

Abstract
Chromatin cross-linking is widely used for mapping the distribution of chromosomal proteins by immunoprecipitation, but our knowledge of the physical properties of chromatin complexes remains rudimentary. Density gradients have been long used to separate fragments of cross-linked chromatin with their bound proteins from free protein or free DNA. We find that the association of DNA fragments with very-high-molecular-weight protein complexes shifts their buoyant density to values much lower then that of bulk chromatin. We show that in a CsCl gradient, Polycomb response elements, promoters of active genes, and insulator or boundary elements are found at buoyant densities similar to those of free protein and are depleted from the bulk chromatin fractions. In these regions, the low density is associated with the presence of large protein complexes and with high sensitivity to sonication. Our results suggest that separation of different chromatin regions according to their buoyant density may bias chromatin immunoprecipitation results. Density centrifugation of cross-linked chromatin may provide a simple approach to investigate the properties of large chromatin complexes in vivo.
Information about the precise localization of proteins and protein complexes in their chromatin context is essential to understand not only the mechanisms that regulate gene expression but also the complex interactions that are responsible for chromatin architecture in the nucleus. This information is difficult to obtain. Chromatin complexes are dynamic and very sensitive to experimental procedures. One way to circumvent this problem and preserve the native molecular composition of chromatin for subsequent biochemical analysis is to cross-link it. Formaldehyde is the most common cross-linking agent because its small size and high reactivity allow it to act almost instantly inside live cells (6, 25). Chromatin cross-linked in such a way has been widely used as a substrate for mapping the distribution of chromosomal proteins in chromatin immunoprecipitation (ChIP) experiments (13, 17). Despite the wide use of this technique, the physical and chemical properties of cross-linked chromatin are not well known.
The early studies of chromatin cross-linking used ultracentrifugation in equilibrium density gradients to study its physical properties. These studies showed that bulk cross-linked chromatin fragments have a buoyant density of 1.42 to 1.39 g/cm3, which is distinct from the density of free DNA (∼1.69 g/cm3) or cross-linked protein (∼1.25 g/cm3) (3, 29). Estimations of protein-to-DNA ratios in bulk cross-linked chromatin fragments prepared from cells grown on media supplemented with radioactively labeled amino acids and nucleotides suggested that this ratio is a major determinant of its density (10, 29). This conclusion was supported by experiments that determined the density of bulk cross-linked chromatin after treatment with DNase or proteinase (3).
In these early experiments, the coding region of the heat shock protein 70 gene was found to have the same density as bulk chromatin (25), leading to the general conclusion that the densities of different chromatin regions are very similar. Based on this conclusion Solomon et al. proposed the use of density centrifugation as a preparative method to purify cross-linked chromatin fragments from free DNA and free protein for subsequent use in ChIP (25). Later, however, some specific chromatin regions were found to have a density lower than that of a bulk chromatin. Thus Ip and coauthors (11) noticed that the chromatin of the phosphoenolpyruvate carboxykinase gene acquires a lower density upon induction with dexamethasone. More interestingly, Reneker and Brotherton (20) reported that chromatin regions containing the coding part of the chicken β-globin gene and its 3′ tissue-specific enhancer show a distinctly lower density than bulk chromatin when isolated from cells in which these elements are functionally active.
In the present study we took a closer look at the behavior of different chromatin regions in CsCl density equilibrium gradients. After sonication to produce presumably random chromatin fragments, we found to our surprise that the buoyant density of chromatin varies dramatically among different chromatin regions. Some regions known to be associated with high-molecular-weight protein complexes appear to have a density as low as that of free protein and could be effectively separated from bulk chromatin. In a CsCl gradient, Drosophila Polycomb response elements (PREs), transcriptionally engaged promoters, and scs and scs′ boundary/insulator elements are all found at buoyant densities similar to that of free protein. We have also found that the low density of these chromatin regions correlates with their increased sensitivity to sonication. The latter observation fits with the idea that the low buoyant density is caused by unusually high protein/DNA ratios. We speculate that density centrifugation of cross-linked chromatin may provide a simple means for mapping binding sites of high-molecular-weight protein complexes in vivo. Our results also point out that the use of density gradient purification of cross-linked chromatin may bias the results of ChIP.
MATERIALS AND METHODS
Cross-linking, sonication, and density centrifugation.
Schneider L2 cells were grown to a density of ∼5 × 106 cells/ml in Schneider's Drosophila medium (Gibco). To cross-link cells, 36% (wt/vol) formaldehyde (Fluka) was added directly to the medium to a final concentration of 1% and the culture was typically incubated for 10 more minutes at room temperature. The reaction was stopped by addition of glycine solution (pH 7.0) to a final concentration of 125 mM. Cells were washed once with 1× phosphate-buffered saline and once each with washing buffers A (10 mM HEPES [pH 7.6], 10 mM EDTA, 0.5 mM EGTA, 0.25% [vol/vol] Triton X-100) and B (10 mM HEPES [pH 7.6], 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.01% [vol/vol] Triton X-100). The cells were resuspended in sonication buffer (10 mM HEPES [pH 7.6], 1 mM EDTA, 0.5 mM EGTA) to a concentration of ∼5 × 107 cells/ml.
To disrupt the fixed cells, 5 ml of final cell suspension in a 14-ml Falcon tube was subjected to sonication with a Branson 250 sonifier equipped with a microtip. Four 30-s bursts of sonication at maximum constant power (“microtip limit” setting) were applied with 1-min pauses to avoid overheating. Power was gradually increased to avoid foaming. The samples were kept in ice-ethanol during the whole procedure. The supernatant was transferred to a new 14-ml tube, and N-lauroylsarcosine (Fluka) was added to a final concentration of 0.5% (vol/vol). The lysate was incubated for 10 min at +4°C and then cleared by a 5-min centrifugation at + 4°C in a microcentrifuge.
The clear lysate was mixed with 5.68 g of CsCl (molecular biology grade; Fluka), and the sample volume was adjusted to 10 ml with sonication buffer supplemented with 0.5% (vol/vol) N-lauroylsarcosine, to a final sample density of 1.42 g/cm3. The sample was divided between two 5-ml Beckman Ultra-Clear centrifuge tubes and spun for 60 to 72 h at +20°C (195,000 × g [44,000 rpm in a Sorvall S52-ST rotor]). Four-hundred-microliter fractions (NN 2 to 12) were collected with a peristaltic pump after the centrifuge tube was punctured with an 18-gauge needle just above the junction between the round bottom and the straight parts of the tube. The very bottom fraction (N1) was then collected with a 21-gauge, 80-mm needle and a 1-ml insulin syringe. In some cases (for example, see Fig. 2) the top fraction (N12) acquired a small amount of contamination from the high-density solution at the very bottom of the gradient during the collection, thus shifting the overall density of the top fraction towards somewhat higher values. The refraction index (Ri) of the gradient fractions was measured, and the respective fraction densities (ρ) were calculated using the following formula (24): ρ = (10.8601 · Ri) −13.4974.
FIG. 2.

Cross-linked chromatin of the bxd PRE core has a low buoyant density. Distribution of DNA fragments from the bxd PRE region (A to H) along the same CsCl density gradient was compared to that of a DNA fragment from the coding region of the white gene (I). Here and in Fig. 3 the amount of DNA fragment in each gradient fraction (diamonds and solid line) was determined by real-time PCR using specific oligonucleotide primers and expressed as a fraction of the amount of this DNA fragment present in the entire gradient. The dotted line indicates distribution of bulk DNA estimated by gel electrophoresis and expressed in the same way. (J) Distribution of the BP fragment in a different CsCl density gradient made using very lightly cross-linked chromatin (0.3% formaldehyde).
The fractions were dialyzed against buffer containing 4% (vol/vol) glycerol, 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, and 0.5 mM EGTA, and the cross-links were reversed by incubation at 65°C as described previously (4). To isolate the DNA, samples were extracted with equal volumes of phenol-chloroform and then chloroform and the DNA was precipitated with 3 volumes of absolute ethanol at −70°C in the presence of 1/10 volume 3 M sodium acetate (pH 5.0) and 40 μg of glycogen. The resulting DNA pellet was washed with 70% ethanol and dissolved in 250 μl of deionized water. For use in real-time PCR, an aliquot of each DNA sample was further diluted 50 times.
To analyze the protein content of fractions after reversal of cross-links, 10 to 15 μl of each sample was mixed with 3 μl of sodium dodecyl sulfate (SDS) loading buffer and applied to a 4 to 18% gradient SDS-polyacrylamide gel.
ChIP.
For immunoprecipitations, the dialyzed material from the bulk chromatin fractions (densities, 1.42 to 1.37g/cm3) and the three top fractions (densities, 1.33 to1.30g/cm3) of the equilibrium density gradient was adjusted with radioimmunoprecipitation assay buffer (10 mM Tris-HCl [pH 8.0], 140 mM NaCl, 1 mM EDTA, 1% [vol/vol] Triton X-100, 0.1% [wt/vol] SDS, 0.1% [wt/vol] sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride) and precleared by incubation with Sepharose beads conjugated to protein A (Sigma). The control reaction with a comparable aliquot of the chromatin (corresponding to approximately 5 × 107 cells) sampled prior to the density gradient separation was run in parallel. Clear lysates were further incubated overnight at 4°C with 6 μg of antibodies against Polycomb (PC) (19). The antibody complexes were precipitated with protein A-Sepharose beads (Sigma). The beads were washed five times with 1 ml of radioimmunoprecipitation assay buffer, once with 1 ml of LiCl buffer (250 mM LiCl, 10 mM Tris-HCl [pH 8.0], 1 mM EDTA, 0.5% NP-40, 0.5% sodium deoxycholate), and twice with 1 ml of TE (10 mM Tris-HCl [pH 8.0], 1 mM EDTA). The DNA was recovered as described previously (4). Immunoprecipitated DNA was dissolved in 150 μl of water. Control mock immunoprecipitations were done essentially in the same way except that no antibodies were added to the reaction mixture. The yields of the reactions were quantified by real-time PCR.
Real-time PCR and data analysis.
DNA from 5 μl of solution was amplified by real-time PCR in 20 μl of reaction mixture containing 10 μl of 2× SYBR Green PCR Master Mix (Bio-Rad) and a 0.5 μM concentration of the corresponding primer. For sequences of oligonucleotide primers and their respective annealing temperatures, see Table S1 in the supplemental material. PCR was performed in 96-well plates with the iCycler real-time PCR detection system controlled by iCycler iQ software v3.0A (Bio-Rad). The molar amount of the specific DNA fragment present in the reaction mixture was derived from the comparison of the threshold cycle value obtained in PCR to the appropriate six-point standard curve. Standard curves were made by amplification of serial dilutions of genomic DNA isolated from SL2 cells.
To estimate the distribution of total DNA or protein in a gradient, 2.5% of each fraction was analyzed by electrophoresis on agarose or SDS-polyacrylamide gels, respectively. The gel images were quantified with a FluorChem 8800 imaging system (Alpha Innotech).
General methods.
All general DNA methods (22) and general protein procedures (1) were described previously. The probes for Southern blot hybridization were generated by PCR using the same specific oligonucleotide primers as for quantitative amplification of the corresponding regions from the DNA of the density gradient fractions.
RESULTS AND DISCUSSION
PREs migrate with low-density fractions in CsCl density gradients.
In attempts to prepare cross-linked chromatin of SL2 cells for immunoprecipitation, we were surprised to obtain very low yields of DNA sequences containing the bxd PRE after CsCl density gradient purification. To understand how the loss of PRE sequences occurred, we scanned the gradient fractions by using real-time PCR and pairs of oligonucleotide primers specific for DNA fragments from this region (Fig. 1A). Surprisingly, although a small amount of BglI-PstI (BP) and FM5 fragments, which correspond to the core of the bxd PRE (9), are present in the position of bulk chromatin, the majority of the core PRE DNA is found at the top of the gradient, at a density corresponding to that of free protein (Fig. 1C and 2E and F). A less prominent effect is also seen for the FM4 fragment positioned just next to the PRE core (Fig. 2D). In contrast to the PRE, the coding region of the white gene, which is transcriptionally inert in SL2 cells (5), as well as the PRE flanking sequences, coincides with the bulk chromatin fractions of the same gradient (Fig. 1 and 2). An analysis of the protein content of the gradient fractions shows that little protein is found at densities corresponding to free DNA (1.59 g/cm3 and higher), as expected (Fig. 1B and C). A peak of histones corresponds to the fractions containing bulk chromatin (Fig. 1B and C). Most of the protein in the gradient is found near the top, at a density of 1.30 g/cm3.
FIG. 1.

Separation of cross-linked chromatin in a CsCl density gradient. (A) Map of the bxd PRE and adjoining region of the Ubx gene. Sites for the following endonucleases are indicated: BamHI (B') AvaII (A), StyI (S), HinfI (F), EcoRI (R), BglII (B), PstI (P), KpnI (K), and HindIII (H). The coordinates of BamHI and HindIII sites according to the sequence given in reference 14 are shown below. Note the cluster of DNase I-hypersensitive sites (arrows) in the PRE core region. The positions of the DNA fragments, whose migration in the equilibrium density gradients was analyzed by real-time PCR, are indicated below the map. Typical pictures of bulk DNA (B) and protein (C) distributions along the density gradient as assayed by gel electrophoresis. The positions of H1 and core histones are marked by black circles. The fraction densities in grams per cubic centimeter are indicated above the gels.
The bxd PRE chromatin reproducibly displays low buoyant density under a very broad range of cross-linking conditions. No significant differences in the distribution of the bxd PRE DNA could be seen between the gradients prepared from chromatin cross-linked for as little as 2 min at +25°C with 1% formaldehyde solution and chromatin cross-linked with 5% formaldehyde solution for 10 min at the same temperature. Only in the gradients prepared from visibly under-cross-linked material (2 min, 0.3% formaldehyde, +25°C) is the peak of the BP fragment at the low density no longer visible (Fig. 2J). We have noticed, however, that under those conditions the migration of the bxd PRE chromatin is still different from that of the bulk chromatin. While the peak of the bulk chromatin is shifted to higher buoyant density, indicative of poor cross-linking (compare Fig. 2E and J), the PRE DNA appears to be smeared along the entire gradient, with a maximum corresponding to the position of the free DNA. We believe that the reason for this is that the core PRE region is largely devoid of nucleosomes (T. Kahn, Y. Schwartz, G. Dellino, and V. Pirrotta, unpublished results).
Surprised by the unusual behavior of bxd PRE fragments, we then asked if DNA of other PREs can also be found preferentially at the top of the gradient. As shown in Fig. 3C, chromatin of the iab7 PRE behaves essentially in the same way as chromatin of the bxd PRE.
FIG. 3.

Migration of various chromatin regions in a CsCl density gradient. Fractions of a single density gradient, different from the one used in the experiment for which results are shown in Fig. 2, were scanned by real-time PCR using oligonucleotide primers specific for different chromatin regions (diamonds and solid line). To facilitate the comparison between the two sets of experiments, the distributions of BP fragment (A) and white coding region (B) are included. The dotted line indicates the distribution of bulk DNA estimated by gel electrophoresis.
Chromatin fragments associated with large protein complexes have inherently low density.
We reasoned that, since the molecular size of the Polycomb complexes associated with the PRE is reputed to be very large (16, 23, 27), the density contribution of the protein might greatly outweigh that of the DNA, resulting in a much lower overall density of the bxd PRE chromatin than that of bulk chromatin, in which the nucleosomal histones constitute the primary protein component. We therefore determined the positions in the density gradient of several regions that are believed to be stably associated with protein complexes of high molecular weight. The promoters of the hsp26 and β1-tubulin genes are known to contain a transcriptionally active, but paused, high-molecular-weight RNA polymerase II complex as well as other promoter factors (21). The promoter region of the actin 5C gene is also found in association with the RNA polymerase II transcription complex; however, the rate of RNA polymerase binding to and initiation of transcription from this promoter closely approximates the rate at which polymerase escapes the promoter and enters into productive transcription (21). Despite differences in their regulation, these three promoter regions are found in the low-density fractions at the top of the gradient (Fig. 3D, E, and F). In contrast, the promoters of the white and Ubx genes, which are inactive in SL2 cells (2, 5), are found in the same fractions as bulk chromatin (Fig. 3G and H). We also looked at other types of chromatin regions that are thought to be associated with chromatin complexes of potentially high molecular weight. The insulator elements scs and scs′ bind complexes containing, respectively, Zw5 and BEAF32 proteins (8, 30). These complexes are thought to anchor the insulator elements to other elements and produce topological constraints. Consistent with this idea, scs and scs′ sequences are found in the low-density fractions at the top of the gradient made from cross-linked chromatin of SL2 cells (Fig. 3G and G).
The scs and scs′ insulators are both also in close proximity to transcriptional promoters. Whether the density shift is caused by the promoter complexes or by the insulator complexes, these results support the interpretation that the binding of high-molecular-weight protein complexes is a necessary prerequisite for the chromatin region to acquire a low buoyant density. Is it the only requirement?
Chromatin regions with low buoyant density are hypersensitive to sonication.
Sonication has been most frequently used to generate soluble chromatin fragments because of the widely held belief that it most nearly approximates a random fragmentation. The cross-linked chromatin used in our analysis had been solubilized by sonication, producing fragments whose DNA ranged in length from 20 to 0.2 kb, with the majority of the fragments having a length of 2 to 3 kb. If we assume that the partial volume of a DNA-protein complex is equal to the sum of the partial volumes of DNA (VD) and protein (VP), we can write:
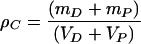 |
(1) |
where ρC is the buoyant density of a particular chromatin fragment, mD is the mass of the DNA, and mP is the mass of the protein bound to that fragment. If we ignore the differences in hydration of proteins and DNA due to variation in the activity of water at different CsCl concentrations and estimate VD and VP from the buoyant densities of free DNA (ρD) and free protein (ρP) from equation 1, we obtain:
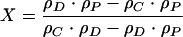 |
(2) |
where X = mP/mD, the weight ratio of protein to DNA in the chromatin complex (for the complete derivation, see also the supplemental material). Though equation 2 is not entirely correct, it is accurate enough for the present purpose (for a confirmation, see reference 3). Using buoyant densities of 1.69 g/cm3 for the DNA and 1.25 g/cm3 for the protein in equation 2, we can calculate that a complex with a buoyant density of 1.30 g/cm3 would have a ratio X of ∼6. Therefore, in order to acquire a density of 1.30 g/cm3, a 2-kb DNA fragment should be associated with a protein complex of about 8 MDa. Even allowing for the weight of a histone component that may be associated with the 2-kb DNA fragment, the molecular weight of the protein complex that has to be associated, for example, with the PRE region would greatly exceed the weight of any known Polycomb complex (16, 23, 27). These speculations formally suggest two possibilities. Either the molecular masses of the complexes bound to the PRE, promoter, and insulator regions are much larger than usually envisioned or the average length of the DNA fragments associated with low-buoyant-density chromatin is much shorter than 2 kb. It follows from equation 2 that in order to outweigh the DNA a Polycomb repressive complex 1 with a reported molecular mass of around 2 MDa (23) should be associated with DNA fragments shorter than 500 bp.
To discriminate between these two possibilities, we determined the size distribution of fragments containing the bxd PRE, the hsp26 promoter, or the white gene coding regions in the SL2 chromatin preparation that was afterwards analyzed by ultracentrifugation in the CsCl density gradient. As shown in Fig. 4, the DNA fragments containing the white gene coding sequences vary in size from 20 to 0.2 kb, with an average size around 2 kb; i.e., they have a size range similar to that of bulk chromatin (Fig. 1B). In contrast DNA fragments containing the bxd PRE or the hsp26 promoter regions are much smaller, about 200 to 500 bp. We conclude that chromatin regions with low buoyant density are hypersensitive to sonication.
FIG. 4.
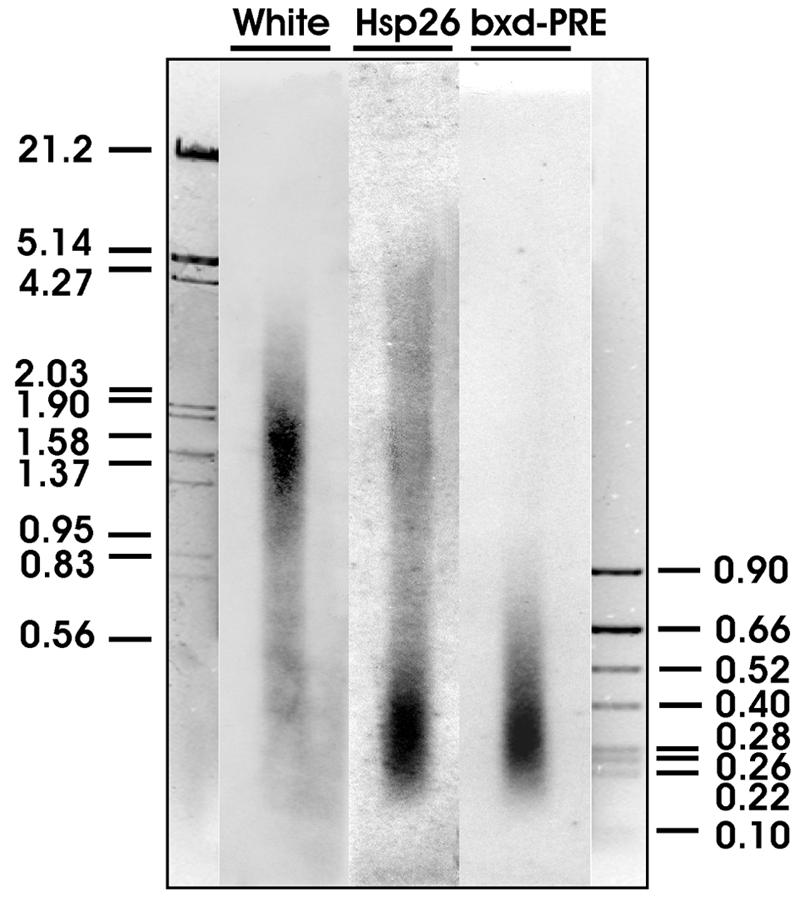
DNA fragment sizes of different regions in sonicated chromatin. The total DNA isolated from cross-linked sonicated SL2 chromatin was separated by electrophoresis, transferred to a nylon membrane, and hybridized with radioactively labeled probes from the coding region of the white gene, the bxd PRE, and the hsp26 promoter region. The positions of marker DNA fragments (sizes in kilobases) are indicated.
It is clear that sonication hypersensitivity by itself could not account for the low density. The smallest fragments in our chromatin preparations are approximately 200 bp in length. That is about the size of the DNA fragment incorporated in a single nucleosome (12). The ratio of protein to DNA mass in the nucleosome is very close to 1, which is approximately the ratio calculated for the bulk chromatin fraction from equation 2. Therefore, in order to reach a density of 1.30 g/cm3, even the most sonication-sensitive chromatin regions must be associated with protein complexes at least five times heavier then a nucleosome, i.e., with a complex of more than 550 kDa.
It is interesting that Reneker and Brotherton (20) reported that discrete regions of the chicken β-globin gene cluster have tissue-specific hypersensitivity to sonication and, more intriguingly, that chromatin fragments from those regions appeared to have a low buoyant density. Although it is difficult to compare directly our density centrifugation experiments with those of Reneker and Brotherton (20) since they used CsCl-guanidine hydrochloride gradients instead of CsCl gradients supplemented with N-lauroylsarcosine, the two sets of observations look strikingly similar.
Many, but not all, of the tissue-specific sonication-hypersensitive sites of the chicken β-globin locus coincided with DNase I-hypersensitive sites (20). Similarly, the bxd PRE, iab7 PRE, hsp26 promoter, scs, and scs′ chromatin regions that displayed lower density in our experiments have been shown previously to contain multiple DNase I-hypersensitive sites (7, 15, 26, 28). It is possible that DNase-hypersensitive sites might often be sites sensitive to mechanical breakage of the DNA. More likely, we suspect that, in the general case, the discontinuities in mass distribution are principally responsible for sonication sensitivity near the binding sites of high-molecular-weight protein complexes.
In the experiment for which results are presented in Fig. 4 the different sensitivities to sonication between cross-linked chromatin of the white gene coding region and the chromatin of the bxd PRE or hsp26 promoter region are highlighted by the relatively large average size of the DNA fragments (about 2 kb) in the bulk chromatin preparation. We would like to emphasize that the distinct buoyant densities of these regions are not restricted to preparations of mildly fragmented cross-linked chromatin. As shown in Fig. 5, the DNA of the bxd PRE core could still be found in the low-density fractions of the CsCl gradient, although the chromatin in this preparation was more intensely sonicated, yielding an average DNA fragment size below 1 kb (compare Fig. 5A and 1B). We should also note that in our hands any well-cross-linked chromatin, i.e., chromatin retaining a full complement of nucleosomal histones covalently attached to the DNA and thus banding at a density of about 1.40g/cm3 in the CsCl gradient, appeared to be resistant to shearing to DNA fragments much smaller then 1 kb. Higher sonication sensitivity of bulk chromatin was always associated with poor cross-linking, indicated by the shift of the chromatin band to higher densities. Currently we do not know if more intensive fragmentation of well-cross-linked chromatin by more powerful sonifiers is possible without affecting the integrity of large protein complexes.
FIG. 5.

Cross-linked chromatin of the bxd PRE core retains its low buoyant density after intensive sonication. The chromatin cross-linked as described in the Materials and Methods was subjected to prolonged shearing (16 30-s bursts of sonication as compared to the usual 4). (A) As demonstrated by electrophoresis in a 1% agarose gel, the sizes of the DNA fragments isolated from chromatin treated in such a way (lane 2) range from 4 kbp to 100 bp, with an average size below 1 kb. Lanes 1 and 3 contain DNA size markers. (B) Real-time PCR with DNA from the fractions of the density gradient prepared from this chromatin shows that the bxd PRE (diamonds and solid line) still has a low density compared to that of the bulk chromatin or the coding region of the white gene (empty squares and dotted line).
Density centrifugation biases ChIP results.
It has been generally assumed that the buoyant densities of different regions of cross-linked chromatin are fairly constant and that equilibrium density centrifugation would be a useful purification step before immunoprecipitation (25). Though many of the ChIP protocols have now deleted this time-consuming step, it is still found in all published versions of the procedure designed for Drosophila cultured cells and embryos (4, 17, 18). More importantly, many of the published ChIP data on the in vivo distribution of Drosophila chromatin proteins were obtained using a CsCl density gradient purification step. Since our results demonstrate that some regions appear to be almost quantitatively excluded from the fractions of bulk cross-linked chromatin, we decided to test directly if equilibrium density centrifugation changes the outcome of ChIP.
Equal volumes of dialyzed material from the bulk chromatin fractions (densities, 1.42 to1.37g/cm3) and from the top three fractions (densities, 1.33 to 1.30g/cm3) of the same gradient were subjected to ChIP with antibodies against PC protein. A control reaction with an equivalent amount of the same chromatin, which was sampled prior to density gradient centrifugation, was run in parallel. The amount of BP fragment immunoprecipitated in the presence or absence of anti-PC antibodies was quantified by real-time PCR and compared to that of the white coding region and the Ubx promoter. Consistent with expectations, the absolute amount of BP DNA precipitated with anti-PC antibodies from the low-density fractions is about 10 times higher than that from the bulk chromatin fractions (Fig. 6A). This observation indicates that a substantial fraction of the bxd PRE DNA from the top of the CsCl gradient is indeed associated with the PC complex.
FIG. 6.

Immunoprecipitation of chromatin from different parts of the CsCl equilibrium density gradient. Two density gradients made from the same chromatin were run in parallel. Both were analyzed for the distribution of bxd PRE, white coding, and Ubx promoter regions as well as bulk DNA. The results of the analysis for one of the gradients are shown in Fig. 2. ChIP was carried out using equal volumes from the gradient fractions containing the bulk DNA peak (bulk chromatin) and the low-density material (top fraction) as well as equivalent amounts of input chromatin before ultracentrifugation (total chromatin). The absolute amounts of specific DNA fragments precipitated with anti-PC and without antibody were quantified by real-time PCR. (A) The results from the experiments with the two density gradients were averaged and plotted with standard deviations shown. (B) The same data are represented after normalization to the initial amounts of each DNA fragment in the corresponding ChIP reaction.
If we compare the absolute amounts of immunoprecipitation obtained with the bulk chromatin fractions, we would conclude that more PC is associated with the Ubx promoter and white coding region than with the PRE. It is clear from Fig. 2 and 5, however, that the reason for this is that the bulk fractions are specifically depleted of PRE sequences. When we calculate the amount of precipitation as a function of the amount of input sequence present in the different fractions (Fig. 6B), the PRE sequences appear always to be the primary binding site for PC. The Ubx promoter precipitation, though significantly higher than background, is severalfold lower.
The presence of PRE sequences in the bulk fractions remains to be accounted for. Three factors may be involved in the explanation. One is the possible heterogeneity in the complexes present at the PRE: if the assembly of the Polycomb group (PcG) complex is a dynamic process, at any one time a fraction of the cells might contain an incompletely assembled complex. Another factor is the degree of sonication. Even with a homogeneous complex population, some chromatin fragments might survive the sonication treatment with a substantially larger DNA fragment. Third is the degree of cross-linking. Again, in a population of chromatin fragments, some fractions might remain incompletely cross-linked such that some of the components of the PcG complex might be lost from the fragment during the CsCl gradient fractionation. ChIP is clearly a complex procedure with many variables that need to be taken into consideration for a meaningful evaluation of the results. At any rate it is clear that purification of cross-linked chromatin by density centrifugation can introduce a strong bias in the ChIP results. Thus, much published data may need to be reexamined.
In conclusion, we would like to note that the separation of different chromatin regions according to their buoyant densities seems to provide a simple approach to test if a region is associated with a large protein complex in vivo. In principle such an approach requires no prior information about protein complex composition and is very sensitive since little starting material is needed. In its current version the approach is limited to regions hypersensitive to sonication. At present we do not know whether binding of large protein complexes necessarily leads to sonication sensitivity. We predict, however, that if bulk chromatin can be fragmented to less than 500 bp in DNA length without affecting the integrity of large protein complexes, the in vivo binding sites of such complexes could be identified and the occupied sites could be separated from nonoccupied sites by density centrifugation. This could be used to detect fractions of a population of sites in transient states of a dynamic equilibrium.
Supplementary Material
Acknowledgments
We are grateful to Sergei Demakov for helpful discussions.
This work was supported by grants to V.P. from the Swiss National Science Foundation and from the “Frontiers in Genetics” Pôle de Recherche National of the Swiss National Science Foundation.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Bollag, D. M., M. D. Rozycki, and S. J. Edelstein. 1996. Protein methods. Wiley-Liss, Inc., New York, N.Y.
- 2.Breiling, A., B. M. Turner, M. E. Bianchi, and V. Orlando. 2001. General transcription factors bind promoters repressed by Polycomb group proteins. Nature 412:651-655. [DOI] [PubMed] [Google Scholar]
- 3.Brutlag, D., C. Schlehuber, and J. Bonner. 1969. Properties of formaldehyde-treated nucleohistone. Biochemistry 8:3214-3218. [DOI] [PubMed] [Google Scholar]
- 4.Cavalli, G., V. Orlando, and R. Paro. 1999. Mapping DNA target sites of chromatin-associated proteins by formaldehyde cross-linking in Drosophila embryos, p. 20-30. In W. A. Bickmore (ed.), Chromosome structural analysis: a practical approach. Oxford University Press, Oxford, United Kingdom.
- 5.de Cock, J. G., E. C. Klink, W. Ferro, P. H. Lohman, and J. C. Eeken. 1991. Repair of UV-induced pyrimidine dimers in the individual genes Gart, Notch and white from Drosophila melanogaster cell lines. Nucleic Acids Res. 19:3289-3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dedon, P. C., J. A. Soults, C. D. Allis, and M. A. Gorovsky. 1991. A simplified formaldehyde fixation and immunoprecipitation technique for studying protein-DNA interactions. Anal. Biochem. 197:83-90. [DOI] [PubMed] [Google Scholar]
- 7.Dellino, G. I., C. Tatout, and V. Pirrotta. 2002. Extensive conservation of sequences and chromatin structures in the bxd polycomb response element among Drosophilid species. Int. J. Dev. Biol. 46:133-141. [PubMed] [Google Scholar]
- 8.Gaszner, M., J. Vazquez, and P. Schedl. 1999. The Zw5 protein, a component of the scs chromatin domain boundary, is able to block enhancer-promoter interaction. Genes Dev. 13:2098-2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horard, B., C. Tatout, S. Poux, and V. Pirrotta. 2000. Structure of a Polycomb response element and in vitro binding of polycomb group complexes containing GAGA factor. Mol. Cell. Biol. 20:3187-3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ilyin, Y. V., and G. P. Georgiev. 1969. Heterogeneity of deoxynucleoprotein particles as evidenced by ultracentrifugation of cesium chloride density gradient. J. Mol. Biol. 41:299-303. [DOI] [PubMed] [Google Scholar]
- 11.Ip, Y. T., V. Jackson, J. Meier, and R. Chalkley. 1988. The separation of transcriptionally engaged genes. J. Biol. Chem. 263:14044-14052. [PubMed] [Google Scholar]
- 12.Kornberg, R. D., and Y. Lorch. 1999. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98:285-294. [DOI] [PubMed] [Google Scholar]
- 13.Kuo, M. H., and C. D. Allis. 1999. In vivo cross-linking and immunoprecipitation for studying dynamic protein:DNA associations in a chromatin environment. Methods 19:425-433. [DOI] [PubMed] [Google Scholar]
- 14.Martin, C. H., C. A. Mayeda, C. A. Davis, C. L. Ericsson, J. D. Knafels, D. R. Mathog, S. E. Celniker, E. B. Lewis, and M. J. Palazzolo. 1995. Complete sequence of the bithorax complex of Drosophila. Proc. Natl. Acad. Sci. USA 92:8398-8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mishra, R. K., J. Mihaly, S. Barges, A. Spierer, F. Karch, K. Hagstrom, S. E. Schweinsberg, and P. Schedl. 2001. The iab-7 Polycomb response element maps to a nucleosome-free region of chromatin and requires both GAGA and pleiohomeotic for silencing activity. Mol. Cell. Biol. 21:1311-1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng, J., C. M. Hart, K. Morgan, and J. A. Simon. 2000. A Drosophila ESC-E(Z) protein complex is distinct from other Polycomb group complexes and contains covalently modified ESC. Mol. Cell. Biol. 20:3069-3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orlando, V. 2000. Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitation. Trends Biochem. Sci. 25:99-104. [DOI] [PubMed] [Google Scholar]
- 18.Orlando, V., H. Strutt, and R. Paro. 1997. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods 11:205-214. [DOI] [PubMed] [Google Scholar]
- 19.Poux, S., D. McCabe, and V. Pirrotta. 2001. Recruitment of components of Polycomb Group chromatin complexes in Drosophila. Development 128:75-85. [DOI] [PubMed] [Google Scholar]
- 20.Reneker, J. S., and T. W. Brotherton. 1991. Discrete regions of the avian beta-globin gene cluster have tissue-specific hypersensitivity to cleavage by sonication in nuclei. Nucleic Acids Res. 19:4739-4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rougvie, A. E., and J. T. Lis. 1990. Postinitiation transcriptional control in Drosophila melanogaster. Mol. Cell. Biol. 10:6041-6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 23.Shao, Z., F. Raible, R. Mollaaghababa, J. R. Guyon, C. T. Wu, W. Bender, and R. E. Kingston. 1999. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell 98:37-46. [DOI] [PubMed] [Google Scholar]
- 24.Sober, H. A. 1973. Selected data for molecular biology, p. J-296. In H.A. Sober (ed.), Handbook of biochemistry. CRC Press, Cleveland, Ohio.
- 25.Solomon, M. J., P. L. Larsen, and A. Varshavsky. 1988. Mapping protein-DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell 53:937-947. [DOI] [PubMed] [Google Scholar]
- 26.Thomas, G. H., and S. C. Elgin. 1988. Protein/DNA architecture of the DNase I hypersensitive region of the Drosophila hsp26 promoter. EMBO J. 7:2191-2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tie, F., J. Prasad-Sinha, A. Birve, A. Rasmuson-Lestander, and P. J. Harte. 2003. A 1-megadalton ESC/E(Z) complex from Drosophila that contains Polycomblike and RPD3. Mol. Cell. Biol. 23:3352-3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Udvardy, A., E. Maine, and P. Schedl. 1985. The 87A7 chromomere. Identification of novel chromatin structures flanking the heat shock locus that may define the boundaries of higher order domains. J. Mol. Biol. 185:341-358. [DOI] [PubMed] [Google Scholar]
- 29.Varshavsky, A. J., V. V. Bakayev, Y. V. Ilyin, A. A. Bayev, Jr., and G. P. Georgiev. 1976. Studies on chromatin. Free DNA in sheared chromatin. Eur. J. Biochem. 66:211-223. [DOI] [PubMed] [Google Scholar]
- 30.Zhao, K., C. M. Hart, and U. K. Laemmli. 1995. Visualization of chromosomal domains with boundary element-associated factor BEAF-32. Cell 81:879-889. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.