neuronal nitric-oxide synthase by regulating nitric oxide feedback inhibition. J Biol Chem. 1999;274:26907–26911. doi: 10.1074/jbc.274.38.26907. [DOI] [PubMed] [Google Scholar]
- 46.Panda K, Adak S, Aulak KS, Santolini J, McDonald JF, Stuehr DJ. Distinct influence of N-terminal elements on neuronal nitric-oxide synthase structure and catalysis. J Biol Chem. 2003;278:37122–37131. doi: 10.1074/jbc.M304456200. [DOI] [PubMed] [Google Scholar]
- 47.Adak S, Stuehr DJ. A proximal tryptophan in NO synthase controls activity by a novel mechanism. J Inorg Biochem. 2001;83:301–308. doi: 10.1016/s0162-0134(00)00176-8. [DOI] [PubMed] [Google Scholar]
- 48.Konas DW, Takaya N, Sharma M, Stuehr DJ. Role of Asp(1393) in catalysis, flavin reduction, NADP(H) binding, FAD thermodynamics, and regulation of the nNOS flavoprotein. Biochemistry. 2006;45:12596–12609. doi: 10.1021/bi061011t. [DOI] [PubMed] [Google Scholar]
- 49.Tejero J, Biswas A, Haque MM, Wang ZQ, Hemann C, Varnado CL, Novince Z, Hille R, Goodwin DC, Stuehr DJ. Mesohaem substitution reveals how haem electronic properties can influence the kinetic and catalytic parameters of neuronal NO synthase. Biochem J. 2011;433:163–174. doi: 10.1042/BJ20101353. [DOI] [PubMed] [Google Scholar]
- 50.Fukushima T, Nixon JC. Analysis of Reduced Forms of Biopterin in Biological Tissues and Fluids. Analytical Biochemistry. 1980;102:176–188. doi: 10.1016/0003-2697(80)90336-x. [DOI] [PubMed] [Google Scholar]
- 51.Griffith OW, Stuehr DJ. Nitric oxide synthases: properties and catalytic mechanism. Annu Rev Physiol. 1995;57:707–736. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]