

ABSTRACT
During the process of oxidative phosphorylation, protons are pumped into the mitochondrial intermembrane space to establish a mitochondrial membrane potential (MMP). The electrochemical gradient generated allows protons to return to the matrix through the ATP synthase complex and generates ATP in the process. MitoQ is a lipophilic cationic drug that is adsorbed to the inner mitochondrial membrane; however, the cationic moiety of MitoQ remains in the intermembrane space. We found that the positive charges in MitoQ inhibited the activity of respiratory chain complexes I, III, and IV, reduced proton production, and decreased oxygen consumption. Therefore, a pseudo-MMP (PMMP) was formed via maintenance of exogenous positive charges. Proton backflow was severely impaired, leading to a decrease in ATP production and an increase in AMP production. Excess AMP activates AMP kinase, which inhibits the MTOR (mechanistic target of rapamycin) pathway and induces macroautophagy/autophagy. Therefore, we conclude that MitoQ increases PMMP via proton displacement with exogenous positive charges. In addition, PMMP triggered autophagy in hepatocellular carcinoma HepG2 cells via modification of mitochondrial bioenergetics pathways.
KEYWORDS: autophagy, energy metabolism, MitoQ, proton displacement, pseudo-MMP
Introduction
The mitochondrion is a double-membrane-bound organelle. Protein complexes in the inner membrane (NADH dehydrogenase, cytochrome c reductase, and cytochrome c oxidase) transfer protons (H+), and the incremental production of energy is used to pump protons back into the intermembrane space.1 A strong electrochemical gradient is established across the inner membrane as the proton concentration increases in the intermembrane space. Protons can return to the matrix through the ATP synthase complex, and their potential energy is used to synthesize ATP from ADP and inorganic phosphate.2 Therefore, the MMP is used to maintain the cellular energy supply, which is essential for normal physiological processes.3,4
MitoQ consists of a triphenylphosphonium (TPP+) cation moiety covalently attached to the ubiquinone moiety of the endogenous antioxidant CoQ10 via a 10-carbon aliphatic carbon chain.5 The positive charge of TPP+ allows cation accumulation within the mitochondria; cation transport is driven by the large transmembrane potential (ΔΨm) (150–180 mV), this transmembrane potential is generated by the proton gradient formed during the transfer of an electron to oxygen. Recent studies have shown that MitoQ and several structural and functional analogs of MitoQ (MitoCP, TPP+ conjugated to 3-carboxyproxylnitroxide; mitovitamin E, TPP+ conjugated to vitamin E) selectively accumulate in the mitochondria and consequently induce greater toxicity in cancer cells and inhibit tumor growth in vitro and vivo.6-8 However, the mechanisms associated with the toxic effect of MitoQ in cancer cells are unclear.
Autophagy is the natural, tissue-destructive mechanism that degrades unnecessary or dysfunctional cellular components. PRKAA/AMPK (AMP-activated protein kinase) is involved in the regulation of autophagy.9 Under normal physiological conditions, binding of ATP to AMPK keeps the kinase activity low. When energy depletion occurs in cells, the AMP:ATP ratio increases, the AMP levels increase, and the exchange of AMP for ATP increases the kinase activity 5-fold.10 The activated AMPK inhibits MTOR via phosphorylation, and activates TSC2 (tuberous sclerosis 2), a negative regulator of MTOR,11 leading to increased formation of autophagosomes and autophagy flux. AMPK positively regulates autophagy in mammalian cells, which is consistent with the adverse effect of AMPK on MTOR signaling.12 Autophagy controls various physiological processes, including adaptation to starvation, degradation of aberrant structures, turnover of damaged organelles, tumor suppression, and programmed cell death.13,14 The aim of this study was to investigate the induction of autophagy by MitoQ by generating exogenous positive charges and interfering with MMP, identify the relationship between changes in mitochondrial bioenergetics and cytotoxicity in cancer cells, and evaluate the effect of MitoQ on autophagy.
Results
MitoQ induces autophagy in HepG2 cells and inhibits cellular proliferation
Autophagy can be seen by an increase of autophagosomes in cells. As shown in Figure 1A, after 2 h treatment, 1 µM MitoQ caused the accumulation of large cytoplasmic vacuoles in HepG2 cells. Cyto-ID, a recently developed cationic amphiphilic tracer dye, labels autophagic compartments with minimal staining of lysosomes, suggesting that it is a specific autophagy marker. As shown in Figure 1B, no or very weak Cyto-ID fluorescence was detected in the control group whereas treatment with MitoQ for 2 h increased the signal of Cyto-ID (green fluorescence). The strength of induction of autophagy by MitoQ was similar to that of rapamycin (an autophagy inducer). In addition, the redistribution of GFP-LC3 from a diffusive cytosolic to a punctate autophagosome-associated pattern was also observed after treatment with MitoQ (Fig. S1). The effect of MitoQ on the proliferation of HepG2 cells was determined for up to 84 h using an RT-CES system (real-time cell electronic sensing). MitoQ (1 µM) significantly inhibited cell growth (Fig. 1C), but not induce obvious apoptosis (Fig. S2). This observation demonstrated that the inhibition of proliferation was not apoptosis-dependent, but potentially involved the induction of autophagy.
Figure 1.
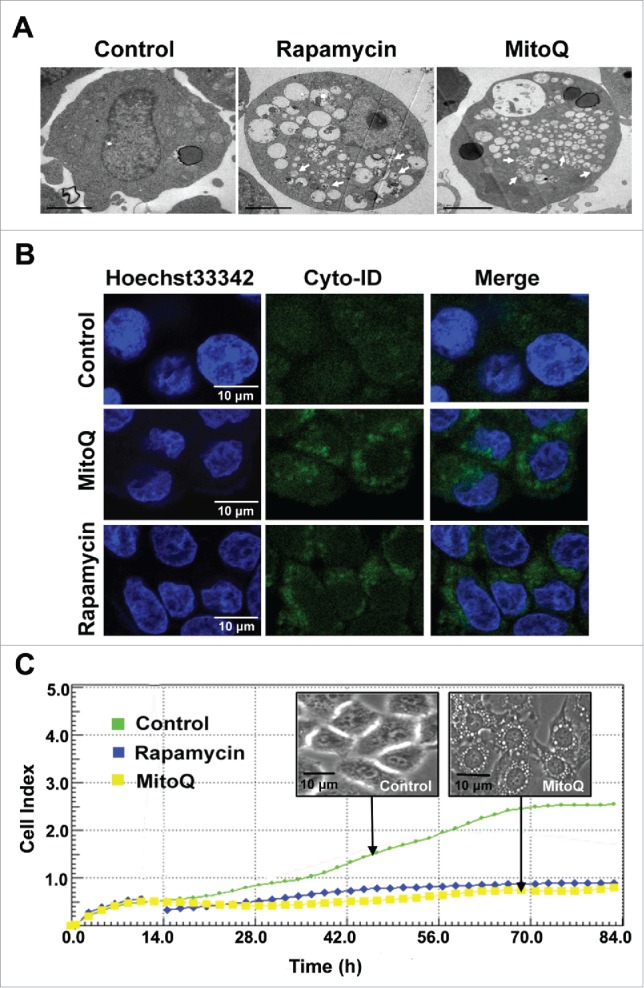
Effects of MitoQ on autophagy and proliferation in HepG2 cells. (A) Transmission electron microscopy was used to study the formation of autophagic vacuoles after 2 h of treatment with MitoQ (scale bar: 2 μm). The arrows point to autophagosomes. (B) The Cyto-ID dye specifically labeled the autophagic compartments with minimal staining of lysosomes and endosomes. (C) Cell index values were determined every 2 h using an RT-CES system for up to 84 h.
MitoQ is selectively enriched in mitochondria of HepG2 cells
To assess the targeting of MitoQ to mitochondria, the concentrations of MitoQ were respectively measured in whole cells and mitochondria isolated from HepG2 cells by high performance liquid chromatography-tandem mass spectrometry (HLPC-MS). The HPLC-MS calibration curves were obtained using MitoQ standards at the concentrations of 1, 10, 100, and 1000 ng/ml (Fig. 2B). Treatment of HepG2 cells with MitoQ for 1 h resulted in a >90% enrichment of MitoQ in mitochondria (MitoQ in isolated mitochondria: 10.18 ng/ml; MitoQ in whole cells: 10.34 ng/ml) (Fig. 2A and C).
Figure 2.
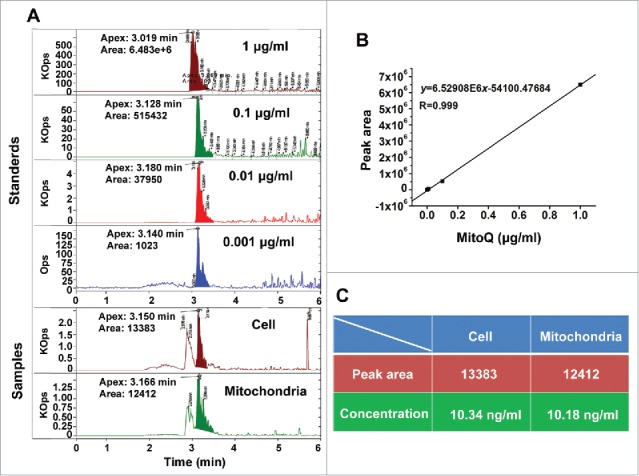
Quantitative analysis of the targeted effect of MitoQ in mitochondria. (A) HPLC-MS chromatograms corresponding to MitoQ. (B) Standard curve of MitoQ in the concentration range of 0.001–1.000 μg/ml. (C) List of sample statistics. Cell group: after treatment of HepG2 cells with MitoQ, the lysis solution of whole cells was detected. Mitochondria group: after treatment of HepG2 cells with MitoQ, the lysis solution of isolated mitochondria was detected.
MitoQ induces a pseudo-mitochondrial membrane potential (PMMP)
As shown in Figure 3D, MitoQ was primarily adsorbed to the inner mitochondrial membrane, whereas its cationic moiety remained in the intermembrane space.15,16 After treatment of HepG2 cells with MitoQ for 30 min, the red fluorescence intensity of the cyanine dye JC-1 (a probe for mitochondrial membrane potential) increased by 11.3% whereas the green fluorescence intensity of JC-1 (corresponding to the monomeric dispersed form) decreased by 6.5% compared with the control group (Fig. 3A). The results demonstrated that the positive charges of MitoQ enhanced MMP (ΔΨm). Meanwhile, the activities of respiratory chain complexes I, III, and IV (Fig. 3B) and the proton pumping rates (Fig. 3C) were inhibited by MitoQ. Furthermore, ΔΨm returned to a steady-state when the cells were pretreated with MitoQ for 60 min (Fig. 3A). At this point, considering the inhibition of the respiratory chain complexes, we propose that ΔΨm was maintained by the positive charges of MitoQ but not by mitochondrial protons. To verify this hypothesis, 10 μM carbonyl cyanide m-chlorophenyl hydrazone (CCCP), a proton ionophore,17 was used to dissipate ΔΨm via transport of protons across the inner mitochondrial membrane. We observed that pretreatment with MitoQ for 1 h followed by CCCP significantly increased MMP compared with treatment with CCCP alone (Fig. 3A). Therefore, a PMMP was formed by maintaining positive charges corresponding to MitoQ in the intermembrane space.
Figure 3.
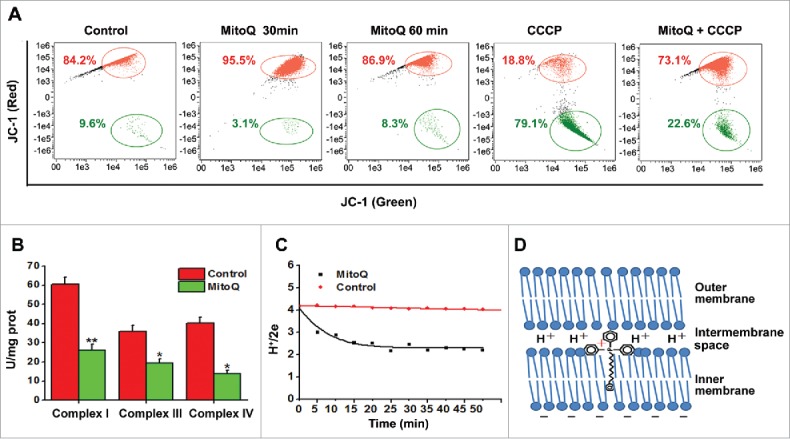
Generation of pseudo-mitochondrial membrane potential (PMMP) by MitoQ. (A) Fluorescence of HepG2 cells stained with JC-1 was determined by flow cytometry. (B) The activities of respiratory chain complexes related to proton production were detected using commercial kits. (C) After different treatments with MitoQ, the rates of proton pumping were measured with a fast-responding pH electrode system. (D) Schematic diagram of MitoQ on the mitochondrial membrane. All data are presented as mean ± SEM from 3 independent experiments. *P < 0.05, **P < 0.01 vs. control.
PMMP induced the abnormal energy supply in HepG2 cells
To further investigate the effect of MitoQ-induced PMMP on energy metabolism, mitochondrial respiration and ATP levels were measured using a Clark oxygen electrode and HPLC. The ratio of state 3 and state 4 (respiratory control ratio [RCR]) is an index used to assess respiratory chain function and oxidative phosphorylation. Treatment of HepG2 cells with MitoQ for 1 h decreased the RCR from 4.81 to 1.04 (Fig. 4A). Moreover, this treatment increased the AMP peak area and markedly decreased the ATP peak area compared with a control group (Fig. 4B). These results indicate that PMMP impairs mitochondrial respiratory function and blocks the energy supply.
Figure 4.
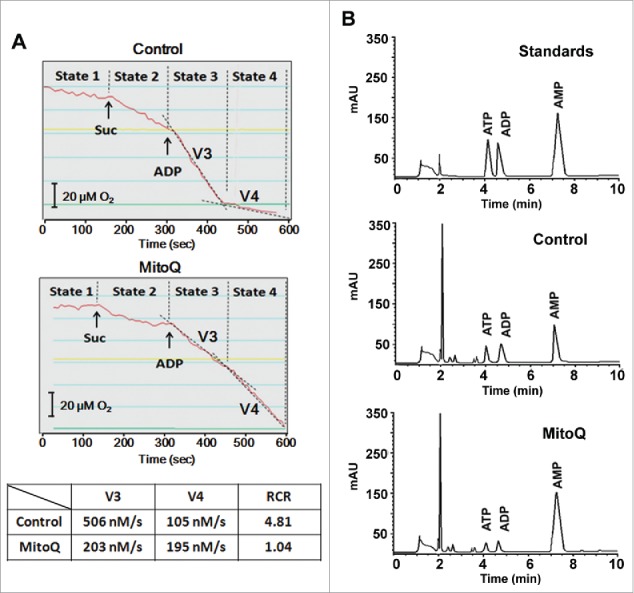
Effects of PMMP induced by MitoQ on energy metabolism in HepG2 cells. (A) After treatment with 1 μM MitoQ, a Clark oxygen electrode was used to measure oxygen consumption. (B) The adenylates AMP, ADP, and ATP were determined by HPLC. The AMP, ADP, and ATP standards were used to calibrate the system. The retention time was expressed in minutes and signal intensity was measured at 260 nm and expressed in arbitrary units of absorption.
MitoQ promotes autophagic flux via the AMPK-MTOR pathway
AMPK is an energy sensor, and therefore its activity is associated with energy metabolism levels and autophagy. The western blotting results indicated that 1 µM MitoQ treatment significantly enhanced the phosphorylation of AMPK in HepG2 cells and effectively activated AMPK (Fig. 5A). In addition, the phosphorylation of MTOR was markedly downregulated (Fig. 5A). MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) is directed to the phagophore during autophagosome formation and has been used as an autophagy marker.18 The SQSTM1/p62 protein serves as a link between LC3 and ubiquitinated substrates. SQSTM1 becomes incorporated within the completed autophagosome and is degraded in autolysosomes;19 SQSTM1 accumulates when autophagic flux is inhibited, and decreased levels can be observed when autophagic flux is induced. Figure 5B showed that MitoQ resulted in the conversion of LC3-I to LC3-II (corresponding to autophagy induction), and reduced the level of SQSTM1 in a time-dependent manner. Furthermore, the combination of MitoQ and 10 μM chloroquine (an alkalinizing agent that blocks autophagosome fusion with the lysosome) increased the Cyto-ID fluorescence in HepG2 cells compared with treatment with MitoQ alone (Fig. 5C), indicating that MitoQ promoted autophagic flux via the AMPK-MTOR pathway. Finally, compound C (an AMPK inhibitor) could effectively suppress AMPK activation, the conversion of LC3-I to LC3-II and autophagic flux induced by MitoQ (Fig. S3).
Figure 5.
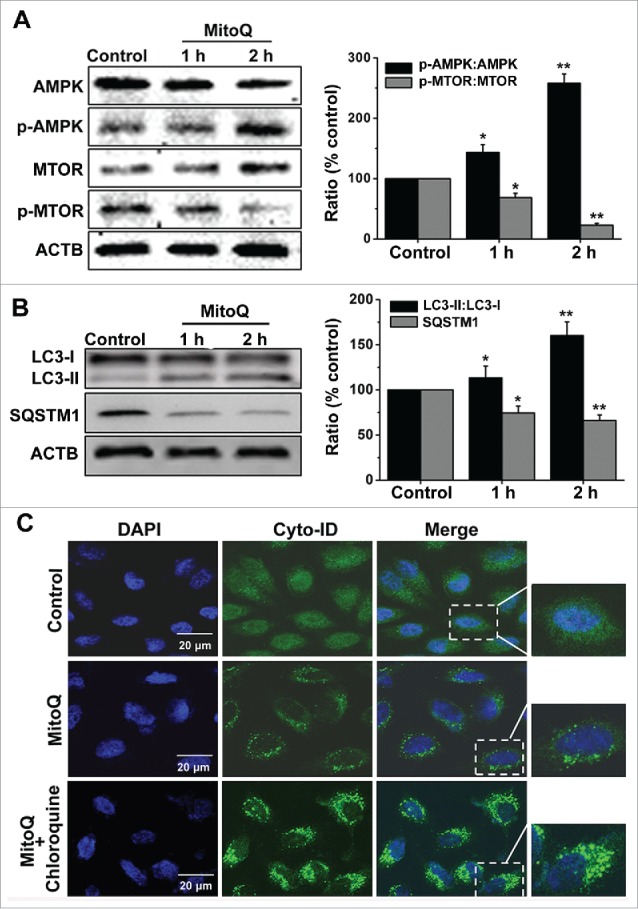
Effects of PMMP induced by MitoQ on the AMPK-MTOR-autophagy pathway. (A) The phosphorylation of AMPK and MTOR were analyzed by western blotting. (B) The conversion of LC3-I to LC3-II was measured by western blotting. ACTB was used as a loading control. (C) The Cyto-ID assay measured changes in autophagic flux. All data are presented as mean ± SEM from 3 independent experiments. *P < 0.05, **P < 0.01 vs. control.
Discussion
As the energy power plants of cells, mitochondria generate ATP by using the proton electrochemical gradient potential, which is generated by serial reduction of electrons through the respiratory electron transport chain.20 The reductive transfer of electrons through respiratory chain complexes I–IV in the inner mitochondrial membrane provides the energy necessary to transport protons against their concentration gradient across the inner mitochondrial membrane, resulting in a net accumulation of H+ in the intermembrane space, and the transported protons return to the mitochondrial matrix through the F1-Fo ATP-synthase complex, thus producing ATP.2 The total force that transports protons into the mitochondria is the MMP (Δψm).21 The results of the present study indicate that MitoQ is adsorbed to the inner mitochondrial membrane; however, its cationic moiety remains in the intermembrane space, resulting in the addition of a large number of positive charges. The balance of Δψm was maintained by MitoQ attenuating the activity of respiratory chain complexes I, III, and IV (which are associated with proton generation) (Fig. 3B) and decreasing the proton pumping rate (Fig. 3C). Furthermore, the positive charges in MitoQ replaced protons in the establishment of the electrochemical gradient across the inner membrane. In this instance, however, proton backflow was severely impaired, resulting in the decreased production of ATP (Fig. 4B). This distinct MMP could not maintain a normal energy supply and therefore was designated “pseudo-MMP” (PMMP). This study is the first to hypothesize about PMMP and helped elucidate the mechanisms used by lipophilic cation drugs to induce PMMP.
Mitochondria as an antitumor target have attracted considerable attention in recent years. Mitochondrial inhibitors have been reported to possess antitumor activity. Interestingly, most mitochondrial inhibitors are lipophilic cation drugs. For example, a report in Science indicates that the mitochondrial dye Rho123, which inhibits mitochondrial oxidative phosphorylation, exhibits anti-carcinoma activity in mice.22 In 2014, a paper in Nature Communications also reported that VLX600 (a new lipophilic cation molecule) reduced mitochondrial respiration, leading to bioenergetic catastrophe and tumor cell death.23 Metformin is a commonly prescribed antidiabetic drug that inhibits mitochondrial complex I. However, metformin also inhibits tumor formation in diabetic patients.24,25 Metformin exists as a cation at physiological pH and weakly targets mitochondria.23 Thus, the inhibitory effects of these drugs on mitochondria may be related to PMMP. By using the PMMP hypothesis, the antitumor mechanism of lipophilic cation drugs could be revealed more deeply.
MitoQ, as a mitochondrial targeting antioxidant, has attracted much attention by researchers. The approximately 1000-fold greater concentration of MitoQ within mitochondria increases its effectiveness in preventing mitochondrial oxidative damage compared with untargeted antioxidants such as coenzyme Q10. Cases of Parkinson disease are being treated with MitoQ in phase II clinical trials; however, in these trials MitoQ was generally not beneficial likely due to irreversible neuronal damage that had occurred by the time the patients were diagnosed and entered into the trials.26 The mechanisms associated with MitoQ toxicity are unclear. In the present study, we found that MitoQ blocked mitochondrial respiratory function (Fig. 4A) and impaired energy metabolism via induction of PMMP (Fig. 4B). Starvation induced excessive autophagy via the AMPK-MTOR signaling pathway (Fig. 5) and effectively inhibited cell proliferation (Fig. 1C), and this mechanism might be the cause of cytotoxicity of MitoQ. Furthermore, autophagy and PMMP induced by MitoQ also have been preliminarily proved in human promyelocytic leukemia cells (HL-60) in addition to HepG2 cells (Fig. S4).
Current chemotherapies often cause significant morbidity and enhance toxic side effects. Many of the chemotherapeutic drugs are potently cytotoxic to neoplastic and normal cells, although newer targeted therapies developed for specific cancer phenotypes may potentially increase the efficacy and decrease toxic side effects. A major objective in cancer chemotherapy is to enhance tumor cell cytotoxicity without causing undue cytotoxicity in normal cells. In this context, Rao et al. found that MitoQ was 30-fold more cytotoxic to breast cancer cells than to healthy mammary cells.8 Similarly, Cheng reported that Mito-ChM and Mito-ChMAc (structural and functional analogs of MitoQ) exhibit selective antiproliferative and cytotoxic effects in multiple cancer cells but not in noncancerous cells.7 Studies that investigated different types of cancer cells found a higher mitochondrial transmembrane potential in these cells compared with normal cells.27,28 Therefore, the targeting of cancer cell mitochondria with MitoQ should be prioritized; the cytotoxicity induced by PMMP in cancer cells is stronger than in normal cells. If this hypothesis is confirmed, MitoQ will be a promising anticancer drug. In addition, PMMP may be a potential target of selective antitumor agents. These investigations are currently underway in our laboratory.
Methods and materials
Cell culture
Human liver cancer cell line HepG2 cells were cultured in Dulbecco's modified Eagle's medium (Gibco, 11995065), with 1% penicillin/streptomycin (Gibco, 15140122) and 10% fetal bovine serum (TransGen Biotechnology, FS101–02). Cell cultures were performed in a 5% CO2 atmosphere at 37°C. MitoQ was synthesized by Vosun Chemical (444890–41–9). Rapamycin was purchased from Calbiochem (553210), compound C was from Abcam (ab120843) and CCCP was from Sigma-Aldrich (C2759).
Confocal microscopy
To visualize autophagic vacuoles and monitor autophagic flux in live cells, confocal microscopy was used. HepG2 cells were grown in glass-bottom dishes (NEST, 801001), and then treated with 1 µM MitoQ for 2 h, with cells treated with 50 nM autophagy inducer rapamycin for 6 h as positive controls. Thereafter, cells were treated with the nuclear dye Hoechst 33342 (Beyotime Biotechnology, C1022) and the autophagy detection kit Cyto-ID® green dye (Enzo Life Sciences, ENZ-51031-K200) at 37°C for 15 min.29 Confocal images were acquired using a Zeiss LSM-700 confocal microscope (Carl Zeiss, Jena, Germany) equipped with a Plan-Apo 40 × 1.3 NA oil-immersion objective. Images were edited with Photoshop.
Transmission electron microscopy
The ultrastructural analysis of autophagy was performed with transmission electron microscopy.30 Briefly, cells were fixed with 4% glutaraldehyde (Sigma-Aldrich, G5882) and post-fixed in 1% osmium tetroxide (Sigma-Aldrich, 75633) at 4°C. The samples were then washed again, dehydrated with graded alcohol and embedded in Epon-Araldite resin (Electron Microscopy Sciences, 14900). Ultrathin sections with 50-nm thickness were obtained. Sections were then stained with uranyl acetate and lead citrate. A JEM-1220 TEM (JEOL, Tokyo, Japan) was used to observe autophagosomes.
In vitro cell proliferation analysis
The rate of cellular proliferation was analyzed with an RT-CES system (ACEA Bioscience, San Diego, CA, USA). Cells were grown on the surfaces of microelectronic sensors, which are composed of circle-online electrode arrays and are integrated into the bottom surfaces of the microtiter plate. Changes in cell number were monitored and quantified by detecting sensor electrical impedance. Cell index values obtained on the RT-CES system were quantitatively correlated with the cell numbers.31 HepG2 cells were harvested after different treatments and seeded into a 16-well strip at a density of 1 × 103 cells/well. The sensor devices were placed into the 5% CO2 incubator and the cell index value was determined every 2 h automatically by the RT-CES system for up to 84 h.32
Detection of respiratory chain complexes activities
After treatment with 1 µM MitoQ for 1 h, HepG2 cells were collected and washed twice with cold phosphate-buffered saline (HyClone, SH30256.01). Cells were immediately pulverized with an ultrasonic cell crusher (VCX130PB, SONICS, Newtown, CT, USA). After determining the amount of total protein in the mixture, we detected intracellular respiratory chain complex I (Suzhou Comin Biotechnology, FHTA-2-Y), complex III (Suzhou Comin Biotechnology, FHTC-2-Y), and complex IV (Suzhou Comin Biotechnology, FHTD-2-Y) according to the instructions for the reagent kits.
Determination of mitochondrial membrane potential (ΔΨm)
Changes in the mitochondrial membrane potential were studied by staining with the cationic dye JC-1. After incubation with 10 μM JC-1 (Sigma-Aldrich, T4069) staining solution at 37°C in the cell incubator for 15 min, HepG2 cells were washed with JC-1 staining buffer 2 times and then analyzed using a flow cytometer (FlowSight, Amnis, Seattle, WA, USA). In the normal mitochondria, JC-1 aggregates to form a polymer in the mitochondrial matrix, and the polymer emits a strong red fluorescence (Ex = 585 nm, Em = 590 nm). In contrast, in the presence of unhealthy mitochondria, due to the decline or loss of the mitochondrial membrane potential, JC-1 monomers just can be present in the cytoplasm, resulting in a green fluorescence (Ex = 514 nm, Em = 529 nm).33
Measurement of H+/2e
Mitochondria were extracted from HepG2 cells using an Isolation kit (Beyotime Biotechnology, C3601), according to the manufacturer's instructions. Purified mitochondria were suspended in 0.25 M sucrose (Sangon Biotech, A610498), 1 mM EGTA, 5 mM Tris, pH 7.4 at 0°C.
Proton pump rate and H+/2e determinations were carried out using the K3Fe(CN)6 pulse method.34,35 Mitochondria (2 mg/ml) were suspended in 6 ml of 220 mM mannitol (Sangon Biotech, A100122), 70 mM sucrose, 2 mM HEPES, 50 mM KCl, 4 mM MgCl2, 2 µM rotenone (Sigma-Aldrich, R8875), 2 mM KCN, 5 mM succinate (Sangon Biotech, A100165), pH ∼7.35–7.40, 25°C. Then, 0.1 mM K3Fe(CN)6 (Sigma-Aldrich, 244023) used as an electron acceptor was added to the mixture to start the chemical reaction. Electron transport rates were measured in a 557 double-beam spectrophotometer (PerkinElmer, Waltham, MA, USA). Proton ejection was measured with a PHM84 fast-responding pH electrode system (Radiometer Medical, Copenhagen, Denmark). 100 nM of standard HCI was used to calibrate the system.
HPLC-MS analysis
The enrichment of MitoQ in mitochondria was performed using an EVOQ Qube LC-TQ system (Bruker Daltonics, Fremont, CA, USA). The cells were treated with MitoQ, then collected and divided into 2 equal parts, one for whole cell's homogenate, the other for isolation of mitochondria that were homogenized; the 2 homogenates had equal volumes and were extracted twice with the mixture of methylene chloride and methanol (the ratio of methylene chloride to methanol was 2:1), containing 2 mM butylated hydroxytoluene (Sigma-Aldrich, W218405). After drying, the residue was dissolved with cold methanol containing 2 mM butylated hydroxytoluene.
The chromatographic separation was performed with a Bruker liquid chromatography system, using an injection volume of 10 µL, and a reverse phase C18 column (2.1 mm × 150 mm, 3.5-µm particle size, WAT106005, Waters, Milford, MA, USA) maintained at 33°C. The separation followed gradient elution where solvent A was water containing 0.1% formic acid, and solvent B was acetonitrile also containing 0.1% formic acid. The gradient elution time was 10 min per sample with MitoQ showing a retention time close to 3 min.7 The mass spectrometer used electrospray ionization in the positive mode.
Mitochondrial respiration
Oxygen consumption was determined in HepG2 cells at 37°C with a Clark oxygen electrode (Oxytherm, Hansatech Instruments, Norfolk, UK).36 Briefly, Cells were resuspended in 1 ml of buffer (20 mM HEPES, 10 mM MgCl2, 250 mM sucrose, pH 7.4) and then permeabilized with digitonin (0.01%; Sigma-Aldrich, D141). After incubation for 1 min at room temperature, the cell suspension was diluted with 8 ml of buffer. The cells were pelleted and resuspended in respiration buffer (20 mM HEPES, 250 mM sucrose, 2 mM KH2PO4, 10 mM MgCl2, pH 7.4). 10 mM succinate was added as substrate to start state 2 respiration. Then, 1 mM ADP was added in respiration buffer to start state 3 respiration. When ADP was exhausted, mitochondria entered state 4 respiration. Respiratory control ratio corresponded to the ratio of state 3 and state 4 oxygen uptake rate.
Adenylates determination
Samples were pulverized with an ultrasonic cell crusher and extracted with 0.4 M HClO4. The system was kept cold in an ice bath and after 20 min samples were centrifuged at 15,000 g at 4°C for 10 min. The supernatant was immediately neutralized with 4 M K2CO3 and incubated on ice for 30 min. The resulting supernatant was centrifuged again under the same conditions, and the final supernatant was kept on ice to be injected into the HPLC equipment within the subsequent 3 h.
The adenylates (AMP, ADP and ATP) were determined by HPLC with a reverse phase C18 column (4.6 × 250 mm, 5-μm particle size, 990967–902, Agilent Technologies, Santa Clara, CA, USA). The quantifications were performed using gradient elution with a mobile phase containing 35 mM NaH2PO4, 6 mM tetrabutylammonium; mobile phase:acetonitrile gradient 95:5 to 75:25. Detection: 210 nm for 0–10 min.37 The analytical concentration of adenylates was estimated by comparison of the area of the sample with that of standards (10 µM).
Western blot analysis
Cells were lysed in RIPA buffer (Beyotime Biotechnology, P0013C). Proteins were separated by 10% SDS-PAGE and transferred to a methanol-activated PVDF membrane (Bio-Rad, 1620177). The antibodies used were as follows: anti-PRKAA/AMPKα (Cell Signaling Technology, 2532), anti-p-PRKAA/AMPKα (Cell Signaling Technology, 2535), anti-MTOR (Cell Signaling Technology, 2972), anti-p-MTOR (Cell Signaling Technology, 2971), anti-LC3B (Cell Signaling Technology, 2755), anti-SQSTM1/p62 (Cell Signaling Technology, 5144) and ACTB/β-actin (Cell Signaling Technology, 4967) antibody at 4°C overnight. After 1-h incubation with HRP-conjugated secondary antibody (Proteintech, SA00001–2), membranes were washed 3 times for 15 min each with phosphate-buffered saline. Data were presented as relative protein level normalized to ACTB, and the ratio of control samples was taken as 100%.
Statistical analysis
Data are presented as means ± SEM from at least 3 independent experiments and evaluated by analysis of variance (ANOVA) followed by Student Newman-Keuls test. Values of P < 0.05 were considered statistically significant.
Supplementary Material
Abbreviations
- AMPK
AMP-activated protein kinase
- CCCP
carbonyl cyanide m-chlorophenyl hydrazone
- GFP
green fluorescent protein
- HLPC-MS
liquid chromatography-tandem mass spectrometry
- MMP
mitochondrial membrane potential
- MTOR
mechanistic target of rapamycin
- PMMP
pseudo-mitochondrial membrane potential
- RCR
respiratory control ratio
- RT-CES
real-time cell electronic sensing
- SQSTM1
sequestosome 1
- TPP+
triphenylphosphonium
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the Key Program of National Natural Science Foundation of China (U1432248), National Key projects of Research and Development (2016YFC0904600),the National Natural Science Foundation of China (11505245), the Western Talents Program of the Chinese Academy of Sciences (Y460040XB0).
References
- [1].Henze K, Martin W. Evolutionary biology: essence of mitochondria. Nature 2003; 426:127-8; PMID:14614484; http://dx.doi.org/ 10.1038/426127a [DOI] [PubMed] [Google Scholar]
- [2].St-Pierre J, Brand MD, Boutilier RG. Mitochondria as ATP consumers: cellular treason in anoxia. Proc Natl Acad Sci USA 2000; 97:8670-4; PMID:10890886; http://dx.doi.org/ 10.1073/pnas.140093597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schumacker PT, Gillespie MN, Nakahira K, Choi AM, Crouser ED, Piantadosi CA, Bhattacharya J. Mitochondria in lung biology and pathology: more than just a powerhouse. Am J Physiol Lung Cell Mol Physiol 2014; 306:962-74; http://dx.doi.org/ 10.1152/ajplung.00073.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol 2006; 16:551-60; http://dx.doi.org/ 10.1016/j.cub.2006.06.054 [DOI] [PubMed] [Google Scholar]
- [5].Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, Smith RA, Murphy MP. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem 2001; 276:4588-96; PMID:11092892; http://dx.doi.org/ 10.1074/jbc.M009093200 [DOI] [PubMed] [Google Scholar]
- [6].Cheng G, Zielonka J, McAllister D, Hardy M, Ouari O, Joseph J, Dwinell MB, Kalyanaraman B. Antiproliferative effects of mitochondria-targeted cationic antioxidants and analogs: Role of mitochondrial bioenergetics and energy-sensing mechanism. Cancer Lett 2015; 365:96-106; PMID:26004344; http://dx.doi.org/ 10.1016/j.canlet.2015.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cheng G, Zielonka J, McAllister DM, Mackinnon AC Jr, Joseph J, Dwinell MB, Kalyanaraman B. Mitochondria-targeted vitamin E analogs inhibit breast cancer cell energy metabolism and promote cell death. BMC Cancer 2013; 13:285-99; PMID:23764021; http://dx.doi.org/ 10.1186/1471-2407-13-285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rao VA, Klein SR, Bonar SJ, Zielonka J, Mizuno N, Dickey JS, Keller PW, Joseph J, Kalyanaraman B, Shacter E. The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone. J Biol Chem 2010; 285:34447-59; PMID:20805228; http://dx.doi.org/ 10.1074/jbc.M110.133579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Moruno F, Pérez-Jiménez E, Knecht E. Regulation of autophagy by glucose in Mammalian cells. Cells 2012; 1:372-95; PMID:24710481; http://dx.doi.org/ 10.3390/cells1030372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang W, Yang X, López de Silanes I, Carling D, Gorospe M. Increased AMP:ATP ratio and AMP-activated protein kinase activity during cellular senescence linked to reduced HuR function. J Biol Chem 2003; 278:27016-23; PMID:12730239; http://dx.doi.org/ 10.1074/jbc.M300318200 [DOI] [PubMed] [Google Scholar]
- [11].Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell 2006; 124: 471-84; PMID:16469695; http://dx.doi.org/ 10.1016/j.cell.2006.01.016 [DOI] [PubMed] [Google Scholar]
- [12].Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett 2010; 584:1287-95; PMID:20083114; http://dx.doi.org/ 10.1016/j.febslet.2010.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/ 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9:1004-10; PMID:18971948; http://dx.doi.org/ 10.1038/nrm2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ross MF, Prime TA, Abakumova I, James AM, Porteous CM, Smith RA, Murphy MP. Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells. Biochem J 2008; 411:633-45; PMID:18294140; http://dx.doi.org/ 10.1042/BJ20080063 [DOI] [PubMed] [Google Scholar]
- [16].James AM, Sharpley MS, Manas AR, Frerman FE, Hirst J, Smith RA, Murphy MP. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J Biol Chem 2007; 282:14708-18; PMID:17369262; http://dx.doi.org/ 10.1074/jbc.M611463200 [DOI] [PubMed] [Google Scholar]
- [17].Li N, Zhang G, Yi FX, Zou AP, Li PL. Activation of NAD(P)H oxidase by outward movements of H+ ions in renal medullary thick ascending limb of Henle. Am J Physiol Renal Physiol 2005; 289:1048-56; http://dx.doi.org/ 10.1152/ajprenal.00416.2004 [DOI] [PubMed] [Google Scholar]
- [18].Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S, Sun X, Yoon G, Kang Y, Zhong W, et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 2014; 516:108-11; PMID:25383523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151-75; PMID:18188003; http://dx.doi.org/ 10.4161/auto.5338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Perry SW, Norman JP, Barbieri J, Brown EB, Gelbard HA. Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques 2011; 50:98-115; PMID:21486251; http://dx.doi.org/ 10.2144/000113610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Reid RA, Moyle J, Mitchell P. Synthesis of adenosine triphosphate by a protonmotive force in rat liver mitochondria. Nature 1966; 212:257-8; PMID:5970114; http://dx.doi.org/ 10.1038/212257a0 [DOI] [PubMed] [Google Scholar]
- [22].Bernal SD, Lampidis TJ, McIsaac RM, Chen LB. Anticarcinoma activity in vivo of rhodamine 123, a mitochondrial-specific dye. Science 1983; 222:169-72; PMID:6623064; http://dx.doi.org/ 10.1126/science.6623064 [DOI] [PubMed] [Google Scholar]
- [23].Zhang X, Fryknäs M, Hernlund E, Fayad W, De Milito A, Olofsson MH, Gogvadze V, Dang L, Påhlman S, Schughart LA, et al. Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nat Commun 2014; 5:3295; PMID:24548894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sadeghi N, Abbruzzese JL, Yeung SC, Hassan M, Li D. Metformin use is associated with better survival of diabetic patients with pancreatic cancer. Clin Cancer Res 2012; 18:2905-12; PMID:22465831; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Emami RA, Fisel P, Nies AT, Schaeffeler E, Schwab M. Metformin and cancer: from the old medicine cabinet to pharmacological pitfalls and prospects. Trends Pharmacol Sci 2013; 34:126-35; PMID:23277337; http://dx.doi.org/ 10.1016/j.tips.2012.11.005 [DOI] [PubMed] [Google Scholar]
- [26].Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov Disord 2010; 25:1670-4; PMID:20568096; http://dx.doi.org/ 10.1002/mds.23148 [DOI] [PubMed] [Google Scholar]
- [27].Armstrong JS. Mitochondria: a target for cancer therapy. Br J Pharmacol 2006; 147:239-48; PMID:16331284; http://dx.doi.org/ 10.1038/sj.bjp.0706556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Beckham TH, Lu P, Jones EE, Marrison T, Lewis CS, Cheng JC, Ramshesh VK, Beeson G, Beeson CC, Drake RR, et al. LCL124, a cationic analog of ceramide, selectively induces pancreatic cancer cell death by accumulating in mitochondria. J Pharmacol Exp Ther 2013; 344:167-78;PMID:23086228;http://dx.doi.org/ 10.1124/jpet.112.199216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Guo S, Liang Y, Murphy SF, Huang A, Shen H, Kelly DF, Sobrado P, Sheng Z. A rapid and high content assay that measures cyto-ID-stained autophagic compartments and estimates autophagy flux with potential clinical applications. Autophagy 2015; 11:560-72; PMID:25714620; http://dx.doi.org/ 10.1080/15548627.2015.1017181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Swanlund JM, Kregel KC, Oberley TD. Investigating autophagy: quantitative morphometric analysis using electron microscopy. Autophagy 2010; 6:270-7; PMID:19923921; http://dx.doi.org/ 10.4161/auto.6.2.10439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Solly K, Wang X, Xu X, Strulovici B, Zheng W. Application of real-time cell electronic sensing (RT-CES) technology to cell-based assays. Assay Drug Dev Technol 2004; 2:363-72; PMID:15357917; http://dx.doi.org/ 10.1089/adt.2004.2.363 [DOI] [PubMed] [Google Scholar]
- [32].Sun C, Wang Z, Liu Y, Liu Y, Li H, Di C, Wu Z, Gan L, Zhang H. Carbon ion beams induce hepatoma cell death by NADPH oxidase-mediated mitochondrial damage. J Cell Physiol 2014; 229:100-7; PMID:23804302; http://dx.doi.org/ 10.1002/jcp.24436 [DOI] [PubMed] [Google Scholar]
- [33].Bineta MT, Doyleb CJ, Williamsonc JE, Schlegelc P. Use of JC-1 to assess mitochondrial membrane potential in sea urchin sperm. J Exp Mar Bio Ecol 2014; 453:91-100; http://dx.doi.org/ 10.1016/j.jembe.2013.12.008 [DOI] [Google Scholar]
- [34].Price BD, Brand MD. Chemical modification of the mitochondrial bc1 complex by N, N'-dicyclohexylcarbodiimide inhibits proton translocation. Eur J Biochem 1983; 132:595-601; PMID:6303780; http://dx.doi.org/ 10.1111/j.1432-1033.1983.tb07405.x [DOI] [PubMed] [Google Scholar]
- [35].Jones RW, Lamont A, Garland PB. The mechanism of proton translocation driven by the respiratory nitrate reductase complex of Escherichia coli. Biochem J 1980; 190:79-94; PMID:6255943; http://dx.doi.org/ 10.1042/bj1900079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tendi EA, Bosetti F, Dasgupta SF, Stella AM, Drieu K, Rapoport SI. Ginkgo biloba extracts EGb 761 and bilobalide increase NADH dehydrogenase mRNA level and mitochondrial respiratory control ratio in PC12 cells. Neurochem Res 2002; 27:319-23; PMID:11958534; http://dx.doi.org/ 10.1023/A:1014963313559 [DOI] [PubMed] [Google Scholar]
- [37].von Papen M, Gambaryan S, Schütz C, Geiger J. Determination of ATP and ADP Secretion from Human and Mouse Platelets by an HPLC Assay. Transfus Med Hemother 2013; 40:109-16; PMID:23652982; http://dx.doi.org/ 10.1159/000350294 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.