

ABSTRACT
The vacuolar-type H+-translocating ATPase (v-H+-ATPase) has been implicated in the amino acid-dependent activation of the mechanistic target of rapamycin complex 1 (MTORC1), an important regulator of macroautophagy. To reveal the mechanistic links between the v-H+-ATPase and MTORC1, we destablilized v-H+-ATPase complexes in mouse liver cells by induced deletion of the essential chaperone ATP6AP2. ATP6AP2-mutants are characterized by massive accumulation of endocytic and autophagic vacuoles in hepatocytes. This cellular phenotype was not caused by a block in endocytic maturation or an impaired acidification. However, the degradation of LC3-II in the knockout hepatocytes appeared to be reduced. When v-H+-ATPase levels were decreased, we observed lysosome association of MTOR and normal signaling of MTORC1 despite an increase in autophagic marker proteins. To better understand why MTORC1 can be active when v-H+-ATPase is depleted, the activation of MTORC1 was analyzed in ATP6AP2-deficient fibroblasts. In these cells, very little amino acid-elicited activation of MTORC1 was observed. In contrast, insulin did induce MTORC1 activation, which still required intracellular amino acid stores. These results suggest that in vivo the regulation of macroautophagy depends not only on v-H+-ATPase-mediated regulation of MTORC1.
KEYWORDS: ATP6AP2, autophagy, lysosome, MTORC1, ribosomal protein S6 kinase, transcription factor EB, v-H+-ATPase complex
Introduction
Cells digest intracellular components in a process called macroautophagy, which helps to meet the cellular demand for essential nutrients and turnover of damaged organelles. In this process, the autophagosomes that have enclosed their targets mature and obtain degradative properties upon fusion with lysosomes.1 The switch between cell growth and cellular degradation processes is controlled by the mechanistic target of rapamycin complex 1 (MTORC1). The conserved serine-threonine kinase MTOR represents the core of the 2 independently formed complexes MTORC1 and MTORC2,2-4 which are composed of both shared and complex-specific subunits.5 MTORC1-specific subunits comprise the kinase inhibitor AKT1S1/PRAS40 (AKT1 substrate 1 [proline-rich]) and RPTOR/RAPTOR (regulatory associated protein of MTOR, complex 1), which influences MTORC1 localization and substrate binding. MTORC1 dysregulation is associated with a range of important diseases, such as type 2 diabetes, cancer, and neurodegeneration.6
Activated MTORC1 inhibits autophagy and proteolytic digestion within lysosomes.7-10 During this inhibition the autophagy-initiating ULK (unc-51 like kinase) complex is directly phosphorylated and thereby inactivated by MTORC1.7-9 The transcription of autophagosomal as well as lysosomal genes containing CLEAR (coordinated lysosomal expression and regulation) motifs11 is also inhibited by active MTORC1. Finally, TFEB (transcription factor EB), a helix-loop-helix protein containing a leucine zipper motif, is phosphorylated by MTORC1, which prevents its nuclear translocation.12-15
The activation of MTORC1 can be triggered by high intracellular amino acid pools or by extracellular-initiated signaling of growth factors and peptide hormones (Fig. S1). The presence of essential amino acids is sensed both in the cytoplasm, and in the lumen of lysosomes. Recognition of arginine and glutamine occurs in the lysosome and requires the amino acid transporter SLC38A9 and/or the vacuolar-type H+-ATPase (v-H+-ATPase) complex.16-19 Lysosomal amino acids stimulate MTORC1 by activating the lysosome-associated Ragulator complex and the RRAGs (Ras related GTP binding proteins) RRAGA through RRAGD. This machinery then recruits MTORC1 to the cytosolic surface of the lysosomes, a process that is mediated by specific interactions between the MTORC1 component RPTOR/RAPTOR and the RRAGs.20 The translocation of MTORC1 is necessary for subsequent activation of the MTOR kinase by the lysosome-resident RHEB (Ras homolog enriched in brain).21-23
Both the SLC38A9 and the v-H+-ATPase are directly involved in this signaling cascade. v-H+-ATPases are multiprotein complexes belonging to a family of ATP-dependent proton pumps and are responsible for lumenal acidification. They consist of a core machinery of 14 different subunits that are arranged in distinct copy numbers and in 2 main sectors. The peripheral V1 sector contains 8 subunits, and the membrane-integral V0 sector is comprised of 6 different subunits.,24, 25 To complement the structure of the v-H+-ATPase, 2 accessory subunits exist in mammalian complexes that are attached to the V0 sector: ATP6AP1/AC45 (ATPase, H+ transporting, lysosomal accessory protein 1) and ATP6AP2/(pro)renin receptor. Deletion of the Atp6ap2 gene in cardiomyocytes and podocytes results in a significant decrease of the v-H+-ATPase integral membrane proteins26-28 implying a chaperone function of ATP6AP2 for the v-H+-ATPase V0 sector assembly.
The role of the v-H+-ATPase in MTORC1-function is poorly understood. Subunits of the v-H+-ATPase complex interact with the Ragulator complex members LAMTOR1/p18 and LAMTOR2/p14 in an amino acid-dependent manner.16 Both the v-H+-ATPase complex and Ragulator complex are required to anchor MTORC1 to the lysosome.16,29 Knocking down the ATP6V0C subunit of the v-H+-ATPase complex is associated with a reduced activation of MTORC1 upon addition of amino acids.16,17 Until recently, the v-H+-ATPase complex was assumed to be the key factor responsible for amino acid sensing. However, current data argue that the amino acid transporter SLC38A9 is the most critical sensing component. SLC38A9 also interacts with the v-H+-ATPase subunits ATP6V0D1 and ATP6V1B219 suggesting a tight coupling of the amino acid transporter and the v-H+-ATPase complex in amino acid sensing.
To better understand how the v-H+-ATPase complex contributes to amino acid sensitivity and subsequent MTORC1 activation we took advantage of the availability of inducible v-H+-ATPase Atp6ap2 knockout mice and cell lines. Our in vivo experiments show that the lack of the v-H+-ATPase leads to massive accumulation of autophagic vacuoles in hepatocytes despite normal TFEB nuclear translocation. Our data support a critical role for the v-H+-ATPase complex in amino acid sensing but also demonstrate that additional amino acid-dependent pathways can modulate the activation state of MTORC1.
Results
ATP6AP2-deficiency in hepatocytes leads to an accumulation of autophagosomes and endocytic vesicles
Lysosomal v-H+-ATPase is a central element regulating MTORC1 signaling and macroautophagy9,16,30 (Fig. S1). Because hepatocytes are very active in macroautophagy31 we set out to study the in vivo consequences of depletion of the v-H+-ATPase complex on macroautophagy and MTORC1 function. We generated conditional knockout (cKO) mice specifically lacking the v-H+-ATPase-accessory subunit ATP6AP2, which is needed for assembly of the v-H+-ATPase V0 sector (Fig. 1A). The genetic deletion of Atp6ap2 was induced as described previously for Mx1-Cre-Atp6ap2 floxed mice32 after treatment of the mice with synthetic double stranded RNA (poly [I:C]) for 5 d. The livers of these animals were analyzed after another 10 d (Fig. S2A). Already after this relatively short period it became obvious that the livers of the Atp6ap2 cKOs were enlarged by 60% (Fig. S2B, Fig. 1B). Hepatocytes appeared hypertrophic as indicated by hematoxylin and eosin staining (Fig. S2C). Using ROSA26EYFP reporter mice crossed with the Mx1-Cre transgenic mice we confirmed that the Mx1-Cre-mediated recombination occurred in hepatocytes and Kupffer cells (Fig. S2D). Deletion of Atp6ap2 was successful, as revealed by immunoblotting of total liver lysates (Fig. 1C). Residual levels of ATP6AP2 that were evident are likely due to nontargeted liver endothelial cells (Fig. S2D). We further observed the concomitant reduction in the concentrations of V0 subunits TCIRG1/ATP6V0A3 (29 ± 9% remaining), ATP6V0A1 (41 ± 26%), ATP6V0D1 (50 ± 18%) and ATP6V0C (51 ± 12%) (Fig. 1C). Close examination by electron microscopy revealed an accumulation of abnormal vesicles containing partially degraded cytoplasmic material in the Atp6ap2 cKO hepatocytes (Fig. 1D). The morphology of these vesicles was similar to autophagic vacuoles.
Figure 1.
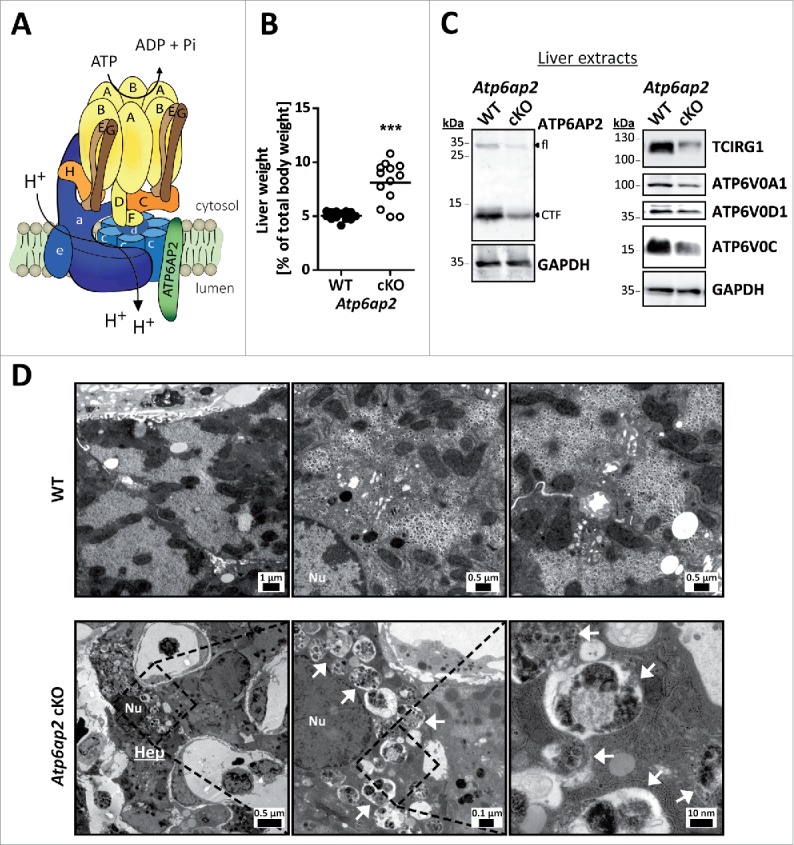
Altered ultrastructure of ATP6AP2-depleted liver. (A) Schematic illustration of the v-H+-ATPase complex composition. The v-H+-ATPase V1 sector (yellow and brown subunits, upper case letters) transfers energy released by ATP hydrolysis into a rotational force that triggers the transport of protons through the V0 sector (blue subunits, lower case letters). ATP6AP2 is an accessory subunit that acts as a chaperone for V0 sector assembly. (B) Livers from Atp6ap2 cKO mice weigh more than those of wild-type (WT) animals. (C) Immunoblot analysis of liver lysates confirms reduced concentrations of ATP6AP2 full length (fl) and C-terminal fragments (CTF) in ATP6AP2-depleted samples that come with lower v-H+-ATPase V0 sector concentrations. GAPDH detection was used as a control for equal protein load. (D) Transmission electron micrographs reveal ultrastructural changes in Atp6ap2 cKO liver as compared with wild-type samples. Affected hepatocytes (Hep) accumulate vesicles (arrows) that contain partially degraded cytoplasmic material. The boxed areas are resolved with higher magnification, Nu, nucleus.
To better define the nature of these vacuoles, immunofluorescence staining of liver sections was performed. MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) was detected as a marker of autophagosomes and autophagic vacuoles, and SQSTM1/p62 (sequestosome 1) as a marker of autophagic cargo. The numbers of LC3-positive vesicles and SQSTM1 staining were massively increased in the knockout hepatocytes (Fig. 2A). Similarly, staining of the late endosomal/lysosomal marker LAMP1 (lysosomal-associated membrane protein 1) was much stronger in the ATP6AP2-depleted cells (Fig. 2A). Ubiquitin, which is an indicator for impaired proteasomal and autophagic degradation, also accumulated in the knockout samples (not shown). It is of note that LC3 but not SQSTM1 was found in LAMP1-containing autolysosomes (Fig. 2A). The profound accumulation of autophagic and endocytic structures was also observed by immunoblotting with antibodies to LC3, SQSTM1 and LAMP1 (Fig. 2B). These data conclusively demonstrate that the lack of the v-H+-ATPase complex in hepatocytes leads to dramatic alterations of the endocytic and autophagic systems. The increase in LC3-II and SQSTM1 indicate that autophagy is dysregulated, leading to accumulation of immature and mature autophagic vacuoles.
Figure 2.
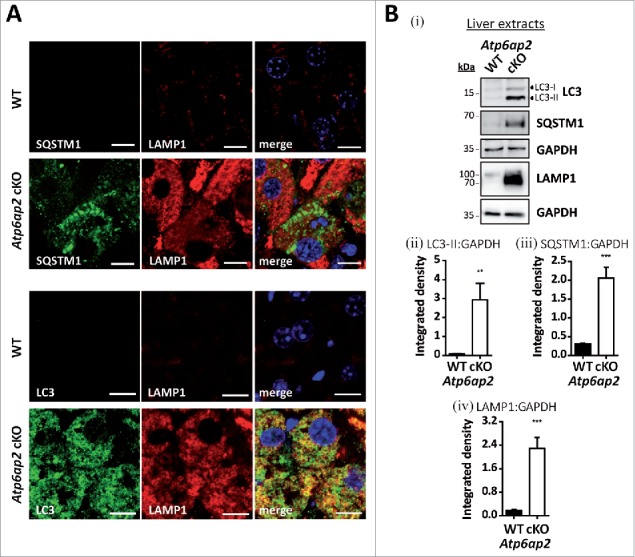
Accumulation of autophagic vacuoles and lysosomes in Atp6ap2 conditional knockout liver. (A) Immunofluorescence analyses of liver sections show an increase in LC3-, SQSTM1- and LAMP1-positive vesicles in ATP6AP2-depleted hepatocytes (cKO) in comparison to control tissue (WT). Scale bars: 10 µm. (Bi) Immunoblot analysis of autophagic marker proteins LC3-I, LC3-II and SQSTM1 and of lysosomal LAMP1 in liver lysates (15 μg per lane) from Atp6ap2 cKO and wild-type mice. All protein concentrations were increased in the knockout samples. (Bii-iv) Quantification of the immunoblots was carried out in relation to GAPDH levels. Shown are mean values ± standard errors from 6 to 7 independent animal preparations per genotype. **P < 0.01, ***P < 0.001 according to unpaired, 2-tailed Student t test.
Decreased autophagic clearance but normal acidification and delivery of endocytic cargo to lysosomes and autolysosomes in cultured ATP6AP2-deficient hepatocytes
Triggered by our observation that SQSTM1 and LAMP1 did not colocalize in ATP6AP2-deficient hepatocytes (Fig. 2A) and by the reports that a lack of the v-H+-ATPase complex leads to a deficit in acidification in fibroblasts and podocytes,27,28 we asked whether autophagic flux and lysosomes and endocytic cargo transport were fully functional in Atp6ap2 cKO hepatocytes.
To this end, we isolated primary hepatocytes from both control and Atp6ap2 cKO mice 5 d after the last poly (I:C)-injection (Fig. S2A). These hepatocytes almost completely lacked ATP6AP2 and the TCIRG1/ATP6V0A3, ATP6V0D1 and ATP6V0C subunits of the V0 sector of the v-H+-ATPase (Fig. S3A). Similar to the situation with liver lysates, we observed increased levels of LC3, SQSTM1 and LAMP1 in the ATP6AP2-deficient hepatocytes. Analyzing early markers of autophagy, we also noted an increase in autophagy-related proteins ATG12–ATG5 and WIPI2 (WD repeat domain, phosphoinositide interacting 2) (Fig. S3A). ATG5 immunostaining revealed more punctate staining in the knockout cells (Fig. S3B). To further investigate the autophagic degradation, we applied bafilomycin A1 (BafA1) to block lysosomal protein degradation and compared the levels of LC3-II in control and knockout hepatocytes under basal and starved (without amino acids and serum) conditions. We observed that BafA1 treatment increased LC3-II levels in wild-type hepatocytes 3.5-fold under fed conditions and 4.2-fold under starved conditions (Fig. S3Ci,ii). In BafA1-treated knockout hepatocytes only a very mild increase of 1.1-fold and 1.3-fold was found under fed and starved conditions, respectively (Fig. S3Ci,ii). Much less LC3 was present in wild-type hepatocytes as compared with knockout cells (Fig. S3Ci,ii). Our data suggest that the accumulation of autophagic vacuoles was due to decreased autophagic degradation. In both genotypes, application of BafA1 significantly decreased LysoTracker® Red signals that were used to visualize acidic organelles (Fig. S3Ciii).
The Atp6ap2 cKO hepatocytes were further characterized by functional lysosomal proteolysis, as monitored by DQ™ Red-BSA fluorescence (Fig. S3D). Similar to the situation in ATP6AP2-depleted liver, SQSTM1 and LAMP1 did only partially colocalize in immunofluorescence stainings (Fig. S3D). To monitor if the reduced number of v-H+-ATPase complexes affected endosome-to-lysosome fusion, we offered the cultured hepatocytes BSA-coated 10-nm gold particles in culture medium for 1 h. The delivery of BSA-gold to endocytic and autophagic compartments was monitored by electron microscopy after a 2-h chase period. These experiments revealed that in the Atp6ap2 cKO hepatocytes gold particles reached late endosomes, lysosomes and autophagic vacuoles (Fig. S4A,B), as well as LAMP2-positive compartments (Fig. S4C) with similar efficiency as in control samples. However, this quantification is affected by the much higher abundance, and likely longer half-life, of autophagic and LAMP2-positive compartments in the Atp6ap2 cKO hepatocytes. The autophagic vacuoles that contained the endocytosed gold particles contained partially degraded cytoplasmic material including ribosomes and endoplasmic reticulum. As judged by morphology, the cytoplasmic cargo was less degraded in the cKO hepatocytes compared with the wild-type cells (Fig. S4A), which is in agreement with the LC3-II blots showing decreased autophagic clearance.
These results revealed that newly ingested endocytic cargo has access to the autophagic and LAMP2-positive compartments in the Atp6ap2 cKO hepatocytes. Further, the colocalization of LC3 and LAMP1 (Fig. 2A) suggests that autophagosome-lysosome fusion occurs normally in these cells. In summary, isolated hepatocytes lacking ATP6AP2 display intracellular vesicle fusion along the endocytic and autophagic pathways as well as lysosomal proteolysis by acidic hydrolases. However, the kinetic of LC3-II degradation was apparently reduced. Our data are in support of the hypothesis that the cellular pathology observed in the liver sections of the Atp6ap2 cKO mice was not caused by impaired fusion or a general defect in acidification but by a decreased autophagic clearance.
Increased MTORC1 activity in Atp6ap2 cKO livers
Because MTORC1 tightly controls macroautophagy and as we have observed an accumulation of autophagic vacuoles and lysosomes in the Atp6ap2 cKO hepatocytes, we predicted that the kinase activity of MTORC1 would be lower in Atp6ap2 cKO livers. To address this question, we analyzed MTORC1 activity after poly (I:C)-mediated induction of ATP6AP2-deficiency (Fig. S2A). For this purpose phosphorylation of the MTORC1 substrates ULK1 (Ser757), a modulator of early steps of macroautophagy, RPS6KB (ribosomal protein S6 kinase [Thr389]) and the MTORC1 repressor AKT1S1 (Thr246) were determined (Fig. 3A). Surprisingly, MTORC1 activity remained constant in ATP6AP2-depleted liver and even increased at d 15 after the first induction, whereas ATP6AP2 full length and C-terminal fragment as well as V0 subunit ATP6V0C were clearly reduced in Atp6ap2 cKO livers (Fig. 3A). In parallel, we monitored the accumulation of lysosome and autophagosome marker proteins. Already at d 7, LAMP1 and LAMP2 concentrations were increased, followed by augmented levels of LC3-I and LC3-II as well as SQSTM1 beginning on d 9 after induction.
Figure 3.
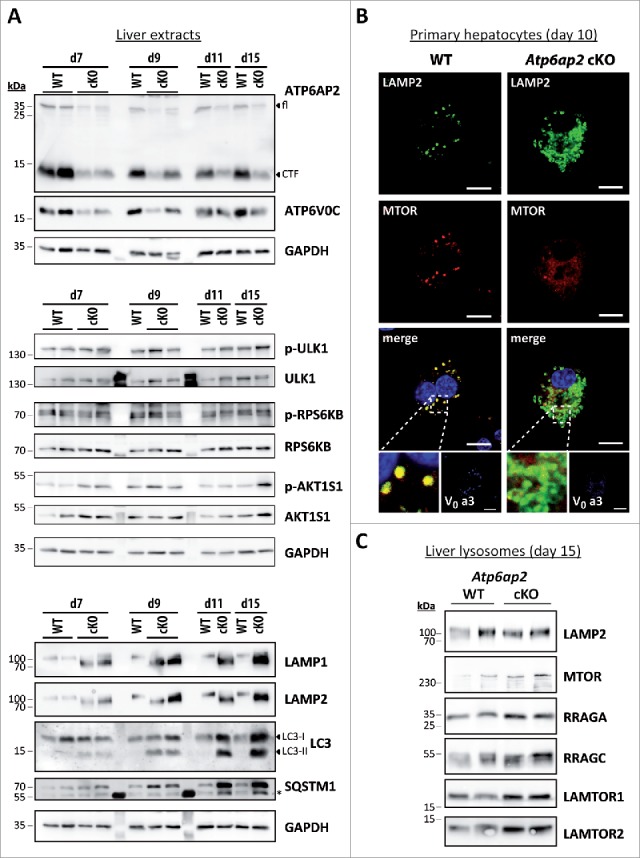
Activation of MTORC1 and lysosomal MTOR recruitment are late responses in ATP6AP2-depleted liver. (A) Time course of the expression levels of selected proteins in wild-type and Atp6ap2 conditional knockout (cKO) liver following the first application of poly (I:C) at d 1. Phosphorylated (p-) and total protein were detected for the MTORC1 targets RPS6KB (Thr389) and ULK1 (Ser757) as well as of the MTORC1 repressor AKT1S1 (Thr246). Detection of GAPDH was used to visualize equal protein loading. * indicates nonspecific antibody binding. CTF, C-terminal fragments. (B) Fluorescence microscopy analysis of MTOR localization in fixed wild-type and Atp6ap2 cKO hepatocytes (d 10). A co-staining of LAMP2 and TCIRG1/ATP6V0A3 (V0 a3) was used to assess lysosomes and the Atp6ap2 knockout efficiency, respectively. Boxed areas are resolved with higher magnification. Scale bars: 10 µm. (C) Liver lysosomes (tritosomes) were isolated from 2 wild-type and 2 Atp6ap2 cKO mice (d 15) and processed for immunoblotting. Atp6ap2 cKO tritosome preparations display increased levels of the kinase MTOR as well as of the MTORC1 recruiting proteins RRAGA, RRAGC, LAMTOR1 and LAMTOR2. LAMP2 levels in the isolated lysosomes were not changed between the genotypes. The experiment was reproduced using a second set of 2 wild-type and 2 Atp6ap2 cKO tritosome preparations (not shown).
We observed that a major fraction of MTOR still localized to lysosomes in the hepatocytes of ATP6AP2-deficient mice (Fig. 3B). MTOR could furthermore be detected in isolated liver lysosomes from these animals (Fig. 3C). In agreement with the increased MTORC1 activity, we found increased levels of the MTOR recruitment factors LAMTOR1, LAMTOR2, RRAGA and RRAGC in the knockout lysosomes (Fig. 3C). Translocation of MTORC1 to lysosomes allows the kinase to be activated by the lysosomal RHEB protein.
The increased phosphorylation of the MTOR substrates RPS6KB, AKT1S1 and ULK1 in the ATP6AP2-depleted liver extracts was accompanied by augmented total cellular levels of each of the 3 proteins (Fig. 4A, B). The overall expression of AKT and MAPK1/ERK2-MAPK3/ERK1 was also increased to 3.7- and 4.8-fold, respectively (Fig. 4A). This suggests a more general effect on intracellular signaling pathways in the absence of ATP6AP2. TFEB (transcription factor EB) was analyzed as another direct substrate of MTORC1. Surprisingly, increased transcription of the TFEB downstream targets Hexa, Atp6v0d1 and Sqstm1 indicated an elevated activity of TFEB in ATP6AP2-depleted liver (Fig. 4C). Because increased MTORC1 activity should result in phosphorylation of the transcription factor and its exclusion from the nucleus, we analyzed the cellular distribution of TFEB in control and Atp6ap2 cKO liver. The ratio of nuclear to cytosolic TFEB was slightly elevated in knockout liver (Fig. 4Di,ii). The nuclear presence of TFEB and an overall increased TFEB expression in Atp6ap2 knockout samples might explain the elevated TFEB-dependent gene transcription. Two TFEB independent autophagy-relevant genes, Atg10 and Atg9a, were downregulated, possibly as a compensatory response of the cells (Fig. 4C). Our experiments are in support of the hypothesis that in vivo MTORC1 and TFEB are regulated by multiple upstream factors. The absence of ATP6AP2 may lead to dysregulation of different signaling events.
Figure 4.
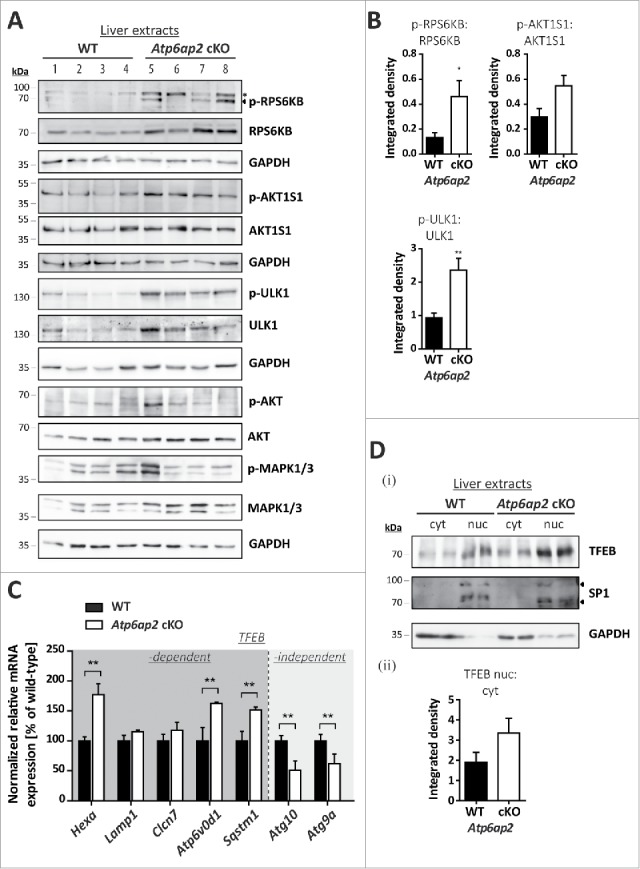
MTORC1 is active in liver bearing decreased v-H+-ATPase levels. (A) Phosphorylation of the MTORC1 targets RPS6KB and ULK1 as well as of the MTORC1 repressor AKT1S1 and the insulin-responsive kinases MAPK1/ERK2-MAPK3/ERK1 (Thr202, Tyr204) and AKT (Ser473) was analyzed by immunoblotting of 4 different wild-type control (WT, lanes 1–4) and 4 Atp6ap2 cKO liver extracts (lanes 5–8) and revealed increased signaling activity in the Atp6ap2 cKO samples. GAPDH staining was used to control for equal protein loads. Irrelevant bands according to the antibody datasheet are labeled with *. (B) Signal intensities from (A) were quantified in regard to the extent of protein phosphorylation and compared between both genotypes. Shown are means ± standard errors from 4 independent liver preparations per genotype. * P < 0.05, ** P < 0.01 according to unpaired, 2-tailed Student t test. (C) Transcription of TFEB-dependent and -independent genes involved in autophagy and lysosomal biogenesis was analyzed by qRT-PCR using whole liver RNA extracts of wild-type and Atp6ap2 cKO animals. Expression levels were normalized to the most stable housekeeping genes and related to the wild-type expression rate. Bars represent mean values of 6 independent RNA preparations per genotype ± standard errors (** P < 0.01 according to unpaired, 2-tailed Student t test between genotypes). (Di) Fractionation of cytosolic (cyt) and nuclear (nuc) proteins from wild-type and Atp6ap2 cKO livers. TFEB displays prominent nuclear localization in both genotypes. SP1 and GAPDH were used as specific nuclear or cytosolic positive control, respectively. (Dii) Quantification of data obtained in (Di) regarding the ratio of nuclear to cytosolic TFEB. Shown are mean values ± standard errors of 6 independent preparations per genotype.
Unaltered autophagy in v-H+-ATPase-depleted fibroblasts
To better understand the role of the v-H+-ATPase in MTORC1 signaling and to avoid possible secondary effects due to the strong hepatic phenotype, we generated fibroblasts specifically lacking the v-H+-ATPase-accessory subunit ATP6AP2. Mice with loxP sites flanking exon 2 of the Atp6ap2 gene26 were used to generate Atp6ap2flox/flox mouse embryonic fibroblasts. These cells and Atp6ap2+/y control cells were stably transfected with a plasmid encoding Cre-recombinase. The successful generation of ATP6AP2-deficient cells was confirmed by immunoblotting (Fig. 5A). Lack of ATP6AP2 led to a significantly reduced expression of other essential v-H+-ATPase subunits, i.e. ATP6V0D1 (22 ± 5% remaining signal), ATP6V0C (26 ± 17%), ATP6V0A1 (22 ± 13%) and TCIRG1/ATP6V0A3 (4 ± 2%). A peripheral subunit of the V1 sector, ATP6V1B2, was not affected (Fig. 5A). In the Atp6ap2 knockout cells, the TCIRG1/ATP6V0A3 subunit, which is the predominant a-subunit in lysosomal membranes, was almost undetectable in LAMP2-positive compartments using immunofluorescence staining (Fig. 5B). Despite the lack of ATP6AP2, lysosome acidification, HEX/acid β-hexosaminidase activity and lysosomal protein degradation were unaltered (Fig. S5). Re-acidification of isolated and alkalinized lysosomes was also not changed in the knockout lysosomes (Fig. S5B).
Figure 5.
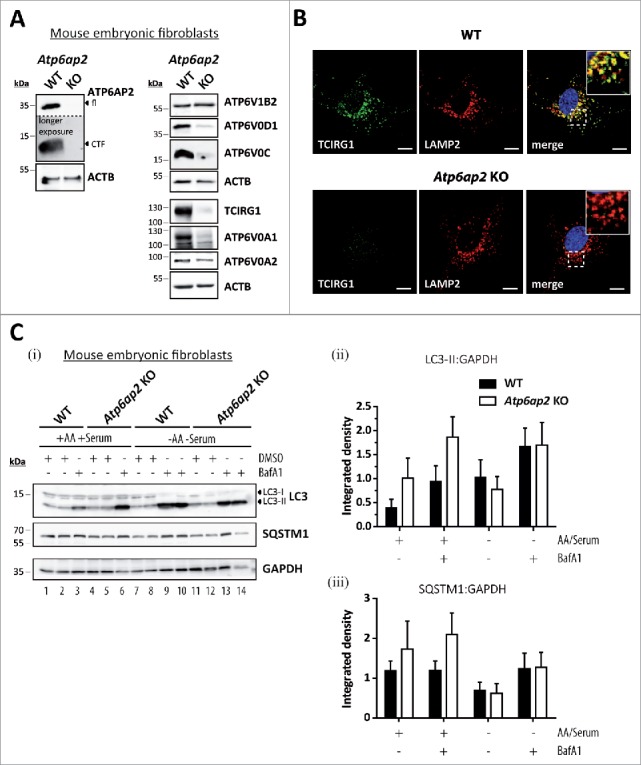
Reduced v-H+-ATPase concentration does not impair autophagic degradation in Atp6ap2 knockout MEFs. (A) Lysates of Atp6ap2 knockout (KO) mouse embryonic fibroblasts lack ATP6AP2 full length (fl) and C-terminal fragments (CTF) in immunoblot analyses. Concomitant probing for the indicated v-H+-ATPase V0 and V1 subunits revealed reduced V0 sector level upon deletion of Atp6ap2. ACTB served as a loading control. (B) Immunofluorescence staining of control and ATP6AP2-deficient MEFs confirmed reduced levels of TCIRG1/ATP6V0A3 that shows a colocalization with the lysosomal marker LAMP2 in wild-type cells. Scale bar: 10 µm. (C) Fibroblasts of both genotypes were cultivated in the presence or absence of free amino acids (AA) and serum for 2 h. Where indicated, cells were pre-treated with 100 nM BafA1 for 1 h to inhibit lysosomal degradation and the treatment continued throughout the experimental setup. Control fibroblasts were incubated in the presence of DMSO (i). Levels of LC3-I, LC3-II and SQSTM1 were detected by immunoblot analyses. Labeling of GAPDH was used to control for an equal protein load. (ii, iii) Quantification of (i) revealed a starvation-induced elevation in the ratio LC3-II to GAPDH and a parallel decrease in the amount of SQSTM1 that appeared independent of the expression of ATP6AP2. Shown are means ± standard errors from 4 independent measurements.
In contrast to the strong accumulation of autophagic vacuoles in liver tissue and isolated hepatocytes, ATP6AP2-deficient mouse embryonic fibroblasts (MEFs) were characterized by an only slightly increased basal level of autophagy. This was revealed by an increased presence of LC3-II in ATP6AP2-deficient cells under conditions of BafA1 treatment (Fig. 5C, lanes 1–6). This accumulation of LC3-II indicates increased formation of autophagosomes in ATP6AP2-deficient cells under basal conditions. However, we observed a similar turnover of the autophagosomal LC3-II, and a drop in the level of the autophagy cargo protein SQSTM1, in control and ATP6AP2-deficient MEFs, when autophagy was induced by starving the cells for 2 h (Fig. 5C, lane 7–14). Under conditions of sufficient nutrient supply (culture medium containing serum, glucose and amino acids) we detected no differences in the degree of autophagy between control cells and ATP6AP2-deficient cells at the ultrastructural level (not shown). These data suggest that in fibroblasts a significant decrease in the v-H+-ATPase level only mildly affects lysosomal acidification and macroautophagy under basal conditions, while no effects were observed under serum and amino acid starvation.
Amino acid-dependent MTORC1 activation requires the presence of ATP6AP2
Our genetically modified cells allowed us to ask whether the downregulation of the v-H+-ATPase complex affects the inactivation and reactivation of MTORC1. In either genotype, withdrawal of both serum and amino acids resulted in an almost complete loss of the phosphorylation of the MTORC1 substrate RPS6KB, as well as a reduction in the phosphorylation of the inhibitory MTORC1 subunit AKT1S1 (Fig. S1, Fig. 6Ai,ii). Adding back amino acids for 2 h led to the reappearance of phosphorylated RPS6KB whereas the AKT-dependent phosphorylation of AKT1S1 did not increase (Fig. 6Ai,ii). It should be noted that phosphorylation of AKT1S1 is not triggered by addition of amino acids only. Insulin and growth factors also contribute to its phosphorylation at threonine 246 after activation of AKT (Fig. S1ii).33 Torin1, a MTORC1 and MTORC2 inhibitor,34 was able to completely suppress the phosphorylation of RPS6KB, even in the presence of amino acids. In Atp6ap2 knockout cells, the inactivation of MTORC1 was similar to what was seen in control cells. However, the phosphorylation of RPS6KB was clearly reduced upon amino acid stimulation (Fig. 6Ai,ii).
Figure 6.
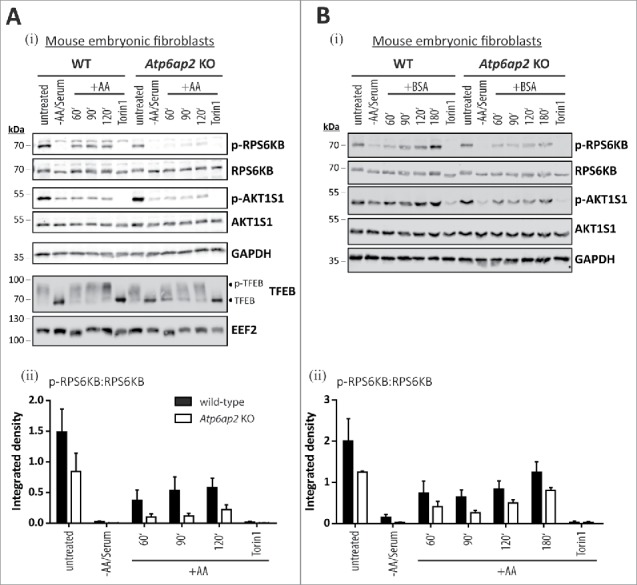
Reduction of the v-H+-ATPase V0 sector level interferes with amino acid-dependent activation of MTORC1. (A) Wild-type control and Atp6ap2 knockout MEFs were cultivated for 1 h in the absence of amino acids and serum to silence MTORC1 signaling. Then, the MTORC1 complex was activated in amino acid-containing DMEM for up to 2 h. Simultaneous treatment with Torin 1 (250 nM) was used to block MTORC1 activation. Basal MTORC1 activity was assessed under fed conditions (untreated). (i) Detection of the total and phosphorylated MTORC1 substrate RPS6KB and AKT1S1 in control and ATP6AP2-deficient MEFs following the stimulation with amino acids. TFEB phosphorylation correlated with the activation of RPS6KB and was decreased in amino acid-stimulated Atp6ap2 knockout cells. GAPDH and EEF2 served as loading controls. (ii) Ratios of p-RPS6KB to total RPS6KB were quantified from 3 independent experiments and are shown as means ± standard errors. (B) Reactivation of MTORC1 by extracellular application of BSA. Fibroblasts of both genotypes were starved for amino acids and serum for 1 h before cells were incubated with 3% (w:v) BSA for the indicated time periods to reactivate MTORC1 following intracellular digestion. (i) Addition of BSA was sufficient to trigger phosphorylation of RPS6KB as well as AKT1S1 regardless of the ATP6AP2 expression. Control samples were left untreated or incubated with the MTOR inhibitor Torin 1. (ii) Quantification of the signal intensities of p-RPS6KB to total RPS6KB depicted as means ± standard errors from 3 independent experiments.
Our observations using the Atp6ap2 knockout cells argue strongly that the MTORC1 activity depends on the presence of the v-H+-ATPase complex.16,17 In the same experiment, we analyzed the MTORC1-dependent phosphorylation status of TFEB. Starvation or Torin1 treatment of wild-type MEFs led to dephosphorylation and presumably nuclear localization of TFEB.13-15,35 Refeeding with amino acids triggered phosphorylation of TFEB in both wild-type and Atp6ap2 knockout MEF cells. However, in the latter cells the level of dephosphorylated TFEB was still higher after 60 and 90 min of refeeding, indicating a reduced MTORC1 activity (Fig. 6AI).
We further addressed the question of whether the MTORC1 activation depends on the mere presence of the v-H+-ATPase complex, its assembly state or its ability to acidify vesicles. To this end, we treated control cells with BafA1, which blocks proton pump activity and favors the disassembly of the complex,36-38 or with saliphenylhalamide A, which inactivates and promotes assembly of the complex.32,39 We performed experiments in the presence of a constant concentration of either of these drugs analyzing MTORC1 reactivation at different times after re-addition of amino acids (Fig. S6A, B). We also investigated MTORC1 reactivation after 2 h of amino acid application using different concentrations of either inhibitor (Fig. S6C, D). These experiments (Fig. S6A-D) revealed that in wild-type fibroblasts, MTORC1 activation by amino acids is independent of the proton pumping and assembly state of the v-H+-ATPase complex.
We observed that when v-H+-ATPase expression was reduced, addition of free amino acids did not promote phosphorylation of RPS6KB (Fig. 6A). This finding prompted us to ask whether a similar block in MTORC1 activation is induced in the absence of ATP6AP2 when intracellular amino acids are generated by protein degradation. We therefore offered bovine serum albumin (BSA) to the MEF cells after starvation (Fig. 6B) and monitored the RPS6KB phosphorylation in control and ATP6AP2-deficient cells. In comparison to the experiment where free amino acids were added, we observed a delayed phosphorylation of the MTORC1 substrate RPS6KB in ATP6AP2-deficient cells, possibly reflecting the uptake and intracellular processing of BSA. In contrast to the free amino acid experiment, the AKT1S1 phosphorylation increased over time, independent of the genotype. This indicates that the intracellular degradation of BSA also affects the AKT-dependent phosphorylation of AKT1S1, a response, which is normally triggered by the activation of receptor tyrosine kinases (Fig. S1ii).
Insulin-dependent MTORC1 activation occurs independently of the v-H+-ATPase complex
Because insulin and growth factors trigger MTORC1 activation through the AKT pathway (Fig. S1ii)40 we wondered if the presence or absence of the v-H+-ATPase complex also affects this signaling pathway. When MTORC1 was inactivated by starvation in control and Atp6ap2 knockout cells, phosphorylation of RPS6KB and AKT1S1 was significantly reduced (Fig. 7A). Addition of 150 nM insulin for up to 1 h led to the appearance of phosphorylated RPS6KB and AKT1S1 in control fibroblasts (Fig. 7A). Surprisingly, a similar response was observed in the cells lacking ATP6AP2. In these experiments, inhibition of MTORC1 activity by Torin 1 was used as a negative control, and as expected RPS6KB and AKT1S1 phosphorylation was abolished after Torin 1 treatment. Activation of AKT and MAPK1/ERK2-MAPK3/ERK1 via the insulin receptor was also demonstrated (Fig. 7AI). In a similar experimental setup, we tested the ability of insulin to trigger MTORC1 activity also in wild-type and ATP6AP2-deficient primary hepatocytes (Fig. 7B). Again, treatment of serum- and amino acid-deprived cells with insulin activated MTORC1 in hepatocytes from both genotypes. These data suggest that the v-H+-ATPase complex is dispensable for the stimulation of MTORC1 by insulin.
Figure 7.
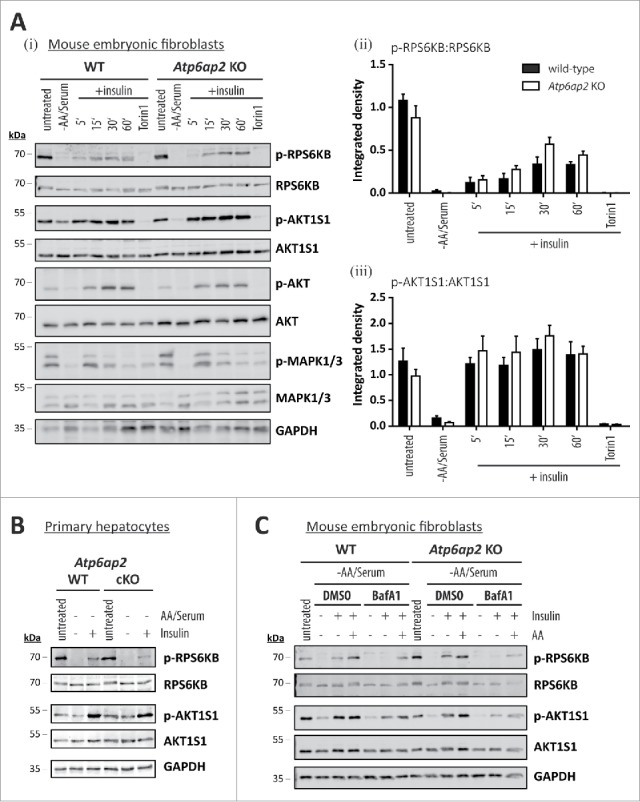
Activation of MTORC1 by insulin depends on amino acids but not on ATP6AP2 expression. (A) Activation of MTORC1 was triggered in wild-type control (WT) and Atp6ap2 knockout (KO) MEFs by 1 h starvation of serum and amino acids and subsequent administration of insulin (150 nM) for 5 to 60 min. Control samples were cultivated in the presence of serum and amino acids (untreated) or co-cultivated with insulin and Torin 1 (250 nM) to interfere with MTORC1 stimulation. (i) Insulin treatment led to elevated signals for phosphorylated AKT1S1 (Thr246) in both genotypes and a concomitant increase in RPS6KB phosphorylation (Thr389). Phosphorylated and total MAPK1/ERK2-MAPK3/ERK1 as well as AKT were assessed to verify insulin receptor activation. Mean ratios of p-RPS6KB to RPS6KB (ii) and p-AKT1S1 to AKT1S1 (iii) as detected in (i) were quantified from 3 independent experiments. Error bars display standard errors. (B) Insulin-dependent activation of MTORC1 in primary wild-type and Atp6ap2 conditional knockout (cKO) hepatocytes as described above (A). Cells were starved for amino acids and serum for 1 h before MTORC1 activity was triggered by application of 150 nM insulin for 15 min. (C) Control and Atp6ap2 knockout fibroblast were maintained in the presence of 50 nM BafA1 for 16 h to block lysosomal proteolysis or autophagy, respectively. MTORC1 activation was then triggered by starvation and stimulation with insulin for 15 min as described in (A), but under continued inhibitor treatment. Simultaneous reactivation of MTORC1 by amino acids and insulin was included to replenish intracellular amino acid pools. Cells were kept in DMEM containing serum and amino acids (untreated) as a control for the steady-state activity of MTORC1 or processed with DMSO as solvent control.
The full activity of MTORC1 requires activation by lysosomal RHEB (Fig. S1).41,42 Because amino acids are involved in recruiting MTORC1 to the lysosomal surface, we tested if amino acids are also needed for the stimulation of MTORC1 by insulin. We therefore depleted the level of intracellular amino acids by treatment with the lysosomal acidification inhibitor BafA1 and followed the phosphorylation of RPS6KB and AKT1S1 in wild-type and Atp6ap2 knockout cells (Fig. 7C). In both cell types, depletion of serum and amino acids resulted in inactivation of MTORC1. Addition of insulin led to a reappearance of MTORC1 activity that was blocked in the presence of BafA1. A similar effect was seen when control cells were treated with the proteasomal inhibitors epoxomicin and MG132 (Fig. S7). Importantly, if amino acids and insulin were added together to inhibitor-treated cells, MTORC1 activity was partially restored (Fig. 7C).
Our data reveal that insulin could activate MTORC1 independently of the v-H+-ATPase complex. However, this activation depends on intracellular amino acids, possibly derived from degradation by lysosomes and the proteasome.
Discussion
The v-H+-ATPase complex, an integral part of the lysosome and endosome membranes, is traditionally regarded as the proton translocation machinery of these organelles.25,43,44 However, in recent years many additional functions have been revealed, including a possible participation in membrane fusion catalysis,45-47 acidification-dependent budding of endosomal carrier vesicles,48 modulation of actin polymerization49-51 and sensing of amino acids resulting in the activation of MTORC1.16,17 MTORC1 activation is necessary to regulate the level of macroautophagy in response to nutrient availability.1,52
Our study addressed in detail how the v-H+-ATPase complex affects the function of MTORC1 in controlling autophagy. We observed that depletion of the v-H+-ATPase complex in liver caused an accumulation of autophagic vacuoles. Hepatocytes derived from the ATP2AP2-deficient livers show a strong upregulation of early autophagic marker proteins accompanied by decreased degradation of LC3-II. However, electron microscopy studies and colocalization of LC3 and LAMP1 indicated that fusion of lysosomes with autophagosomes was still functional. Lack of the v-H+-ATPase complex was much better tolerated by fibroblasts than by liver cells, since the level of autophagy was only mildly or not at all affected. The difference between the 2 ATP6AP2-deficient cell types may be due to a different rate of basal autophagy, an altered role of the accessory subunit or the developmental state of the cells.
Although cells lacking the proton pump were unable to signal amino acid availibility to MTORC1, the response to insulin stimulation was still functional. Our conclusion is that in cultured cells and liver tissue the activation state of MTORC1 is controlled by multiple pathways, that include both insulin and growth factor signaling pathways.
Hepatocytes lacking ATP6AP2 displayed an impressive accumulation of autophagic vacuoles, characterized by LC3 upregulation and buildup of SQSTM1. The increased expression of the early autophagy marker proteins WIPI2 and ATG5 suggests higher rates of autophagosome formation in Atp6ap2 knockout hepatocytes. The accumulation of autophagosomes may also imply that protein degradation is inhibited and that undegraded material accumulates in large vesicles similar to what happens in lysosomal storage disorder cells. However, ATP6AP2-deficient hepatocytes in fact showed increased lysosomal hydrolase activities and elevated expression of the LAMPs. In contrast to previously described conditional Atp6ap2 knockout mice with disruption in cardiomyocytes and podocytes26-28 an acidification defect was not detected in hepatocytes. As discussed,32 a minor amount of the v-H+-ATPase complex can be sufficient to maintain vesicular acidification. Further, autophagosomes still acquired externally added endocytic tracers and showed partial degradation (as judged by electron microscopy morphology) of cytoplasmic content in the absence of the v-H+-ATPase. Because BafA1 only mildly affected the LC3-II levels in knockout hepatocytes we reasoned that the kinetics of autolysosome formation and autolysosome degradation might be altered when Atp6ap2 was deleted. Together with the increased formation of autophagosomes, this may lead to the observed buildup of autophagic vacuoles in knockout hepatocytes.
We speculated that in the absence of functional v-H+-ATPase MTORC1 activity is inhibited,7,9,53 leading to the increased production of autophagosomes. In accordance with this view, we observed that the expression of selected TFEB-downstream genes was upregulated and TFEB was still found in nuclear extracts of ATP6AP2-deficient livers. This might reflect reduced MTORC1-mediated phosphorylation of TFEB. However, in contrast to the hypothesis that MTORC1 is inactive we found normal levels of phosphorylated ULK1, RPS6KB and AKT1S1 in ATP6AP2-deficient mice. The phosphorylation of these substrates suggests a comparable MTORC1 activity in wild-type and Atp6ap2 knockout liver. The decreased phosphorylation of TFEB may be explained by PPP3/calcineurin-dependent dephosphorylation54 of TFEB or reduced TFEB phosphorylation by other kinases55,56 such as recently shown for TFEB-phosphorylation by PRKC/PKC (protein kinase C).57 Next to the regulation of TFEB nuclear translocation, the increased overall expression of TFEB is one likely reason for the elevated expression of TFEB downstream genes in Atp6ap2 cKO tissue. Our experiments demonstrate that the accumulation of autophagosomes and lysosomes in ATP6AP2-deficient hepatocytes occurs despite normal or even increased activity of MTORC1 and its substrates. This finding is in favor of the view that in liver MTORC1-mediated control of autophagy is not always the dominant factor.
It could well be that—in addition to MTORC1—alternative pathways, such as PDPK1 (phosphoinositide dependent protein kinase 1)-phosphatidylinositol 3-kinase (PtdIns3K) signaling, exist in vivo, which allow MTORC1-independent phosphorylation of RPS6KB.58 The phosphorylation state of RPS6KB, widely accepted as an reliable readout to monitor MTORC1 activity should therefore be considered with caution. Similarly, an augmented AKT activity may cause the increased phosphorylation of AKT1S1.33
The liver pathology after ATP6AP2-depletion was very pronounced and secondary effects may have contributed to the observed accumulation of endocytic vesicles in these cells. It seems likely that cytokine- and hormone-mediated signaling40 became altered due to the changed morphology and function of hepatocytes. In contrast, the link between v-H+-ATPase and MTORC1 signaling could be analyzed without secondary effects in the response to different stimuli in control and ATP6AP2-deficient fibroblasts. In control fibroblasts, lysosome acidification and assembly state of the v-H+-ATPase complex did not affect the amino acid-dependent MTORC1 activation, suggesting that sensing of amino acids requires neither the proton pumping activity nor the dissassembly of the V0-V1 complex of v-H+-ATPase. These observations in MEF cells are in contrast to earlier results in HEK cells16 where similar concentrations of salicylihalamide A blocked amino acid-dependent MTORC1 reactivation. This discrepancy may reflect cell-type specific responses to the drugs, different off-target effects of the used drugs or small variations in the experimental conditions. It should be noted that cell-based siRNA experiments in which the ATP6V0C v-H+-ATPase subunit16 was depleted, are in agreement with our findings in MEF cells. Similar to the observations made in HEK cells, activation of MTORC1 was reduced after feeding amino acids or albumin as external amino acids sources, in fibroblasts lacking ATP6AP2. These data support the view that the presence of the v-H+-ATPase complex is critical for MTORC1 activation by amino acid stimulation.16,20 MTORC1 activity was not completely abolished by addition of amino acids to starved Atp6ap2 knockout cells. This can be explained by the presence of amino acid sensing mechanisms, occurring independently of v-H+-ATPases, as have recently been revealed for leucine and arginine.59-61 Also, extracellular glucose contributes to MTORC1 activation.59,62,63 It remains open how much the recently discovered amino acid transporter SLC38A917-19 can signal without the v-H+-ATPase complex.
Surprised by the unaltered basal activity of MTORC1 in the absence of ATP6AP2, we analyzed whether the PtdIns3K-dependent pathway compensated for the decreased direct response to amino acids. We observed that in the ATP6AP2-deficient fibroblasts insulin triggered an increased activation of MTORC1. When the available intracellular amino acids were reduced by inhibiting intracellular protein degradation in fibroblasts, insulin signaling was disturbed. This supports the view of a crosstalk between the input signals of amino acid concentrations and growth factors.40,64 Moreover, MTORC1 activity in Atp6ap2 cKO hepatocytes was also responsive to insulin treatment. This might explain the ability of the knockout cells to maintain MTORC1 in an active state even when v-H+-ATPase-mediated amino acid sensing is impaired. The view of a complex regulation of MTORC1 by different stimuli is supported by the recent observation that TSC2 as an inhibitor of MTORC1 activity is recruited to lysosomes by different conditions of stress or starvation.65 Only the combined absence of these stimulants will allow the MTORC1 inhibitor to leave its lysosomal localization and permit MTORC1 activity.
Taken together our findings show that the role of the v-H+-ATPase complex in the regulation of autophagy is not exclusively dependent on lysosome-based pathways. The response of the MTORC1 pathway to a lack of ATP6AP2 in liver occurs only after accumulation of autophagomes. This suggests that the proton pump is a central element regulating autophagy in murine cells, whereas PtdIns3K-dependent pathways may be upregulated to provide an effective option for cell survival.
Material and methods
Antibodies
The following proteins were detected using the denoted antibodies: Phosphorylated (p)-AKT (Ser473; 4060), AKT (4691), p-AKT1S1 (Thr246; 2997), AKT1S1 (2691), ATP6V1B2 (14617), p-MAPK1/ERK2-MAPK3/ERK1 (Thr202, Tyr204; 4370), MAPK1/ERK2-MAPK3/ERK1 (4695), LAMTOR1 (8975), LAMTOR2 (8145), MTOR (2983), p-RPS6KB (Thr389; 9205), RPS6KB (9202), p-ULK1 (Ser757; 14202), ULK1 (8054), all from Cell Signaling Technology, ACTB/β-Actin (Sigma-Aldrich, A2066), ATP6AP2,32 ATP6V0A1 (kindly provided by Dr. Beth S. Lee, Ohio State University College of Medicine, Columbus, OH, USA)66 ATP6V0A2 (Abcam, ab96803), TCIRG1/ATP6V0A3 (generous gift from Dr. T. Jentsch, FMP/MDC, Berlin, Germany), ATP6V0C,32 ATP6V0D1 (Proteintech group, 18274–1-AP), GAPDH/glyceraldehyde-3-phosphate-dehydrogenase (Santa Cruz Biotechnology, sc-25778), EEF2/eukaryotic translation elongation factor 2 (Abcam, ab33523), LAMP1 (DSHB, 1D4B), LAMP2 (DSHB, Abl93), LC3 (MBL International, PM036), SP1 (Merck Millipore, ABE135), SQSTM1 (Enzo Life Sciences, BML-PW-9860), TFEB (Bethyl Laboratories, A303–673A-M). Secondary antibodies conjugated to horseradish peroxidase, Alexa Fluor® 488, Alexa Fluor® 594 or Alexa Fluor® 647 were purchased from Dianova and Life Technologies (Dianova, 111–035–144, 11–035–143, 706–035–148; Life Technologies, A11006, A21247, A11034, A11037, A11073). IRDye® 800CW secondary antibody was from LI-COR (926–32213).
Experimental animals
Mice bearing LoxP-sites surrounding exon 2 of Atp6ap2 and expressing the Cre recombinase under the control of the Mx1 promotor have been described previously.32 Atp6ap2flox/flox and Atp6ap2flox/Y mice with Mx1-Cretg/+ alleles were referred to as conditional knockout animals. Homo- or hemizygous floxed littermates negative for the Mx1-Cre transgene served as wild-type controls. Three intraperitoneal injections of 250 µg polyinosinic–polycytidylic acid (poly [I:C]; Sigma-Aldrich, P9582) were applied within 5 d to induce expression of the Cre recombinase in 6-wk-old cKO and control animals. After a further 5 to 10 d in housing, mice were killed for experimental analyses. All animal experiments were conducted in agreement with local guidelines for the use of animals and their care.
Cell lines
Cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Life Technologies, 41965) containing 4 mM L-glutamine and 4.5 g/l glucose and supplemented with 10% (v:v) fetal bovine serum (Biochrom AG, S0115) in a humidified 5% CO2 atmosphere at 37°C.
Mouse embryonic fibroblast lines were generated from embryos with floxed Atp6ap2 alleles at embryonic d 13.5. Single cells were obtained by trypsin digest upon decapitation of the embryos and removal of all inner organs. MEFs were immortalized by transfection of the SV40 large T antigen. To induce a knockout of Atp6ap2, the Cre recombinase was introduced by additional cotransfection of pGK-hygro67 and pCAG-Cre:GFP (Addgene, 18782 and 13776) and selection for stable hygromycin B resistance (500 µg/ml; Invivogen, ant-hg-1).
Modulation of MTORC1 activity
Stimulation with amino acids – For initial MTORC1 inactivation cells were rinsed with phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) and incubated for 1 h in Earle's balanced salt solution (EBSS; Sigma-Aldrich, E2888). Reactivation was initiated following a washing step by incubation in DMEM or 3% (w:v) BSA (Roth, T844.4) in EBSS for 1 to 3 h as indicated. Treatment with 250 nM Torin 1 (Cayman Chemical, 10997) during the reactivation period was used to silence MTOR kinase activity even in the presence of amino acids.
Stimulation with insulin – MTORC1 activity was diminished as described above. Cells were rinsed with PBS and stimulated with 150 nM insulin (SAFC Biosciences, 91077C) in EBSS for up to 1 h as noted. Induction of MTORC1 activity was further conducted in the presence of 250 nM Torin 1 as a negative control. For concomitant inhibition of lysosomal proteolysis or macroautophagy, cells were incubated for 16 h with 50 nM BafA1 (Calbiochem, 196000) or 5 mM 3-methyladenine (Sigma-Aldrich, M9281), respectively. Inhibitor treatment was continued throughout the MTORC1 inactivation-reactivation procedure. Control samples were handled with the respective solvent. Combined activation by insulin and amino acids was achieved by stimulation with 150 nM insulin in DMEM for 15 min.
Immunoblotting and quantification
Cell lysates were prepared as described previously.32 Proteins were separated on 12.5% SDS-PAGE gels and transferred to nitrocellulose membranes (Whatman, GE Healthcare, 10426994) by semi-dry blotting. The Amersham ECL Advanced Western Blotting Detection kit (GE Healthcare, RPN2135) was used in combination with ImageJ software to detect and quantify antibody signals. Immunoblots concerning MTORC1 regulation (AKT1S1, RPS6KB) were analyzed and quantified using a LI-COR Odyssey® Fc imager and the Image Studio™ software.
Fractionation of cytosolic and nuclear proteins
Liver samples were homogenized in a dounce homogenizer with 10 strokes in fractionation buffer (10 mM HEPES, pH 7.4, 250 mM sucrose [Carl Roth, 4621.2], 1 mM EDTA). Non-broken down aggregates were excluded by filtration through a 100-µm nylon cell strainer (Corning, 734–0004) and the homogenate centrifuged at 600 x g at 4°C for 10 min to separate nuclei (pellet) from the post nuclear supernatant. Nuclei were washed in fractionation buffer and centrifuged as above for re-collection. Cytoplasmic proteins were further enriched from the post nuclear supernatant by centrifugation at 600 x g at 4°C for 10 min. The supernatants containing cytosolic proteins and the nuclear fractions were sonicated for 30 sec using a Branson Sonifier 450 (level 7 in a cup horn, Emerson Industrial Automation), before protein content was measured using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, 23225). Samples of 20 µg protein per lane were analyzed by immunoblotting.
Immunofluorescence analysis
Cultured cells – Semi-confluent cultures grown on coverslips were rinsed 3 times with PBS, fixed with 4% (w:v) paraformaldehyde (Carl Roth, 0335) solution and permeabilized in 0.2% (w:v) saponin in PBS. Nonspecific binding sites were saturated with 10% (v:v) fetal bovine serum, 0.2% (w:v) saponin in PBS (blocking solution). For antibody staining, antibodies were diluted in blocking solution and applied to the coverslips overnight at 4°C (primary antibody) or for 1 h at room temperature (secondary antibody) in a humidified chamber. Coverslips were mounted with 17 % (w:v) Mowiol 4–88 (Calbiochem, 475904), 33% (v:v) glycerol, 20 mg/ml DABCO (1,4-diaza-bicyclo-[2,2,2]-octane; Sigma-Aldrich, D2522) and 5 μg/ml DAPI (4-,6-diamidino-2-phenylindole; Sigma-Aldrich, 32670) for nucleus staining. Confocal images were acquired with an Olympus FV1000 confocal laser scanning microscope.
Liver sections – Small pieces of liver were fixed overnight in 4% (w:v) paraformaldehyde solution and transferred to 30% (w:v) sucrose in 0.1 M phosphate buffer (PB) pH 7.4 (16 mM KH2PO4, 84 mM Na2HPO4). Using a dry ice-cooled sledge microtome, 35-µm-thick slices were generated, rinsed 3 times in 0.1 M PB and blocked in blocking solution (4% [v:v] goat serum [Thermo Fisher Scientific, 16210064], 0.5% [v:v] Triton X-100 [Sigma, X100] in 0.1 M PB). Primary antibody staining was conducted in blocking solution overnight at 4°C, sections rinsed and incubated with secondary antibody in 0.1 M PB containing 0.25% (v:v) Triton X-100 for 2 h at room temperature. Before embedding in 17% (w:v) Mowiol 4–88, 33% (v:v) glycerol and 20 mg/ml DABCO, liver sections were rinsed, stained with DAPI (5 μg/ml) and washed again.
Transmission electron microscopy
Mice of the indicated genotypes were perfused transcardially with 6% (v:v) glutaraldehyde (Polysciences, 00216A) in 0.1 M PB. Liver samples were post-fixed in 2% (w:v) OsO4, dehydrated and embedded in Araldite® M/dodecenylsuccinic anhydride (Sigma-Aldrich, 10951). Ultrathin sections were cut on an EM UC6 ultramicrotome (Leica), collected on Formvar-coated copper grids (Agar Scientific, G2020C) and stained according to standard protocols. Images were acquired with an EM900 transmission electron microscope (Zeiss).
RNA extraction, reverse transcription and quantitative real-time PCR (qRT-PCR)
cDNA was prepared from 0.5–2 µg total liver RNA extracts and qRT-PCR assays designed and performed as described previously.32 Relative mRNA expression was calculated by normalizing Cp values to the logarithmic average Cp of the most stable housekeeping genes (Tuba1a, Hprt/Hprt1 and Actb). Resulting DCp were compared between genotypes for statistical analyses. To visualize normalized mRNA expression, primer efficiencies (E) were determined for each PCR by co-measurement of a set of serial cDNA dilutions to obtain E(−DCp) plots. The normalized mRNA expression rates were further related to the wild-type expression levels.
Purification of liver lysosomes (tritosomes)
Mice were treated with an intraperitoneal injection of 4 µl 17% (v:v) tyloxapol (Triton WR-1339; Sigma-Aldrich, T0307) per gram body weight, 3 d before sacrifice. The purification of tritosomes was performed as described previously68,69 using differential centrifugation and separation in a discontinuous sucrose gradient.
Statistical analysis
Statistical significance between genotypes was analyzed via unpaired Student t tests using GraphPad Instat 3 software. Error bars indicate the standard deviation/standard error of mean values (*p < 0.05, **p < 0.01, ***p < 0.001).
Supplementary Material
Abbreviations
- AKT1S1
AKT1 substrate 1 (proline-rich)
- ATP6AP2
ATPase H+ transporting lysosomal accessory protein 2
- ATG
autophagy related
- BafA1
bafilomycin A1
- cKO
conditional knockout
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- LAMP1/2
lysosomal-associated membrane protein 1/2
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MTORC1
mechanistic target of rapamycin (MTOR) complex 1
- PtdIns3K
phosphatidylinositol 3-kinase
- RHEB
Ras homolog enriched in brain
- RPS6KB
ribosomal protein S6 kinase
- RRAG
Ras-related GTP binding
- SQSTM1
sequestosome
- TFEB
transcription factor EB
- v-H+-ATPase
vacuolar-type H+-translocating ATPase
Disclosure of potential conflicts of interest
The authors declare no conflict of interest.
Acknowledgment
We are grateful to Urska Repnik and Gareth Griffiths for some of the immunoelectron microscopy experiments. We thank the Electron Microscopy Unit at the Institute of Biotechnology, University of Helsinki, Finland, for technical help and availability of instruments.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft SPP1580 (to PS and AH) and the GRK1459 (to PS and JH). ELE was supported by the Academy of Finland. JKDB acknowledges support for the Robert A. Welch Foundation (grant I-1422).
References
- [1].Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol 2015; 25:354-63; PMID:25759175; http://dx.doi.org/ 10.1016/j.tcb.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994; 369:756-8; PMID:8008069; http://dx.doi.org/ 10.1038/369756a0 [DOI] [PubMed] [Google Scholar]
- [3].Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994; 78:35-43; PMID:7518356; http://dx.doi.org/ 10.1016/0092-8674(94)90570-3 [DOI] [PubMed] [Google Scholar]
- [4].Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, Abraham RT. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem 1995; 270:815-22; PMID:7822316; http://dx.doi.org/ 10.1074/jbc.270.2.815 [DOI] [PubMed] [Google Scholar]
- [5].Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274-93; PMID:22500797; http://dx.doi.org/ 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dazert E, Hall MN. mTOR signaling in disease. Curr Opin Cell Biol 2011; 23:744-55; PMID:21963299; http://dx.doi.org/ 10.1016/j.ceb.2011.09.003 [DOI] [PubMed] [Google Scholar]
- [7].Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992-2003; PMID:19225151; http://dx.doi.org/ 10.1091/mbc.E08-12-1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284:12297-305; PMID:19258318; http://dx.doi.org/ 10.1074/jbc.M900573200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al.. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20:1981-91; PMID:19211835; http://dx.doi.org/ 10.1091/mbc.E08-12-1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol 2013; 15:555-64; PMID:23728461; http://dx.doi.org/ 10.1038/ncb2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 2011; 20:3852-66; PMID:21752829; http://dx.doi.org/ 10.1093/hmg/ddr306 [DOI] [PubMed] [Google Scholar]
- [12].Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al.. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429-33; PMID:21617040; http://dx.doi.org/ 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et al.. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012; 31:1095-108; PMID:22343943; http://dx.doi.org/ 10.1038/emboj.2012.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012; 8:903-14; PMID:22576015; http://dx.doi.org/ 10.4161/auto.19653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 2012; 5:ra42; PMID:22692423; http://dx.doi.org/ 10.1126/scisignal.2002790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011; 334:678-83; PMID:22053050; http://dx.doi.org/ 10.1126/science.1207056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, Tagliabracci VS, Guan KL. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 2015; 347:194-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rebsamen M, Pochini L, Stasyk T, de Araujo ME, Galluccio M, Kandasamy RK, Snijder B, Fauster A, Rudashevskaya EL, Bruckner M, et al.. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 2015; 519:477-81; PMID:25561175; http://dx.doi.org/ 10.1038/nature14107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, Yuan ED, Jones TD, Chantranupong L, Comb W, et al.. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015; 347:188-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol 2014; 24:400-6; PMID:24698685; http://dx.doi.org/ 10.1016/j.tcb.2014.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol 2003; 5:566-71; PMID:12766776; http://dx.doi.org/ 10.1038/ncb996 [DOI] [PubMed] [Google Scholar]
- [22].Stocker H, Radimerski T, Schindelholz B, Wittwer F, Belawat P, Daram P, Breuer S, Thomas G, Hafen E. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol 2003; 5:559-65; PMID:12766775; http://dx.doi.org/ 10.1038/ncb995 [DOI] [PubMed] [Google Scholar]
- [23].Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol 2005; 15:702-13; PMID:15854902; http://dx.doi.org/ 10.1016/j.cub.2005.02.053 [DOI] [PubMed] [Google Scholar]
- [24].Toei M, Saum R, Forgac M. Regulation and isoform function of the V-ATPases. Biochemistry 2010; 49:4715-23; PMID:20450191; http://dx.doi.org/ 10.1021/bi100397s [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cotter K, Stransky L, McGuire C, Forgac M. Recent Insights into the Structure, Regulation, and Function of the V-ATPases. Trends Biochem Sci 2015; 40:611-22; PMID:26410601; http://dx.doi.org/ 10.1016/j.tibs.2015.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kinouchi K, Ichihara A, Sano M, Sun-Wada GH, Wada Y, Kurauchi-Mito A, Bokuda K, Narita T, Oshima Y, Sakoda M, et al.. The (pro)renin receptor/ATP6AP2 is essential for vacuolar H+-ATPase assembly in murine cardiomyocytes. Circ Res 2010; 107:30-4;PMID:20570919;http://dx.doi.org/ 10.1161/CIRCRESAHA.110.224667 [DOI] [PubMed] [Google Scholar]
- [27].Oshima Y, Kinouchi K, Ichihara A, Sakoda M, Kurauchi-Mito A, Bokuda K, Narita T, Kurosawa H, Sun-Wada GH, Wada Y, et al.. Prorenin receptor is essential for normal podocyte structure and function. J Am Soc Nephrol 2011; 22:2203-12; PMID:22052048; http://dx.doi.org/ 10.1681/ASN.2011020202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Riediger F, Quack I, Qadri F, Hartleben B, Park JK, Potthoff SA, Sohn D, Sihn G, Rousselle A, Fokuhl V, et al.. Prorenin receptor is essential for podocyte autophagy and survival. J Am Soc Nephrol 2011; 22:2193-202; PMID:22034640; http://dx.doi.org/ 10.1681/ASN.2011020200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010; 141:290-303; PMID:20381137; http://dx.doi.org/ 10.1016/j.cell.2010.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pena-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, Wolff NC, Tran TA, Zou L, Xie XJ, Corey DR, Brugarolas J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J 2011; 30:3242-58; PMID:21804531; http://dx.doi.org/ 10.1038/emboj.2011.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Czaja MJ, Ding WX, Donohue TM Jr., Friedman SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, et al.. Functions of autophagy in normal and diseased liver. Autophagy 2013; 9:1131-58; PMID:23774882; http://dx.doi.org/ 10.4161/auto.25063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kissing S, Hermsen C, Repnik U, Nesset CK, von Bargen K, Griffiths G, Ichihara A, Lee BS, Schwake M, De Brabander J, et al.. Vacuolar ATPase in phagosome-lysosome fusion. J Biol Chem 2015; 290:14166-80; PMID:25903133; http://dx.doi.org/ 10.1074/jbc.M114.628891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 2007; 25:903-15; PMID:17386266; http://dx.doi.org/ 10.1016/j.molcel.2007.03.003 [DOI] [PubMed] [Google Scholar]
- [34].Liu Q, Chang JW, Wang J, Kang SA, Thoreen CC, Markhard A, Hur W, Zhang J, Sim T, Sabatini DM, et al.. Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benz o[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. J Med Chem 2010; 53:7146-55; PMID:20860370; http://dx.doi.org/ 10.1021/jm101144f [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Settembre C, Ballabio A. TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy 2011; 7:1379-81; PMID:21785263; http://dx.doi.org/ 10.4161/auto.7.11.17166 [DOI] [PubMed] [Google Scholar]
- [36].Bowman BJ, McCall ME, Baertsch R, Bowman EJ. A model for the proteolipid ring and bafilomycin/concanamycin-binding site in the vacuolar ATPase of Neurospora crassa. J Biol Chem 2006; 281:31885-93; PMID:16912037; http://dx.doi.org/ 10.1074/jbc.M605532200 [DOI] [PubMed] [Google Scholar]
- [37].Poea-Guyon S, Ammar MR, Erard M, Amar M, Moreau AW, Fossier P, Gleize V, Vitale N, Morel N. The V-ATPase membrane domain is a sensor of granular pH that controls the exocytotic machinery. J Cell Biol 2013; 203:283-98; PMID:24165939; http://dx.doi.org/ 10.1083/jcb.201303104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wang D, Hiesinger PR. The vesicular ATPase: a missing link between acidification and exocytosis. J Cell Biol 2013; 203:171-3; PMID:24165933; http://dx.doi.org/ 10.1083/jcb.201309130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Xie XS, Padron D, Liao X, Wang J, Roth MG, De Brabander JK. Salicylihalamide A inhibits the V0 sector of the V-ATPase through a mechanism distinct from bafilomycin A1. J Biol Chem 2004; 279:19755-63; PMID:14998996; http://dx.doi.org/ 10.1074/jbc.M313796200 [DOI] [PubMed] [Google Scholar]
- [40].Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol 2015; 25:545-55; PMID:26159692; http://dx.doi.org/ 10.1016/j.tcb.2015.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 2003; 17:1829-34; PMID:12869586; http://dx.doi.org/ 10.1101/gad.1110003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol 2003; 13:1259-68; PMID:12906785; http://dx.doi.org/ 10.1016/S0960-9822(03)00506-2 [DOI] [PubMed] [Google Scholar]
- [43].Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 2007; 8:917-29; PMID:17912264; http://dx.doi.org/ 10.1038/nrm2272 [DOI] [PubMed] [Google Scholar]
- [44].Nishi T, Forgac M. The vacuolar (H+)-ATPases–nature's most versatile proton pumps. Nat Rev Mol Cell Biol 2002; 3:94-103; PMID:11836511; http://dx.doi.org/ 10.1038/nrm729 [DOI] [PubMed] [Google Scholar]
- [45].Vavassori S, Mayer A. A new life for an old pump: V-ATPase and neurotransmitter release. J Cell Biol 2014; 205:7-9; PMID:24733582; http://dx.doi.org/ 10.1083/jcb.201403040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Strasser B, Iwaszkiewicz J, Michielin O, Mayer A. The V-ATPase proteolipid cylinder promotes the lipid-mixing stage of SNARE-dependent fusion of yeast vacuoles. EMBO J 2011; 30:4126-41; PMID:21934648; http://dx.doi.org/ 10.1038/emboj.2011.335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bayer MJ, Reese C, Buhler S, Peters C, Mayer A. Vacuole membrane fusion: V0 functions after trans-SNARE pairing and is coupled to the Ca2+-releasing channel. J Cell Biol 2003; 162:211-22; PMID:12876274; http://dx.doi.org/ 10.1083/jcb.200212004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gu F, Gruenberg J. ARF1 regulates pH-dependent COP functions in the early endocytic pathway. J Biol Chem 2000; 275:8154-60; PMID:10713138; http://dx.doi.org/ 10.1074/jbc.275.11.8154 [DOI] [PubMed] [Google Scholar]
- [49].Vitavska O, Merzendorfer H, Wieczorek H. The V-ATPase subunit C binds to polymeric F-actin as well as to monomeric G-actin and induces cross-linking of actin filaments. J Biol Chem 2005; 280:1070-6; PMID:15525650; http://dx.doi.org/ 10.1074/jbc.M406797200 [DOI] [PubMed] [Google Scholar]
- [50].Chen SH, Bubb MR, Yarmola EG, Zuo J, Jiang J, Lee BS, Lu M, Gluck SL, Hurst IR, Holliday LS. Vacuolar H+-ATPase binding to microfilaments: regulation in response to phosphatidylinositol 3-kinase activity and detailed characterization of the actin-binding site in subunit B. J Biol Chem 2004; 279:7988-98; PMID:14662773; http://dx.doi.org/ 10.1074/jbc.M305351200 [DOI] [PubMed] [Google Scholar]
- [51].Holliday LS, Lu M, Lee BS, Nelson RD, Solivan S, Zhang L, Gluck SL. The amino-terminal domain of the B subunit of vacuolar H+-ATPase contains a filamentous actin binding site. J Biol Chem 2000; 275:32331-7; PMID:10915794; http://dx.doi.org/ 10.1074/jbc.M004795200 [DOI] [PubMed] [Google Scholar]
- [52].Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32:2-11; PMID:22025673; http://dx.doi.org/ 10.1128/MCB.06159-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Settembre C, Medina DL. TFEB and the CLEAR network. Methods Cell Biol 2015; 126:45-62; PMID:25665440 [DOI] [PubMed] [Google Scholar]
- [54].Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, et al.. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 2015; 17:288-99; PMID:25720963; http://dx.doi.org/ 10.1038/ncb3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Huck B, Duss S, Hausser A, Olayioye MA. Elevated protein kinase D3 (PKD3) expression supports proliferation of triple-negative breast cancer cells and contributes to mTORC1-S6K1 pathway activation. J Biol Chem 2014; 289:3138-47; PMID:24337579; http://dx.doi.org/ 10.1074/jbc.M113.502633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Moore SF, Hunter RW, Hers I. Protein kinase C and P2Y12 take center stage in thrombin-mediated activation of mammalian target of rapamycin complex 1 in human platelets. J Thromb Haemost 2014; 12:748-60; PMID:24612393; http://dx.doi.org/ 10.1111/jth.12552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Li Y, Xu M, Ding X, Yan C, Song Z, Chen L, Huang X, Wang X, Jian Y, Tang G, et al.. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat Cell Biol 2016; 18:1065-77; PMID:27617930; http://dx.doi.org/ 10.1038/ncb3407 [DOI] [PubMed] [Google Scholar]
- [58].Dufner A, Thomas G. Ribosomal S6 kinase signaling and the control of translation. Exp Cell Res 1999; 253:100-9; PMID:10579915; http://dx.doi.org/ 10.1006/excr.1999.4683 [DOI] [PubMed] [Google Scholar]
- [59].Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, Sabatini DM. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016; 351:43-8; PMID:26449471; http://dx.doi.org/ 10.1126/science.aab2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Carroll B, Maetzel D, Maddocks OD, Otten G, Ratcliff M, Smith GR, Dunlop EA, Passos JF, Davies OR, Jaenisch R, et al.. Control of TSC2-Rheb signaling axis by arginine regulates mTORC1 activity. Elife 2016; 5, e11058; http://dx.doi.org/ 10.7554/eLife.11058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, Wang T, Harper JW, Gygi SP, Sabatini DM. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 2016; 165:153-64; PMID:26972053; http://dx.doi.org/ 10.1016/j.cell.2016.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Efeyan A, Zoncu R, Chang S, Gumper I, Snitkin H, Wolfson RL, Kirak O, Sabatini DD, Sabatini DM. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 2013; 493:679-83; PMID:23263183; http://dx.doi.org/ 10.1038/nature11745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Mahimainathan L, Das F, Venkatesan B, Choudhury GG. Mesangial cell hypertrophy by high glucose is mediated by downregulation of the tumor suppressor PTEN. Diabetes 2006; 55:2115-25; PMID:16804083; http://dx.doi.org/ 10.2337/db05-1326 [DOI] [PubMed] [Google Scholar]
- [64].Zheng X, Liang Y, He Q, Yao R, Bao W, Bao L, Wang Y, Wang Z. Current models of mammalian target of rapamycin complex 1 (mTORC1) activation by growth factors and amino acids. Int J Mol Sci 2014; 15:20753-69; PMID:25402640; http://dx.doi.org/ 10.3390/ijms151120753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Demetriades C, Plescher M, Teleman AA. Lysosomal recruitment of TSC2 is a universal response to cellular stress. Nat Commun 2016; 7:10662; PMID:26868506; http://dx.doi.org/ 10.1038/ncomms10662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Manolson MF, Yu H, Chen W, Yao Y, Li K, Lees RL, Heersche JN. The a3 isoform of the 100-kDa V-ATPase subunit is highly but differentially expressed in large (>or = 10 nuclei) and small (<or = nuclei) osteoclasts. J Biol Chem 2003; 278:49271-8; PMID:14504271; http://dx.doi.org/ 10.1074/jbc.M309914200 [DOI] [PubMed] [Google Scholar]
- [67].Mortensen RM, Zubiaur M, Neer EJ, Seidman JG. Embryonic stem cells lacking a functional inhibitory G-protein subunit (alpha i2) produced by gene targeting of both alleles. Proc Natl Acad Sci U S A 1991; 88:7036-40; PMID:1908087; http://dx.doi.org/ 10.1073/pnas.88.16.7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kowalewski B, Lubke T, Kollmann K, Braulke T, Reinheckel T, Dierks T, Damme M. Molecular characterization of arylsulfatase G: expression, processing, glycosylation, transport, and activity. J Biol Chem 2014; 289:27992-8005; PMID:25135642; http://dx.doi.org/ 10.1074/jbc.M114.584144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wattiaux R, Wibo M, Baudhuin P. [Effect of the injection of Triton WR 1339 on the hepatic lysosomes of the rat]. Arch Int Physiol Biochim 1963; 71:140-2; PMID:13999241 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.