

ABSTRACT
Atg5-null mice are neonatal lethal. We have revealed in our recent paper that these mice die due to neuronal dysfunction resulting in suckling failure. Our new mouse model, atg5–/–;Eno2/Nse-Atg5 mice, where Atg5 is deficient in the whole body except for neurons, enables us to analyze the consequences of macroautophagy/autophagy-deficiency in the whole body of adult mice.
KEYWORDS: ATG5, autophagy deficiency, hypogonadism, iron deficiency, neonatal lethality, neuronal dysfunction, suckling failure, systemic analysis
Mice deficient for factors in the ATG conjugation systems (e.g. ATG3, ATG5, ATG7, ATG12 and ATG16L1), which function at a late step of autophagosome formation and maturation, die within 1 d after birth. In general, mammalian neonates undergo sudden starvation and activate autophagy when transplacental nutrition is terminated by birth. Autophagy-deficient neonates show lower tissue and plasma amino acid levels than wild-type neonates, suggesting that autophagy is important for maintaining the nutritional status during the neonatal starvation period. However, the exact cause of death of ATG conjugation-deficient animals remained elusive. Although their neonates appear anatomically normal, there is one potentially lethal abnormality observed; they fail to suckle breast milk. However, the suckling defect alone cannot explain the premature death because Atg5-null mice show even lower amino acid levels and die earlier than nonsuckling wild-type neonates. Thus, we hypothesized that the early lethal phenotype is due to a combination of a metabolic defect and the suckling failure, and that Atg5-null neonates may be able to survive if they can suckle milk.
We suspected that the suckling defect of Atg5-null neonates was caused by neuronal dysfunction; therefore, we introduced exogenous GFP-Atg5 to these Atg5-null mice under the control of the neuron-specific Eno2/Nse promoter. The resulting atg5–/–;Eno2-Atg5 mice express ATG5 almost exclusively in neurons (Fig. 1). These atg5–/–;Eno2-Atg5 mice can successfully suckle milk and most of them survive for more than 2 mo. Several individuals even survived beyond half a year. These data suggest that Atg5-deficient neonates die due to neuronal dysfunction that leads to suckling failure and that an autophagy-deficiency in other organs is not fatal for at least several months.
Figure 1.
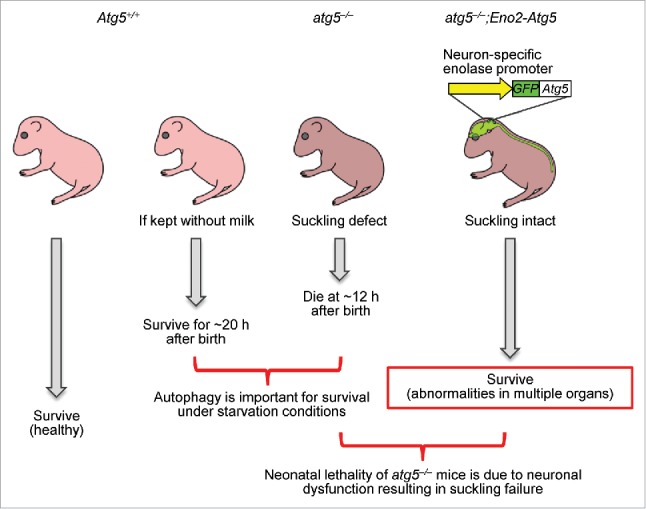
A schematic model of atg5–/–;Eno2-Atg5 mice. GFP-ATG5 is expressed under the regulation of the neuron-specific Eno2 (enolase 2, gamma neuronal) promoter in atg5–/– mice (atg5–/–;Eno2-Atg5). These mice are whole body Atg5-deficient except for the neurons. atg5–/– mice die at approximately 12 h after birth, which is significantly shorter than wild-type mice kept without milk feeding, suggesting that nutrients supplied by autophagy are important for survival under starvation conditions. Whereas atg5–/– mice cannot suckle breast milk, atg5–/–;Eno2-Atg5 mice successfully suckle milk and survive until adulthood, suggesting that the neonatal lethality of atg5–/– mice is due to neuronal dysfunction resulting in suckling failure. atg5–/–;Eno2-Atg5 mice survive for several months, and abnormalities are observed in multiple organs.
Survival of atg5–/–;Eno2-Atg5 mice enabled us to analyze organs and tissues from adult autophagy-deficient mice. As this mouse model is based on Atg5-null mice, complete knockout efficiency would be expected compared with Cre-mediated conditional knockout models. This model is also useful when an appropriate tissue-specific Cre-expressing mouse line is not available.
The body size of atg5–/–;Eno2-Atg5 mice is smaller than wild-type mice. The liver and spleen are significantly enlarged, whereas the skeletal muscles, pituitary gland, and male and female reproductive organs are significantly smaller in atg5–/–;Eno2-Atg5 mice. atg5–/–;Eno2-Atg5 mice demonstrate accumulation of ubiquitinated proteins and SQSTM1/p62 in many tissues, but not uniformly. These proteins highly accumulate especially in the liver, heart, pancreas, and muscle, suggesting that dependency on autophagy in quality control differs among organs and tissues.
Among the multiple abnormalities observed in atg5–/–;Eno2-Atg5 mice, we further focused on 2 prominent ones, namely hypogonadism and hypochromic anemia. atg5–/–;Eno2-Atg5 mice have small reproductive organs and a severe defect in spermatogenesis. In addition, the serum testosterone level is reduced. Spermatogenesis is regulated by the hypothalamus-pituitary-gonadal axis through hormonal regulation. Indeed, pituitary hormones (luteinizing hormone and follicle-stimulating hormone) and LEP (leptin), an adipose-derived hormone that affects the hypothalamic-pituitary-gonadal axis, are also lower in these mice. Thus, the low testosterone levels and spermatogenetic failure might be due to defects in the pituitary gland, the adipose tissue, and the testis itself.
Anemia was also observed in our atg5–/–;Eno2-Atg5 mice. Although previous studies have suggested that autophagy deficiency could cause anemia due to defects in mitochondria elimination during differentiation or haematopoietic stem cell function, we found a different mechanism, iron deficiency. Indeed, anemia in atg5–/–;Eno2-Atg5 mice is at least partially rescued by iron supplementation. Further study revealed that absorption of iron in the intestine is reduced, which is caused by transcriptional dysregulation of iron absorption-related proteins (e.g., CYBRD1/duodenal cytochrome b, SLC11A2/divalent metal transporter 1, and SLC40A1/ferroportin). Conversely, intestinal epithelium-specific Atg5-deficient mice (Atg5flox/flox;Vil/Villin-Cre) do not show iron-deficiency or anemia, while they have similar transcriptional dysregulation of iron-related genes, suggesting the possible involvement of multiple organs in the iron-deficiency of atg5–/–;Eno2-Atg5 mice.
We have listed a broad range of abnormalities observed in the whole body of atg5–/–;Eno2-Atg5 mice in our recent paper, which will hopefully be an important resource for further understanding the physiologic roles of autophagy.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by JST PRESTO (to A.K.), a Grant-in-Aid for JSPS Fellows (Grant Number 25*7082) (to S.R.Y), and a JSPS KAKENHI Grant-in-Aid for Scientific Research on Innovative Areas (Grant Number 25111005) (to N.M.).