

Abstract
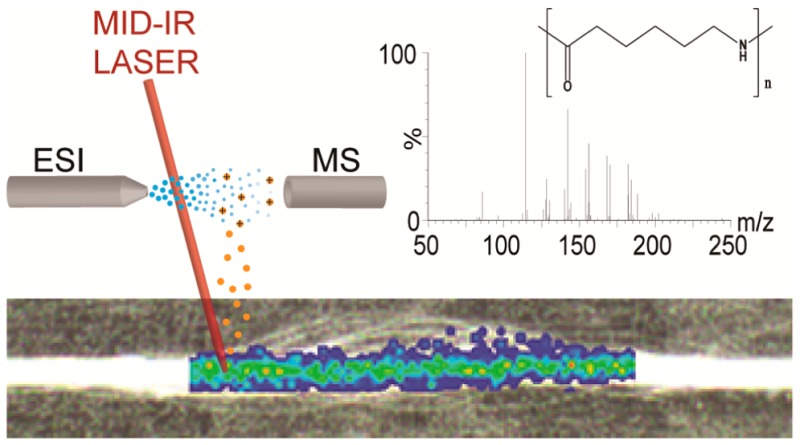
Direct analysis of synthetic fibers under ambient conditions is highly desired to identify the polymer, the finishes applied and irregularities that may compromise its performance and value. In this paper, laser ablation electrospray ionization ion mobility time-of-flight mass spectrometry (LAESI-IMS-TOF-MS) was used for the analysis of synthetic polymers and fibers. The key to this analysis was the absorption of laser light by aliphatic and aromatic nitrogen functionalities in the polymers. Analysis of polyamide (PA) 6, 46, 66, and 12 pellets and PA 6, 66, polyaramid and M5 fibers yielded characteristic fragment ions without any sample pretreatment, enabling their unambiguous identification. Synthetic fibers are, in addition, commonly covered with a surface layer for improved adhesion and processing. The same setup, but operated in a transient infrared matrix-assisted laser desorption electrospray ionization (IR-MALDESI) mode, allowed the detailed characterization of the fiber finish layer and the underlying polymer. Differences in finish layer distribution may cause variations in local properties of synthetic fibers. Here we also show the feasibility of mass spectrometry imaging (MSI) of the distribution of a finish layer on the synthetic fiber and the successful detection of local surface defects.
Synthetic fibers such as polyamide and polyester are widely used in many industrial materials, fabrics, clothes, etc., and their importance can hardly be overemphasized. High-performance fibers such as polyaramid are heat resistant and stronger than steel on an equal weight basis. Because of these properties, they can be used in, for example, ballistic vests, cables, optical fiber reinforcement, as well as in rubber reinforcement such as in tires, high-pressure hoses, conveyer belts, etc. The properties of synthetic fibers and their interaction with surrounding materials are strongly influenced by the chemical treatment of their surface.1−4 A large number of different types of surface layers can be used to adapt the fiber properties to the desired applications. Surface layers are used for processing, e.g., to reduce friction, electrostatic charging, and abrasion5 or to allow better adhesion to other polymers in blends.6−8 Differences in the amount and distribution of the surface layer lead to variations in properties of the fiber.9 Such surface defects might result in weak spots leading to abrasion or localized reduced adhesion,10 which could be detrimental in high-performance applications.
Mass spectrometry (MS) is an excellent tool for the characterization of polymers as well as polymer surfaces and additives. The use of MS in polymer analysis has been reviewed recently.11,12 Secondary ion mass spectrometry (SIMS) is commonly used for the analysis of polymers and synthetic fibers.13−26 SIMS typically produces small fragment ions for solid polymer samples, e.g., CN– in PA fibers.18,20,21 Oligomers of PA of 6 up to 24 repeating units were observed after dissolution of the fiber in trifluoroacetic acid, deposition on a silver substrate, and SIMS analysis.13 Also, matrix-assisted laser desorption ionization MS (MALDI-MS) is frequently used for the characterization of polymers.27−33 Applications range from mass spectrometry imaging of polymer membranes, showing polymer distributions and contaminations on membrane surfaces,32 to the characterization of branching in polyaramid fibers.33 The analysis of fibers by SIMS or MALDI-MS is, however, hampered by sample pretreatment requirements and ionization under vacuum conditions. Analysis under ambient conditions without any sample pretreatment would be very useful for synthetic fibers in order to quickly identify the material, finish layers, and defects without introducing any pretreatment-induced bias.
Direct analysis in real time (DART) is an ambient MS technique first introduced in 2005 by Cody.34 DART has been used for the detection of additives in different polymer materials such as softeners and stabilizers or degradation products,35−40 but the polymer material itself was not detected since DART relies on thermal desorption. Some progress has been made to fingerprint insoluble polymers under ambient conditions using thermal-assisted atmospheric pressure glow discharge (TA-APGD) following fixation of the sample on a heated stage.41 Other plasma-based techniques such as plasma-assisted desorption ionization (PADI) and flowing afterglow atmospheric pressure glow discharge (FA-APGD) were reported to successfully identify different insoluble polymers.42,43 While certainly useful, the main drawbacks of these techniques relate to long stabilization times and relatively low spatial resolution that precludes mass spectrometry imaging (MSI) of fibers. Another ambient ionization MS technique used for identification of polymer species is desorption electrospray ionization (DESI).44−47 DESI relies on the solubility of sample material for desorption and subsequent ESI-like ionization mechanisms.48 For the analysis of hydrophobic polymers, such as poly(methyl methacrylate) and polymethylstyrene, methanol solutions containing formic acid or salts such as lithium bromide or silver nitrate were used.49 Without water in the solution, however, DESI is less able to create higher charge states which limits polymer analysis applications.
Alternatively, laser-based techniques are fast and offer much smaller spot sizes. Electrospray-assisted laser desorption ionization (ELDI) was used to characterize dried polymer standard solutions of PPG 1000, PMMA 1300, and PEG 1500 on the surface of a steel sample plate.50 Tuning of the laser wavelength to specific polymer absorption bands removed the necessity of an external matrix and allowed successful ablation of bulk polymer material, as demonstrated with a free electron laser operated at a wavelength of 3.43 μm to interact with the aliphatic CH stretch vibration of polystyrene.51 Ablated material can subsequently be postionized by different techniques such as electrospray ionization, chemical ionization, or photoionization.52−54
Laser ablation electrospray ionization (LAESI) is an ambient (imaging) MS technique first introduced by Nemes and Vertes in 2007.52 It uses a mid-infrared laser producing a wavelength of 2.94 μm that addresses hydroxyl functionalities, such as endogenous water molecules in biomaterials. In principle, the same wavelength can also be absorbed by aliphatic or aromatic nitrogen functionalities in polymers. The absorbed energy could break the polymer chains and ablate characteristic fragments under ambient conditions for subsequent mass spectrometric analysis. In the absence of laser-absorbing functional groups, a small amount of water vapor can be condensed on a sample surface to absorb the laser energy, as was shown for the detailed structure elucidation of dyes on fabrics by infrared matrix-assisted laser desorption electrospray ionization (IR-MALDESI).55 Detailed characterization along the length of a 1 mm wide synthetic fiber was feasible. IR-MALDESI-MSI of dyes on textile fibers within a forensic context even showed single filament images as small as 10 μm in diameter.56 When combining MSI with ion mobility separation (IMS), an additional separation dimension becomes available to identify different polymer distributions and to separate ions with the same mass to charge (m/z) ratio but having different collisional cross sections.57
In this research, we demonstrate the detailed MS characterization of different synthetic fibers such as PA 6, 46, 66, 12, polyaramid, and M5 by direct LAESI-MS. In addition, we provide full finish characterization, show the feasibility of mass spectrometry imaging of the fiber, the distribution of the finish layer, and the detection of local surface defects by LAESI-IMS-TOF-MSI operated in a transient (ice) matrix-assisted laser ablation mode.
Experimental Section
Materials
Ultrapure water (H2O) 18.2 MΩ cm at 25 °C was freshly produced daily with a Millipore (Molsheim, France) integral 3 system. Methanol (MeOH) LC–MS grade was purchased from VWR (Leuven, Belgium). Formic acid (FA) LC–MS grade was bought from Fisher Scientific (Geel, Belgium). Leucine-enkephalin (leu-enk) for lock mass-corrected mass calibration was purchased from Waters (Manchester, U.K.). Para-aramid trimer-NH2 (structure is show in Figure S1) and Twaron para-aramid yarn were provided by Teijin Aramid (Arnhem, The Netherlands). One standard yarn type (1680 dtex, f1000) was produced without applying a basic spin finish. In a second step, the “naked” yarn was treated with a finish, consisting of an aqueous solution (4.25%) of Lansurf OA10 (polyethylene glycol (PEG) 400 monooleate, structure is shown in Figure S2), from Lankem (Dukinfield, United Kingdom). After application of this finish using a slit applicator, the yarn was dried by passing through a hot air oven (3.6 s at 180 °C). The estimated (final) finish amount was 0.5% Lansurf OA10 (finish weight on yarn weight). Poly[2,6-diimidazo (4,5-β-4′,5′-ε)pyridinylene-1,4-(2,5-dihydroxy)phenylene] (M5) fiber, polyethylene terephthalate (PET) fiber, and the PA fibers PA 6 (Mw unknown), PA 66 (Mw 27 × 103), PA 66 (Mw 46 × 103), and PA 66 (Mw 33 × 103) were from laboratory stock, as were poly(methyl methacrylate) (PMMA) (Mw unknown) pellets and the PA pellets PA 6 (Mw 30 × 103), PA 6 (Mw 31 × 103), PA 6 (Mw 15 × 103), PA 12 (Mw unknown), PA 66 (Mw 22 × 103), and PA 46 (Mw unknown). Chemical structures of fibers and pellets are given in Figure 1.
Figure 1.

Chemical structures of (A) PA 6, (B) PA 12, (C) PA 46, (D) PA 66, (E) polyaramid, and (F) M5.
LAESI-MS of Polymers and Fibers
A Protea Biosciences (Morgantown, WV) LAESI DP-1000 system was coupled to a Waters (Manchester, U.K.) Synapt G2S traveling wave ion mobility time-of-flight mass spectrometer and used for the analysis of all sample materials. Both polymer pellets and synthetic fibers were directly mounted with Tesa double-sided tape (Hamburg, Germany) to the sample stage that was kept at 10 °C. LAESI desktop software v.2.0.1.3 (Protea Biosciences) was used to control experimental parameters of the LAESI system. The Nd:YAG optical parametric oscillator mid infrared laser (2.94 μm) was set to 100% laser power (Φ 3.2 J/cm2) and 10 pulses with a specified pulse length of 5 ns were acquired on every spot (diameter 200 μm) at a frequency of 10 Hz with a between spot interval of 200 μm. A solution of MeOH–H2O (1:1) with 0.1% FA and 40 ng/mL leu-enk was used as electrospray solvent at a flow rate of 1 μL/min. Electrospray voltage was set at ∼3.5 kV in order to have a stable Taylor cone. Nitrogen was used as LAESI bath gas at 20 L/h. The Synapt G2S was controlled by Masslynx v4.1 SCN 883 (Waters) and operated in positive ion TOF-MS resolution mode, m/z range 50–1200 Da, scan time 1 s, and source and interface temperatures were both set at 150 °C. For ion mobility TOF-MS the IMS wave velocity was set to 650 m/s and the transfer velocity at 1200 m/s. Background-subtracted mass spectra were created using the “combine spectrum” function in Masslynx: five scans, each corresponding to 10 laser pulses were averaged, and 50 scans of the electrospray background were subtracted. Proteaplot v2.0.8.5 (Protea Biosciences) was used to create maximum intensity ion maps. Driftscope v2.7 (Waters) was used to select the different polymer distributions within the m/z versus drift time space and to clean the spectral background.
IR-MALDESI and LAESI Mass Spectrometry Imaging of Fibers with a Finish Layer
For MSI of the finish layers on the fibers, the same instrument and experimental conditions were used as described in the previous section, with the exception of the sample stage temperature which was set to −19 °C, starting 15 min before analysis in order to achieve a thin layer of ice on the sample. Consequently, instead of LAESI, ice-assisted LAESI or IR-MALDESI55,56 occurred for typically the first two or three (out of ten) laser pulses applied. Optical images of the 10 cm × 7 cm sample stage were obtained and used to superimpose the ion maps. A 60 × 6 pattern (350 sample locations) with an interval of 400 μm was acquired around the synthetic fiber area on the optical image. The MSI analysis, including the recording of electrospray background, had a total runtime of 45 min.
Results and Discussion
LAESI Fingerprinting of Synthetic Polymers
LAESI experiments conventionally use a mid-infrared laser (2.94 μm) to efficiently transfer laser energy to the strong OH stretch vibration of water-containing (mostly biological) samples in order to ablate sample material prior to ionization by electrospray for subsequent MS analysis. Besides the strong OH absorption band, also NH stretch vibrations of amines and amides absorb at this wavelength. So initially, we examined LAESI-TOF-MS as a tool for rapid ambient identification of solid NH-containing polymer materials without any sample pretreatment. Figure 2 presents the background-subtracted mass spectra of different polyamides. The mass spectra obtained show characteristic ions and confirm the capability of LAESI fingerprinting of polyamides. The mass accuracies corresponding to the proposed elemental compositions are provided in Table S1. The mass spectrum of PA 6 (Figure 2A) shows a high abundance ion at m/z 114.091 having the elemental composition [C6H12NO]+, which represents residual caprolactam monomer and ions formed by cleavage of the amide bond. This high abundance ion differentiates PA 6 from the other polyamides and was also observed by Klun after acid hydrolysis of PA 6 and subsequent electrospray MS analysis.58 In LAESI analysis, although at lower intensity, m/z values 128.105 [C7H14NO]+, 142.121 [C8H16NO]+, 156.138 [C9H18NO]+, 170.153 [C10H20NO]+, and 184.168 [C11H22NO]+ were additionally detected. Two extra PA 6 samples were analyzed providing similar results, the background-subtracted mass spectra are presented in Figures S3 and S4. The LAESI mass spectrum of PA 12 (Figure 2B) shows a single ion at m/z 198.188 having the elemental composition of [C12H24NO]+. Like PA 6, this ion represents residual monomer and/or cleavage of the amide bond yielding the ion for the repeating unit of PA 12. In contrast to PA 6 and 12, PA 46 and PA 66 are synthesized from two different monomers; therefore, their mass spectra will be more complex. Cleavage of the amide bond will lead to fragments containing at least either two nitrogen atoms or two oxygen atoms, and furthermore cleavage of carbon–carbon bonds could yield fragments that contain the intact amide bond. The LAESI mass spectrum of PA 46 is presented in Figure 2C and shows ions at m/z values 128.105 [C7H14NO]+, 142.126 [C8H16NO]+, and 156.138 [C9H18NO]+ that are consistent with the intact amide bond and carbon–carbon bond cleavages at different positions of the polymer backbone. In addition, the ion at m/z 115.089 [C5H11N2O]+ is formed, which is characteristic for polyamides containing 1,4-butanediamine, like PA 46. This fragment contains the amide group, the four carbon atoms, and the final nitrogen yielding an elemental composition unique for PA 46. Finally, the mass spectrum of PA 66 is depicted in Figure 2D. Like PA 46 this spectrum contains ions at m/z values 128.105 [C7H14NO]+, 142.121 [C8H16NO]+, and 156.138 [C9H18NO]+. However, in PA 66 this ion series continues with the m/z values 170.153 [C10H20NO]+ and 184.168 [C11H22NO]+ that could not be found in PA 46. In addition, the absence of m/z values 115.089 (PA 46), 198.188 (PA 12), and high intensity m/z 114.091 (PA 6) makes this spectrum unique for identification of PA 66 polymer. It should be noted that MS/MS data acquisition following precursor ion selection of specific LAESI induced fragment ions may add additional evidence for the identity of the polymers but was not further investigated here.
Figure 2.

LAESI-TOF-MS background-subtracted mass spectra of polyamide pellets (A) PA 6, (B) PA 12, (C) PA 46, and (D) PA 66.
In the MS analysis of polyamides with other ionization techniques like SIMS, MALDI and sequential pyrolysis field desorption, PA distributions of multiple intact oligomers have been detected.13,59−61 Differentiation between, e.g., PA 6 and PA 66 could not be achieved. With FA-APGD, the cyclic monomer ion of PA 66 at m/z 227 could be detected.43 In LAESI, we detect characteristic polymer fragments in the low mass range. This is in contrast with the suggestion that LAESI is a low energy ionization technique comparable to ESI.52 Note that in the present situation the NH stretching vibration within the amide bond is addressed in the absence of water and, as a result, the laser energy is not dissipated by an excess of hydroxyl moieties from the sample matrix.
LAESI Fingerprinting of Synthetic Fibers
Similarly, LAESI-MS can be used to identify the polymer used in synthetic fibers. Different NH-containing fibers, such as the polyamides PA 6 and PA 66 and the aromatic fibers polyaramid and M5, were analyzed by LAESI-TOF-MS. Additionally, the obtained data were used to generate spatial ion maps superimposed onto the camera images of the fibers in order to explore LAESI-MSI possibilities. Figure 3 shows ion maps and background-subtracted mass spectra of (A) PA 6, (B) PA 66, (C) polyaramid, and (D) M5 fibers. The LAESI mass spectrum of PA 6 (Figure 3A) shows the same fragment ion at m/z 114.091 as obtained with the pellets, and a range of ions formed by breaking of carbon–carbon bonds in the polymer backbone. Most likely, PA 6 fiber contains less residual caprolactam monomer than the PA 6 polymer sample analyzed in Figure 2A. The additional ions in Figure 3A were also observed (but at much lower relative intensity) in the LAESI mass spectrum of the PA 6 polymer sample shown in Figure 2A. The LAESI mass spectra obtained from PA 66 fibers (Figure 3B and Figures S5 and S6) show similar fragment ions as seen for the PA 66 pellets in Figure 2D, strengthening our claim on the identification of PA polymers, regardless of processing into fibers or as raw polymer materials. The LAESI mass spectrum of the aromatic polyaramid fiber shows characteristic fragment ions at m/z 107.059 and m/z 135.057, which belong to the elemental compositions [C6H7N2]+ and [C7H7N2O]+, respectively. These fragments originate from the aromatic ring containing the two (1,4-) amine groups [C6H7N2]+ and one additional carbonyl group from the amide [C7H7N2O]+. The second aromatic fiber analyzed, M5, does not contain an amide bond (cf. Figure 1); nevertheless, the laser energy could be absorbed by either or both the secondary amine and hydroxyl groups. The background-subtracted LAESI mass spectrum of M5 given in Figure 3D shows fragment ions at m/z 120.058, m/z 143.059, and m/z 144.053. The elemental compositions of these fragments are [C6H6N3]+, [C9H7N2]+, and [C8H6N3]+, respectively. Although these elemental compositions are difficult to explain and require multiple bond cleavages, these ions are unique for M5 among the NH-containing fibers such as polyamides and polyaramides. Therefore, these ions provide the ability to quickly identify this fiber under ambient conditions. In comparison with, and in contrast to other ionization techniques like pyrolysis MS,62 MALDI and IR-MALDESI,33,55,56 LAESI exclusively provides fragments in the low mass range. In SIMS, oxygen containing fragment ions from PA 6 filaments with m/z values 31, 45, 114, and 227 were detected in positive ion mode by Yip et al.20 In comparison, the fragment ions m/z 31 and 45 were outside the mass range of our mass spectrometer, but m/z value 114 corresponds to the mass found in this study for the repeating unit fragment of PA 6.
Figure 3.

LAESI background-subtracted mass spectra and ion maps of (A) PA 6 fiber, (B) PA 66 fiber, (C) polyaramid fiber, and (D) M5 fiber. Ion maps show the highest intensity ion: (A) m/z 114.091, (B) m/z 156.138, (C) m/z 135.057, and (D) m/z 143.059.
In addition to straightforward polymer identification, the feasibility of LAESI-MSI was explored in the same experiments. Reconstructed ion maps, superimposed on the optical images, show the distribution of the most abundant fragment ion in Figure 3A–D. In all cases the ion follows nicely the 1 mm wide fiber pattern on the sample stage. At 200 μm spatial resolution, approximately five locations were analyzed over the fiber diameter. These results are very encouraging for imaging of finish layers on fibers.
LAESI Induced Fragmentation of Polymers
In order to support our hypothesis that laser-induced fragmentation, initiated by absorption at NH functionalities, is the primary cause of the observed polymer characteristic fragment ions, we performed a range of complementary experiments. First, we extracted polyaramid fiber with MeOH (as described in the Supporting Information) to find out whether unreacted monomers, dimer, trimer, etc. could be present and contribute to the observed characteristic ions. The obtained ESI-MS background subtracted mass spectrum is depicted in Figure S7. Evidently, the finish is detected, but the fiber polymer fragments at m/z 107.059 and 135.057 (or dimers/trimers) were not.
To investigate the possibility of acid hydrolysis due to the close proximity of the formic acid-containing electrospray plume, we placed 10 cm of polyaramid fiber into 4 mL of electrospray solution for 30 min. Subsequently, we analyzed this solution by ESI-MS and, as expected, did not detect any of the characteristic polymer fragments (data not shown). It should be kept in mind however that the pH experienced in electrospray droplets may be lower (due to charging and concentration effects of the spray) than that of the bulk spray solution.
In addition to the analysis of polyaramid material with LAESI in the absence of water, aramid trimer-NH2 was also investigated. Figure S8A presents the ESI background subtracted mass spectrum of a 1 μg/mL trimer-NH2 solution in MeOH–H2O (1:1) + 0.1% FA, showing ions at m/z 347.149 [M + H]+, 369.126 [M + Na]+, and 174.080 [M + 2H]2+. Also, the LAESI background subtracted mass spectrum of a 10 μg/mL trimer-NH2 solution in MeOH–H2O (1:1) is depicted (Figure S8B), showing a single ion at m/z 347.149 [M + H]+. In contrast, LAESI analysis of trimer-NH2 powder (mass spectrum is shown in Figure S8C) did not display any of these masses, just the fragment ions m/z 107.059 and 135.057, as with the LAESI analysis of polyaramid fiber. This demonstrates that the observed fragmentation of the solid polymer pellets and fibers is a result of the dissipation of the laser energy. To examine the importance of absorption by NH functionalities within the polymer material, two materials without the NH functionality were measured: PMMA pellets and PET fiber. In accordance with our hypothesis, no ions from these polymers were detected (data not shown).
To further explore the effect of laser energy on the fragmentation of the polymer materials, PA 66 pellets were analyzed at different amounts of laser energy: 20, 40, 60, 80, and 100%. Only at 100% laser power fragment ions, e.g., m/z 142.121 and 156.138 were detected. Most likely, this is a result of the laser ablation threshold, as is reported for IR laser ablation of other polymeric materials in the literature.51,63 Kappes et al. proposed an ablation mechanism well below decomposition temperature that is photomechanical, i.e., a stress due to the thermal expansion of the polymer.63 Here we hypothesize that in case the laser energy cannot be dissipated by the solvent (or endogenous water), the NH-functionalities in the solid material will absorb the energy. As the stretch vibrations are unable to dissipate the excess of energy, multiple bond cleavages in the polymer backbone will occur, resulting in ablated polymer fragments.
IR-MALDESI and LAESI MSI of Polyaramid Fiber with Finish
A PEG 400 monooleate finish was applied to polyaramid fiber at a fiber treatment line as described in the Experimental Section. Investigations of the fiber containing this finish layer were conducted with transient IR-MALDESI and LAESI IMS-TOF-MSI in a single setup. The background-subtracted mass spectrum of polyaramid fiber containing 0.5% (m/m) finish is presented in Figure S9A and shows several polymer distributions. The characterization of this finish was supported by extracting the different polymer distributions from the m/z versus ion mobility map provided in Figure 4. Three main distributions were assigned: (1) a [(PEG)n + NH4]+ distribution (Figure S9B), (2) a [(PEG)n monooleate + NH4]+ distribution (Figure S9C), and (3) a [(PEG)n dioleate + NH4]+ distribution (Figure S9D). In addition to these polymer distributions, m/z 283.264 and m/z 309.282 are present, which are tentatively assigned to protonated oleic acid and an elimination product of the PEG ester of oleic acid (structure is shown in Figure S10), respectively. These results directly obtained from a fiber surface are, apart from the different cations and lower intensities, in excellent agreement with the LAESI data obtained from a 1% solution of the finish in MeOH (Figure S11A–F). In the latter, [M + Na]+ ions dominated over [M + NH4]+ ions. In other words, the transient ice-assisted LAESI provided the same low-energy ablation of the finish polymers from the surface as from solution. Similar cationized ethylene glycol oligomers were also found in the analysis of PEG 3000 with DESI;45 however, in our study no multiple charged polymers were observed due to a lower degree of PEG polymerization.
Figure 4.

IR-MALDESI-IMS m/z versus ion mobility map of 0.5% (m/m) PEG 400 monooleate applied as described in the Experimental Section on polyaramid fiber. Mass spectra of selected areas are shown for 1 in Figure S9B, for 2 in Figure S9C, and for 3 in Figure S9D.
The distribution of the finish layer along the fiber and thus any surface defects, i.e., the areas without finish, could be visualized by reconstructing ion maps of the finish ions. An artificial defect of approximately 5 mm along the fiber length was produced by local heating of the fiber with 0.5% finish for 3 s with a soldering iron. Characteristic ions from both the fiber and the finish ion, e.g., [(PEG)10 monooleate + NH4]+ were superimposed on the optical image to create the ion maps, depicted in Figure 5. These ion maps show the polyaramid fiber (Figure 5A) and the finish layer (Figure 5B) with surface defects, demonstrating the ability of ambient imaging to investigate finish layers and the detection of defects in finish layers by transient IR-MALDESI and LAESI-IMS-TOF-MSI in a single experiment.
Figure 5.

LAESI and IR-MALDESI ion maps of polyaramid fiber with 0.5% (m/m) PEG 400 monooleate finish applied as described in the Experimental Section and an artificial fabricated surface defect, indicated with a cyan line. Part A displays the ion map of one of the fiber fragments (m/z 135.057 ± 0.001), while part B displays an ion from the finish distribution [(PEG)10 monooleate + NH4]+ (m/z 740.551 ± 0.001).
Conclusion
This study investigated polymer fingerprinting, finish characterization, and the detection of surface defects by mass spectrometry imaging (MSI) under ambient conditions without any sample pretreatment. It was shown that the laser at a wavelength of 2.94 μm ablates solid polymer material containing aliphatic and aromatic nitrogen groups, providing fragments to successfully identify PA 6, PA 46, PA 66, PA 12, polyaramid, and M5 polymer species. It was also shown that the finish layer could be fully characterized and, by MSI, localized, which provides detailed insight into surface defects and thus weak spots in the material. Further research might explore possibilities of other laser wavelengths, such as 3.43 μm light to interact with the aliphatic CH stretch vibration to broaden the polymer application range.
Acknowledgments
This research receives funding from The Netherlands Organisation for Scientific Research (NWO) in the framework of the Technology Area TA-COAST2 of the Fund New Chemical Innovations (Project No. 053.21.111) with Wageningen University, Maastricht University, RIKILT, Omics2Image, Waters, and DSM Resolve as partners and Teijin Aramid as an associated partner.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.6b04641.
Chemical structures of aramid trimer-NH2 and PEG 400 monooleate; table with mass accuracy for proposed elemental compositions in Figure 2; additional LAESI-MS background subtracted mass spectra of PA 6 pellets and PA 66 fibers; method of extraction of aramid fiber and ESI-MS parameters; ESI-MS background subtracted mass spectrum of MeOH extract of polyaramid fiber; ESI and LAESI-MS background subtracted mass spectra of aramid trimer-NH2; IR-MALDESI-IMS-MS background subtracted mass spectra of PEG 400 monooleate on polyaramid fiber; proposed chemical structure of m/z 309.282; LAESI-IMS-MS background subtracted mass spectra and 2D map of a 1% solution of PEG 400 monooleate in MeOH (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Luo S.; Van Ooij W. J. J. Adhes. Sci. Technol. 2002, 16, 1715–1735. 10.1163/156856102320396102. [DOI] [Google Scholar]
- Drzal L. T.; Madhukar M. J. Mater. Sci. 1993, 28, 569–610. 10.1007/BF01151234. [DOI] [Google Scholar]
- Zhang X.; Chen P.; Yu Q.; Ma K.; Ding Z.; Zhu X. Vacuum 2013, 97, 1–8. 10.1016/j.vacuum.2013.03.005. [DOI] [Google Scholar]
- Chen J.; Zhu Y.; Ni Q.; Fu Y.; Fu X. Appl. Surf. Sci. 2014, 321, 103–108. 10.1016/j.apsusc.2014.09.196. [DOI] [Google Scholar]
- Niestegge R. Chem. Fibers Int. 2001, 51, 450–451. [Google Scholar]
- de Lange P. J.; Akker P. G.; Maas A. J. H.; Knoester A.; Brongersma H. H. Surf. Interface Anal. 2001, 31, 1079–1084. 10.1002/sia.1147. [DOI] [Google Scholar]
- de Lange P. J.; Akker P. G. J. Adhes. Sci. Technol. 2012, 26, 827–839. [Google Scholar]
- Peng T.; Cai R.; Chen C.; Wang F.; Liu X.; Wang B.; Xu J. J. Macromol. Sci., Part B: Phys. 2012, 51, 538–550. 10.1080/00222348.2011.609777. [DOI] [Google Scholar]
- de Lange P. J.; Akker P. G.; Mäder E.; Gao S.-L.; Prasithphol W.; Young R. J. Compos. Sci. Technol. 2007, 67, 2027–2035. 10.1016/j.compscitech.2006.11.018. [DOI] [Google Scholar]
- Weigel J. Chem. Fibers Int. 2010, 60, 230–233. [Google Scholar]
- Paine M. R. L.; Barker P. J.; Blanksby S. J. Anal. Chim. Acta 2014, 808, 70–82. 10.1016/j.aca.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Crotty S.; Gerişlioğlu S.; Endres K. J.; Wesdemiotis C.; Schubert U. S. Anal. Chim. Acta 2016, 932, 1–21. 10.1016/j.aca.2016.05.024. [DOI] [PubMed] [Google Scholar]
- Bletsos I. V.; Hercules D. M.; Greifendorf D.; Benninghoven A. Anal. Chem. 1985, 57, 2384–2388. 10.1021/ac00289a049. [DOI] [Google Scholar]
- Reddy S. S.; Dong X.; Murgasova R.; Gusev A. I.; Hercules D. M. Macromolecules 1999, 32, 1367–1374. 10.1021/ma9807670. [DOI] [Google Scholar]
- Burrell M. C.; Chao H. S. I.; Meerman T. P.; Peterson G. S. Surf. Interface Anal. 1997, 25, 799–803. 10.1002/(SICI)1096-9918(199709)25:10<799::AID-SIA302>3.0.CO;2-M. [DOI] [Google Scholar]
- Kang H. M.; Yoon T. H.; van Ooij W. J. J. Adhes. Sci. Technol. 2006, 20, 1155–1169. 10.1163/156856106777920802. [DOI] [Google Scholar]
- Valantin C.; Benoit R.; D M. P.; Lacroix F.; Gomez E.; Phalip P.; Morcel J.; Tricoche D.; Hocine N. A. Appl. Surf. Sci. 2016, 360 (Part B), 623–633. 10.1016/j.apsusc.2015.11.027. [DOI] [Google Scholar]
- Zhou C.; Li M.; Garcia R.; Crawford A.; Beck K.; Hinks D.; Griffis D. P. Anal. Chem. 2012, 84, 10085–10090. 10.1021/ac3025569. [DOI] [PubMed] [Google Scholar]
- de Lange P. J.; Mahy J. W. G. Fresenius' J. Anal. Chem. 1995, 353, 487–493. 10.1007/BF00321308. [DOI] [Google Scholar]
- Yip J.; Chan K.; Sin K. M.; Lau K. S. Appl. Surf. Sci. 2003, 205, 151–159. 10.1016/S0169-4332(02)01015-2. [DOI] [Google Scholar]
- Biganska O.; Darque-Ceretti E.; Giulieri F.; Combarieu R. Surf. Interface Anal. 2001, 31, 847–855. 10.1002/sia.1114. [DOI] [Google Scholar]
- Malshe P.; Mazloumpour M.; El-Shafei A.; Hauser P. Surf. Coat. Technol. 2013, 217, 112–118. 10.1016/j.surfcoat.2012.11.081. [DOI] [Google Scholar]
- Yip J.; Chan K.; Sin K. M.; Lau K. S. Polym. Int. 2004, 53, 627–633. 10.1002/pi.1420. [DOI] [Google Scholar]
- Wu S. R.; Sheu G. S.; Shyu S. S. J. Appl. Polym. Sci. 1996, 62, 1347–1360. 10.1002/(SICI)1097-4628(19961128)62:9<1347::AID-APP5>3.0.CO;2-H. [DOI] [Google Scholar]
- Lineton W. B.; Nunn N. S.; Bishop D. P. J. Text. Inst. 1999, 90, 385–394. [Google Scholar]
- Pelster A.; Körsgen M.; Kurosawa T.; Morita H.; Arlinghaus H. F. Anal. Chem. 2016, 88, 9638–9646. 10.1021/acs.analchem.6b02415. [DOI] [PubMed] [Google Scholar]
- Nielen M. W. F. Mass Spectrom. Rev. 1999, 18, 309–344. 10.1002/(SICI)1098-2787(1999)18:5<309::AID-MAS2>3.0.CO;2-L. [DOI] [Google Scholar]
- Scarff C. A.; Snelling J. R.; Knust M. M.; Wilkins C. L.; Scrivens J. H. J. Am. Chem. Soc. 2012, 134, 9193–9198. 10.1021/ja2118656. [DOI] [PubMed] [Google Scholar]
- Shan L.; Murgasova R.; Hercules D. M.; Houalla M. J. Mass Spectrom. 2001, 36, 140–144. 10.1002/jms.110. [DOI] [PubMed] [Google Scholar]
- Hilton G. R.; Jackson A. T.; Thalassinos K.; Scrivens J. H. Anal. Chem. 2008, 80, 9720–9725. 10.1021/ac801716c. [DOI] [PubMed] [Google Scholar]
- Montaudo G.; Samperi F.; Montaudo M. S. Prog. Polym. Sci. 2006, 31, 277–357. 10.1016/j.progpolymsci.2005.12.001. [DOI] [Google Scholar]
- Krueger K.; Terne C.; Werner C.; Freudenberg U.; Jankowski V.; Zidek W.; Jankowski J. Anal. Chem. 2013, 85, 4998–5004. 10.1021/ac4002063. [DOI] [PubMed] [Google Scholar]
- Gies A. P.; Kliman M.; McLean J. A.; Hercules D. M. Macromolecules 2008, 41, 8299–8301. 10.1021/ma8019464. [DOI] [Google Scholar]
- Cody R. B.; Laramée J. A.; Durst H. D. Anal. Chem. 2005, 77, 2297–2302. 10.1021/ac050162j. [DOI] [PubMed] [Google Scholar]
- Rothenbacher T.; Schwack W. Rapid Commun. Mass Spectrom. 2009, 23, 2829–2835. 10.1002/rcm.4194. [DOI] [PubMed] [Google Scholar]
- Rothenbacher T.; Schwack W. Rapid Commun. Mass Spectrom. 2010, 24, 21–29. 10.1002/rcm.4350. [DOI] [PubMed] [Google Scholar]
- Mess A.; Vietzke J.-P.; Rapp C.; Francke W. Anal. Chem. 2011, 83, 7323–7330. 10.1021/ac2011608. [DOI] [PubMed] [Google Scholar]
- Ackerman L. K.; Noonan G. O.; Begley T. H. Food Addit. Contam., Part A 2009, 26, 1611–1618. 10.1080/02652030903232753. [DOI] [PubMed] [Google Scholar]
- Haunschmidt M.; Klampfl C. W.; Buchberger W.; Hertsens R. Analyst 2010, 135, 80–85. 10.1039/B911040B. [DOI] [PubMed] [Google Scholar]
- Fouyer K.; Lavastre O.; Rondeau D. Anal. Chem. 2012, 84, 8642–8649. 10.1021/ac301759q. [DOI] [PubMed] [Google Scholar]
- Zhang N.; Zhou Y.; Zhen C.; Li Y.; Xiong C.; Wang J.; Li H.; Nie Z. Analyst 2012, 137, 5051–5056. 10.1039/c2an35770d. [DOI] [PubMed] [Google Scholar]
- Salter T. L.; Gilmore I. S.; Bowfield A.; Olabanji O. T.; Bradley J. W. Anal. Chem. 2013, 85, 1675–1682. 10.1021/ac302677m. [DOI] [PubMed] [Google Scholar]
- Jecklin M. C.; Gamez G.; Zenobi R. Analyst 2009, 134, 1629–1636. 10.1039/b819560a. [DOI] [PubMed] [Google Scholar]
- Takáts Z.; Wiseman J. M.; Gologan B.; Cooks R. G. Science 2004, 306, 471–473. 10.1126/science.1104404. [DOI] [PubMed] [Google Scholar]
- Nefliu M.; Venter A.; Cooks R. G. Chem. Commun. 2006, 888–890. 10.1039/b514057a. [DOI] [PubMed] [Google Scholar]
- Williams J. P.; Hilton G. R.; Thalassinos K.; Jackson A. T.; Scrivens J. H. Rapid Commun. Mass Spectrom. 2007, 21, 1693–1704. 10.1002/rcm.3008. [DOI] [PubMed] [Google Scholar]
- Friia M.; Legros V.; Tortajada J.; Buchmann W. J. Mass Spectrom. 2012, 47, 1023–1033. 10.1002/jms.3057. [DOI] [PubMed] [Google Scholar]
- Fenn J. B.; Mann M.; Meng C. K.; Wong S. F.; Whitehouse C. M. Mass Spectrom. Rev. 1990, 9, 37–70. 10.1002/mas.1280090103. [DOI] [Google Scholar]
- Jackson A. T.; Williams J. P.; Scrivens J. H. Rapid Commun. Mass Spectrom. 2006, 20, 2717–2727. 10.1002/rcm.2660. [DOI] [PubMed] [Google Scholar]
- Huang M.-Z.; Hsu H.-J.; Wu C.-I.; Lin S.-Y.; Ma Y.-L.; Cheng T.-L.; Shiea J. Rapid Commun. Mass Spectrom. 2007, 21, 1767–1775. 10.1002/rcm.3011. [DOI] [PubMed] [Google Scholar]
- Johnson S. L.; Bubb D. M.; Haglund R. F. Jr Appl. Phys. A: Mater. Sci. Process. 2009, 96, 627–635. 10.1007/s00339-009-5290-3. [DOI] [Google Scholar]
- Nemes P.; Vertes A. Anal. Chem. 2007, 79, 8098–8106. 10.1021/ac071181r. [DOI] [PubMed] [Google Scholar]
- Herdering C.; Reifschneider O.; Wehe C. A.; Sperling M.; Karst U. Rapid Commun. Mass Spectrom. 2013, 27, 2595–2600. 10.1002/rcm.6727. [DOI] [PubMed] [Google Scholar]
- Vaikkinen A.; Shrestha B.; Koivisto J.; Kostiainen R.; Vertes A.; Kauppila T. J. Rapid Commun. Mass Spectrom. 2014, 28, 2490–2496. 10.1002/rcm.7043. [DOI] [PubMed] [Google Scholar]
- Cochran K. H.; Barry J. A.; Muddiman D. C.; Hinks D. Anal. Chem. 2013, 85, 831–836. 10.1021/ac302519n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran K. H.; Barry J. A.; Robichaud G.; Muddiman D. C. Anal. Bioanal. Chem. 2015, 407, 813–820. 10.1007/s00216-014-8042-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanucara F.; Holman S. W.; Gray C. J.; Eyers C. E. Nat. Chem. 2014, 6, 281–294. 10.1038/nchem.1889. [DOI] [PubMed] [Google Scholar]
- Klun U.; Andrenšek S.; Kržan A. Polymer 2001, 42, 7095–7099. 10.1016/S0032-3861(01)00132-X. [DOI] [Google Scholar]
- Montaudo G.; Montaudo M. S.; Puglisi C.; Samperi F. J. Polym. Sci., Part A: Polym. Chem. 1996, 34, 439–447. 10.1002/(SICI)1099-0518(199602)34:3<439::AID-POLA13>3.0.CO;2-Q. [DOI] [Google Scholar]
- Bahr U.; Lüderwald I.; Müller R.; Schulten H. R. Angew. Makromol. Chem. 1984, 120, 163–175. 10.1002/apmc.1984.051200110. [DOI] [Google Scholar]
- Schulten H.-R.; Plage B. J. Polym. Sci., Part A: Polym. Chem. 1988, 26, 2381–2394. 10.1002/pola.1988.080260911. [DOI] [Google Scholar]
- Schulten H. R.; Plage B.; Ohtani H.; Tsuge S. Angew. Makromol. Chem. 1987, 155, 1–20. 10.1002/apmc.1987.051550101. [DOI] [Google Scholar]
- Kappes R. S.; Schönfeld F.; Li C.; Golriz A. A.; Nagel M.; Lippert T.; Butt H.-J.; Gutmann J. S. SpringerPlus 2014, 3, 489. 10.1186/2193-1801-3-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.