

ABSTRACT
Constitutive activation of the MTOR pathway is a key feature of defects in the tuberous sclerosis complex and other genetic neurodevelopmental diseases, collectively referred to as MTORopathies. MTORC1 hyperactivity promotes anabolic cell functions such as protein synthesis, yet at the same time catabolic processes such as macroautophagy/autophagy are suppressed. Mitochondria are major substrates of autophagy; however, their role in MTORopathies remains largely undefined. Here, we review our recent study showing that several aspects of mitochondrial function, dynamics and turnover are critically impaired in neuronal models of TSC. We discuss the relevance of these findings to neurological manifestations associated with TSC and speculate on autophagy as a novel treatment target for MTORopathies.
KEYWORDS: autism, autophagy, axonal transport, carbamazepine, lysosome, mitochondria, mTOR, mTORC1, rapamycin, synapse
Tuberous sclerosis complex (TSC) is an autosomal-dominant disease that affects many different organ systems including the central nervous system. In many cases, TSC leads to disabling neurological manifestations such as epilepsy, intellectual disability and autism spectrum disorder. Loss-of-function mutations in TSC1 or TSC2 are the genetic cause of the disease and both genes encode critical upstream suppressors of MTORC1, a key signaling complex that controls many aspects of cellular metabolism. Whereas MTORC1 inhibitors have emerged as therapeutics, their role in the treatment of complex neurological manifestations in TSC remains uncertain.
Numerous TSC1/2- and MTORC1-related molecular pathways have been explored in nonneuronal cell types, but have not been probed in neurons. Elucidating the downstream biology of loss of TSC1/2 in neurons may thus provide more precise therapeutic targets for neurological manifestations in TSC. One conundrum in the MTORopathy field is that constitutive activation of MTORC1 promotes anabolic metabolism, while repressing catabolic processes including autophagy. Mitochondria play a crucial role in supporting this anabolic metabolism, yet at the same time their turnover depends on autophagic flux. How these circumstances, an increased demand for healthy mitochondria on the one hand and a blockade of their turnover on the other, go together remains unknown. Neuropathological studies in TSC have documented impaired autophagic flux and accumulation of organelles, including mitochondria, which may lead to elevated levels of oxidative stress. Despite this, however, relatively little is known about the impact of MTORC1 on the turnover of mitochondria in neurons.
Using TSC as a genetically tractable model, we investigated the impact of aberrant MTOR signaling on mitochondrial dynamics, function and turnover in neurons in vitro and in vivo (Fig. 1). In summary, TSC-deficient neurons:
Progressively accumulate mitochondria in cell bodies but are depleted of functional mitochondria in axons, including those that support presynaptic sites
Show a shift toward increased retrograde transport of axonal mitochondria and a disorganized transport pattern promoting a progressive depletion of mitochondria from axons
Demonstrate impaired respiratory chain function and a decrease in mitochondrial membrane potential
Show impaired local axonal and global mitophagic flux following induction of mitophagy trough mitochondrial depolarization
Display impaired autophagosome turnover following mitophagy induction, thus localizing the defect to the late and autophagy-dependent stages of the mitophagy pathway
Show restoration of mitochondrial homeostasis after stimulation of autophagy through MTOR-dependent (rapamycin) or MTOR-independent (carbamazepine) mechanisms
Figure 1.
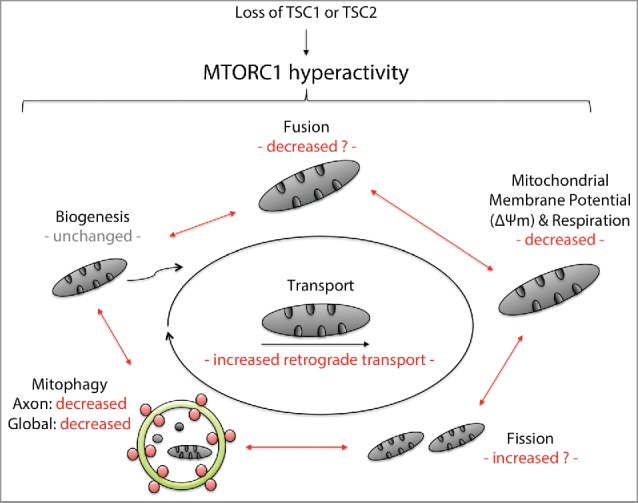
Synopsis of mitochondrial dynamics and turnover in neuronal models of TSC. TSC is a prototypical MTORopathy and a genetically-tractable model that enables us to study the downstream consequences of a constitutive activation of MTORC1 in neurons. Using a variety of methods in neuronal models of TSC, including induced pluripotent stem cell-derived neurons from TSC patients and neuron-specific Tsc1 knockout mice, we were able to show that loss of TSC1/2 leads to impaired mitophagy both in the axon and cell body. This leads to an increase in mitochondrial mass and mitochondrial dysfunction signified by mitochondrial fragmentation as well as loss of the mitochondrial membrane potential and respiratory chain capacity in a significant subset of organelles. In the axon, retrograde transport of mitochondria is enhanced, which contributes to a progressive depletion of mitochondria from axons including from presynaptic sites.
Our first set of experiments asked the simple question: Do TSC neurons contain more mitochondria? Using a flow-cytometry-based approach we confirmed that indeed the mitochondrial mass in Tsc2-deficient neurons is significantly increased. Looking at this in greater detail using microscopy, we were surprised to find that while there is a significant increase in mitochondria in the cell body, axons of Tsc2-deficient neurons are depleted of mitochondria. Interestingly, we also found that mitochondria in these neurons are smaller and display a rounder configuration, which suggests mitochondrial fragmentation. Confirming these observations in vivo using electron microscopy, we found that callosal projection axons in Tsc1cc;Syn1-Cre+ mice contain fewer and smaller mitochondria. To test the functional implications of these findings, we probed the respiratory function of mitochondria in cultured Tsc2-deficient neurons. We found deficits in all domains of respiratory function and a significant decrease in the mitochondrial membrane potential, particularly in mitochondria that localize to the dendritic and axonal compartments. These findings were confirmed in induced pluripotent stem cell-derived human cortical neurons from TSC patients, which interestingly show a more prominent phenotype when we introduced a second loss-of-function mutation in TSC2 using transcription activator-like effector nucleases/TALEN.
Is this increase in mitochondrial mass a result of an increase in mitochondrial biogenesis or a reduction in mitochondrial turnover? While we did not observe significant changes in factors associated with mitochondrial biogenesis, we found that the turnover of mitochondria through autophagy, or mitophagy, is critically impaired both in the axon and globally. In the axon, we found that mitochondrial depolarization in Tsc2-deficient neurons leads to an impaired recruitment of LC3-positive phagophores (the precursors to autophagosomes) compared with controls. Again using a flow cytometry-based approach to probe the mitochondrial mass in a large number of neurons, we found that stimulation of mitophagy with agents that depolarize the mitochondrial membrane leads to the degradation of a small fraction of mitochondria in controls, while no such turnover is observed in Tsc2-deficient neurons. Corroborating a block in mitophagy, we found that several components of the mitophagy pathway accumulate in Tsc2-deficient neurons, including markers of mitophagy initiation such as PARK2/parkin but also autophagosome markers such as LC3-II. Mapping the deficit to the late stages of the pathway, we found that the recruitment of phagophores to damaged mitochondria as well as the fusion of autophagosomes with lysosomes is impaired.
Probing the therapeutic significance of our findings, we treated Tsc2-deficient neurons and Tsc1cc;Syn1-Cre+ mice with the MTOR inhibitor rapamycin and found a restoration of mitochondrial phenotypes including mitochondrial mass, transport and mitophagy. To test whether this was secondary to a correction of autophagy, we also explored MTOR-independent enhancers of autophagy. We found that treatment with carbamazepine similarly restores mitochondrial homeostasis in TSC and importantly replenishes functional mitochondria at synapses.
These results provide evidence for a previously unappreciated role of impaired neuronal mitophagy in TSC and identify mitochondrial turnover as a novel therapeutic target in MTORopathies. With the emergence of novel compounds that modulate autophagy independently of MTOR, we hope that future studies will help develop autophagy-based treatments and biomarkers in addition to approaches that target MTOR.
Abbreviations
- MTOR
mechanistic target of rapamycin
- MTORC1
mechanistic target of rapamycin complex 1
- TSC
tuberous sclerosis complex
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
The authors acknowledge support by the Graduate Academy (D.E.-F.) and the Young Investigator Award Program at Heidelberg University (D.E.-F., L.W.), the Daimler & Benz Foundation (D.E.-F.), the Reinhard-Frank Foundation (D.E.-F.), the German National Academic Foundation (A.S.), the National Institutes of Health (NIH) (U54HD090255), the Nancy Lurie Marks Family Foundation and the Boston Children's Hospital Translational Research Program (M.S.).