

ABSTRACT
Autophagy is an essential component of host immunity and used by viruses for survival. However, the autophagy signaling pathways involved in virus replication are poorly documented. Here, we observed that rabies virus (RABV) infection triggered intracellular autophagosome accumulation and results in incomplete autophagy by inhibiting autophagy flux. Subsequently, we found that RABV infection induced the reduction of CASP2/caspase 2 and the activation of AMP-activated protein kinase (AMPK)-AKT-MTOR (mechanistic target of rapamycin) and AMPK-MAPK (mitogen-activated protein kinase) pathways. Further investigation revealed that BECN1/Beclin 1 binding to viral phosphoprotein (P) induced an incomplete autophagy via activating the pathways CASP2-AMPK-AKT-MTOR and CASP2-AMPK-MAPK by decreasing CASP2. Taken together, our data first reveals a crosstalk of BECN1 and CASP2-dependent autophagy pathways by RABV infection.
KEYWORDS: autophagy, BECN1, CASP2-dependent pathway, RABV, viral protein P
Introduction
Autophagy is a cellular regulatory process that removes unnecessary or dysfunctional cytoplasmic material by delivering it to the lysosome for degradation.1,2 Autophagy includes microautophagy, macroautophagy, and chaperone-mediated autophagy. During macroautophagy, a phagophore forms around a substrate to generate an autophagic vesicle surrounded by 2 membranes.3 After maturation, such autophagosomes fuse with late endosomes and lysosomes leading to degradation of the sequestered material.4 Identification of ATG (autophagy related) genes has led to the elucidation of the molecular mechanisms of autophagy.5-7 While the complete function of many ATGs and their protein products is still unknown, 2 ubiquitin-like systems, ATG12–ATG5 and MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3)–phosphatidylethanolamine, have been shown to mediate the formation of the autophagosome.8 These systems are required for the elongation and the closure of the phagophore (the precursor to the autophagosome) membranes. Finally, the fusion of the autophagosome with the lysosome requires LAMP2 (lysosomal-associated membrane protein 2) and the GTPase RAB7.9-13 These ubiquitin-like conjugation reactions are initiated by class III phosphatidylinositol 3-kinase (PtdIns3K) complexes, which include the BECN1/Beclin 1 protein. Interestingly, BECN1-positive complexes that contain ATG14 seem to promote autophagosome formation, while others that contain the Uvrag (UV radiation resistance associated) gene14 may facilitate the fusion between autophagosomes and lysosomes.14-16
As a highly conserved process, autophagic regulation is complex.17 Over the past decade, host proteins that regulate autophagy have been reported to do so by different mechanisms and pathways, such as those involving PtdIns3K, AKT, MTOR, MAPK, AMPK,7,18 CASP2/caspase 219 and the proteins encoded by the ATG genes. Recently, autophagy has been shown to be modulated by intracellular pathogens. Tobacco mosaic virus induces autophagosome formation by inducing endoplasmic reticulum stress.20 Herpes simplex virus 1 induces autophagy via the EIF2AK2/PKR (eukaryotic translation initiation factor 2 α kinase 2)-mediated phosphorylation of EIF2S1 (eukaryotic translation initiation factor 2 subunit α),21 and also inhibits autophagy by BECN1 binding to viral protein ICP34.5.22 The vesicular stomatitis virus envelope glycoprotein mediates virus entry and induces autophagy, likely by inhibiting the AKT.23 Avibirnavirus capsid protein VP2 binding to host heat shock protein 90 kDa α induces autophagy by inactivating the AKT-MTOR pathway.24 Hepatitis C virus induces autophagy through the RAB5 and PtdIns3K (whose catalytic subunit is termed PIK3C3) pathway.25 Human immunodeficiency virus 1 inhibits autophagy through the SRC-AKT and STAT3 (signal transducer and activator of transcription 3) pathway.26 Human papillomavirus-host cell interaction stimulates the phosphoinositide 3-kinase-AKT-MTOR pathway and inhibits autophagy.27
Rabies virus (RABV), a fatal neurotropic virus, is a prototypical virus in the genus Lyssavirus genus, family Rhabdoviridae. RABV has a single-stranded negative sense RNA genome encoding the nucleoprotein (N), the RNA-dependent RNA polymerase (L), the nonenzymatic polymerase cofactor phosphoprotein (P), the matrix protein (M), and the glycoprotein (G). During RABV infection, viral transcription and replication are performed in intracellular Negri bodies (NBs).28 In the present study, we address for the first time that incomplete autophagy can be induced during virulent and attenuated RABV infections in vivo and in vitro. We found that RABV infection activated the CASP2-AMPK-MAPK1/3/11-AKT1-MTOR dependent autophagy pathways by BECN1. Our study highlights the role of viral P protein in modulating the BECN1-linked, CASP2-dependent incomplete autophagy pathways.
Results
RABV infection triggers autophagosome accumulation in vitro and in vivo
To investigate whether cellular macroautophagy is altered in response to RABV infection, N2a cells were transiently transfected with green fluorescent protein (GFP)-LC3B, and mice were infected with attenuated RABV HEP-Flury or virulent RABV CVS-11. The expression of LC3-II, SQSTM1/p62 (sequestosome 1) and RABV viral protein in N2a cells and brains of mice was analyzed at different time points postinfection. In comparison to mock-infected cells, GFP-LC3B has a colocalization with viral protein N (Fig. 1A) in the HEP-Flury virus-infected cells and GFP-LC3B autophagosomes increased visibly at 36 and 48 h postinfection (hpi). The level of endogenous lipidated LC3-II in cells and mice infected with RABV, which correlates with an increased number of autophagosomes, was detected by western blotting using anti-LC3A/B antibody (P < 0.01 or 0.001, Fig. 1B and C). However, the autophagosome cargo SQSTM1, which is one of the autophagy markers,29 was not increased (P > 0.05, Fig. 1B and C). Similar autophagosome accumulation and LC3-II increases were detected in N2a cells and mice infected with virulent RABV, however SQSTM1 were not increased (Fig. S1). Collectively, these data demonstrate that both attenuated and virulent RABV induce autophagosome accumulations after in vitro and in vivo infection, and that components of the macroautophagy signaling cascade are upregulated during RABV infection.
Figure 1.
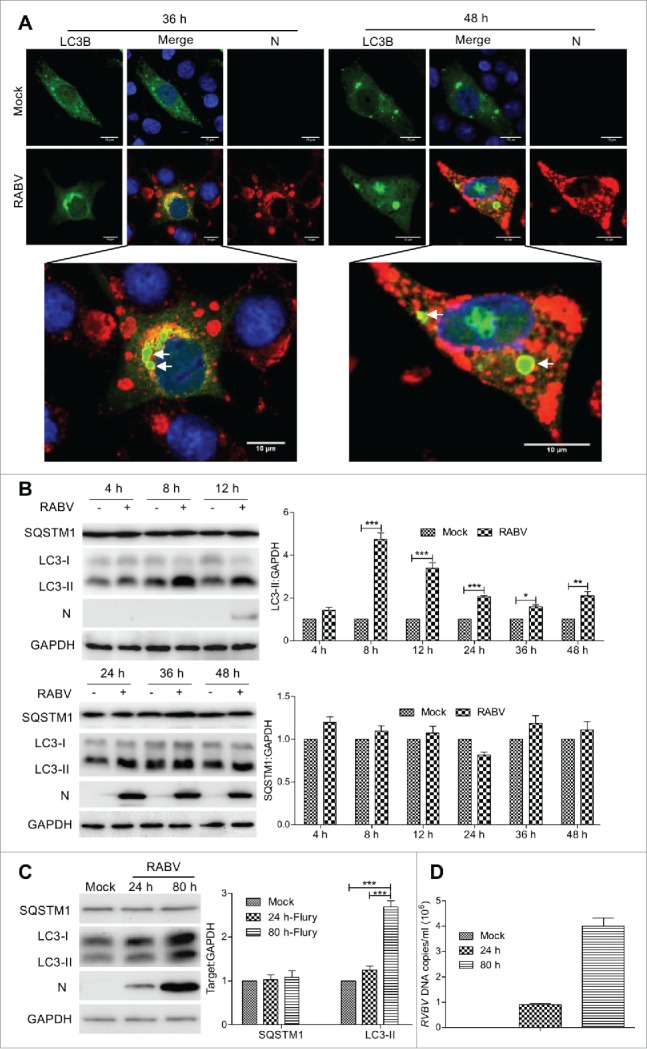
RABV infection induces autophagosome accumulation in N2a cells and brain of mice. (A) N2a cells transfected with GFP-LC3B for 12 h, and then infected with RABV strain Flury at MOI = 2 for 36 h and 48 h. The cells were fixed, immunostained with mouse anti-N mAb (red), and autophagosomes (green) were observed under a confocal microscopy. DAPI (blue) was used to stain nuclear DNA. Scale bar: 10 μm. (B) N2a cells were infected with RABV strain Flury at MOI = 2 or mock-infected for 4 h, 8 h, 12 h, 24 h, 36 h and 48 h. (C) 3-d-old-ICR mice were injected intracerebrally with RABV strain Flury at a dose of 100 TCID50 or PBS (100 μl per mouse) for 24 hpi and 80 hpi, and the brains of the mice were isolated. The cellular and cerebullar samples in (B) and (C) were then analyzed by western blotting with mouse anti-N mAb, and rabbit anti-LC3A/B, anti-SQSTM1 and anti-GAPDH antibodies. The ratio of SQSTM1, LC3-II to GAPDH was normalized to control conditions. Error bars: Mean ± SD of 3 independent tests. Two-way ANOVA; *P < 0.05; **P < 0.01; ***P < 0.001. (D) Quantification of viral DNA by real-time quantitative PCR. The quantification results were expressed as viral DNA copy numbers per ml of genomic DNA from the brain of RABV-infected mice. All experiments were performed in triplicate.
RABV infection inhibits autophagy flux
Autophagosomes are transient vesicles that fuse with lysosomal vesicles and deliver their cargo for degradation by lysosomal hydrolysis. Therefore, the accumulation of autophagosomes could result from increased formation or decreased degradation of these vesicles.29 To investigate autophagosome accumulation during RABV infection, cells were infected with HEP-Flury strain and labeled with LysoTracker Red, which allows for identification of acidic compartments or organelles in live cells. As expected, GFP-LC3B and LysoTracker Red colocalized in N2a cells with and without HEP-Flury infection upon rapamycin (Rapa) treatment. However, in RABV-infected cells without Rapa treatment, essentially no colocalization between autophagosomes and LysoTracker Red was observed, and most of the large autophagosomes were devoid of LysoTracker Red staining (Fig. 2A and Fig. S2). This data suggests that autophagosomes do not fuse with acidic compartments after RABV infection. To rule out the possibility that autophagosomes fuse with lysosomes but were not efficiently acidified in infected cells, we investigated the colocalization of GFP-LC3B with the LAMP1 in RABV-infected cells. Similarly, GFP-LC3B had a colocalization with LAMP1 in Rapa-treated cells with and without RABV infection. In contrast, GFP-LC3B did not colocalize with LAMP1 in RABV-infected cells without Rapa treatment and the large GFP-LC3B vesicles were LAMP1-negative (Fig. 2B and Fig. S2). These data suggest that autophagosomes do not efficiently fuse with lysosomes in RABV-infected cells.
Figure 2.
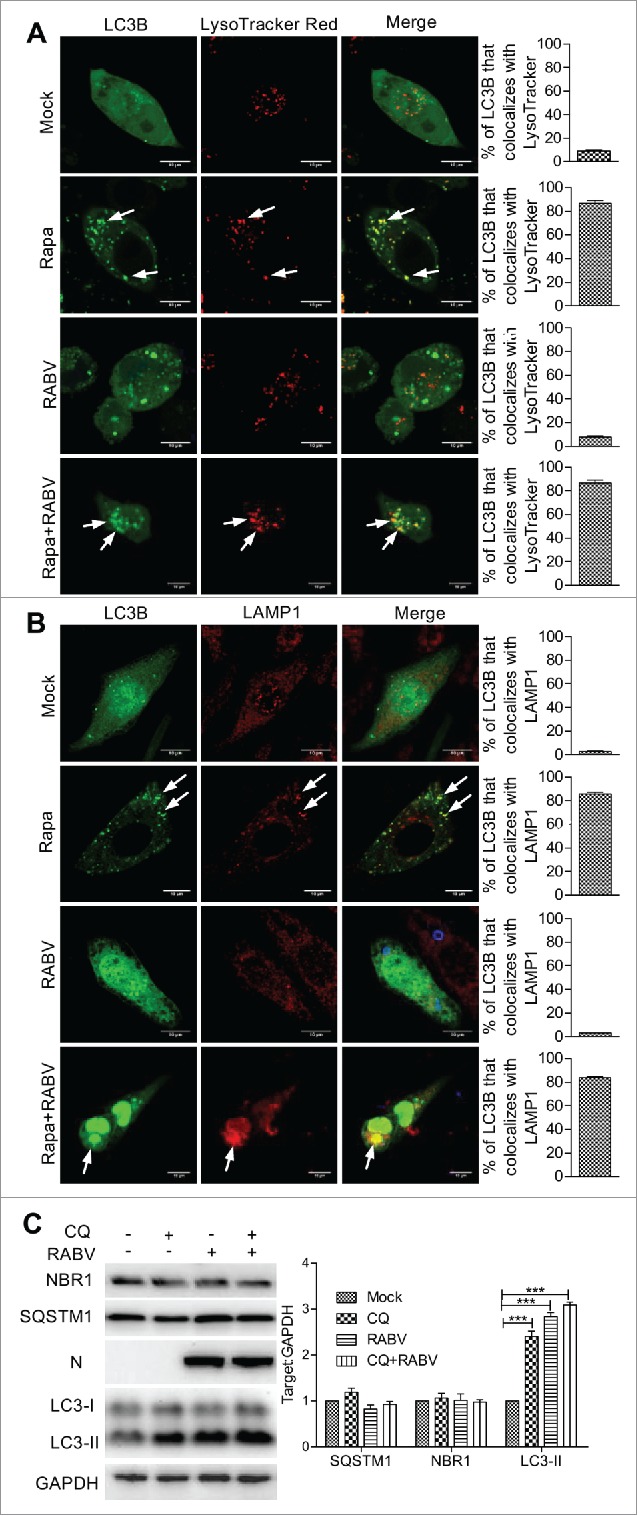
Autophagosomes fail to fuse with lysosomes in RABV-infected cells. N2a cells transfected with a plasmid encoding GFP-LC3B for 12 h, and then infected with RABV HEP-Flury strain at MOI = 2 for 24 h. (A) Cells were incubated with LysoTracker Red (50 nM) for 15 min; GFP-LC3B-labeled autophagosome (green) colocalization with LysoTracker Red-stained acidified vesicles (red) were observed by confocal microscopy (white arrows). (B) Cells were fixed, and immunostained with the lysosome marker rabbit anti-LAMP1 (red) and mouse anti-N mAb (blue), and observed by confocal microscopy to analyze fusion of autophagosomes with lysosomes (white arrows). Scale bars: 10 μm. (C) RABV-infected N2a cells with 5.6 μM chloroquine treatment of 24 h, and tested by western blotting using mouse anti-N mAb, and rabbit anti-LC3A/B, anti-SQSTM1 anti-NBR1 and anti-GAPDH antibodies. The ratio of SQSTM1, NBR1 and LC3-II to GAPDH was normalized to control conditions. Error bars: Mean ± SD of 3 independent tests. Two-way ANOVA; *P < 0.05; **P < 0.01; ***P < 0.001.
To confirm the blockage of autophagosome fusion with lysosomes in RABV-infected cells, we analyzed the turnover of autophagosomes. The 24 hpi RABV-infected and mock-infected cells were treated with chloroquine (CQ), a lysosomal proteolysis inhibitor, for 6 h, and the levels of LC3-II, SQSTM1 and NBR1 (NBR1, autophagy cargo receptor), a member of sequestosome-like receptors30,31 were analyzed by western blotting. In mock-infected cells, lipidated LC3-II accumulated upon CQ treatment, indicating that turnover of autophagosomes efficiently occurred by lysosomal proteolysis (P < 0.001, Fig. 2C). In contrast, in RABV-infected cells, no further accumulation of LC3-II could be observed in response to CQ treatment, demonstrating that turnover of autophagosomes did not occur at a significant rate by lysosomal proteolysis in RABV-infected cells. In addition, SQSTM1, a common constituent of protein aggregates widely used to monitor autophagy flux, and NBR1 were then determined in RABV-infected cells upon CQ treatment. However, there was no significant increase (P < 0.05, Fig. 2C) in SQSTM1 and NBR1, revealing that degradation of macroautophagy substrates did not occur during RABV infection. Taken together, these data suggest that RABV infection inhibits the fusion of autophagosomes with lysosomes and that autophagy flux is inhibited.
Autophagic induction enhances RABV replication
To further determine the effect of autophagy activity on RABV replication, we investigated the autophagosome induction. N2a cells were treated with different concentration of Rapa or with Earle's balanced salt solution (EBSS) or with 3-methyladenine (3-MA) or with wortmannin. Following Rapa or EBSS starvation-induced autophagy, conversion of LC3-I to LC3-II increased significantly in RABV HEP-Flury strain-infected cells (P < 0.001, Fig. 3A and B) and the RABV titer increased significantly (P < 0.01, Fig. 3F), indicating that autophagic induction enhanced RABV replication. Next, 3-MA, an inhibitor with activity against class III PtdIns3K that is widely used for suppression of autophagy by blocking the formation of autophagosomes, and wortmannin, an inhibitor of autophagosome initiation, were used to inhibit autophagy. Figure 3C and G showed that the inhibition of autophagy induced by 3-MA or wortmannin reduced the level of viral N protein (P < 0.001) and RABV progeny (P < 0.01 or 0.05). Similarly Atg5 knockdown, which is an essential factor for activation of autophagosome formation and maturation,32 exhibited a significant decrease of viral N protein expression and lower yield of RABV progeny, compared with the cells transfected with nontargeting short hairpin RNA (shRNA) (P < 0.001, Fig. 3D and H). In addition, to exclude the possibility that Atg5 knockdown caused innate antiviral immune responses,33 we further knocked down Lc3b RNA message with shRNA. Similar results were observed (P < 0.001, Fig. 3E and H) compared with Atg5-knockdown cells. Taken together, these findings suggest that cellular autophagy benefits RABV replication.
Figure 3.
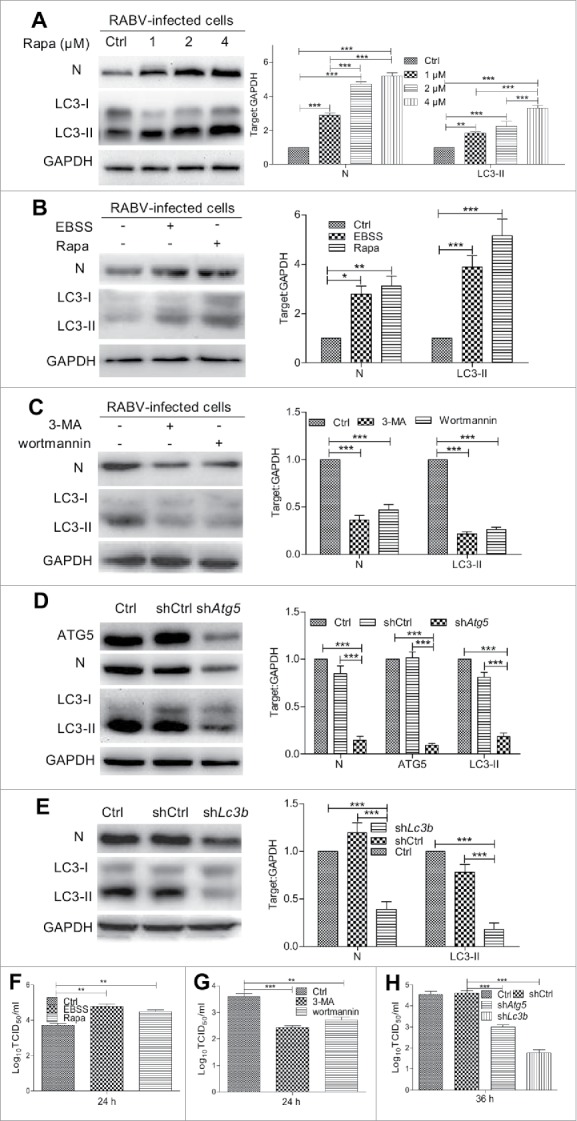
Effect of autophagy on RABV replication. (A) N2a cells were pretreated in DMEM containing 1 μM, 2 μM, 4 μM Rapa for 2 h, then were infected with RABV HEP-Flury strain at MOI = 2 and cultured in DMEM containing Rapa for 24 h. (B) N2a cells were pretreated with 1 μM of Rapa or EBSS for 2 h, then were infected with RABV HEP-Flury strain at MOI = 2 for 1 h and further incubated in DMEM containing Rapa or EBSS for 24 h. (C) N2a cells were pretreated with 5 mM 3-MA or 1 μM wortmannin for 2 h, then were infected with RABV HEP-Flury strain at MOI = 2 and incubated in the absence or presence of 5 mM 3-MA or 1 μM wortmannin for 24 h. (D) and (E) N2a cells were transfected with nontargeting shCtrl and shRNA for Atg5 (shAtg5) or Lc3b (shLc3b) for 12 h, then were infected with RABV and incubated for 36 h. Cellular supernatant and lysates in (A) to (E) were harvested for virus titer detection and for western blotting with mouse anti-N mAb, rabbit anti-LC3A/B and anti-GAPDH antibodies. Virus titers in N2a cells were determined by TCID50 assay. The ratio of LC3-II or N to GAPDH was normalized to control conditions. (F) RABV titer of EBSS or Rapa treated cells. (G) RABV titer of 3-MA or wortmannin treated cells. (H) RABV titer of shAtg5 or shLc3b treated cells. Error bars: Mean ± SD of 3 independent tests. Data were analyzed with 2-way ANOVA in (A) to (E) and one-way ANOVA in (F) to (H); *P < 0.05; **P < 0.01; ***P < 0.001.
The AKT-MTOR signaling pathway is activated in RABV-induced incomplete autophagy
To investigate the incomplete autophagy pathway regulating RABV infection, we next determined the MTOR-dependent signaling pathway. Compared to mock-infected cells, the levels of phosphorylated AKT (P < 0.001) and MTOR (P < 0.01) in RABV-infected cells was significantly increased (P < 0.001, Fig. 4A and C), suggesting that AKT and MTOR was relevant to RABV-mediated incomplete autophagy. Subsequently, to analyze whether the AKT-MTOR pathway was an essential step in RABV-induced incomplete autophagy, the AKT-MTOR pathway was knocked down by shRNA targeting Akt1 or Mtor before RABV infection. The results showed that the MTOR phosphorylation decreased remarkably in cells treated with shAkt1 interference compared with the RABV-infected cells without shRNA interference (P < 0.001, Fig. 4Aand C). In contrast, the AKT phosphorylation was not interfered with significantly in shMtor-treated cells in comparison with RABV-infected cells without shRNA (P > 0.05, Fig. 4B and C). However, the level of LC3-II significantly diminished in RABV-infected cells treated with shAkt1 or shMtor (P < 0.001), indicating that the autophagic induction was inhibited by shAkt1 and shMtor during RABV infection. Correspondingly, titer of RABV showed a slight decrease in shAkt1- and shMtor-treated cells (Fig. 4D). Moreover, RABV infection did not alter significantly the phosphorylation and expression of AKT and MTOR in cells treated with shAkt1 or shMtor (Fig. 4C). Collectively, these observations indicate that RABV infection stimulates incomplete autophagy by activating the AKT-MTOR pathway, but does not directly affect the phosphorylation of AKT and MTOR.
Figure 4.
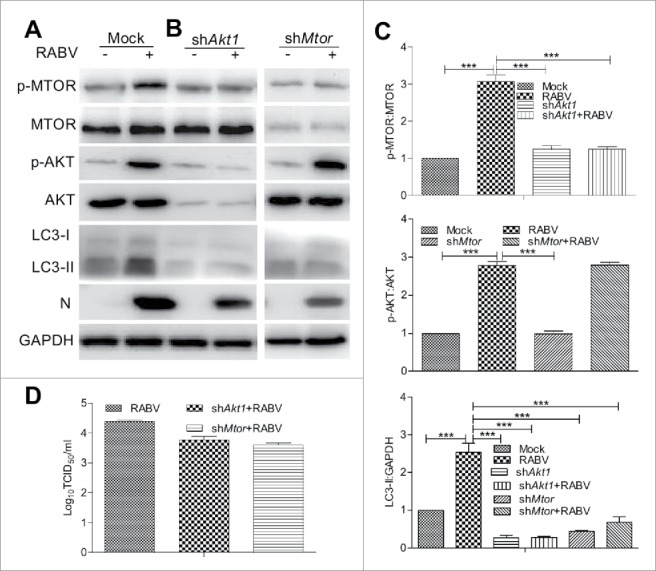
The MTOR and AKT signaling pathway is activated during RABV infection. (A) N2a cells were infected with RABV HEP-Flury strain at MOI = 2 for 24 h. (B) N2a cells were transfected with shRNA targeting Akt1 or Mtor for 24 h, then cells were infected with RABV HEP-Flury strain at MOI = 2 for 36 h. Infected cells were then harvested for further western blotting analysis with mouse anti-N mAb, and rabbit anti-p-MTOR, anti-MTOR, anti-p-AKT, anti-AKT, anti-LC3A/B, and anti-GAPDH antibodies. (C) The ratio of p-MTOR:MTOR, p-AKT:AKT and LC3-II:GAPDH was normalized to control conditions. (D) Virus titers in N2a cells were determined by TCID50 assay. Error bars: Mean ± SD of 3 independent tests. One-way ANOVA; *P < 0.05; **P < 0.01; ***P < 0.001.
The AMPK-MAPK pathway is involved in incomplete autophagy induction during RABV infection
The AMPK and MAPK proteins are reported to play a key role in regulating autophagy. Also, AMPK positively regulates MAPK activity.34-37 Thus, we examined whether PRKAA/AMPKα and MAPK (MAPK1/ERK2, MAPK3/ERK1, MAPK11/p38b) were involved in inducing incomplete autophagy in RABV-infected N2a cells. Figure 5 showed that the phosphorylation level of PRKAA, MAPK1/3, and MAPK11 were significantly upregulated in RABV-infected cells compared with mock-infected cells (P < 0.001). To further investigate their role in incomplete autophagy, we inactivated PRKAA by small interfering RNA (siRNA) targeting Prkaa1 before RABV infection. The results showed that the AKT, MTOR, MAPK1/3, and MAPK11 phosphorylation levels were significantly decreased in RABV-infected cells treated with siRNA against Prkaa1 (P < 0.001, Fig. 5A and C) compared with the RABV-infected cells without siRNA treatment, suggesting that the Prkaa1 silencing suppresses the phosphorylation of AKT, MTOR, MAPK1/3, and MAPK11. To further explore the relationship between MAPK and AKT-MTOR, the Mapk1 gene was knocked down by the siRNA specific for Mapk1; we found no significant alteration in the AKT, MTOR and PRKAA phosphorylation levels in RABV-infected cells with siMapk1 treatment in comparison with RABV-infected cells without siMapk1 treatment (Fig. 5), showing that the MAPK inhibition did not disturb the phosphorylation of AKT, MTOR and PRKAA. Similarly, the conversion of LC3-I to LC3-II in cells treated with siRNA targeting Prkaa1 or Mapk1 was significantly downregulated compared with that in RABV-infected cells without siRNA treatment (P < 0.001, Fig. 5), demonstrating that the knockdown of the genes Prkaa1and Mapk1 downregulates incomplete autophagic induction. Moreover, RABV infection did not significantly alter the phosphorylation and expression of AKT, MTOR, PRKAA, MAPK1/3 and MAPK11 in cells treated with siPrkaa1 or siMapk1 (Fig. 5C). These results suggest AMPK is the upstream regulator of MAPK, AKT and MTOR in RABV-mediated incomplete autophagy and RABV infection not directly affects the activation of AMPK and MAPK.
Figure 5.
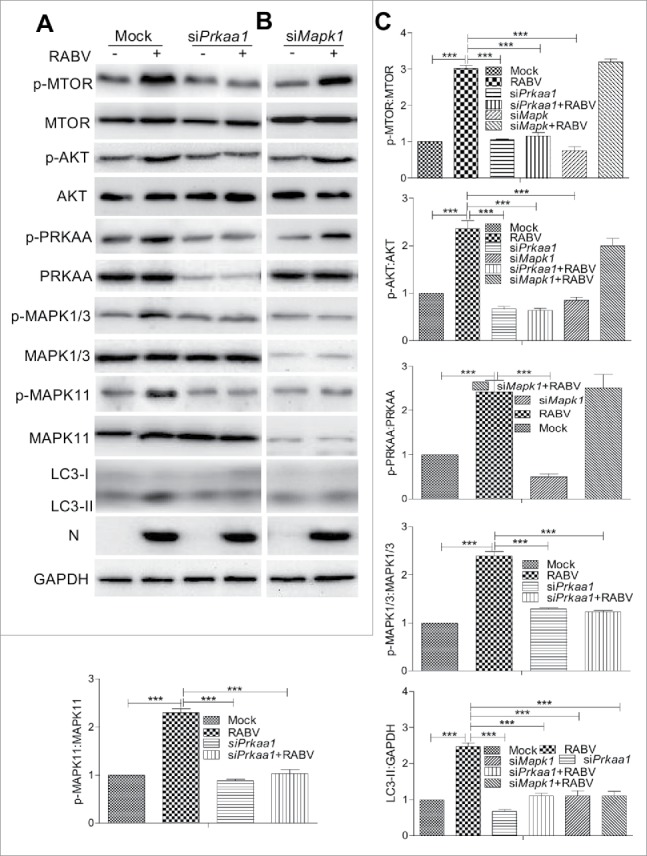
RABV infection induces autophagy via activating the AMPK-MAPK pathway. (A, B) N2a cells were transfected with siRNA targeting Prkaa1or Mapk1 for 24 h, then the cells were infected with RABV HEP-Flury strain at MOI = 2 and subjected to further incubation for 36 h. Infected cells were then harvested for further western blotting analysis with mouse anti-N mAb, and rabbit anti-AKT, anti-p-AKT, anti-p-MTOR, anti-MTOR, anti-p-PRKAA, anti- PRKAA, anti-p-MAPK1/3, anti-MAPK1/3, anti-p-MAPK11, anti-MAPK11, anti-LC3A/B and anti-GAPDH antibodies. (C) The ratio of p-MTOR:MTOR, p-AKT:AKT, p-PRKAA:PRKAA, p-MAPK1/3:MAPK1/3, p-MAPK11:MAPK11and LC3-II:GAPDH was normalized to control conditions. Error bars: Mean ± SD of 3 independent tests. One-way ANOVA; *P < 0.05; **P < 0.01; ***P < 0.001.
CASP2 is inhibited in RABV-mediated incomplete autophagy
During the experiment we also observed that RABV infection significantly downregulated CASP2 expression and increased markedly the LC3-II level (P < 0.001, Fig. 6B). CASP2 has previously been reported as a negative autophagy regulator of AMPK upstream.19 To do this, the Prkaa1 were knocked down by siRNA, respectively. The data showed that the expression profile of CASP2 had no significant alteration in siPrkaa1 treated cells infected with RABV compared with RABV-infected cells without siPrkaa1 treatment (Fig. 6A), manifesting that RABV suppressed CASP2 expression was not activated obviously by knocking down Prkaa1. To further detect the role of CASP2 in PRKAA activity, cells were treated with shCasp2. As shown in Figure 6B and C, in comparison with RABV-infected cells, Casp2 knockdown significantly downregulated the phosphorylation of MTOR, AKT, and PRKAA and the LC3-II level (P < 0.001) but not total amount of these proteins, validating that CASP2 located at the upstream regulator of AMPK, and Casp2 knockdown inhibited autophagic induction and did not perceptibly affect CASP3, 8 or 9 expression. However, the PRKAA phosphorylation and LC3-II level had no significant difference in Casp2 knockdown cells with or without RABV infection, indicating that RABV infection did not play the role in stimulating autophagy under the condition of Casp2 knockdown. These observations suggest that RABV infection stimulates incomplete autophagy by decreasing CASP2.
Figure 6.
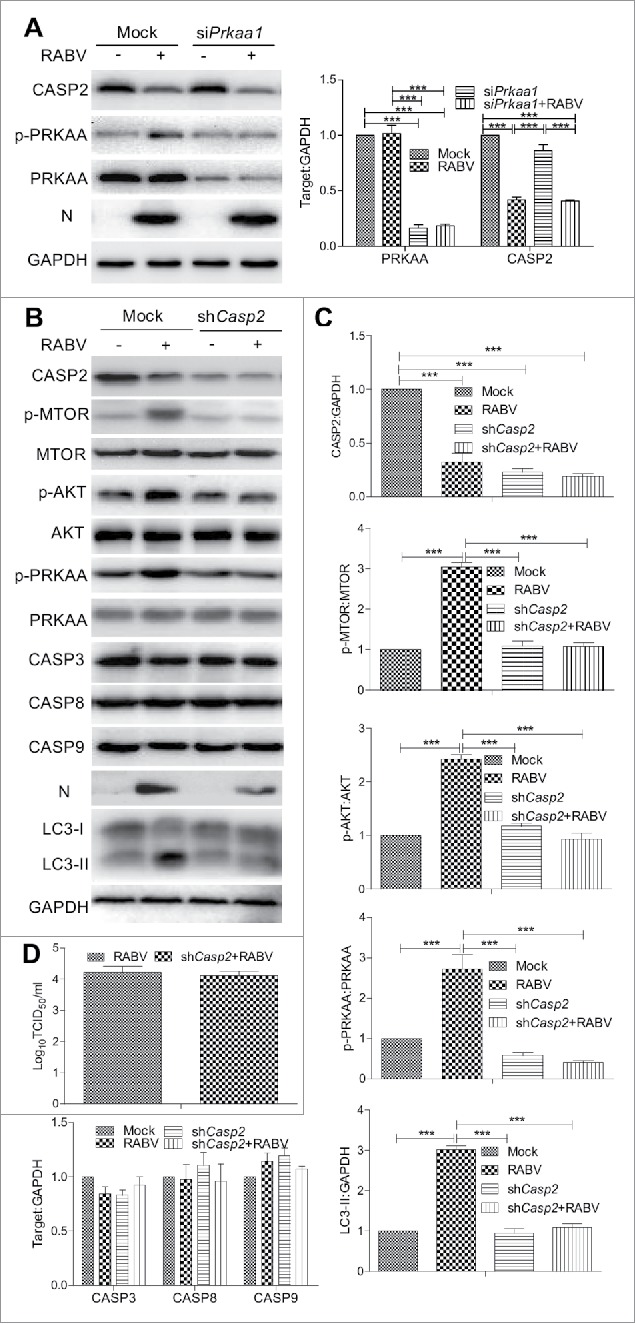
CASP2 is the upstream regulator of AMPK in RABV-mediated autophagy. N2a cells were transfected with siRNA targeting Prkaa1 (A) or shRNA specific for Casp2 (B) for 24 h, then the cells were infected with RABV at MOI = 2 and subjected to further incubation for 36 h. Infected cells were then harvested for further western blotting analysis with mouse anti-N mAb, and rabbit anti-CASP2, anti-p-MTOR, anti-MTOR, anti-p-AKT, anti-AKT, anti-p-PRKAA, anti- PRKAA, anti-CASP3, anti-CASP8, anti-CASP9, anti-LC3A/B and anti-GAPDH antibodies. (C) The ratio of p-MTOR:MTOR, p-AKT:AKT, p- PRKAA:PRKAA, and CASP2, CASP3, CASP8, CASP9, and LC3-II to GAPDH was normalized to control conditions. (D) Virus titers in N2a cells were determined by TCID50 assay. Error bars: Mean ± SD of 3 independent tests. One-way ANOVA; *P < 0.05; **P < 0.01; ***P < 0.001.
RABV N/P protein promotes incomplete autophagic responses by inhibiting CASP2 expression
As the main structural protein of RABV, viral proteins N and P are important components of NBs and are involved in viral transcription and replication.28 The RABV genomic RNA is encapsidated by the viral protein N, forming the N-RNA template for transcription and replication by the viral RNA-dependent RNA polymerase whose major components are L and P protein. To identify viral proteins that are responsible for autophagic activity in RABV-infected cells, N2a cells were cotransfected with GFP-LC3B and with Flag-tagged plasmids pCMV-N-Flag (Flag) encoding viral N (Flag-N) or P (Flag-P) or L (Flag-L) or G (Flag-G) or M (Flag-M) gene. The results revealed that the N or P-transfected cells but not L, G, M-transfected cells had a strong accumulation of GFP-LC3B by confocal microscopy analysis (Fig. 7A and Fig. S3). Consistent with this, the levels of LC3-II were dramatically increased in N or P transfected cells compared with the empty vector transfected cells by western blotting, and only viral protein P revealed a dose-dependency for increasing LC3-II level (P < 0.001, Fig. 7B). However, all viral proteins of RABV did not caused significant increases in SQSTM1 levels (P > 0.05, Fig. 7B and Fig. S3). In addition, further investigation showed that both viral proteins N and P dramatically upregulated the phosphorylation level of AKT, MTOR, PRKAA, MAPK1/3 and MAPK11 (P < 0.001, Fig. 7C), and reduced CASP2 level (P < 0.001, Fig. 7C), These data indicate that the RABV N and/or P proteins are sufficient for activating incomplete autophagy by decreasing CASP2.
Figure 7.
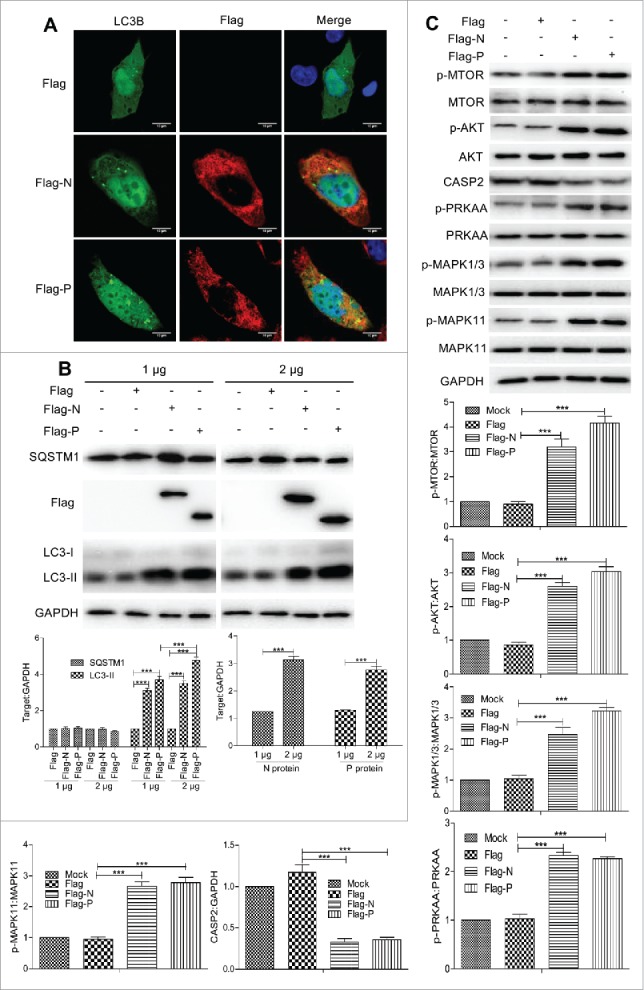
RABV N/P inhibited CASP2 expression and were sufficient for inducing autophagy by the phosphorylation of AKT, MTOR, AMPK and MAPK. (A) N2a cells cotransfected with Flag-N or P and GFP-LC3B (green) for 24 h, fixed, and immunostained with mouse anti-Flag antibody (red), and observed under confocal microscopy. DAPI (blue) was used to stain nuclear DNA. Scale bar: 10 μm. (B) Cells were transfected with the vector Flag-N or P at a dose of 1 or 2 μg for 48 h, harvested and analyzed by western blotting using mouse anti-Flag mAb, rabbit anti-LC3A/B, anti-SQSTM1, and anti-GAPDH antibodies. The ratio of SQSTM1 and LC3-II to GAPDH was normalized to control conditions. (C) Cells were transfected with the vector Flag-N/P for 48 h, harvested and analyzed by western blotting using mouse anti-Flag mAb, and rabbit anti-p-MTOR, anti-MTOR, anti-p-AKT, anti-AKT, anti-CASP2, anti-p-PRKAA, anti- PRKAA, anti-p-MAPK1/3, anti-MAPK1/3, anti-p-MAPK11, anti-MAPK11 and anti-GAPDH antibodies. The ratio of p-MTOR:MTOR, p-AKT:AKT, p-PRKAA:PRKAA, p-MAPK1/3:MAPK1/3, p-MAPK11:MAPK11 and CASP2:GAPDH was normalized to control conditions. Error bars: Mean ± SD of 3 independent tests. Two-way ANOVA in (B) and one-way ANOVA in (C); *P < 0.05; **P < 0.01; ***P < 0.001.
BECN1 binding to viral protein P is crucial for incomplete autophagy induction by the CASP2-mediated signaling pathway
To further investigate the role that viral N and P proteins in inducing incomplete autophagy, we examined whether BECN1, PRKAA, CASP2 and MAPK11 had an interaction with viral proteins N and P in CASP2-mediated autophagic induction, respectively. Confocal analysis showed that BECN1 strongly colocalized with viral proteins N and P in cells cotransfected with the genes N, P and Becn1 (Fig. 8A). Meanwhile, by coimmunoprecipitation (CoIP) analysis, only viral protein P had a strong interaction with host BECN1 (Fig. 8B) in cells cotransfected with genes Becn1 and P but not N. Regrettably, the interaction of PRKAA, CASP2 and MAPK11 with viral proteins N and P was not detected in confocal and CoIP analyses (Fig. 8B). Moreover, we knocked down cellular Becn1 by siBecn1 to further show whether the viral protein P or BECN1 affected the expression of CASP2, CASP3, CASP8 and CASP9 (Fig. S4). The results showed that there was a detectable downregulation of CASP2 and phosphorylated PRKAA (P < 0.01, Fig. 8C), and an insignificant alteration of CASP3, CASP8 and CASP9 (Fig. S4) in Becn1-knockdown cells with viral gene P transfection or infected with RABV (data not shown). To verify an upstream activity of BECN1, we further detected the dynamics of PIK3C3 (phosphatidylinositol 3-kinase catalytic subunit type 3) and ULK1 (unc-51 like autophagy activating kinase 1) as the components of the BECN1 complex. The results showed that the expression of PIK3C3 and the phosphorylation of ULK1 protein were upregulated in cells infected with RABV and transfected with gene P (Fig. 8D), indicating that viral protein P stimulates ULK1 phosphorylation and PIK3C3 expression. Collectively, these data demonstrate that viral protein P binding to BECN1 induces incomplete autophagy by reducing CASP2.
Figure 8.
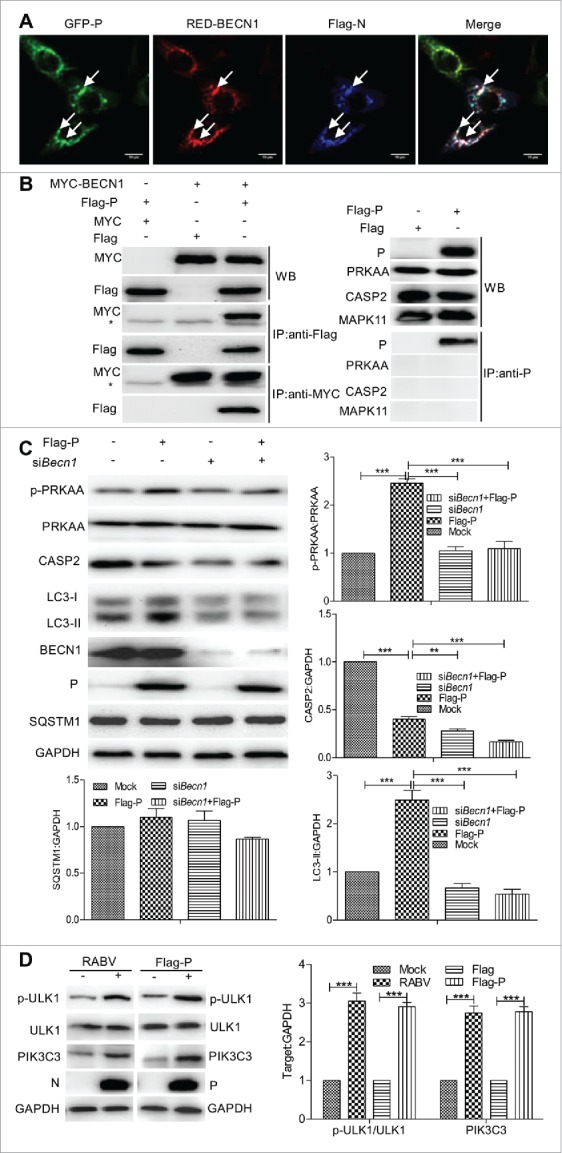
Viral protein P has an interaction with BECN1 in the CASP2-mediated signaling pathway. (A) N2a cell were cotransfected with Flag-N, GFP-P, RED-BECN1 plasmids for 24 h, and BECN1 (red) colocalization with viral protein N (blue) or P (green) was detected by using the indicated antibodies in confocal microscopy. White arrows indicated the colocalization sites. Scale bar: 10 μm. (B) N2a cells were cotransfected with Flag-P and Myc-BECN1 for 48 h, and the interactions between P and BECN1 was determined by using the indicated antibodies. * indicates immunoglobulin heavy chain of the antibodies used for immunoprecipitation. WB, western blotting; IP, immunoprecipitation. (C) N2a cells were cotransfected with Flag-P and siRNA targeting Becn1 for 48 h, and then harvested for western blotting analysis with mouse anti-P and anti-BECN1 mAbs, rabbit anti-CASP2, anti-p-PRKAA, anti-PRKAA, anti-LC3A/B, anti-SQSTM1 and anti-GAPDH antibodies. The ratio of p-PRKAA:PRKAA, and CASP2, LC3-II and SQSTM1 to GAPDH was normalized to control conditions. (D) ULK1 phosphorylation and PIK3C3 expression. N2a cells were infected with RABV Flury strain at MOI = 2 or transfected with Flag-P for 24 h, harvested and detected ULK1 phosphorylation and PIK3C3 expression by western blotting with mouse anti-Flag antibody, rabbit anti-p-ULK1, anti-ULK1, anti-PIK3C3 and anti-GAPDH antibodies. Error bars: Mean ± SD of 3 independent tests. One-way ANOVA; *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
Autophagy is known to be an important pathway involved in the removal of intracellular pathogens, including bacteria and viruses. In addition to its role in innate immunity, autophagy also contributes to the adaptive immune response, participating in the presentation of pathogen antigens on major histocompatibility complex molecules.38 Many viruses have been shown to subvert the autophagic machinery to enhance viral replication.24,39 In this study, we demonstrate accumulation of autophagosomes and no change in SQSTM1 levels in RABV-infected cells (Fig. 1). Also, GFP-LC3B colocalization with LAMP1 (a lysosome marker) cannot be detectable in RABV-infected cells (Fig. 2). These results suggest that in vitro and in vivo RABV infection induces an incomplete autophagy in which autophagosomes do not efficiently fuse with lysosomes.
In all eukaryotes, MTOR leads to a swift response to various environmental cues, regulating cell metabolism and immune responses by regulating the kinase AKT.40,41 Recent data also demonstrate that the AKT-TSC-MTOR pathway was inactivated by hepatitis C virus for inducing autophagy,42 avian influenza viruses result in autophagic cell death by inhibiting MTOR,43 and vesicular stomatitis virus induces autophagy by regulating the phosphoinositide 3-kinase-AKT signaling pathway.23 However, the relationship between pathogen and the autophagy signaling pathway was poorly understood.
CASP2, a well-known regulator of apoptosis, has been shown to be an endogenous negative regulator of autophagy upstream of the AMPK-MTOR and AMPK-MAPK pathways during oxidative stress, and loss of CASP2 upregulates endogenous autophagy under normal conditions.19 In this report, we demonstrate that RABV infection induces autophagy by increasing the phosphorylation of AMPK, MAPK, AKT and MTOR, and decreasing CASP2 (Figs. 4 and 7). Moreover, either P-transfected cells or Becn1 knockdown cells showed the reduction of CASP2 (Figs. 7 and 8). Interestingly, BECN1 was confirmed to have an interaction with the RABV P protein. Therefore our data demonstrates for the first time a connection between BECN1 and the CASP2-mediated autophagy pathway and that RABV induces incomplete autophagy by activating the pathways BECN1-CASP2-AMPK-AKT-MTOR and BECN1-CASP2-AMPK-MAPK by BECN1 binding to viral protein P. Moreover, recent data also show an incomplete autophagic response stimulated by the viral protein M of the wild-type virulent RABV strain GD-SH-01in NA cells,44 implying that the different components of RABV participate probably in the induction of incomplete autophagy during infection.
BECN1, as an interacting partner for the mammalian class III PtdIns3K PIK3C3, is required for macroautophagy in nutrient-starved cells,45-47 for normal lysosomal enzyme sorting and for cell cycling.48,49 In contrast with its documented role in macroautophagy, the possible role of BECN1 as an essential chaperone or adaptor for PIK3C3 in normal trafficking pathways has received little attention.47 Some reports show that the interaction between BECN1 and the antiapoptotic protein BCL2 is the regulating switch between the autophagic and apoptotic machinery.50,51 Binding of BECN1 to BCL2 inhibits BECN1-mediated autophagy via sequestration of BECN1 away from class III PtdIns3K.51-53 Autophagy initiation signaling requires both the ULK1 kinase and the BECN1- PIK3C3 core complex to generate autophagosomes,54 and the recruitment of PIK3C3 to the phagophore requires the activity of ULK kinase.55,56 In our experiment, we observed the upregulation of PIK3C3 and phosphorylated ULK1, and the interaction between BECN1 and viral protein P. These results imply that RABV protein P binding to BECN1 may form a P-BECN1-ULK1-PIK3C3 complex to regulate the autophagy pathway and that BECN1 may serve as an interaction platform for a bridge for the RABV protein P and PIK3C3.
In conclusion, RABV P binding to BECN1 can induce incomplete autophagy through the pathways BECN1-CASP2-AMPK-MAPK and BECN1-CASP2-AMPK-AKT-MTOR and RABV-induced incomplete autophagy provides the scaffolds for the replication of RABV genome (Fig. 9). Our study provides evidence that incomplete autophagy is induced in response to RABV infection through CASP2-mediated AMPK-MAPK1/3/11-AKT1-MTOR pathways activation triggered by the BECN1-P complex. Our study provides the possibility that controlling the CASP2-mediated AMPK-MAPK-AKT-MTOR pathways might serve as a strategy for antiviral autophagy.
Figure 9.
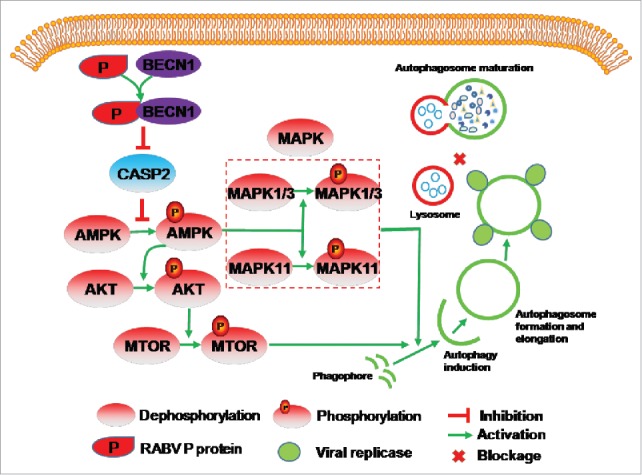
Proposed model of RABV-induced incomplete autophagy via the CASP2-AMPK- MAPK1/3/11-AKT1-MTOR pathways. RABV-encoded P protein leads to a CASP2 decrease by binding to BECN1. CASP2 reduction activates the phosphorylation of AMPK. The phosphorylated AMPK then activates the phosphorylation of AKT, MAPK11 and MAPK1/3. The phosphorylated AKT results in phosphorylation of MTOR. The phosphorylated MAPK11, MAPK1/3 and MTOR activates autophagosome formation. The autophagosome engulfs RABV virions, and does not deliver them to lysosomes.
Materials and methods
Antibodies and reagents
Rabbit anti-LC3A/B (4108), anti-p-AKT (Ser473) (4060), anti-AKT (4691), and anti-p-MTOR (Ser2448; 5536), anti-MTOR (2983), anti-p-PRKAA (4185), anti-PRKAA (2532), anti-p-MAPK1/3 (4370), anti-MAPK1/3 (4695), anti-p-MAPK11 (4511), and anti-MAPK11 (9212), NBR1 (9891) antibodies were purchased from Cell Signaling Technology. Rabbit anti-CASP2 (ab179520), anti-PIK3C3 (ab124095), anti-p-ULK1 (ab133747), anti-ULK1 (ab128859), anti-CASP3 (ab179517), anti-CASP8 (ab25901), anti-CASP9 (ab2014) and anti-ATG5 (ab108327) antibodies were purchased from Abcam. Anti-BECN1 (sc-48341) antibody was purchased from Santa Cruz Biotechnology. Rabbit anti-SQSTM1 (3340–1), anti-MYC (R1208–1) and anti-GAPDH/glyceraldehyde-3-phosphate dehydrogenase (2251–1) antibodies were purchased from Epitomics. Mouse monoclonal antibodies (mAbs) to N and P proteins of RABV were produced in our laboratory.57 Dulbecco's modified Eagle's medium (DMEM, 11995), Donkey anti-mouse IgG secondary antibody, Alexa Fluor® 647 (A-31571) and 546 (A10036) conjugate were obtained from Thermo Scientific. Rapa (R8781), wortmannin (W1628) and 3-MA (M9281) were purchased from Sigma-Aldrich. EBSS (14155–063) was purchased from Gibco. CQ (50–63–5[tlrl-chq]) was purchased from Invivogen. EXFectTM Transfection Reagent (T101–01/02) was purchased from Vazyme Biotech and LysoTracker Red DND-99 (L-7528) was purchased from Invitrogen.
Cells culture and viruses
Mouse neuroblastoma N2a cells (CCL−131) and human 293T cells (CRL-3216) from ATCC were maintained in DMEM supplemented with 10% fetal bovine serum, 100 U of penicillin mg/ml, and 100 μg of streptomycin/ml.
RABV strains HEP-Flury and CVS-11 were propagated in N2a cells. Briefly, cells were infected with RABV at a multiplicity of infection (MOI) of 2. After 1 h absorption, the supernatant was removed, and the cells were washed 3 times with sterile phosphate-buffered saline (PBS: 8.4 mM Na2HPO4, 1.5 mM KH2PO4, 136.9 mM NaCl, 2.7 mM KCl, pH 7.2) to remove unbound viruses. The infected cells were grown in fresh medium at 37°C and 5% CO2 for the indicated times. For autophagy alteration, cells were pretreated with Rapa, EBSS, wortmannin, or 3-MA for 2 h before virus infection.
Construction of expression plasmids and transfection
The GFP-P, Flag-N and Flag-P plasmids were constructed and stored in our laboratory.58 The mouse Lc3b gene (GenBank accession number: NM026160.4) was PCR-amplified from total cellular RNA of N2a cells with gene-specific primers (upstream primer: 5′CCCAAGCTTGGGAT GCCGTCCGAGAAGACCT 3′, downstream primer: 5′GGGGTACCCCTTACACAGCCATTGC TGTC3′) by Mastercycler® pro (Eppendorf AG, 6321XQ201070, Hamburg, Germany) and cloned into the GFP vector (Clontech, 6082–1). Mouse Becn1 was amplified from N2a genomic cDNA using the 2 pairs of specific primers (upstream primer 5′CGGAATTCCGATGGAGGGGTCTAAGGCGTCC3′ and downstream primer 5′CCGCT CGAGTCACTTGTTATAGAACTGTGAG3′ for the plasmid pCMV-N-Myc [Myc, Clontech, 635689], upstream primer 5′CGGAATTCCGATGGAGGGGTCTAAGGCGTCC3′, downstream primer 5′ CGGGATCCCGCTTGTTATAGAACTGTGAGGAC3′ for the plasmid pDsRed2-N1 [RED, Clontech, 632406]) and cloned into the vector Myc and RED, respectively. The full-length G and M genes as well as the truncated L (951 and 1524bp) gene of RABV were amplified via PCR from the cDNA of HEP-Flury and then cloned into Flag (Clontech, 635688), respectively. The PCR specific primers used are as follows: upstream primer 5′ CCCAAGCTTGGGATGGTTCCTCAGGTTCTTTTG 3′ and downstream primer 5′ GGGGTACCCCTCACAGTCTGGTCTCGCC3′ for the G gene, upstream primer 5′ CCCAAGCTTGGGATGAACTTTCTATGTAAG 3′ and downstream primer 5′ GGGGTACCCCTTATTCTAAAAGCAGAGAAG 3′ for the M gene, upstream primer 5′ CCCAAGCTTGGGATGCTGGATCCGGGAGAG 3′ and downstream primer 5′ GGGGTACCCCTTACCTAAACTTCTCTGC 3′ for the L (951bp), and upstream primer 5′ CCCAAGCTTGGGATGCTGGATCCGGGAGAG 3′ and downstream primer 5′ GGGGTACCCCTTAAATGACCTTCTCGCTAGG 3′ for the L (1524bp). Transfection of these plasmids DNA was performed with ExFectTM Transfection Reagent.
The following shRNAs targeting mouse (Mus musculus, abbreviated as Mm) MmAtg5, MmLc3b, MmAkt1, MmMtor and MmCasp2-shRNA in a pGPU6/GFP/Neo vector, were purchased from Genepharma. siRNA targeting MmBecn1 (5′GGAGUGGAAUGAAAUCAAUTT3′), MmPrkaa1 (5′GCGUGUACGAAGGAAGAAUTT3′), MmMapk1 (5′GUGCUCUGCUUAUGAUAAUTT3′) and negative control (5′UUCUCCGAACGUGUCACGUT3′) were also synthesized by Genepharma. The shRNA and siRNA were transfected into N2a cells with EXFectTM Transfection Reagent according to the manufacturer's instructions, respectively. The cells were infected with RABV at 24 h post-transfection and incubated at 37°C for 36 h. Infected cells were then harvested for further analysis. The efficiency of shRNA and siRNA knockdown was evaluated by western blotting using the specific antibodies.
Confocal microscopy
N2a cells were transfected with GFP-LC3B for 12 h, Flag-N or treated with Rapa for 24 h, and cotransfected with GFP-LC3B and/or RED-BECN1and Flag-N, or Flag-P, Flag-M, Flag-G, Flag-L (951bp), Flag-L (951bp) for 24 h. The cells were washed with PBS, fixed, and permeabilized with 4% paraformaldehyde in PBS at 4°C for 20 min and then washed again with PBS. The resultant cells were incubated with the monoclonal or polyclonal antibodies to the target protein as the first antibody and the corresponding secondary antibody. Nuclei were stained with 4, 6-diamidino-2-phenylindole (DAPI). The stained cells were washed 5 times with PBS- 0.1% Tween 20 (Sigma-Aldrich, 9005–64–5). For staining of acidic compartments, 50 nM LysoTracker Red DND-99 was added to the medium. The medium was removed after 15 min and the live cells rinsed 3 times with PBS. The stained and live cells were examined under a Zeiss LSM 510 laser confocal microscopy (Zeiss, Oberkochen, Baden-Württemberg, Germany).
Western blotting
Cells were harvested and lysed immediately in RIPA buffer (Beyotime, P0013B). Lysate protein quantification was performed by bicinchoninic acid assay kit (Pierce, 23225). An equal volume of each sample was separated by sodium dodecyl sulfate polyacrylamide-gel electrophoresis and the protein bands were transferred onto polyvinylidene fluoride membranes (Millipore, ISEQ00010). After blocking with 5% nonfat dry milk containing 0.1% Tween 20 for 1 h at 37°C, the membranes were incubated with primary antibody for at 4°C overnight, followed by horseradish peroxidase-conjugated anti-mouse/rabbit IgG (Kirkegaard & Perry Laboratories, 074–1806/074–1506) and visualized using a SuperSignal West Femto Substrate Trial Kit (Thermo Scientific, 34096). Three independent biological experiments were performed for western blotting quantification analysis. ImageJ software (National Institutes of Health, Bethesda, MD, USA) was used to quantify the intensity of protein bands.
Coimmunoprecipitation
CoIP was performed as described previously, with some modifications.59 Briefly, the infected or transfected N2a cells were washed twice with cold PBS, and lysed for 3 h in cell lysis buffer with NP-40 (Beyotime, P0013F) with 1 mM phenylmethylsulfonyl fluoride protease inhibitor (Beyotime, ST505) at 4°C. Cell lysates were centrifuged at 12,000 × g for 10 min at 4°C to remove insoluble fractions. The soluble fractions were pretreated with protein A/G agarose beads (Santa Cruz Biotechnology, sc-2003) for 1 h at 4°C. Pretreated supernatants were further incubated with IP antibody overnight at 4°C. Fresh protein A/G agarose was then added at 4°C for 8 h before washing with PBS. The bound proteins were eluted by boiling in 4 × SDS-PAGE loading buffer and subjected to western blotting analysis.
Drug treatment and viral titers detection
N2a cells grown in 6-well culture plates (Corning, 3506) were pretreated with DMSO (control), Rapa, 3-MA, or wortmannin, or EBSS for 2 h, or transfected with control shRNA (shCtrl), MmAtg5 shRNA or Mm Lc3b shRNA for 12 h. The cells were then infected with RABV at a MOI of 2. At various time postinfection, cell culture supernatants were collected, and the cells were subjected to 3 freeze–thaw cycles for detecting 50% tissue culture infectious doses (TCID50). Extracellular viruses were determined by inoculating 10-fold dilutions of the cell culture supernatant onto confluent N2a cells in 96-well culture plates (Corning, 3596). After 48 h of incubation, supernatant was removed, and the cells were fixed with 80% cold acetone and viral antigen was detected using immunofluorescence. Virus titers were calculated using the Reed-Muench method and expressed as TCID50/ml.
Animal experiments
Animal experiments were conducted in the animal facility at the Institute of Basic Medical Sciences, Zhejiang University, in accordance with governmental and institutional guidelines. Kunming 3-d-old Institute of Cancer Research (ICR) mice were purchased from the experimental animal center of Zhejiang Province. The mice were housed in a pathogen-free facility in groups of 5 or fewer mice and were fed ad libitum. Three-d-old Kunming mice were intracerebrally injected with either vehicle control (DMEM) or RABV strain HEP-Flury or CVS-11 (100 TCID50 per mouse). The mice that died within 3 d were not used for data analysis. At 24 h and 80 h postinfection, mice were killed and brain tissues were collected from each mouse, homogenated, and subjected to western blotting. All experimental protocols involving animals have been approved by the Scientific Ethical Committee of the Zhejiang University (No. ZJU2013105002), China.
Statistical analysis
Statistically significant differences between groups and treatments were determined by 2-way analysis of variance (ANOVA) and between groups were determined by one-way with the Tukey Multiple Comparison Test and using GraphPad Prism 5 software. A P value of less than 0.05 was considered statistically significant.
Supplementary Material
Abbreviations
- AMPK
AMP-activated protein kinase
- ANOVA
analysis of variance
- ATG
autophagy related; BECN1, Beclin 1
- CASP
caspase
- CoIP
coimmunoprecipitation
- CQ
chloroquine
- DAPI
4, 6-diamidino-2-phenylindole
- DMEM
Dulbecco's modified Eagle's medium
- EBSS
Earle's balanced salt solution
- Flag
pCMV-N-Flag
- G
glycoprotein
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GFP
green fluorescent protein
- hpi
hours postinfection
- ICR
Institute of Cancer Research
- L
the RNA-dependent RNA polymerase
- LAMP
lysosomal-associated membrane protein
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- M
matrix protein
- 3-MA
3-methyladenine
- MAPK
mitogen-activated protein kinase
- mAbs
mouse monoclonal antibodies
- MOI
multiplicity of infection
- MTOR
mechanistic target of rapamycin
- Myc
pCMV-N-Myc
- N
nucleoprotein
- NBR1
NBR1, autophagy cargo receptor
- NBs
Negri bodies
- P
phosphoprotein
- PBS
phosphate-buffered saline
- PtdIns3K
phosphatidylinositol 3-kinase (class III)
- RABV
rabies virus
- Rapa
rapamycin
- RED
pDsRed2-N1
- shCtrl
control shRNA
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- SQSTM1
sequestosome 1
- TCID50
50% tissue culture infectious doses
- ULK1
unc-51 like autophagy activating kinase 1
Disclosure of potential conflicts of interest
There were no potential conflicts of interest to be disclosed.
Acknowledgments
We gratefully acknowledge Yunqin Li for technical assistance on laser confocal microscopy.
Funding
This work was supported by grants from National Key Technology R & D Program of China (Grant No. 2016YFD0500400, 2015BAD12B01), the National Special Fund for Public Welfare Industry (Project No. 201103032) of China and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
References
- [1].Esclatine A, Chaumorcel M, Codogno P. Macroautophagy signaling and regulation. Curr Top Microbiol Immunol 2009; 33533-70; PMID:19802559; http://dx.doi.org/ 10.1007/978-3-642-00302-8_2 [DOI] [PubMed] [Google Scholar]
- [2].Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J Cell Sci 2005; 118(Pt 1):7-18; PMID:15615779; http://dx.doi.org/ 10.1242/jcs.01620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dreux M, Chisari FV. Viruses and the autophagy machinery. Cell Cycle 2010; 9(7):1295-307; PMID:20305376; http://dx.doi.org/ 10.4161/cc.9.7.11109 [DOI] [PubMed] [Google Scholar]
- [4].Marino G, Lopez-Otin C. Autophagy: molecular mechanisms, physiological functions and relevance in human pathology. Cell Mol Life Sci 2004; 61(12):1439-54; PMID:15197469; http://dx.doi.org/ 10.1007/s00018-004-4012-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469(7330):323-35; PMID:21248839; http://dx.doi.org/ 10.1038/nature09782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr 2007; 2719-40; PMID:17311494. [DOI] [PubMed] [Google Scholar]
- [7].Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9(10):1102-9; PMID:17909521; http://dx.doi.org/ 10.1038/ncb1007-1102 [DOI] [PubMed] [Google Scholar]
- [8].Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2001; 2(3):211-6; PMID:11265251; http://dx.doi.org/ 10.1038/35056522 [DOI] [PubMed] [Google Scholar]
- [9].Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20(7):1981-91; PMID:19211835; http://dx.doi.org/ 10.1091/mbc.E08-12-1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol 2010; 22(2):140-9; PMID:20097051; http://dx.doi.org/ 10.1016/j.ceb.2010.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature 1998; 395(6700):395-8; PMID:9759731; http://dx.doi.org/ 10.1038/26506 [DOI] [PubMed] [Google Scholar]
- [12].Tanida I. Autophagy basics. Microbiol Immunol 2011; 55(1):1-11; PMID:21175768; http://dx.doi.org/ 10.1111/j.1348-0421.2010.00271.x [DOI] [PubMed] [Google Scholar]
- [13].Trocoli A, Djavaheri-Mergny M. The complex interplay between autophagy and NF-kappaB signaling pathways in cancer cells. Am J Cancer Res 2011; 1(5):629-49; PMID:21994903 [PMC free article] [PubMed] [Google Scholar]
- [14].Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009; 11(4):385-96; PMID:19270696; http://dx.doi.org/ 10.1038/ncb1846 [DOI] [PubMed] [Google Scholar]
- [15].Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol 2009; 11(4):468-76; PMID:19270693; http://dx.doi.org/ 10.1038/ncb1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008; 19(12):5360-72; PMID:18843052; http://dx.doi.org/ 10.1091/mbc.E08-01-0080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yuk JM, Yoshimori T, Jo EK. Autophagy and bacterial infectious diseases. Exp Mol Med 2012; 44(2):99-108; PMID:22257885; http://dx.doi.org/ 10.3858/emm.2012.44.2.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yang YP, Liang ZQ, Gu ZL, Qin ZH. Molecular mechanism and regulation of autophagy. Acta Pharmacol Sin 2005; 26(12):1421-34; PMID:16297339; http://dx.doi.org/ 10.1111/j.1745-7254.2005.00235.x [DOI] [PubMed] [Google Scholar]
- [19].Tiwari M, Sharma LK, Vanegas D, Callaway DA, Bai Y, Lechleiter JD, Herman B. A nonapoptotic role for CASP2/caspase 2: modulation of autophagy. Autophagy 2014; 10(6):1054-70; PMID:24879153; http://dx.doi.org/ 10.4161/auto.28528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li L, Wang L, Xiao R, Zhu G, Li Y, Liu C, Yang R, Tang Z, Li J, Huang W, et al. The invasion of tobacco mosaic virus RNA induces endoplasmic reticulum stress-related autophagy in HeLa cells. Biosci Rep 2012; 32(2):171-86; PMID:21729006; http://dx.doi.org/ 10.1042/BSR20110069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Talloczy Z, HWt Virgin, Levine B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006; 2(1):24-9; PMID:16874088; http://dx.doi.org/ 10.4161/auto.2176 [DOI] [PubMed] [Google Scholar]
- [22].Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell host & microbe 2007; 1(1):23-35; http://dx.doi.org/ 10.1016/j.chom.2006.12.001 [DOI] [PubMed] [Google Scholar]
- [23].Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 2009; 30(4):588-98; PMID:19362021; http://dx.doi.org/ 10.1016/j.immuni.2009.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hu B, Zhang Y, Jia L, Wu H, Fan C, Sun Y, Ye C, Liao M, Zhou J. Binding of the pathogen receptor HSP90AA1 to avibirnavirus VP2 induces autophagy by inactivating the AKT-MTOR pathway. Autophagy 2015; 11(3):503-15; PMID:25714412; http://dx.doi.org/ 10.1080/15548627.2015.1017184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Su WC, Chao TC, Huang YL, Weng SC, Jeng KS, Lai MM. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J Virol 2011; 85(20):10561-71; PMID:21835792; http://dx.doi.org/ 10.1128/JVI.00173-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Van Grol J, Subauste C, Andrade RM, Fujinaga K, Nelson J, Subauste CS. HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PloS One 2010; 5(7):e11733; http://dx.doi.org/ 10.1371/journal.pone.0011733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Surviladze Z, Sterk RT, DeHaro SA, Ozbun MA. Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. J Virol 2013; 87(5):2508-17; PMID:23255786; http://dx.doi.org/ 10.1128/JVI.02319-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lahaye X, Vidy A, Pomier C, Obiang L, Harper F, Gaudin Y, Blondel D. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: Evidence that NBs are sites of viral transcription and replication. J virol 2009; 83(16):7948-58; PMID:19494013; http://dx.doi.org/ 10.1128/JVI.00554-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282(33):24131-45; PMID:17580304; http://dx.doi.org/ 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- [30].von Hoven G, Kloft N, Neukirch C, Ebinger S, Bobkiewicz W, Weis S, Boller K, Janda KD, Husmann M. Modulation of translation and induction of autophagy by bacterial exoproducts. Med Microbiol Immun 2012; 201(4):409-18; http://dx.doi.org/ 10.1007/s00430-012-0271-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009; 8(13):1986-90; PMID:19502794; http://dx.doi.org/ 10.4161/cc.8.13.8892 [DOI] [PubMed] [Google Scholar]
- [32].Matsushita M, Suzuki NN, Obara K, Fujioka Y, Ohsumi Y, Inagaki F. Structure of Atg5.Atg16, a complex essential for autophagy. J Biol Chem 2007; 282(9):6763-72; PMID:17192262; http://dx.doi.org/ 10.1074/jbc.M609876200 [DOI] [PubMed] [Google Scholar]
- [33].Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin KQ, Ishii KJ, Kawai T, Akira S, Suzuki K, et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci USA 2007; 104(35):14050-5; PMID:17709747; http://dx.doi.org/ 10.1073/pnas.0704014104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kim JH, Park JM, Kim EK, Lee JO, Lee SK, Jung JH, You GY, Park SH, Suh PG, Kim HS. Curcumin stimulates glucose uptake through AMPK-p38 MAPK pathways in L6 myotube cells. J Cell Physiol 2010; 223(3):771-8; PMID:20205235; http://dx.doi.org/ 10.1002/jcp.22093 [DOI] [PubMed] [Google Scholar]
- [35].Sajan MP, Bandyopadhyay G, Miura A, Standaert ML, Nimal S, Longnus SL, Van Obberghen E, Hainault I, Foufelle F, Kahn R, et al. AICAR and metformin, but not exercise, increase muscle glucose transport through AMPK-, ERK-, and PDK1-dependent activation of atypical PKC. Am J Physiol Endocrinol Metab 2010; 298(2):E179-92; PMID:19887597; http://dx.doi.org/ 10.1152/ajpendo.00392.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang J, Whiteman MW, Lian H, Wang G, Singh A, Huang D, Denmark T. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J Biol Chem 2009; 284(32):21412-24; PMID:19520853; http://dx.doi.org/ 10.1074/jbc.M109.026013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tang G, Yue Z, Talloczy Z, Hagemann T, Cho W, Messing A, Sulzer DL, Goldman JE. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum Mol Genet 2008; 17(11):1540-55; PMID:18276609; http://dx.doi.org/ 10.1093/hmg/ddn042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol 2007; 7(10):767-77; PMID:17767194; http://dx.doi.org/ 10.1038/nri2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Espert L, Codogno P, Biard-Piechaczyk M. Involvement of autophagy in viral infections: antiviral function and subversion by viruses. J Mol Med (Berl) 2007; 85(8):811-23; PMID:17340132; http://dx.doi.org/ 10.1007/s00109-007-0173-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett 2010; 584(7):1287-95; PMID:20083114; http://dx.doi.org/ 10.1016/j.febslet.2010.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu C, Chapman NM, Karmaus PW, Zeng H, Chi H. mTOR and metabolic regulation of conventional and regulatory T cells. J Leukocyte Biol 2015; PMID:25714803; http://dx.doi.org/ 10.1189/jlb.2RI0814-408R [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Huang H, Kang R, Wang J, Luo G, Yang W, Zhao Z. Hepatitis C virus inhibits AKT-tuberous sclerosis complex (TSC), the mechanistic target of rapamycin (MTOR) pathway, through endoplasmic reticulum stress to induce autophagy. Autophagy 2013; 9(2):175-95; PMID:23169238; http://dx.doi.org/ 10.4161/auto.22791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sun Y, Li C, Shu Y, Ju X, Zou Z, Wang H, Rao S, Guo F, Liu H, Nan W, et al. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci Signal 2012; 5(212):ra16; PMID:22355189; http://dx.doi.org/ 10.1126/scisignal.2001931 [DOI] [PubMed] [Google Scholar]
- [44].Peng J, Zhu S, Hu L, Ye P, Wang Y, Tian Q, Mei M, Chen H, Guo X. Wild-type rabies virus induces autophagy in human and mouse neuroblastoma cell lines. Autophagy 2016; 1-17; PMID:26799652; http://dx.doi.org/ 10.1080/15548627.2016.1196315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 2000; 275(2):992-8; PMID:10625637; http://dx.doi.org/ 10.1074/jbc.275.2.992 [DOI] [PubMed] [Google Scholar]
- [46].Eskelinen EL, Prescott AR, Cooper J, Brachmann SM, Wang L, Tang X, Backer JM, Lucocq JM. Inhibition of autophagy in mitotic animal cells. Traffic 2002; 3(12):878-93; PMID:12453151; http://dx.doi.org/ 10.1034/j.1600-0854.2002.31204.x [DOI] [PubMed] [Google Scholar]
- [47].Zeng X, Overmeyer JH, Maltese WA. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J Cell Sci 2006; 119(Pt 2):259-70; PMID:16390869; http://dx.doi.org/ 10.1242/jcs.02735 [DOI] [PubMed] [Google Scholar]
- [48].Row PE, Reaves BJ, Domin J, Luzio JP, Davidson HW. Overexpression of a rat kinase-deficient phosphoinositide 3-kinase, Vps34p, inhibits cathepsin D maturation. Biochem J 2001; 353(Pt 3):655-61; PMID:11171063; http://dx.doi.org/ 10.1042/bj3530655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Siddhanta U, McIlroy J, Shah A, Zhang Y, Backer JM. Distinct roles for the p110alpha and hVPS34 phosphatidylinositol 3′-kinases in vesicular trafficking, regulation of the actin cytoskeleton, and mitogenesis. J Cell Biol 1998; 143(6):1647-59; PMID:9852157; http://dx.doi.org/ 10.1083/jcb.143.6.1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30(6):678-88; PMID:18570871; http://dx.doi.org/ 10.1016/j.molcel.2008.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes 2013; 62(4):1270-81; PMID:23223177; http://dx.doi.org/ 10.2337/db12-0533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122(6):927-39; PMID:16179260; http://dx.doi.org/ 10.1016/j.cell.2005.07.002 [DOI] [PubMed] [Google Scholar]
- [53].Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 2008; 4(5):600-6; PMID:18497563; http://dx.doi.org/ 10.4161/auto.6260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, Pagliarini V, Matteoni S, Fuoco C, Giunta L, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol 2010; 191(1):155-68; PMID:20921139; http://dx.doi.org/ 10.1083/jcb.201002100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010; 6(6):764-76; PMID:20639694; http://dx.doi.org/ 10.4161/auto.6.6.12709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Koyama-Honda I, Itakura E, Fujiwara TK, Mizushima N. Temporal analysis of recruitment of mammalian ATG proteins to the autophagosome formation site. Autophagy 2013; 9(10):1491-9; PMID:23884233; http://dx.doi.org/ 10.4161/auto.25529 [DOI] [PubMed] [Google Scholar]
- [57].Zhang J, Ruan X, Zan J, Zheng X, Yan Y, Liao M, Zhou J. Efficient generation of monoclonal antibodies against major structural proteins of rabies virus with suckling mouse brain antigen. Monoclon Antib Immunodiagn Immunother 2014; 33(2):94-100; PMID:24746150; http://dx.doi.org/ 10.1089/mab.2013.0087 [DOI] [PubMed] [Google Scholar]
- [58].Zhang J, Wu X, Zan J, Wu Y, Ye C, Ruan X, Zhou J. Cellular chaperonin CCTgamma contributes to rabies virus replication during infection. J Virol 2013; 87(13):7608-21; PMID:23637400; http://dx.doi.org/ 10.1128/JVI.03186-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhang X, Zhou J, Wu Y, Zheng X, Ma G, Wang Z, Jin Y, He J, Yan Y. Differential proteome analysis of host cells infected with porcine circovirus type 2. J Proteome Res 2009; 8(11):5111-9; PMID:19708719; http://dx.doi.org/ 10.1021/pr900488q [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.