

SUMMARY
Congenital abnormalities of the kidney and urinary tract (CAKUT) are one of the leading congenital defects to be identified on prenatal ultrasound. CAKUT represents a broad spectrum of abnormalities, from transient hydronephrosis to severe bilateral renal agenesis. CAKUT is a major contributor to chronic and end stage kidney disease (CKD/ESKD) in children. Both genetic abnormalities and the fetal environment contribute to CAKUT. Monogenic gene mutations identified in human CAKUT have advanced our understanding of molecular mechanisms of renal development. Low nephron number and solitary kidneys are associated with increased risk of adult onset CKD and ESKD. Premature and low birth weight infants represent a high risk population for low nephron number. Additional research is needed to identify modifiable factors to enhance nephron development in premature infants and biomarkers and appropriate follow-up of premature and low birth weight infants into adulthood.
Keywords: Renal development, Prematurity, Congenital abnormalities of the kidney and urinary tract (CAKUT), Fetal ultrasonography, Hydronephrosis, Urinary tract obstruction
1. Introduction
Renal anomalies are a frequent form of congenital abnormality and are often diagnosed prenatally. In addition, perinatal insults and premature birth may impact renal development. Here we review mechanisms of abnormal renal development and discuss the evaluation of fetal and premature infant kidneys.
2. Normal renal development
Human kidney development begins in the first trimester [1]. There are three stages of mammalian kidney development: the pronephros, mesonephros, and metanephros. The pronephros and mesonephros form and then essentially involute [2]. The metanephros develops into the final functional mammalian kidney.
The pronephros consists of simple tubules and forms at three weeks of gestation. Just caudal to the pronephros, the mesonephros forms at four weeks. The mesonephros consists of filtering units (glomeruli with tubules) and as they degenerate the mesonephros forms the mesonephric (or Wolffian) duct. The ureteric bud is an outgrowth of the mesonephric duct that invades the surrounding metanephric mesenchyme during the fifth week of gestation. The ureteric bud undergoes branching, establishing the radial structure of the kidney and the nephron number [2,3]. Signaling from the tips of the branching ureteric bud induces nephron formation, with conversion of metanephric mesenchyme to renal epithelia (renal vesicle). In turn, reciprocal signaling from the metanephric mesenchyme to the ureteric tree stimulates radial renal branching. The ureteric tree forms the renal collecting ducts, pelvis, and ureters. The renal vesicle becomes a comma, then an S-shaped body, and ultimately forms the glomerulus, proximal tubule, loop of Henle, and distal tubules.
The first glomeruli form at 9–10 weeks of gestation [4]. During the late second and third trimester, there is an exponential increase in nephrons between 18 and 32 weeks [1,5]. Nephron development is complete between 32 and 36 weeks [5]. Fetal urine becomes a major contributor to amniotic fluid at about 20 weeks, with production of 300 mL/kg fetal weight/day [6,7].
3. Congenital abnormalities of the kidney and urinary tract (CAKUT)
Congenital abnormalities of the kidney and urinary tract are one of the most frequent major birth defects, representing up to 20–30% of all major birth defects [8]. Whereas the prevalence of abnormalities often depends upon the setting (e.g. tertiary care centers often have a higher prevalence of anomalies), some estimates place the frequency of renal anomalies at about one in 500 live births [9,10]. There is a broad spectrum of renal defects, from simple hydronephrosis to bilateral renal agenesis. In one large population-based study of more than 20,000 fetuses and newborns, about one-third of renal anomalies were detected by prenatal ultrasound [9]. These anomalies were on average detected late in pregnancy, in the third trimester [9]. Time of diagnosis depended upon severity of the abnormality; for example, mean time of diagnosis of bilateral renal agenesis was 24 weeks of gestation, whereas mean time of diagnosis of hydronephrosis was 30 weeks [9]. The most frequently occurring renal anomaly diagnosed on prenatal ultrasound is hydronephrosis [11]. Fetal hydronephrosis may be transient or related to upper or lower urinary tract obstruction or vesicoureteral reflux (VUR) (discussed in detail below). The second most frequent anomaly is renal cysts (either bilateral or unilateral), followed by renal agenesis (unilateral > bilateral) [12]. Renal dysplasia may be observed, associated with hydronephrosis or cysts [12].
4. Cellular mechanisms leading to CAKUT
The most severe renal anomaly, renal aplasia (agenesis), results when the ureteric bud either fails to form or fails to reach/induce the metanephric mesenchyme leading to apoptosis (Figure 1). Unless it receives growth factors released by the ureteric bud (e.g. glial-derived neurotrophic factor or GDNF), the metanephric mesenchyme will undergo apoptosis, leading to renal agenesis [13]. Bilateral renal agenesis results in oligohydramnios, leading to severe oligohydramnios (formerly Potter’s) sequence (pulmonary hypoplasia, club feet, micrognathia) and fetal or perinatal death [14,15].
Fig. 1.
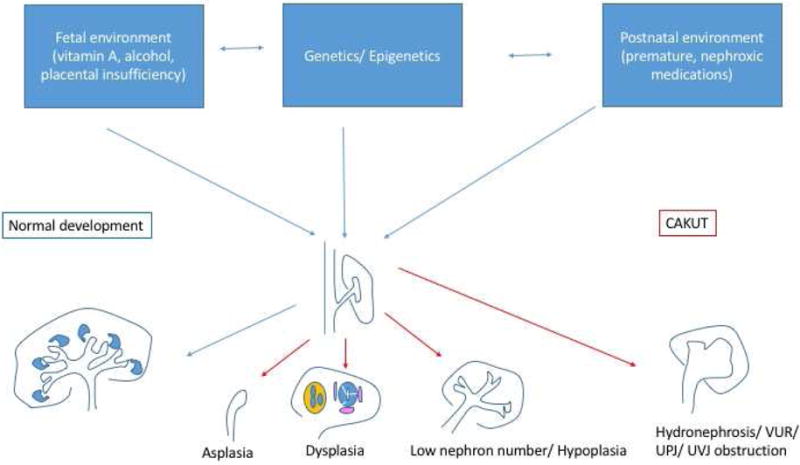
Genetic, epigenetic, and environmental factors interact to modify renal developmental pathways and lead to a spectrum of congenital abnormalities of the kidney and urinary tract (CAKUT). VUR, vesicoureteral reflux; UPJ, ureteropelvic junction; UVJ, ureterovesical junction.
Inhibitory signals restrict and guide the site of ureteric bud outgrowth to a single location [16]. Failure to restrict the site of outgrowth may lead to multiple ureteric buds and duplication of the kidney and/or collecting system. In the most severe cases, there is complete duplication, with an upper pole and lower pole ureter. Duplication is frequently associated with other ureteral anomalies, including VUR and ureterovesical junction (UVJ) obstruction [17]. The classic findings in complete duplications are that the upper ureter connects to the bladder in an ectopic location and is obstructed, whereas the lower pole ureter has VUR [18]. Even in the absence of duplications, defects in ureteric bud outgrowth and branching are associated with VUR, UVJ as well as ureteropelvic junction (UPJ) obstruction [19].
Experiments in fetal sheep have demonstrated that ureteral or urethral obstruction impairs fetal kidney development [20–22]. Postnatal ureteral obstruction also induces tubulointerstitial fibrosis in rodent models [23]. Whereas in humans it is not possible to eliminate the possibility that factors that affected the ureteral development independently lead to defects in renal development, it is clear that both UPJ and UVJ obstructions may result in obstructive uropathies and CKD in childhood [24]. UPJ and UVJ are rarely bilateral and thus do not typically lead to childhood ESKD unless there are associated contralateral renal anomalies [25].
In contrast, urethral obstruction during fetal development (as with posterior urethral valves) is one of the more frequent causes of ESKD in childhood [26,27]. The pathophysiology of PUV is not fully understood, and there are several competing theories on its mechanism [28]. One theory is that the “valves” (which are not truly valves) are mucosal membranes that form and fail to involute during urethral development. Alternatively, they may represent an overgrowth of normally present mucosal folds. A final theory is that they result from abnormal development of the mesonephric or Müllerian duct. A variant of this is that the mesonephric duct has an abnormal anterior insertion into the cloaca. When the rectal–urethral septum forms, the abnormal insertion prevents proper migration of the duct cranially and posteriorly and leads to obstructing anterior–lateral folds. Obstruction of the urethra during bladder development leads to permanent defects in bladder smooth musculature differentiation, with excess fibrotic tissues. PUV is frequently associated with VUR and renal dysplasia, and, as with urethral obstruction, it remains unclear whether the dysplasia results from obstruction/VUR or separate individual defects in renal development (or both).
High grade VUR is associated with renal parenchymal damage, and bilateral VUR contributes to childhood CKD and ESRD [27]. Urinary tract infections in the setting of high grade VUR have significant risk of pyelonephritis [29]. Chronic pyelonephritis and inflammation likely contributes to scarring of renal parenchyma and loss of kidney function [29]. Some studies using mouse models suggest that infection is a key component of renal damage with high grade VUR [30]. However, it remains a source of controversy as, in some cases, high grade VUR in the absence of infection may also lead to progressive renal damage [31]. As with obstructive uropathies, it is also possible that defects that result in VUR may independently lead to renal developmental defects.
Defects in nephron differentiation may also result in CAKUT. In particular, defects in the mesenchyme to epithelial transition results in renal dysplasia [32]. Histologically, renal dysplasia is characterized by immature tubules surrounded by perivascular cuffs [33]. Metaplastic tissues, such as cartilage, may also be present. On renal ultrasound imaging, renal dysplasia most often appears as small echogenic kidneys with poor corticomedullary differentiation [34,35].
Nephrons are induced at the tips of the branching ureteric bud. Thus, the number of ureteric bud branches will determine the nephron number [36,37]. Severely impaired branching manifests as renal hypoplasia [38]. Additional potential mechanism resulting in renal hypoplasia is depletion of renal epithelial progenitors [39–41]. The cap mesenchymal cells that surround the ureteric bud tips represent a unique population of progenitor cells that are programmed to allow for differentiation into renal epithelia cells but also have capacity for self-renewal. There is a tight balance between differentiation and maintenance of the progenitor population [40]. In mouse models, premature differentiation and depletion of nephron progenitors results in renal hypoplasia [41]. Histologically, hypoplastic kidneys have few nephrons but differ from dysplasia in that they are normally formed [42]. Whereas it is difficult to reliably differentiate hypoplasia from dysplasia on ultrasound, classically renal hypoplasia manifests as small kidneys (<2 standard deviations below the mean for age) [42]. Together, defects of renal aplasia, dysplasia and hypoplasia along with reflux and obstructive uropathy are the leading cause of chronic and end stage kidney disease in childhood, accounting for about 40% of children with CKD and 30% of children with ESKD [27].
Whereas severe CAKUT leads to childhood CKD and ESRD, milder variations in ureteric bud branching likely contribute to the broad variation of nephron number observed in humans, from 200,000 to 2,000,000 [43]. Populations with decreased nephron number (such as Australian aboriginals) have higher rates of proteinuria and chronic kidney disease in adulthood (see below) [44]. It is thought that the decreased renal reserve contributes to risk for renal disease. Brenner famously developed the hypothesis that low nephron number leads to hyperfiltration of glomeruli [45]. Hyperfiltration increases glomerular pressures, inducing podocyte damage and loss. This additional glomerular damage exacerbates the nephron deficit and potentially systemic hypertension, leading to a vicious cycle of glomerular damage and scarring and progression to ESRD. In support of Brenner’s hypothesis, experimental models of reduced nephron number (5/6 nephrectomy) in rats lead to progressive renal disease [46,47].
The potential long term effects of nephron deficits are particularly relevant in low birth weight and premature infants. Low birth weight is strongly associated with low nephron number [48]. Prematurity may also result in a low nephron number, as discussed in greater detail below [49]. Multiple epidemiologic studies have identified associations of low birth weight with risk for renal disease, although a few have failed to identify such an effect [50–58].
5. Genetic defects leading to CAKUT
Advances in genomic sequencing and genetic manipulation in mouse and other animal models have led to major advances in our understanding of the gene networks that regulate renal development. The leading genetic syndromes associated with CAKUT are trisomies 21, 18, and 13 [59]. Large scale studies of cohorts of children with non-syndromic CAKUT have demonstrated that between 6% and 17% may have single gene defects [60,61]. Another 10–15% have large copy number variants (CNVs) that may contribute to CAKUT [62–64]. Even in mouse models, the phenotype from genetic deletion varies considerably, suggesting that gene–gene and gene–environment interactions contribute to CAKUT [65–67]. Thus, it is therefore likely that many cases of CAKUT are polygenic, with multiple gene variants contributing. Genetic causes of CAKUT have been reviewed recently [68–72], therefore we will focus here on a subset of mutations identified in humans.
One of the earliest genes to have mutations identified in association with non-syndromic human CAKUT was the tyrosine kinase RET receptor for GDNF. Biallelic inactivating genetic mutations in RET are associated with the most severe manifestation of CAKUT, bilateral renal agenesis [73]. The RET receptor is expressed on the tips of the ureteric buds, whereas GDNF is secreted by the metanephric mesenchyme [74]. GDNF/Ret signaling stimulates ureteric bud outgrowth, and thus defects in this signaling pathway result in failure of the ureteric bud to reach the metanephric mesenchyme, resulting in renal agenesis [75–77]. Some studies suggest that mutations in RET/GDNF are relatively rare in renal agenesis, although genetic variants in GDNF/Ret are associated with sporadic CAKUT [78,79].
Autosomal dominant mutations in HNF1B are the most frequent monogenic etiology of CAKUT [60,80,81]. HNF1B is a transcription factor that contributes to both ureter development and nephron segmentation [82–84]. Likely due to its multiple roles in renal development, HNF1B genetic variants are associated with a broad spectrum of CAKUT, from renal hypoplasia to non-functioning multicystic dysplastic kidneys [81,85–88]. HNF1B variants are also associated with maturity onset diabetes of the young (MODY), as in renal cysts and diabetes syndrome [89,90], as well as hyperuricemia with hypomagnesemia [91]. The hypomagnesemia is likely due to a direct effect of HNF1B on expression of sodium/potassium ATPase subunits that modify renal absorption of magnesium in the distal convoluted tubule [92,93].
Other autosomal dominant mutations that have been associated with sporadic CAKUT are in transcription factors in the renal cells that regulate early renal epithelial differentiation, including EYA1 [61,94,95]. EYA1 binds and regulates multiple other transcription factors that maintain the nephron progenitor population and stimulate GDNF expression (including SIX and PAX2 proteins) [96–99]. Mutations in EYA1/SIX1/SIX5 are associated with branchio-oto-renal syndrome [100–102]. PAX2 is expressed in the differentiating renal vesicle but also in the branching ureteric bud [103]. Autosomal dominant mutations in PAX2 are associated with renal–coloboma syndrome [104]. In large studies, identification of genetic mutations in EYA1 and PAX2 in what appeared to be sporadic cases of CAKUT led to more detailed eye and hearing examination, with identification of subtle deficits [60,61].
Autosomal recessive genetic mutations likely occur at a low frequency in sporadic CAKUT (for example, Kohl et al. identified autosomal recessive mutations in 12 genes in 2.5% of almost 600 children with CAKUT [105]). These included gene defects in FRAS/FREM, which are associated with Fraser syndrome (CAKUT) [105–107]. The protein products of these genes modify heparin sulfate, which bind the majority of growth factors (e.g. HGF, EGF, VEGF) that stimulate UB branching [108–111].
Studies of familial VUR have identified genetic defects in the SLIT-ROBO guidance signaling, and more recently genetic defects in TNXB, which encodes Tenascin XB [112–115]. Mouse models indicate that Slit-Robo signaling likely restricts interactions between the nephrogenic mesenchyme and ureteric bud to facilitate bud outgrowth at a single site [116,117]. Tenascin XB is an extracellular matrix glycoprotein that is expressed by uroepithelia of the UVJ, and thus may contribute to closure of the UVJ during voiding [115].
6. Adverse fetal environment effects on renal development
Together, chromosomal anomalies, copy number variants, and monogenic genetic abnormalities may account for up to 50% of CAKUT (Figure 2). However, environmental factors likely also contribute to CAKUT [118]. In-utero exposure to an adverse fetal environment is associated with CAKUT in humans. A variety of insults including hyperglycemia, vitamin A deficiency (in the developing world), and cocaine and alcohol exposures are associated with a higher risk of renal anomalies [119–121]. The mechanisms of these defects are not fully understood; however, hyperglycemia may affect ureteric bud branching [121]. Similarly, vitamin A (retinoic acid) is required for Ret expression [122]. Thus, vitamin A deficiency may impair ureteric bud branching due to decreased Ret expression [123,124].
Fig. 2.
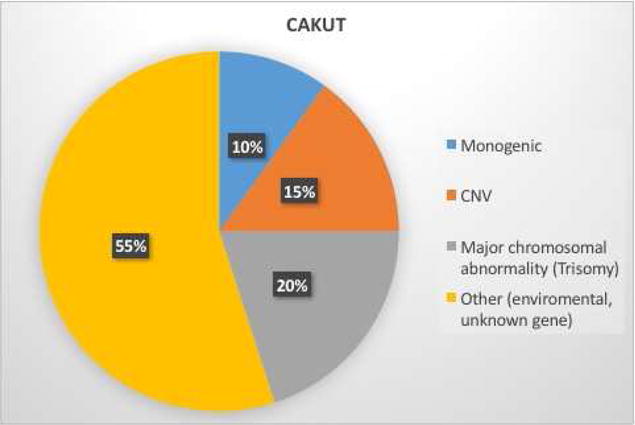
Studies suggest genetic factors, including major chromosomal anomalies, copy number variants (CNV), and monogenic mutations/deletions likely contribute to up to 45% of congenital abnormalities of the kidney and urinary tract (CAKUT). Environmental factors, including an adverse fetal environment, and not yet discovered genetic factors likely contribute to the remainder.
Barker first hypothesized in 1990 that fetal programming has long term effects on risk for adult onset disease [125]. His study of the Dutch population exposed to famine during World War II demonstrated that children from mothers with inadequate caloric intake had higher incidence of cardiovascular disease and diabetes. As discussed above, Brenner extended his hypothesis to the kidney, focusing on the maladaptive effects of hyperfiltration in the setting of low nephron number [46]. Given that nephron number is established at birth, recent studies have focused on the role of the fetal environment on determining nephron number.
Intrauterine growth retardation is a frequent cause of low birth weight.
The effects of ureteroplacental insuffiency, as well as maternal caloric and protein deprivation on fetal renal development have been studied in rodent models [126]. Whereas not all models have had the same manifestations, likely due to differences in precise timing and nutrient content, caloric or protein restriction in utero induces low nephron number and hypertension and proteinuria in the adult offspring.
One of the mechanisms by which in utero exposures may result in long term effects is epigenetic modifications [70,127]. Epigenetic modifications include histone methylation and acetylation that regulate gene transcription by modifying the accessibility of chromatin to transcription factors [128]. Another form of epigenetic modification is DNA methylation. Highly methylated DNA in promotor regions is thought result in repression of gene transcription. Research into epigenetic regulation of renal development is an area of active research, and our understanding of how in-utero exposures may induce epigenetic changes in the kidney is limited. However, there are strong data suggesting that the epigenetic state of the fetal kidney is different than the adult kidney, and that shifts in the epigenetic state likely contribute to regulation of transcription of renal developmental genes [118,128,129]. In addition, human chronic kidney disease is associated with alterations in the epigenetic profiles [130]. Thus, epigenetic changes may provide a mechanistic link whereby in-utero exposures lead to long term increased risk of renal disease in adulthood.
Maternal medication exposures are another source of renal developmental defects. The renin–angiotensin system augments ureteric bud branching, and maternal use of angiotensin-converting enzyme inhibitors or angiotensin receptor blockers is associated with renal agenesis and anomalies [131]. Other medications, including glucocorticoids, mycophenolate mofetil, antiepileptic drugs, and cyclophosphamide may be associated with renal anomalies [132–136].
7. Prematurity effects on renal development
As discussed above, new nephrons form at an exponential rate between 20 and 28 weeks of gestation. Nephron formation ceases between 28 and 36 weeks. Thus, in addition to low birth weight, premature infants born before 36 weeks may not have completed nephron development. Limited data are available about the effects of prematurity on renal development. Unlike in term infants, nephrons may continue to form after birth in premature infants [137,138]. Studies of autopsies indicated that nephron formation may continue after birth in premature infants born prior to completion of nephron development [138,139]. However, premature cessation of nephrogenesis appears to be widespread, leading to a nephron deficit [138,139].
In addition to prematurity, postnatal exposures to nephrotoxins may affect renal development. A study of very low birth weight neonates in the neonatal intensive care unit determined that 90% of the neonates were exposed to at least one nephrotoxic agent and that the average neonate was exposed to almost two weeks of nephrotoxins [140]. Thirty percent of the premature infants received gentamicin after birth and 50% received amphoteracin. Fifty percent also received another nephrotoxin, indomethacin, as a non-surgical approach to close patent ductus arteriosus. Due to high renin–angiotensin levels in the preterm neonate, neonatal renal blood flow critically depends upon prostaglandin-induced vasodilation of afferent arterioles [141]. Thus, indomethacin can markedly reduce renal perfusion pressure and induce acute kidney injury (AKI) in a significant subset of neonates [140,142]. Of note, 26% of the cohort developed AKI, with the smallest infants being at highest risk.
Recently studies have suggested that repeated or severe AKI may result in CKD, and it is unclear what the effect of nephrotoxins are on the developing kidney in premature infants [49]. Low birth weight and prematurity are associated with glomerular disease later in childhood, especially focal glomerulosclerosis [49,143].
Recently a multicenter retrospective chart review of neonatal acute kidney was performed [144]. This and other studies may improve our understanding of the impact of prematurity, low birth weight, and perinatal insults on renal outcomes.
8. Antenatal diagnosis of renal pathology
8.1. Normal prenatal ultrasound findings
Fetal kidneys appear lobular and can be identified in the paraspinal region on prenatal ultrasound as early as 9–12 weeks of gestation. At 12 weeks, kidneys are typically 1 cm in length, and they grow to an average 2.7 cm in length by 20 weeks. In addition, between 15 and 20 weeks corticomedullary differentiation occurs, and an echogenic cortex with the hypoechogenic, dark renal pyramids of the medulla should be apparent by 20 weeks. Typically, the renal cortex is less echogenic than the liver by 20 weeks. Glomerular filtration begins at about nine weeks, but does not contribute significantly to amniotic fluid until skin develops at 19–20 weeks. The bladder may be visualized by ultrasound at 10–14 weeks, and bladder emptying is visible from 15 weeks. Bladder capacity is 10 mL at 30 weeks and increases to 50 mL in term infants. Urine becomes a major contributor to amniotic fluid and increases from 20 weeks until term with an average of 300 mL/kg body weight/day.
8.2. Antenatal evaluation of pyelectasis
The most frequent renal abnormality to be diagnosed by prenatal ultrasound is pyelectasis. Whereas there is no complete consensus about the best definition and grading system for pyelectasis, measurement of the anterior–posterior diameter (APD) of the renal pelvis in the transverse plane is the standard approach for diagnosis [145]. An APD of >15 mm is strongly predictive of uretero-pelvic junction obstruction [146]. However, APD is affected by multiple factors, including gestational age and maternal hydration status (and hence fetal urine output), and may worsen or improve during later development [147]. An APD of >4 mm in the second trimester or >7 mm in the third trimester are indications for follow-up [145,148–150]. Generally, dilation in the second trimester is an indication for follow-up ultrasound in the third trimester. Grading systems incorporate whether the hydronephrosis involves dilation major – or, in more severe cases, minor renal calyces – and whether there is associated thinning of the renal parenchyma [145].
8.3. Other factors predicting outcomes in pyelectasis
Low amniotic fluid (or oligohydramnios) is a major factor that influences postnatal outcomes of pyelectasis [151]. Oligohydramnios is defined as <500 mL of amniotic fluid, and is typically assessed by the amniotic fluid index (AFI, measurements of amniotic fluid pockets ≥2 cm) [152,153]. AFI <5–6 indicates oligohydramnios [152]. Oligohydramnios is most often associated with the severest forms of CAKUT, such as PUVs or bilateral renal agenesis. Oligohydramnios leads to fetal compression and oligohydramnios (Potter’s) sequence [wide set eyes, beaked nose, low set and posteriorly rotated ears, micrognathia, talipes equinovarus (club feet), and pulmonary hypoplasia] [154,155]. Pulmonary hypoplasia leads to increased risk of pneumothorax after birth and is the major determinant of postnatal survival [156]. In the setting of PUV, unilateral VUR may be protective and decrease the risk of mortality and renal failure [156].
There have been attempts to use urinary indices to predict renal outcomes, especially in cases of severe pyelectasis. Cutoffs exist for predicting better renal prognosis for urinary electrolytes (sodium <100 mEq/L and calcium <8 mg/dL) and urinary concentration (osmolarity <200 mOsm/L), and markers of renal damage (β2 microglobulin <4 mg/L and protein <40 mg/dL) have been proposed, but are not without controversy [150,157]. Various measures have been attempted to predict long term outcomes of fetal hydronephrosis. Serum creatinine at one year of life seems to be the best predictor of long term outcomes [158,159], although recently ultrasound assessment of renal parenchymal area has been proposed as a marker [160].
8.4. Postnatal evaluation of fetal pyelectasis
Fetal pyelectasis may represent urethral/bladder outflow obstruction (as discussed above), VUR, UPJ obstruction, non-obstructed dilated ureters (mega-ureter), or transient hydronephrosis. Studies suggest that the majority (up to almost 90%) of patients diagnosed with fetal pyelectasis in the second trimester have mild hydronephrosis, and 80% of these patients will not need surgical intervention [149,161,162]. The majority of these will have transient hydronephrosis that will resolve. An estimated 10–20% of infants with fetal pyelectasis have VUR [163].
There remains some controversy about postnatal evaluation of fetal pyelectasis. In cases where urologic intervention may be required after birth, such as bilateral hydronephrosis, oligohydramnios and/or bladder abnormalities, a renal/bladder ultrasound should be obtained immediately (1–2 days, depending on the situation) after birth [164]. In addition, in cases of fetal pyelectasis where adherence to follow-up is a concern, postnatal ultrasound may be performed after birth. However, due to relatively lower urine output in the initial 24–48 h after birth, a postnatal ultrasound performed in the first days of life may under-represent the degree of hydronephrosis and be falsely reassuring [164]. Thus, except in select cases as above, optimal timing for a postnatal ultrasound to assess for persistence and severity of fetal pyelectasis is between 7 and 10 days after birth [164]. In cases where there is a normal postnatal ultrasound initially, it is recommended to repeat the ultrasound at 4–6 weeks of age [164].
The more severe the postnatal pyelectasis the greater the likelihood of a condition requiring surgical intervention, either UPJ obstruction or VUR [165]. Therefore, antibiotic prophylaxis and a voiding cystourethrogram (VCUG) is generally indicated for infants with persistent moderate or severe hydronephrosis after birth [153,166]. Unfortunately, the severity of dilation does not correlate well with severity of VUR, and even those with resolved hydronephrosis may have VUR. The consensus guidelines for fetal hydronephrosis recommend discussion with families about risk and benefits of antibiotics and the VCUG in these cases [145].
In the absence of reflux, the next step for evaluation of moderate or severe hydronephrosis is assessment for urinary tract obstruction with a Tc99 Mag3 renal scan with furosemide [167]. Infants require bladder catherization to ensure bladder emptying during the study, as bladder retention can delay emptying times. Tc99 Mag3 and furosemide are secreted by renal proximal tubules, and the immaturity of renal proximal tubules after birth can make a Mag3 scan non-diagnostic in the first few weeks of life. Generally, it is preferable to wait until at least 4–6 weeks after birth to obtain a Mag3 renal scan.
9. Conclusions
CAKUT are one of the leading congenital anomalies. CAKUT most often results from defects in ureteric bud outgrowth and branching or from epithelial differentiation from mesenchymal renal progenitors. Both gene and environmental factors likely contribute to CAKUT. Whereas single gene defects are likely associated with the minority of non-syndromic CAKUT, genetic studies of animals and humans have led to advances in the understanding of the cellular and molecular mechanisms regulating renal and genitourinary tract development. Prenatal ultrasound is useful for detection of CAKUT, although not all defects may be identified. Ultrasound findings and amniotic fluid parameters provide some metrics to help predict prognosis. Postnatal imaging is required to confirm the diagnosis and define the defect. Continued research into genetic and environmental influences on renal development, genotype–phenotype correlations, and predictors of fetal outcomes may lead to improved options for diagnosis and prognosis.
In addition, CAKUT represents a broad spectrum of disease. It includes “mild” defects, including low nephron number, to severe bilateral defects that lead to fetal or neonatal death. Even “mild” defects may result in long term risk for CKD in adulthood. Thus, long term prospective studies, for example, of the extremely preterm infants, will be important to better understand the impact of low nephron number on the life-course and risk for adult onset CKD.
Acknowledgments
Funding sources
None.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
None declared.
References
- 1.Potter EL. Normal and abnormal development of the kidney. Chicago: Year Book Medical Publishers; 1972. [Google Scholar]
- 2.Blake J, Rosenblum ND. Renal branching morphogenesis: morphogenetic and signaling mechanisms. Semin Cell Dev Biol. 2014;36:2–12. doi: 10.1016/j.semcdb.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 3.Costantini F. Genetic controls and cellular behaviors in branching morphogenesis of the renal collecting system. Dev Biol. 2012;1:693–713. doi: 10.1002/wdev.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kreidberg JA. Podocyte differentiation and glomerulogenesis. J Am Soc Nephrol. 2003;14:806–14. doi: 10.1097/01.asn.0000054887.42550.14. [DOI] [PubMed] [Google Scholar]
- 5.Hinchliffe SA, Sargent PH, Howard CV, Chan YF, van Velzen D. Human intrauterine renal growth expressed in absolute number of glomeruli assessed by the disector method and Cavalieri principle. Lab Invest. 1991;64:777–84. [PubMed] [Google Scholar]
- 6.Underwood MA, Gilbert WM, Sherman MP. Amniotic fluid: not just fetal urine anymore. J Perinatol. 2005;25:341–8. doi: 10.1038/sj.jp.7211290. [DOI] [PubMed] [Google Scholar]
- 7.Verburg BO, Geelhoed JJ, Steegers EA, et al. Fetal kidney volume and its association with growth and blood flow in fetal life: The Generation R Study. Kidney Int. 2007;72:754–61. doi: 10.1038/sj.ki.5002420. [DOI] [PubMed] [Google Scholar]
- 8.dos Santos Junior AC, de Miranda DM, Simoes e Silva AC. Congenital anomalies of the kidney and urinary tract: an embryogenetic review. Birth Defects Res C, Embryo Today Rev. 2014;102:374–81. doi: 10.1002/bdrc.21084. [DOI] [PubMed] [Google Scholar]
- 9.Queisser-Luft A, Stopfkuchen H, Stolz G, Schlaefer K, Merz E. Prenatal diagnosis of major malformations: quality control of routine ultrasound examinations based on a five-year study of 20,248 newborn fetuses and infants. Prenat Diagn. 1998;18:567–76. [PubMed] [Google Scholar]
- 10.Andrés-Jensen L, Jørgensen FS, Thorup J, et al. The outcome of antenatal ultrasound diagnosed anomalies of the kidney and urinary tract in a large Danish birth cohort. Archs Dis Childh. 2016;101:819–24. doi: 10.1136/archdischild-2015-309784. [DOI] [PubMed] [Google Scholar]
- 11.Kumar S, Walia S, Ikpeme O, et al. Postnatal outcome of prenatally diagnosed severe fetal renal pelvic dilatation. Prenatal Diagn. 2012;32:519–22. doi: 10.1002/pd.2893. [DOI] [PubMed] [Google Scholar]
- 12.Kumar M, Thakur S, Puri A, et al. Fetal renal anomaly: factors that predict survival. J Pediatr Urol. 2014;10:1001–7. doi: 10.1016/j.jpurol.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 13.Perantoni AO, Timofeeva O, Naillat F, et al. Inactivation of FGF8 in early mesoderm reveals an essential role in kidney development. Development (Cambridge, UK) 2005;132:3859–71. doi: 10.1242/dev.01945. [DOI] [PubMed] [Google Scholar]
- 14.Curry CJ, Jensen K, Holland J, Miller L, Hall BD. The Potter sequence: a clinical analysis of 80 cases. Am J Med Genet. 1984;19:679–702. doi: 10.1002/ajmg.1320190408. [DOI] [PubMed] [Google Scholar]
- 15.Dicker D, Samuel N, Feldberg D, Goldman JA. The antenatal diagnosis of Potter syndrome (Potter sequence). A lethal and not-so-rare malformation. Eur J Obstet Gynecol Reprod Biol. 1984;18:17–24. doi: 10.1016/0028-2243(84)90028-5. [DOI] [PubMed] [Google Scholar]
- 16.Michos O. Kidney development: from ureteric bud formation to branching morphogenesis. Curr Opin Genet Dev. 2009;19:484–90. doi: 10.1016/j.gde.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rasouly HM, Lu W. Lower urinary tract development and disease. Systems Biol Med. 2013;5:307–42. doi: 10.1002/wsbm.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hindryckx A, De Catte L. Prenatal diagnosis of congenital renal and urinary tract malformations. Facts Views Vision ObGyn. 2011;3:165–74. [PMC free article] [PubMed] [Google Scholar]
- 19.Avni EF, Dacher JN, Stallenberg B, Collier F, Hall M, Schulman CC. Renal duplications: the impact of perinatal ultrasound on diagnosis and management. Eur Urol. 1991;20:43–8. doi: 10.1159/000471658. [DOI] [PubMed] [Google Scholar]
- 20.Farrugia MK, Long DA, Godley ML, et al. Experimental short-term fetal bladder outflow obstruction: I. Morphology and cell biology associated with urinary flow impairment. J Pediatr Urol. 2006;2:243–53. doi: 10.1016/j.jpurol.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 21.Duncombe GJ, Barker AP, Moss TJ, et al. The effects of overcoming experimental bladder outflow obstruction in fetal sheep. J Maternal Fetal Neonatal Med. 2002;11:130–7. doi: 10.1080/jmf.11.2.130.137. [DOI] [PubMed] [Google Scholar]
- 22.Pringle KC, Zuccollo J, Kitagawa H, Koike J, Delahunt B. Renal dysplasia produced by obstructive uropathy in the fetal lamb. Pathology. 2003;35:518–21. doi: 10.1080/00313020310001619145. [DOI] [PubMed] [Google Scholar]
- 23.Hiatt MJ, Ivanova L, Trnka P, Solomon M, Matsell DG. Urinary tract obstruction in the mouse: the kinetics of distal nephron injury. Lab Invest. 2013;93:1012–23. doi: 10.1038/labinvest.2013.90. [DOI] [PubMed] [Google Scholar]
- 24.Parkhouse HF, Barratt TM. Investigation of the dilated urinary tract. Pediatr Nephrol (Berlin) 1988;2:43–7. doi: 10.1007/BF00870379. [DOI] [PubMed] [Google Scholar]
- 25.Asl AS, Maleknejad S. Clinical outcome and follow-up of prenatal hydronephrosis. Saudi J Kidney Dis Transplant. 2012;23:526–31. [PubMed] [Google Scholar]
- 26.Roth KS, Carter WH, Jr, Chan JC. Obstructive nephropathy in children: long-term progression after relief of posterior urethral valve. Pediatrics. 2001;107:1004–10. doi: 10.1542/peds.107.5.1004. [DOI] [PubMed] [Google Scholar]
- 27.Seikaly MG, Ho PL, Emmett L, Fine RN, Tejani A. Chronic renal insufficiency in children: the 2001 Annual Report of the NAPRTCS. Pediatr Nephrol (Berlin) 2003;18:796–804. doi: 10.1007/s00467-003-1158-5. [DOI] [PubMed] [Google Scholar]
- 28.Kajbafzadeh A. Congenital urethral anomalies in boys. Part I: posterior urethral valves. Urol J. 2005;2:59–78. [PubMed] [Google Scholar]
- 29.Mattoo TK, Chesney RW, Greenfield SP, et al. Renal Scarring in the Randomized Intervention for Children with Vesicoureteral Reflux (RIVUR) Trial. Clin J Am Soc Nephrol. 2016;11:54–61. doi: 10.2215/CJN.05210515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bowen SE, Watt CL, Murawski IJ, Gupta IR, Abraham SN. Interplay between vesicoureteric reflux and kidney infection in the development of reflux nephropathy in mice. Dis Models Mech. 2013;6:934–41. doi: 10.1242/dmm.011650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mattoo TK. Vesicoureteral reflux and reflux nephropathy. Adv Chronic Kidney Dis. 2011;18:348–54. doi: 10.1053/j.ackd.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stark K, Vainio S, Vassileva G, McMahon AP. Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature. 1994;372:679–83. doi: 10.1038/372679a0. [DOI] [PubMed] [Google Scholar]
- 33.Chen RY, Chang H. Renal dysplasia. Archs Pathol Lab Med. 2015;139:547–51. doi: 10.5858/arpa.2013-0660-RS. [DOI] [PubMed] [Google Scholar]
- 34.Caiulo VA, Caiulo S, Gargasole C, et al. Ultrasound mass screening for congenital anomalies of the kidney and urinary tract. Pediatr Nephrol (Berlin) 2012;27:949–53. doi: 10.1007/s00467-011-2098-0. [DOI] [PubMed] [Google Scholar]
- 35.Riccabona M. Renal failure in neonates, infants, and children: the role of ultrasound. Ultrasound Clin. 2006;1:457–69. [Google Scholar]
- 36.al-Awqati Q, Goldberg MR. Architectural patterns in branching morphogenesis in the kidney. Kidney Int. 1998;54:1832–42. doi: 10.1046/j.1523-1755.1998.00196.x. [DOI] [PubMed] [Google Scholar]
- 37.Cullen-McEwen LA, Drago J, Bertram JF. Nephron endowment in glial cell line-derived neurotrophic factor (GDNF) heterozygous mice. Kidney Int. 2001;60:31–6. doi: 10.1046/j.1523-1755.2001.00767.x. [DOI] [PubMed] [Google Scholar]
- 38.Dziarmaga A, Clark P, Stayner C, et al. Ureteric bud apoptosis and renal hypoplasia in transgenic PAX2-Bax fetal mice mimics the renal–coloboma syndrome. J Am Soc Nephrol. 2003;14:2767–74. doi: 10.1097/01.asn.0000094082.11026.ee. [DOI] [PubMed] [Google Scholar]
- 39.Kanda S, Tanigawa S, Ohmori T, et al. Sall1 maintains nephron progenitors and nascent nephrons by acting as both an activator and a repressor. J Am Soc Nephrol. 2014;25:2584–95. doi: 10.1681/ASN.2013080896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kopan R, Chen S, Little M. Nephron progenitor cells: shifting the balance of self-renewal and differentiation. Curr Top Dev Biol. 2014;107:293–331. doi: 10.1016/B978-0-12-416022-4.00011-1. [DOI] [PubMed] [Google Scholar]
- 41.Self M, Lagutin OV, Bowling B, et al. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 2006;25:5214–28. doi: 10.1038/sj.emboj.7601381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cain JE, Di Giovanni V, Smeeton J, Rosenblum ND. Genetics of renal hypoplasia: insights into the mechanisms controlling nephron endowment. Pediatr Res. 2010;68:91–8. doi: 10.1203/PDR.0b013e3181e35a88. [DOI] [PubMed] [Google Scholar]
- 43.Bagby SP. Maternal nutrition, low nephron number, and hypertension in later life: pathways of nutritional programming. J Nutr. 2007;137:1066–72. doi: 10.1093/jn/137.4.1066. [DOI] [PubMed] [Google Scholar]
- 44.Hoy WE, Kincaid-Smith P, Hughson MD, et al. CKD in Aboriginal Australians. Am J Kidney Dis. 2010;56:983–93. doi: 10.1053/j.ajkd.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 45.Brenner BM. The etiology of adult hypertension and progressive renal injury: an hypothesis. Bulletin et memoires de l’Academie royale de medecine de Belgique. 1994;149:121–5. discussion 5–7. [PubMed] [Google Scholar]
- 46.Brenner BM, Mackenzie HS. Nephron mass as a risk factor for progression of renal disease. Kidney Int. 1997;63(Suppl):S124–7. [PubMed] [Google Scholar]
- 47.Hostetter TH, Olson JL, Rennke HG, Venkatachalam MA, Brenner BM. Hyperfiltration in remnant nephrons: a potentially adverse response to renal ablation. J Am Soc Nephrol. 2001;12:1315–25. [Google Scholar]
- 48.Bertram JF, Douglas-Denton RN, Diouf B, Hughson MD, Hoy WE. Human nephron number: implications for health and disease. Pediatr Nephrol (Berlin) 2011;26:1529–33. doi: 10.1007/s00467-011-1843-8. [DOI] [PubMed] [Google Scholar]
- 49.Carmody JB, Charlton JR. Short-term gestation, long-term risk: prematurity and chronic kidney disease. Pediatrics. 2013;131:1168–79. doi: 10.1542/peds.2013-0009. [DOI] [PubMed] [Google Scholar]
- 50.Fagerudd J, Forsblom C, Pettersson-Fernholm K, et al. Low birth weight does not increase the risk of nephropathy in Finnish type 1 diabetic patients. Nephrol Dialysis Transplant. 2006;21:2159–65. doi: 10.1093/ndt/gfl217. [DOI] [PubMed] [Google Scholar]
- 51.Berglund D, MacDonald D, Jackson S, et al. Low birthweight and risk of albuminuria in living kidney donors. Clin Transplant. 2014;28:361–7. doi: 10.1111/ctr.12321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dyck R, Klomp H, Tan L, Stang MR. An association of maternal age and birth weight with end-stage renal disease in Saskatchewan. Sub-analysis of registered Indians and those with diabetes. Am J Nephrol. 2003;23:395–402. doi: 10.1159/000074066. [DOI] [PubMed] [Google Scholar]
- 53.Herrera J, Rodriguez-Iturbe B. End-stage renal disease and acute glomerulonephritis in Goajiro Indians. Kidney Int Suppl. 2003:S22–6. doi: 10.1046/j.1523-1755.63.s83.6.x. [DOI] [PubMed] [Google Scholar]
- 54.Vikse BE, Irgens LM, Leivestad T, Hallan S, Iversen BM. Low birth weight increases risk for end-stage renal disease. J Am Soc Nephrol. 2008;19:151–7. doi: 10.1681/ASN.2007020252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ruggajo P, Svarstad E, Leh S, Marti HP, Reisaether AV, Vikse BE. Low birth weight and risk of progression to end stage renal disease in IgA nephropathy – a retrospective registry-based cohort study. PloS One. 2016;11:e0153819. doi: 10.1371/journal.pone.0153819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fan ZJ, Lackland DT, Kenderes B, Krisher J, Freedman BI. Impact of birth weight on familial aggregation of end-stage renal disease. Am J Nephrol. 2003;23:117–20. doi: 10.1159/000068037. [DOI] [PubMed] [Google Scholar]
- 57.Fan Z, Lipsitz S, Egan B, Lackland D. The impact of birth weight on the racial disparity of end-stage renal disease. Ann Epidemiol. 2000;10:459. doi: 10.1016/s1047-2797(00)00105-8. [DOI] [PubMed] [Google Scholar]
- 58.Jang WS, Kim WH, Choi K, et al. Incidence, risk factors and clinical outcomes for acute kidney injury after aortic arch repair in paediatric patients. Eur J Cardio-Thoracic Surg. 2014;45:e208–14. doi: 10.1093/ejcts/ezu132. [DOI] [PubMed] [Google Scholar]
- 59.Stoll C, Dott B, Alembik Y, Roth MP. Associated nonurinary congenital anomalies among infants with congenital anomalies of kidney and urinary tract (CAKUT) Eur J Med Genet. 2014;57:322–8. doi: 10.1016/j.ejmg.2014.04.014. [DOI] [PubMed] [Google Scholar]
- 60.Weber S, Moriniere V, Knuppel T, et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol. 2006;17:2864–70. doi: 10.1681/ASN.2006030277. [DOI] [PubMed] [Google Scholar]
- 61.Hwang DY, Dworschak GC, Kohl S, et al. Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. 2014;85:1429–33. doi: 10.1038/ki.2013.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Caruana G, Wong MN, Walker A, et al. Copy-number variation associated with congenital anomalies of the kidney and urinary tract. Pediatr Nephrol (Berlin) 2015;30:487–95. doi: 10.1007/s00467-014-2962-9. [DOI] [PubMed] [Google Scholar]
- 63.Sanna-Cherchi S, Kiryluk K, Burgess KE, et al. Copy-number disorders are a common cause of congenital kidney malformations. Am J Hum Genet. 2012;91:987–97. doi: 10.1016/j.ajhg.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Westland R, Verbitsky M, Vukojevic K, et al. Copy number variation analysis identifies novel CAKUT candidate genes in children with a solitary functioning kidney. Kidney Int. 2015;88:1402–10. doi: 10.1038/ki.2015.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Narlis M, Grote D, Gaitan Y, Boualia SK, Bouchard M. Pax2 and pax8 regulate branching morphogenesis and nephron differentiation in the developing kidney. J Am Soc Nephrol. 2007;18:1121–9. doi: 10.1681/ASN.2006070739. [DOI] [PubMed] [Google Scholar]
- 66.Schuchardt A, D’Agati V, Larsson-Blomberg L, Costantini F, Pachnis V. RET-deficient mice: an animal model for Hirschsprung’s disease and renal agenesis. J Intern Med. 1995;238:327–32. doi: 10.1111/j.1365-2796.1995.tb01206.x. [DOI] [PubMed] [Google Scholar]
- 67.Dziarmaga A, Quinlan J, Goodyer P. Renal hypoplasia: lessons from Pax2. Pediatr Nephrol (Berlin) 2006;21:26–31. doi: 10.1007/s00467-005-2039-x. [DOI] [PubMed] [Google Scholar]
- 68.Caruana G, Bertram JF. Congenital anomalies of the kidney and urinary tract genetics in mice and men. Nephrology (Carlton, Victoria) 2015;20:309–11. doi: 10.1111/nep.12402. [DOI] [PubMed] [Google Scholar]
- 69.Nakanishi K, Yoshikawa N. Genetic disorders of human congenital anomalies of the kidney and urinary tract (CAKUT) Pediatr Int. 2003;45:610–16. doi: 10.1046/j.1442-200x.2003.01779.x. [DOI] [PubMed] [Google Scholar]
- 70.Nicolaou N, Renkema KY, Bongers EM, Giles RH, Knoers NV. Genetic, environmental, and epigenetic factors involved in CAKUT. Nature Rev Nephrol. 2015;11:720–31. doi: 10.1038/nrneph.2015.140. [DOI] [PubMed] [Google Scholar]
- 71.Vivante A, Kohl S, Hwang DY, Dworschak GC, Hildebrandt F. Single-gene causes of congenital anomalies of the kidney and urinary tract (CAKUT) in humans. Pediatr Nephrol (Berlin) 2014;29:695–704. doi: 10.1007/s00467-013-2684-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Uy N, Reidy K. Developmental genetics and congenital anomalies of the kidney and urinary tract. J Pediatr Genet. 2016;5:51–60. doi: 10.1055/s-0035-1558423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Skinner MA, Safford SD, Reeves JG, Jackson ME, Freemerman AJ. Renal aplasia in humans is associated with RET mutations. Am J Hum Genet. 2008;82:344–51. doi: 10.1016/j.ajhg.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Costantini F. GDNF/Ret signaling and renal branching morphogenesis: from mesenchymal signals to epithelial cell behaviors. Organogenesis. 2010;6:252–62. doi: 10.4161/org.6.4.12680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Basson MA, Watson-Johnson J, Shakya R, et al. Branching morphogenesis of the ureteric epithelium during kidney development is coordinated by the opposing functions of GDNF and Sprouty1. Dev Biol. 2006;299:466–77. doi: 10.1016/j.ydbio.2006.08.051. [DOI] [PubMed] [Google Scholar]
- 76.Chi X, Michos O, Shakya R, et al. Ret-dependent cell rearrangements in the Wolffian duct epithelium initiate ureteric bud morphogenesis. Dev Cell. 2009;17:199–209. doi: 10.1016/j.devcel.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Costantini F, Shakya R. GDNF/Ret signaling and the development of the kidney. BioEssays. 2006;28:117–27. doi: 10.1002/bies.20357. [DOI] [PubMed] [Google Scholar]
- 78.Jeanpierre C, Mace G, Parisot M, et al. RET and GDNF mutations are rare in fetuses with renal agenesis or other severe kidney development defects. J Med Genet. 2011;48:497–504. doi: 10.1136/jmg.2010.088526. [DOI] [PubMed] [Google Scholar]
- 79.Chatterjee R, Ramos E, Hoffman M, et al. Traditional and targeted exome sequencing reveals common, rare and novel functional deleterious variants in RET-signaling complex in a cohort of living US patients with urinary tract malformations. Hum Genet. 2012;131:1725–38. doi: 10.1007/s00439-012-1181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bockenhauer D, Jaureguiberry G. HNF1B-associated clinical phenotypes: the kidney and beyond. Pediatr Nephrol (Berlin) 2016;31:707–14. doi: 10.1007/s00467-015-3142-2. [DOI] [PubMed] [Google Scholar]
- 81.Bohn S, Thomas H, Turan G, et al. Distinct molecular and morphogenetic properties of mutations in the human HNF1beta gene that lead to defective kidney development. J Am Soc Nephrol. 2003;14:2033–41. doi: 10.1097/01.asn.0000078808.70309.c4. [DOI] [PubMed] [Google Scholar]
- 82.Paces-Fessy M, Fabre M, Lesaulnier C, Cereghini S. Hnf1b and Pax2 cooperate to control different pathways in kidney and ureter morphogenesis. Hum Mol Genet. 2012;21:3143–55. doi: 10.1093/hmg/dds141. [DOI] [PubMed] [Google Scholar]
- 83.Massa F, Garbay S, Bouvier R, et al. Hepatocyte nuclear factor 1beta controls nephron tubular development. Development (Cambridge, UK) 2013;140:886–96. doi: 10.1242/dev.086546. [DOI] [PubMed] [Google Scholar]
- 84.Naylor RW, Przepiorski A, Ren Q, Yu J, Davidson AJ. HNF1beta is essential for nephron segmentation during nephrogenesis. J Am Soc Nephrol. 2013;24:77–87. doi: 10.1681/ASN.2012070756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alvelos MI, Rodrigues M, Lobo L, et al. A novel mutation of the HNF1B gene associated with hypoplastic glomerulocystic kidney disease and neonatal renal failure: a case report and mutation update. Medicine. 2015;94:e469. doi: 10.1097/MD.0000000000000469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C. HNF1B-associated renal and extra-renal disease – an expanding clinical spectrum. Nature Rev Nephrol. 2015;11:102–12. doi: 10.1038/nrneph.2014.232. [DOI] [PubMed] [Google Scholar]
- 87.Hasui M, Kaneko K, Tsuji S, et al. Different phenotypes of HNF1ss deletion mutants in familial multicystic dysplastic kidneys. Clin Nephrol. 2013;79:484–7. doi: 10.5414/cn107136. [DOI] [PubMed] [Google Scholar]
- 88.Verhave JC, Bech AP, Wetzels JF, Nijenhuis T. Hepatocyte nuclear factor 1beta-associated kidney disease: more than renal cysts and diabetes. J Am Soc Nephrol. 2016;27:345–53. doi: 10.1681/ASN.2015050544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Anik A, Catli G, Abaci A, Bober E. Maturity-onset diabetes of the young (MODY): an update. J Pediatr Endocrinol Metab. 2015;28:251–63. doi: 10.1515/jpem-2014-0384. [DOI] [PubMed] [Google Scholar]
- 90.Bingham C, Hattersley AT. Renal cysts and diabetes syndrome resulting from mutations in hepatocyte nuclear factor-1beta. Nephrol Dialysis Transplant. 2004;19:2703–8. doi: 10.1093/ndt/gfh348. [DOI] [PubMed] [Google Scholar]
- 91.Bingham C, Ellard S, van’t Hoff WG, et al. Atypical familial juvenile hyperuricemic nephropathy associated with a hepatocyte nuclear factor-1beta gene mutation. Kidney Int. 2003;63:1645–51. doi: 10.1046/j.1523-1755.2003.00903.x. [DOI] [PubMed] [Google Scholar]
- 92.Adalat S, Woolf AS, Johnstone KA, et al. HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol. 2009;20:1123–31. doi: 10.1681/ASN.2008060633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ferre S, Veenstra GJ, Bouwmeester R, Hoenderop JG, Bindels RJ. HNF-1B specifically regulates the transcription of the gammaa-subunit of the Na+/K+-ATPase. Biochem Biophys Res Commun. 2011;404:284–90. doi: 10.1016/j.bbrc.2010.11.108. [DOI] [PubMed] [Google Scholar]
- 94.Bekheirnia MR, Bekheirnia N, Bainbridge MN, et al. Whole-exome sequencing in the molecular diagnosis of individuals with congenital anomalies of the kidney and urinary tract and identification of a new causative gene. Genet Med. 2016 Sep 22; doi: 10.1038/gim.2016.131. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hoskins BE, Cramer CH, 2nd, Tasic V, et al. Missense mutations in EYA1 and TCF2 are a rare cause of urinary tract malformations. Nephrol Dialysis Transplant. 2008;23:777–9. doi: 10.1093/ndt/gfm685. [DOI] [PubMed] [Google Scholar]
- 96.Gong KQ, Yallowitz AR, Sun H, Dressler GR, Wellik DM. A Hox-Eya-Pax complex regulates early kidney developmental gene expression. Mol Cell Biol. 2007;27:7661–8. doi: 10.1128/MCB.00465-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xu J, Wong EY, Cheng C, et al. Eya1 interacts with Six2 and Myc to regulate expansion of the nephron progenitor pool during nephrogenesis. Dev Cell. 2014;31:434–47. doi: 10.1016/j.devcel.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xu J, Xu PX. Eya-six are necessary for survival of nephrogenic cord progenitors and inducing nephric duct development before ureteric bud formation. Dev Dynam. 2015;244:866–73. doi: 10.1002/dvdy.24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xu PX, Adams J, Peters H, Brown MC, Heaney S, Maas R. Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nature Genet. 1999;23:113–7. doi: 10.1038/12722. [DOI] [PubMed] [Google Scholar]
- 100.Chang EH, Menezes M, Meyer NC, et al. Branchio-oto-renal syndrome: the mutation spectrum in EYA1 and its phenotypic consequences. Hum Mutat. 2004;23:582–9. doi: 10.1002/humu.20048. [DOI] [PubMed] [Google Scholar]
- 101.Hoskins BE, Cramer CH, Silvius D, et al. Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am J Hum Genet. 2007;80:800–4. doi: 10.1086/513322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kochhar A, Orten DJ, Sorensen JL, et al. SIX1 mutation screening in 247 branchio-oto-renal syndrome families: a recurrent missense mutation associated with BOR. Hum Mutat. 2008;29:565. doi: 10.1002/humu.20714. [DOI] [PubMed] [Google Scholar]
- 103.Batchelder CA, Lee CC, Martinez ML, Tarantal AF. Ontogeny of the kidney and renal developmental markers in the rhesus monkey (Macaca mulatta) Anat Rec (Hoboken, NJ) 2010;293:1971–83. doi: 10.1002/ar.21242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bower MA, Schimmenti LA, Eccles MR. Renal coloboma syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, et al., editors. GeneReviews®. Seattle, WA: University of Washington; [Google Scholar]
- 105.Kohl S, Hwang DY, Dworschak GC, et al. Mild recessive mutations in six Fraser syndrome-related genes cause isolated congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol. 2014;25:1917–22. doi: 10.1681/ASN.2013101103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kabra M, Gulati S, Ghosh M, Menon PS. Fraser-cryptophthalmos syndrome. Indian J Pediatr. 2000;67:775–8. doi: 10.1007/BF02723939. [DOI] [PubMed] [Google Scholar]
- 107.Saisawat P, Tasic V, Vega-Warner V, et al. Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Kidney Int. 2012;81:196–200. doi: 10.1038/ki.2011.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pavlakis E, Chiotaki R, Chalepakis G. The role of Fras1/Frem proteins in the structure and function of basement membrane. Int J Biochem Cell Biol. 2011;43:487–95. doi: 10.1016/j.biocel.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 109.Vrontou S, Petrou P, Meyer BI, et al. Fras1 deficiency results in cryptophthalmos, renal agenesis and blebbed phenotype in mice. Nature Genet. 2003;34:209–14. doi: 10.1038/ng1168. [DOI] [PubMed] [Google Scholar]
- 110.Pitera JE, Scambler PJ, Woolf AS. Fras1, a basement membrane-associated protein mutated in Fraser syndrome, mediates both the initiation of the mammalian kidney and the integrity of renal glomeruli. Hum Mol Genet. 2008;17:3953–64. doi: 10.1093/hmg/ddn297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pitera JE, Woolf AS, Basson MA, Scambler PJ. Sprouty1 haploinsufficiency prevents renal agenesis in a model of Fraser syndrome. J Am Soc Nephrol. 2012;23:1790–6. doi: 10.1681/ASN.2012020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bertoli-Avella AM, Conte ML, Punzo F, et al. ROBO2 gene variants are associated with familial vesicoureteral reflux. J Am Soc Nephrol. 2008;19:825–31. doi: 10.1681/ASN.2007060692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hwang DY, Kohl S, Fan X, et al. Mutations of the SLIT2-ROBO2 pathway genes SLIT2 and SRGAP1 confer risk for congenital anomalies of the kidney and urinary tract. Hum Genet. 2015;134:905–16. doi: 10.1007/s00439-015-1570-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lu W, van Eerde AM, Fan X, et al. Disruption of ROBO2 is associated with urinary tract anomalies and confers risk of vesicoureteral reflux. Am J Hum Genet. 2007;80:616–32. doi: 10.1086/512735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gbadegesin RA, Brophy PD, Adeyemo A, et al. TNXB mutations can cause vesicoureteral reflux. J Am Soc Nephrol. 2013;24:1313–22. doi: 10.1681/ASN.2012121148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Grieshammer U, Le M, Plump AS, Wang F, Tessier-Lavigne M, Martin GR. SLIT2-mediated ROBO2 signaling restricts kidney induction to a single site. Dev Cell. 2004;6:709–17. doi: 10.1016/s1534-5807(04)00108-x. [DOI] [PubMed] [Google Scholar]
- 117.Wainwright EN, Wilhelm D, Combes AN, Little MH, Koopman P. ROBO2 restricts the nephrogenic field and regulates Wolffian duct–nephrogenic cord separation. Dev Biol. 2015;404:88–102. doi: 10.1016/j.ydbio.2015.05.023. [DOI] [PubMed] [Google Scholar]
- 118.Stangenberg S, Chen H, Wong MG, Pollock CA, Saad S. Fetal programming of chronic kidney disease: the role of maternal smoking, mitochondrial dysfunction, and epigenetic modfification. Am J Physiol Renal Physiol. 2015;308:F1189–96. doi: 10.1152/ajprenal.00638.2014. [DOI] [PubMed] [Google Scholar]
- 119.Abi Khalil C, Travert F, Fetita S, et al. Fetal exposure to maternal type 1 diabetes is associated with renal dysfunction at adult age. Diabetes. 2010;59:2631–6. doi: 10.2337/db10-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Merlet-Benichou C. Influence of fetal environment on kidney development. Int J Dev Biol. 1999;43:453–6. [PubMed] [Google Scholar]
- 121.Hokke SN, Armitage JA, Puelles VG, et al. Altered ureteric branching morphogenesis and nephron endowment in offspring of diabetic and insulin-treated pregnancy. PloS One. 2013;8:e58243. doi: 10.1371/journal.pone.0058243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gilbert T. Vitamin A and kidney development. Nephrol dialysis Transplant. 2002;17(Suppl 9):78–80. doi: 10.1093/ndt/17.suppl_9.78. [DOI] [PubMed] [Google Scholar]
- 123.Goodyer P, Kurpad A, Rekha S, et al. Effects of maternal vitamin A status on kidney development: a pilot study. Pediatr Nephrol (Berlin) 2007;22:209–14. doi: 10.1007/s00467-006-0213-4. [DOI] [PubMed] [Google Scholar]
- 124.Bhat PV, Manolescu DC. Role of vitamin A in determining nephron mass and possible relationship to hypertension. J Nutr. 2008;138:1407–10. doi: 10.1093/jn/138.8.1407. [DOI] [PubMed] [Google Scholar]
- 125.Barker DJ, Bagby SP. Developmental antecedents of cardiovascular disease: a historical perspective. J Am Soc Nephrol. 2005;16:2537–44. doi: 10.1681/ASN.2005020160. [DOI] [PubMed] [Google Scholar]
- 126.Janot M, Cortes-Dubly ML, Rodriguez S, Huynh-Do U. Bilateral uterine vessel ligation as a model of intrauterine growth restriction in mice. Reprod Biol Endocrinol. 2014;12:62. doi: 10.1186/1477-7827-12-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gautier JF, Porcher R, Abi Khalil C, et al. Kidney dysfunction in adult offspring exposed in utero to type 1 diabetes is associated with alterations in genome-wide DNA methylation. PloS One. 2015;10:e0134654. doi: 10.1371/journal.pone.0134654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Woroniecki R, Gaikwad AB, Susztak K. Fetal environment, epigenetics, and pediatric renal disease. Pediatr Nephrol (Berlin) 2011;26:705–11. doi: 10.1007/s00467-010-1714-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hilliard SA, El-Dahr SS. Epigenetics mechanisms in renal development. Pediatr Nephrol (Berlin) 2016;31:1055–60. doi: 10.1007/s00467-015-3228-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ko YA, Mohtat D, Suzuki M, et al. Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol. 2013;14:R108. doi: 10.1186/gb-2013-14-10-r108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Botelho Lourenco EL, Lima Ribeiro RC, Araujo VO, Martino-Andrade AJ, Dalsenter PR, Gasparotto AJ. Fetopathies associated with exposure to angiotensin converting enzyme inhibitor from Tropaeolum majus L. Drug Chem Toxicol. 2016:1–5. doi: 10.1080/01480545.2016.1212366. [DOI] [PubMed] [Google Scholar]
- 132.Chan SK, Riley PR, Price KL, et al. Corticosteroid-induced kidney dysmorphogenesis is associated with deregulated expression of known cystogenic molecules, as well as Indian hedgehog. Am J Physiol Renal Physiol. 2010;298:F346–56. doi: 10.1152/ajprenal.00574.2009. [DOI] [PubMed] [Google Scholar]
- 133.Perez-Aytes A, Ledo A, Boso V, et al. In utero exposure to mycophenolate mofetil: a characteristic phenotype? Am J Med Genet Part A. 2008;146a:1–7. doi: 10.1002/ajmg.a.32117. [DOI] [PubMed] [Google Scholar]
- 134.Morgan TM, Jones DP, Cooper WO. Renal teratogens. Clin Perinatol. 2014;41:619–32. doi: 10.1016/j.clp.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 135.Uludag S, Aydin Y, Yilmaz O, Aksoy F, Bakkaloglu D, Sen C. Multiple fetal anomalies in association with topiramate and oxcarbezepine treatment. Fetal Pediatr Pathol. 2012;31:154–8. doi: 10.3109/15513815.2012.659378. [DOI] [PubMed] [Google Scholar]
- 136.Koleganova N, Piecha G, Ritz E. Prenatal causes of kidney disease. Blood Purif. 2009;27:48–52. doi: 10.1159/000167008. [DOI] [PubMed] [Google Scholar]
- 137.Black MJ, Sutherland MR, Gubhaju L, Kent AL, Dahlstrom JE, Moore L. When birth comes early: effects on nephrogenesis. Nephrology (Carlton, Victoria) 2013;18:180–2. doi: 10.1111/nep.12028. [DOI] [PubMed] [Google Scholar]
- 138.Sutherland MR, Gubhaju L, Moore L, et al. Accelerated maturation and abnormal morphology in the preterm neonatal kidney. J Am Soc Nephrol. 2011;22:1365–74. doi: 10.1681/ASN.2010121266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rodriguez MM, Gomez AH, Abitbol CL, Chandar JJ, Duara S, Zilleruelo GE. Histomorphometric analysis of postnatal glomerulogenesis in extremely preterm infants. Pediatr Dev Pathol. 2004;7:17–25. doi: 10.1007/s10024-003-3029-2. [DOI] [PubMed] [Google Scholar]
- 140.Rhone ET, Carmody JB, Swanson JR, Charlton JR. Nephrotoxic medication exposure in very low birth weight infants. J Matern Fetal Neonatal Med. 2014;27:1485–90. doi: 10.3109/14767058.2013.860522. [DOI] [PubMed] [Google Scholar]
- 141.Drukker A. The adverse renal effects of prostaglandin-synthesis inhibition in the fetus and the newborn. Paediatr Child Health. 2002;7:538–43. doi: 10.1093/pch/7.8.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lavery AP, Meinzen-Derr JK, Anderson E, et al. Urinary NGAL in premature infants. Pediatr Res. 2008;64:423–8. doi: 10.1203/PDR.0b013e318181b3b2. [DOI] [PubMed] [Google Scholar]
- 143.Hodgin JB, Rasoulpour M, Markowitz GS, D’Agati VD. Very low birth weight is a risk factor for secondary focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2009;4:71–6. doi: 10.2215/CJN.01700408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jetton JG, Guillet R, Askenazi DJ, et al. Assessment of worldwide acute kidney injury epidemiology in neonates: design of a retrospective cohort study. Front Pediatr. 2016;4:68. doi: 10.3389/fped.2016.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nguyen HT, Herndon CD, Cooper C, et al. The Society for Fetal Urology consensus statement on the evaluation and management of antenatal hydronephrosis. J Pediatr Urol. 2010;6:212–31. doi: 10.1016/j.jpurol.2010.02.205. [DOI] [PubMed] [Google Scholar]
- 146.Dias CS, Silva JM, Pereira AK, et al. Diagnostic accuracy of renal pelvic dilatation for detecting surgically managed ureteropelvic junction obstruction. J Urol. 2013;190:661–6. doi: 10.1016/j.juro.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 147.Babcook CJ, Silvera M, Drake C, Levine D. Effect of maternal hydration on mild fetal pyelectasis. J Ultrasound Med. 1998;17:539–44. doi: 10.7863/jum.1998.17.9.539. quiz 45–6. [DOI] [PubMed] [Google Scholar]
- 148.Cohen-Overbeek TE, Wijngaard-Boom P, Ursem NT, Hop WC, Wladimiroff JW, Wolffenbuttel KP. Mild renal pyelectasis in the second trimester: determination of cut-off levels for postnatal referral. Ultrasound Obstet Gynecol. 2005;25:378–83. doi: 10.1002/uog.1840. [DOI] [PubMed] [Google Scholar]
- 149.Ahmad G, Green P. Outcome of fetal pyelectasis diagnosed antenatally. J Obstet Gynaecol. 2005;25:119–22. doi: 10.1080/01443610500041446. [DOI] [PubMed] [Google Scholar]
- 150.Sinha A, Bagga A, Krishna A, et al. Revised guidelines on management of antenatal hydronephrosis. Indian J Nephrol. 2013;23:83–97. doi: 10.4103/0971-4065.109403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Oliveira EA, Diniz JS, Cabral AC, et al. Prognostic factors in fetal hydronephrosis: a multivariate analysis. Pediatr Nephrol (Berlin) 1999;13:859–64. doi: 10.1007/s004670050716. [DOI] [PubMed] [Google Scholar]
- 152.Magann EF, Sandlin AT, Ounpraseuth ST. Amniotic fluid and the clinical relevance of the sonographically estimated amniotic fluid volume: oligohydramnios. J Ultrasound Med. 2011;30:1573–85. doi: 10.7863/jum.2011.30.11.1573. [DOI] [PubMed] [Google Scholar]
- 153.Liu DB, Armstrong WR, 3rd, Maizels M. Hydronephrosis: prenatal and postnatal evaluation and management. Clin Perinatol. 2014;41:661–78. doi: 10.1016/j.clp.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 154.Prouty LA, Myers TL. Oligohydramnios sequence (Potter’s syndrome): case clustering in northeastern Tennessee. Southern Med J. 1987;80:585–92. [PubMed] [Google Scholar]
- 155.Allen RW, Jr, Rehm NE, Scott JR, Kochenour NK. Antepartum diagnosis and intrapartum management of lethal renal defects. Obstet Gynecol. 1981;58:379–82. [PubMed] [Google Scholar]
- 156.Oliveira EA, Rabelo EA, Pereira AK, et al. Prognostic factors in prenatally-detected posterior urethral valves: a multivariate analysis. Pediatr Surg Int. 2002;18:662–7. doi: 10.1007/s00383-002-0877-1. [DOI] [PubMed] [Google Scholar]
- 157.Elder JS, O’Grady JP, Ashmead G, Duckett JW, Philipson E. Evaluation of fetal renal function: unreliability of fetal urinary electrolytes. J Urol. 1990;144:574–8. doi: 10.1016/s0022-5347(17)39526-5. discussion 93–4. [DOI] [PubMed] [Google Scholar]
- 158.Caione P, Villa M, Capozza N, De Gennaro M, Rizzoni G. Predictive risk factors for chronic renal failure in primary high-grade vesico-ureteric reflux. Br J Urol Int. 2004;93:1309–12. doi: 10.1111/j.1464-410X.04866.x. [DOI] [PubMed] [Google Scholar]
- 159.Warshaw BL, Hymes LC, Trulock TS, Woodard JR. Prognostic features in infants with obstructive uropathy due to posterior urethral valves. J Urol. 1985;133:240–3. doi: 10.1016/s0022-5347(17)48899-9. [DOI] [PubMed] [Google Scholar]
- 160.Pulido JE, Furth SL, Zderic SA, Canning DA, Tasian GE. Renal parenchymal area and risk of ESRD in boys with posterior urethral valves. Clin J Am Soc Nephrol. 2014;9:499–505. doi: 10.2215/CJN.08700813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ismaili K, Avni FE, Wissing KM, Hall M. Long-term clinical outcome of infants with mild and moderate fetal pyelectasis: validation of neonatal ultrasound as a screening tool to detect significant nephrouropathies. J Pediatr. 2004;144:759–65. doi: 10.1016/j.jpeds.2004.02.035. [DOI] [PubMed] [Google Scholar]
- 162.Wollenberg A, Neuhaus TJ, Willi UV, Wisser J. Outcome of fetal renal pelvic dilatation diagnosed during the third trimester. Ultrasound Obstetr Gynecol. 2005;25:483–8. doi: 10.1002/uog.1879. [DOI] [PubMed] [Google Scholar]
- 163.Mallik M, Watson AR. Antenatally detected urinary tract abnormalities: more detection but less action. Pediatr Nephrol (Berlin) 2008;23:897–904. doi: 10.1007/s00467-008-0746-9. [DOI] [PubMed] [Google Scholar]
- 164.Choi YH, Cheon J-E, Kim WS, Kim I-O. Ultrasonography of hydronephrosis in the newborn: a practical review. Ultrasonography (Seoul, Korea) 2016;35:198–211. doi: 10.14366/usg.15073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bouzada MC, Oliveira EA, Pereira AK, et al. Diagnostic accuracy of fetal renal pelvis anteroposterior diameter as a predictor of uropathy: a prospective study. Ultrasound Obstet Gynecol. 2004;24:745–9. doi: 10.1002/uog.1764. [DOI] [PubMed] [Google Scholar]
- 166.Oliveira EA, Oliveira MC, Mak RH. Evaluation and management of hydronephrosis in the neonate. Curr Opin Pediatr. 2016;28:195–201. doi: 10.1097/MOP.0000000000000321. [DOI] [PubMed] [Google Scholar]
- 167.Conway JJ, Maizels M. The “well tempered” diuretic renogram: a standard method to examine the asymptomatic neonate with hydronephrosis or hydroureteronephrosis. A report from combined meetings of The Society for Fetal Urology and members of The Pediatric Nuclear Medicine Council – The Society of Nuclear Medicine. J Nuclear Med. 1992;33:2047–51. [PubMed] [Google Scholar]