

Abstract
Lessons Learned.
Despite involvement of PI3K pathway activation in tumorigenesis of solid tumors, single‐agent PI3K inhibitors have shown modest clinical activity.
Preclinical evidence suggests that combining PI3K pathway inhibitors and chemotherapy can enhance antitumor effects.
In patients with solid tumors, the PI3K inhibitor pilaralisib had a favorable safety profile but did not enhance the antitumor activity of paclitaxel plus carboplatin.
Further clinical evaluation is warranted to identify effective combination strategies with PI3K pathway inhibitors.
Background.
Pilaralisib (SAR245408) is an oral, pan‐class I phosphoinositide 3‐kinase (PI3K) inhibitor. This phase I dose‐escalation study evaluated the maximum tolerated dose (MTD), safety, pharmacokinetics (PK), and pharmacodynamics of pilaralisib in capsule and tablet formulations, administered in combination with paclitaxel and carboplatin in patients with advanced solid tumors.
Methods.
A 3 + 3 design was used. Pilaralisib was administered once daily (QD); paclitaxel (up to 175 mg/m2) and carboplatin (up to area under the curve [AUC] of 6) were administered on day 1 of 21‐day cycles. An MTD expansion cohort of patients with endometrial carcinoma was included.
Results.
Fifty‐eight patients were enrolled. Six patients (10.3%) had dose‐limiting toxicities, of which only rash (two patients, 3.4%) occurred in more than one patient. The MTD of pilaralisib tablets in combination with paclitaxel and carboplatin was determined to be 200 mg QD. The most frequently reported adverse events (AEs) of any grade were neutropenia (67.2%) and thrombocytopenia (67.2%). PK data showed no interaction between pilaralisib and paclitaxel/carboplatin. Tumor tissue showed moderate inhibition of PI3K and mitogen‐activated protein kinase (MAPK) pathways. Seven of 52 evaluable patients had a partial response (PR; 13.5%).
Conclusion.
Pilaralisib had a favorable safety profile but did not enhance the antitumor activity of paclitaxel plus carboplatin in solid tumors.
Abstract
经验总结
• 尽管实体瘤的肿瘤发生过程中涉及PI3K通路激活, 但单药PI3K抑制剂的临床活性不佳。
• 临床前证据显示PI3K通路抑制剂联合化疗可增强抗肿瘤作用。
• 在实体瘤患者中, PI3K抑制剂Pilaralisib具有良好的安全性特征, 但未能增强紫杉醇+卡铂的抗肿瘤活性。
• 有必要通过进一步的临床评价来确定PI3K通路抑制剂的有效联合用药策略。
摘要
背景. Pilaralisib(SAR245408)是一种口服给药的泛I类磷脂酰肌醇3‐激酶(PI3K)抑制剂。本I期剂量递增研究在晚期实体瘤患者中评价了Pilaralisib胶囊和片剂与紫杉醇和卡铂联合给药的最大耐受剂量(MTD)、安全性、药代动力学(PK)和药效学。
方法. 采用3 + 3设计。Pilaralisib每日给药一次(QD), 紫杉醇(最高剂量为175mg/m2)和卡铂[最高剂量至曲线下面积(AUC)为6]在每个21天周期的第1天给药。研究中纳入一个子宫内膜癌患者的MTD扩展队列。
结果. 入组了58例患者。6例患者(10.3%)出现剂量限制性毒性, 其中超过1例患者发生的事件仅包括皮疹(2例患者, 3.4%)。Pilaralisib与紫杉醇和卡铂联用时的MTD确定为200mg QD。最常报告的不良事件(AE, 不考虑级别)为中性粒细胞减少症(67.2%)和血小板减少症(67.2%)。PK数据表明Pilaralisib与紫杉醇/卡铂间无相互作用。在肿瘤组织中观察到对PI3K和丝裂原活化蛋白激酶(MAPK)通路的中度抑制作用。52例可评价患者中有7例达到部分缓解(PR;13.5%)。
结论. Pilaralisib具有良好的安全性特征, 但未增强紫杉醇联合卡铂对实体瘤的抗肿瘤活性。
Discussion
Despite the involvement of PI3K pathway activation in tumorigenesis of solid tumors, single‐agent PI3K inhibitors have shown modest clinical activity. Preclinical evidence suggests that combining PI3K pathway inhibitors and chemotherapy can enhance antitumor effects in solid tumors, providing a rationale for clinical evaluation. The pan‐class I PI3K inhibitor pilaralisib showed preliminary antitumor activity in a phase I monotherapy study in advanced solid tumors, which established the MTD of pilaralisib capsules as 600 mg QD. This study aimed to determine the safety and MTD of pilaralisib in capsule and tablet formulations in combination with paclitaxel and carboplatin in patients with advanced solid tumors.
Fifty‐eight patients were enrolled, of which 25 received pilaralisib capsules (100–600 mg) and 33 received pilaralisib tablets. The tablet starting dose was 200 mg, which was shown to provide exposure similar to 400 and 600 mg capsules in preliminary analyses (Sanofi data on file). Most frequent tumors types included endometrial (33%), lung (12%), breast (9%), and ovarian (9%).
The most frequently occurring AEs were consistent with the known safety profiles of carboplatin, paclitaxel, and pilaralisib (Table 1). Pilaralisib PK findings were consistent with previous studies of pilaralisib monotherapy; paclitaxel/carboplatin did not appear to affect pilaralisib PK. In serial tumor biopsies from two patients (with colorectal adenocarcinoma and cervical carcinoma), inhibition of PI3K/mTOR and MAPK pathways was observed (67%–76% reduction in pAKT, 64%–69% reduction in phosphorylated EIF4E‐binding protein‐1 [p4EBP1], and 70%–73% reduction in phosphorylated extracellular signal‐regulated kinase [pERK]) alongside modest reductions in proliferation and induction of apoptosis (Fig. 1). Of 52 evaluable patients, 13.5% had a PR and 42.3% had stable disease or a progression‐free period lasting ≥12 weeks. Median progression‐free survival was 3.2 months. Molecular alterations in the PI3K pathway did not appear to correlate with response. Adding pilaralisib to paclitaxel and carboplatin did not appear to enhance antitumor efficacy in patients with solid tumors, including patients with endometrial cancer. Similarly, previous studies showed modest efficacy for pilaralisib monotherapy in solid tumors and endometrial cancers. The combination of pilaralisib, paclitaxel, and carboplatin is no longer being investigated in solid tumors.
Table 1. Adverse events of any grade occurring in >25% of patients and of grade ≥3 occurring in >10% of patients treated with pilaralisib plus paclitaxel plus carboplatina.
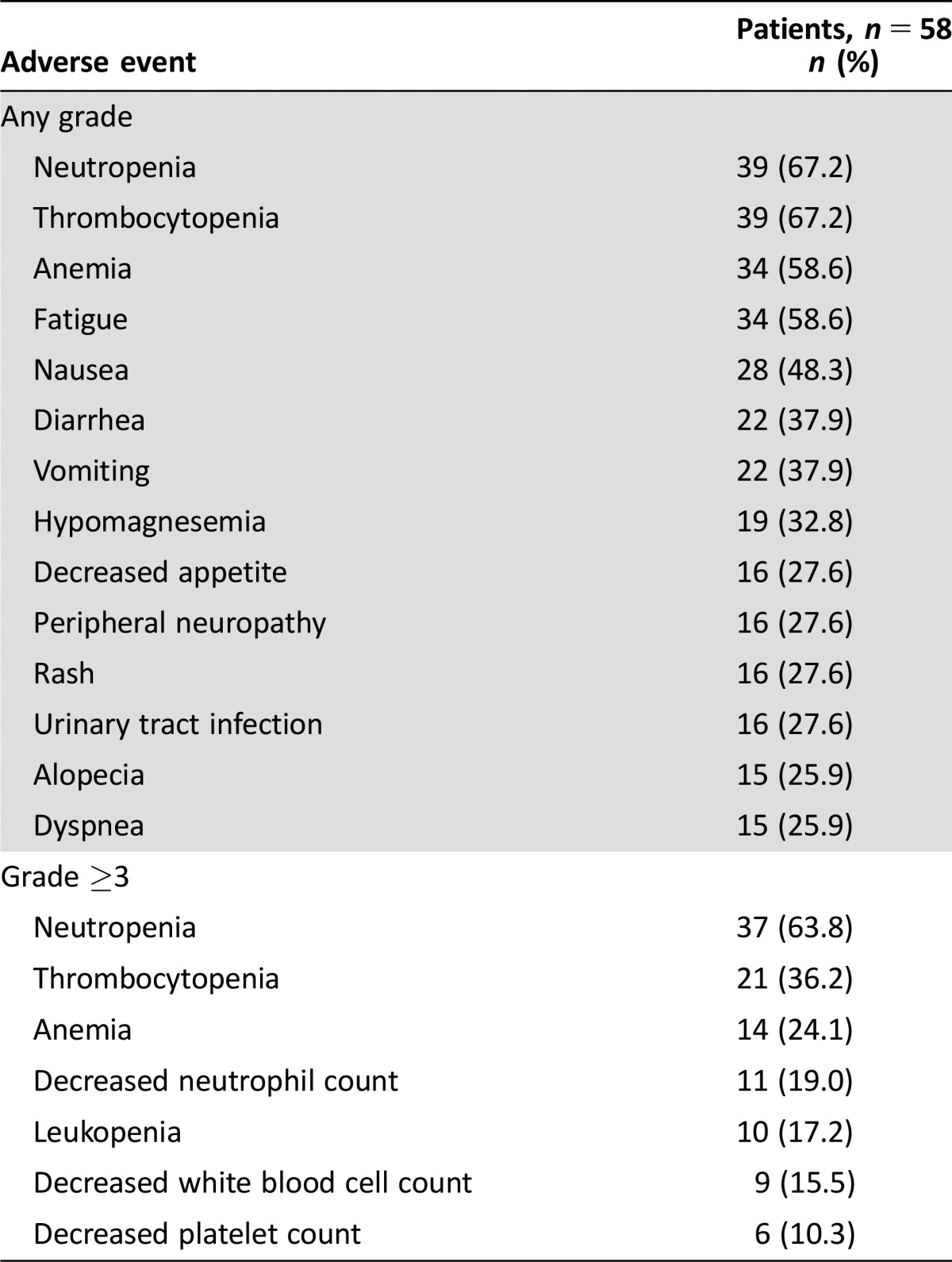
Graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events v3.0.
Figure 1.

PI3K and MAPK pathway inhibition in paired tumor biopsies. Cryopreserved tumor biopsy samples were serially sectioned at 10 microns; representative fields were captured at ×400 magnification. (A): A patient with colon adenocarcinoma (liver metastasis biopsies) receiving 200 mg pilaralisib/150 mg/m2 paclitaxel/AUC 5 carboplatin. Tumor molecular alterations were detected in KRAS, PIK3CA, and TP53 genes. (B): A patient with cervical adenocarcinoma receiving 200 mg pilaralisib/175 mg/m2 paclitaxel/AUC 6 carboplatin. Tumor molecular alteration was detected in PIK3CA gene (I391M polymorphism).
Abbreviations: AUC, area under the curve; EBP1, EIF4E‐binding protein‐1; ERK, extracellular signal‐regulated kinase; MAPK, mitogen‐activated protein kinase; PI3K, phosphoinositide 3‐kinase.
Trial Information
- Disease
Advanced cancer/solid tumor only
- Stage of disease/treatment
Metastatic/Advanced
- Prior Therapy
No designated number of regimens
- Type of study ‐ 1
Phase I
- Type of study ‐ 2
Other
- Primary Endpoint
MTD
- Primary Endpoint
Toxicity
- Additional Details of Endpoints or Study Design
- Phase I, open‐label, nonrandomized, dose‐escalation study. A standard 3 + 3 design was used. Treatment was administered in 21‐day cycles. Pilaralisib (starting dose 200 mg) was administered once daily starting on day 1. Paclitaxel (at doses up to 175 mg/m2) and carboplatin (at doses up to a targeted AUC of 6) were administered on day 1. Patients with advanced solid tumors were enrolled in the dose‐escalation phase. An expansion cohort enrolled patients with endometrial carcinoma. Primary objectives were to evaluate safety and determine the MTD. Secondary objectives were to investigate the relationship between selected biomarkers and efficacy and safety outcomes, to assess PK, and to evaluate preliminary antitumor activity. Eligible patients were aged ≥18 years and had an Eastern Cooperative Oncology Group (ECOG) performance status ≤1 (subjects with performance status 2 were considered following discussion and agreement with the sponsor). In the dose‐escalation phase, patients were required to have a histologically or cytologically confirmed solid tumor that was metastatic or unresectable, and refractory to standard therapy, or for which no known effective therapy existed. An MTD expansion cohort enrolled patients with advanced or recurrent endometrial carcinoma (endometrioid, serous, clear cell adenocarcinoma, adenosquamous carcinoma, or mixed histology, any grade). All patients were required to have adequate organ and bone marrow function and fasting plasma glucose ≤160 mg/dL. Patients who had previously received treatment with a PI3K inhibitor were excluded. All patients provided written informed consent.
- Investigator's Analysis
Evidence of target inhibition but no or minimal antitumor activity
Drug Information
- Drug 1
- Generic/Working name
Pilaralisib
- Drug type
Small molecule
- Drug class
PI3 kinase
- Dose
100–600 mg capsules or 200–300 mg tablets QD
- Route
oral (p.o.)
- Schedule of Administration
100–600 mg capsules or 200–300 mg tablets QD
- Drug 2
- Generic/Working name
Paclitaxel
- Drug type
Small molecule
- Drug class
Microtubule‐targeting agent
- Dose
Doses up to 175 mg/m2 on day 1 of 21‐day cycles
- Route
IV
- Schedule of Administration
Doses up to 175 mg/m2 on day 1 of 21‐day cycles
- Drug 3
- Generic/Working name
Carboplatin
- Drug type
Other
- Drug class
Platinum compound
- Dose
Doses up to a targeted AUC of 6 on day 1 of 21‐day cycles
- Route
IV
- Schedule of Administration
Doses up to a targeted AUC of 6 on day 1 of 21‐day cycles
Patient Characteristics
- Number of patients, male
14
- Number of patients, female
44
- Stage at diagnosis
-
I: 1
II: 1
III: 7
IV: 32
Unknown: 17
- Age
Median (range): 56.5 (25–82)
- Number of prior systemic therapies
Median (range): 3 (1–10)
- Performance Status: ECOG
-
0 — 13
1 — 44
2 —
3 —
unknown —
- Other
Not Collected
- Cancer Types or Histologic Subtypes
-
Endometrium 19
Lung 7
Breast 5
Ovaries 5
Skin 4
Cervix 2
Colon 1
Lymph nodes 1
Other 14
Primary Assessment Method
- Control Arm: Total Patient Population
- Number of patients screened
84
- Number of patients enrolled
58
- Number of patients evaluable for toxicity
58
- Number of patients evaluated for efficacy
52
- Response assessment CR
0
- Response assessment PR
13.5%
- Response assessment SD
48.1%
- Response assessment PD
38.5%
- (Median) duration assessments PFS
3.2 months
- (Median) duration assessments duration of treatment
13 weeks
Adverse Events

Table 3. Treatment‐related adverse events occurring in >10% of patients.

Data is shown as n (%).
Abbreviations: AE, adverse event; AUC, area under the curve.
Assessment, Analysis, and Discussion
- Completion
Study terminated before completion
- Pharmacokinetics/Pharmacodynamics
Not Collected
- Investigator's Assessment
Evidence of target inhibition but no or minimal antitumor activity
Paclitaxel plus carboplatin is a standard treatment for various solid tumors, including first‐line treatment of advanced/recurrent endometrial cancer [1]. However, most patients eventually become resistant to this regimen and experience disease progression. The phosphoinositide 3‐kinase (PI3K) pathway is pivotal for growth in normal cells, and dysregulation of the pathway is involved in tumorigenesis of solid tumors [2], [3], [4], [5], [6], [7]. In endometrial cancers, activation of the PI3K/mTOR pathway is associated with aggressive disease and poor prognosis [3]. Upregulation of PI3K/mTOR pathway signaling has been identified as a mechanism of tumor resistance to paclitaxel and carboplatin. In addition, PI3K/mTOR pathway inhibitors have been shown to augment the cytotoxicity of paclitaxel and carboplatin in cancer cell lines [8], [9], [10]. Therefore, combining paclitaxel plus carboplatin with PI3K inhibition is a rational therapeutic strategy in solid tumors and endometrial cancer.
Pilaralisib is a selective, reversible, pan‐class I PI3K inhibitor that has shown preliminary antitumor activity in a phase I study in advanced solid tumors. Pilaralisib is stable in human hepatocytes in vitro (Sanofi data on file); in patients, <0.1% is excreted unchanged in urine, independent of dose [11]. The maximum tolerated dose (MTD) of pilaralisib capsules was established as 600 mg once daily (QD) [11]. The current study aimed to determine the safety and MTD of pilaralisib capsule and tablet formulations administered in combination with paclitaxel and carboplatin in patients with advanced solid tumors.
Fifty‐eight patients were enrolled. Initially, 25 patients were treated with pilaralisib capsules (100–600 mg). After a protocol amendment, newly enrolled patients (n = 33) received a tablet formulation at 200–300 mg QD. A planned expansion cohort in ovarian cancer was not enrolled due to shortage of pilaralisib supply; the expansion cohort was limited to patients with endometrial cancer (n = 16).
The median duration of pilaralisib treatment was 13 weeks (range 1–80); 51.7% of patients received pilaralisib for >12 weeks. Reasons for treatment discontinuation were disease progression (69.0%), adverse events (AEs)/serious AEs (SAEs, 17.2%), investigator's decision other than AEs (6.9%), patient request (5.2%), and death (1.75%).
Six patients (10.3%) had dose‐limiting toxicities (DLTs): 2/6 receiving 400 mg pilaralisib capsules, 1/4 receiving 600 mg capsules, 1/27 receiving 200 mg tablets, and 2/6 receiving 300 mg tablets. The only DLT occurring in more than one patient was rash (two patients, 3.4%). The MTD of pilaralisib tablets administered in combination with 175 mg/m2 paclitaxel and area under the curve (AUC) 6 carboplatin was determined to be 200 mg QD.
The most frequently occurring AEs were consistent with carboplatin, paclitaxel, and pilaralisib known safety profiles (Tables 1–3). No difference in toxicity was observed between capsule and tablet formulations. Grade 3/4 treatment‐related AEs occurred in 87.9% of patients.
Treatment‐related grade ≥3 events of increases in aspartate aminotransferase and gamma glutamyltransferase were reported for two (3.4%) and one patient (1.7%), respectively; no patient met the criteria for Hy's law. Fifteen patients had treatment‐related AEs in the rash grouping; four (6.9%) had grade ≥3 events. Four patients (6.9%) had treatment‐related AEs in the hyperglycemia grouping; two (3.4%) had grade ≥3 events.
Twenty‐eight patients (48.3%) had an SAE, which was considered treatment related in 12 patients (20.7%). Treatment‐related SAEs occurring in more than one patient were mental status changes, neutropenia, rash, and thrombocytopenia (two patients each, 3.4%). Twelve patients (21.0%) had AEs that led to discontinuation of any study treatment, most commonly fatigue (seven patients, 12.1%), nausea, vomiting, and urinary tract infection (six patients each, 10.3%). Three deaths occurred within 30 days of last study drug dose: one due to disease progression and two due to unknown cause.
Pilaralisib was absorbed with a median tmax of 6–11 hours after repeated doses of 100 to 600 mg pilaralisib QD in combination with paclitaxel and carboplatin (Table 4). On cycle 2 day 1, exposure of pilaralisib was within the range of previous data with pilaralisib monotherapy at steady state (Fig. 2) [11]. There was no apparent impact of coadministration of paclitaxel and carboplatin on pilaralisib pharmacokinetics.
Table 4. Pilaralisib pharmacokinetic parameters after treatment with pilaralisib in combination with paclitaxel and carboplatin on cycle 1 day 1 (C1D1) and cycle 2 day 1 (C2D1).

Median (range)
Abbreviations: AUC, area under the curve; Cmax, maximum concentration; C1D1, cycle 1 day 1; C2D1, cycle 2 day 1; CV%, coefficient of variation; PK, pharmacokinetics, SD, standard deviation; tmax, time to maximum concentration.
Figure 2.
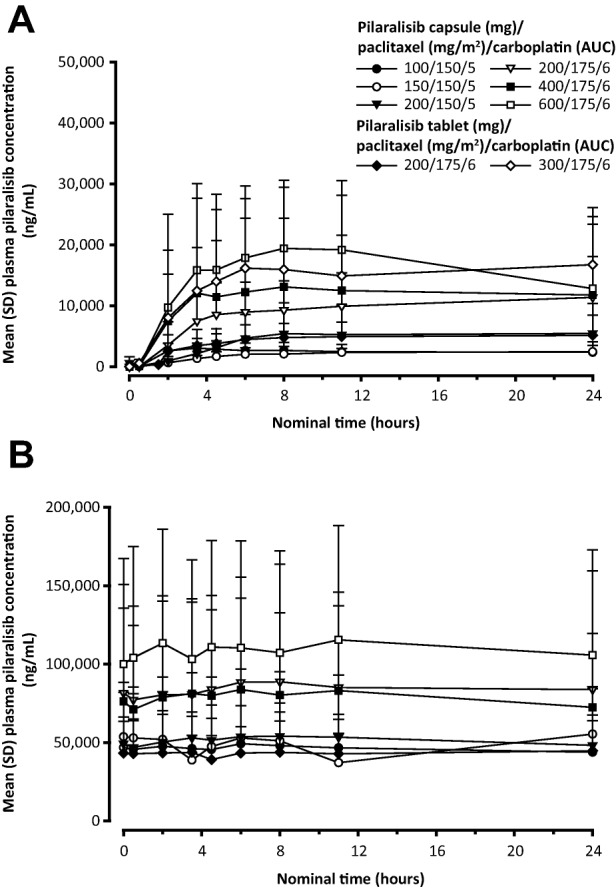
Mean plasma concentration of pilaralisib over time on cycle 1, day 1 (A) and cycle 2, day 1 (B). Abbreviations: AUC, area under the curve; SD, standard deviation.
Modulation of PI3K/mTOR and mitogen‐activated protein kinase (MAPK) pathway biomarkers was assessed in serial tumor biopsies from three patients (Fig. 1 and Table 5). Tumor biopsies from two patients receiving 200 mg pilaralisib capsules showed inhibition of PI3K/mTOR and MAPK pathways and modest reduction of proliferation and induction of apoptosis, whereas no pharmacodynamic impact was observed in biopsies from the third patient tested (200 mg tablets).
Table 5. Summary of pharmacodynamic activity in tumor biopsy sets.

Modulation of PI3K and MAPK pathway biomarkers was assessed in serial tumor biopsies collected from three patients. Levels of PI3K and ERK pathway components (pAKTT308, p4EBP1T70 and pERKT202/Y204) were evaluated using immunofluorescence staining protocols with pixel‐ and intensity‐based quantitative readouts. Proliferation and apoptosis were measured by decrease in Ki67 and fold induction in TUNEL, respectively.
Abbreviations: EBP1, EIF4E‐binding protein‐1; ERK, extracellular signal‐regulated kinase; MAPK, mitogen‐activated protein kinase; PI3K, phosphoinositide 3‐kinase.
Of 52 evaluable patients, best overall response was confirmed partial response (PR) in seven patients (13.5%). Twenty‐two patients (42.3%) had stable disease or a progression‐free period of ≥12 weeks. One of 18 evaluable patients with endometrial cancer had a PR (5.6%). Median progression‐free survival (PFS) was 3.2 months overall and 3.3 months in patients with endometrial cancer. The longest PFS observed was 77.4 weeks in a patient with squamous cell carcinoma of the neck treated with 400 mg capsules. Maximum change in size of target lesions according to prior paclitaxel and carboplatin treatment is shown in Figure 3. No responses occurred in patients who had previously received both paclitaxel and carboplatin.
Figure 3.

Maximum change in target lesions in patients treated with the combination of pilaralisib, paclitaxel, and carboplatin. Bar colors indicate prior treatment with paclitaxel and/or carboplatin. Abbreviations: CR, complete response; PR, partial response.
Despite promising preclinical data, PI3K inhibitors have shown only modest single‐agent activity in solid tumors to date [11], [13], [14], [15], [16]. However, combination regimens could reveal therapeutic utility for these agents. In this study, adding pilaralisib did not appear to enhance the antitumor efficacy of paclitaxel plus carboplatin. This is consistent with the limited efficacy observed with pilaralisib monotherapy in solid tumors [11] and endometrial cancers [12] and findings for other PI3K inhibitors in various solid tumors [13], [14], [15], [16]. Further clinical evaluation is warranted to identify effective combination strategies with PI3K pathway inhibitors.
Figures and Tables
Molecular profiling of tumor tissue was performed in 21 patients in the dose‐escalation phase (Fig. 4A). PIK3CA, KRAS, and BRAF status in cell‐free circulating tumor DNA from peripheral blood sampled at enrollment was also assessed in 17 patients with endometrial cancer (Fig. 4B); four patients (23.54%) had a PIK3CA mutation, including H1047R (two patients), K111E, and R93W (one patient each). No obvious associations between PI3K pathway alterations and responses were observed, consistent with previous studies [11], [12].
Figure 4.

Mutational analysis of tumor tissue and circulating tumor DNA. (A): Dose‐escalation cohort: molecular profiling for gene alterations was performed on archival tumor tissue samples using Sanger sequencing (n = 21), and PTEN protein expression status was evaluated using IHC (n = 12). (B): Endometrial cohort (n = 17). KRAS, PIK3CA, BRAF mutational status at start of pilaralisib treatment (cycle 1, day 1) was assessed in cell‐free circulating tumor DNA obtained from peripheral blood samples using BEAMing assays (Sysmex Inostics). Key: yellow, no alteration detected; blue, gene alteration or altered protein expression (H score <50); grey, status unknown. Abbreviations: EGFR, epidermal growth factor receptor; IHC, immunohistochemistry; PTEN, phosphatase and tensin homolog.
Table 2. Adverse events irrespective of causality occuring in >20% of patients.

Data is shown as n (%).
Abbreviations: AE, adverse event; AUC, area under the curve.
Acknowledgments
This study was funded by Sanofi. The authors received editorial support from Simone Blagg of MediTech Media Ltd., funded by Sanofi. The authors would like to thank Elisa Francesconi for her technical assistance.
Footnotes
ClinicalTrials.gov Identifier: NCT00756847
Sponsor(s): Sanofi
Principal Investigator: Jennifer Wheler
IRB Approved: Yes
Disclosures
Joanne Lager: Sanofi (E); Christelle Castell: Sanofi (E); Li Liu: Sanofi (E); Jason Jiang: Sanofi (E). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Experttestimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1. Goldfinger M, Diaz I, Muggia F. Systemic treatment of endometrial cancer: What is doxorubicin's role? J Clin Oncol 2014;32:2181–2182. [DOI] [PubMed] [Google Scholar]
- 2. Cantley LC. The phosphoinositide 3‐kinase pathway. Science 2002;296:1655–1657. [DOI] [PubMed] [Google Scholar]
- 3. Salvesen HB, Carter SL, Mannelqvist M et al. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc Natl Acad Sci USA 2009;106:4834–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Samuels Y, Wang Z, Bardelli A et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004;304:554. [DOI] [PubMed] [Google Scholar]
- 5. Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene 2008;27:5486–5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3‐kinases as regulators of growth and metabolism. Nat Rev Genet 2006;7:606–619. [DOI] [PubMed] [Google Scholar]
- 7. Liu P, Cheng H, Roberts TM et al. Targeting the phosphoinositide 3‐kinase pathway in cancer. Nat Rev Drug Discov 2009;8:627–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu L, Hofmann J, Lu Y et al. Inhibition of phosphatidylinositol 3'‐kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res 2002;62:1087–1092. [PubMed] [Google Scholar]
- 9. Mondesire WH, Jian W, Zhang H et al. Targeting mammalian target of rapamycin synergistically enhances chemotherapy‐induced cytotoxicity in breast cancer cells. Clin Cancer Res 2004;10:7031–7042. [DOI] [PubMed] [Google Scholar]
- 10. Aissat N, Le Tourneau C, Ghoul A et al. Antiproliferative effects of rapamycin as a single agent and in combination with carboplatin and paclitaxel in head and neck cancer cell lines. Cancer Chemother Pharmacol 2008;62:305–313. [DOI] [PubMed] [Google Scholar]
- 11. Shapiro GI, Rodon J, Bedell C et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan‐class I PI3K inhibitor, in patients with advanced solid tumors. Clin Cancer Res 2014;20:233–245. [DOI] [PubMed] [Google Scholar]
- 12. Matulonis U, Vergote I, Backes F et al. Phase II study of the PI3K inhibitor pilaralisib (SAR245408; XL147) in patients with advanced or recurrent endometrial carcinoma. Gynecol Oncol 2015;136:246–253. [DOI] [PubMed] [Google Scholar]
- 13. Bendell JC, Rodon J, Burris HA et al. Phase I, dose‐escalation study of BKM120, an oral pan‐Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 2012;30:282–290. [DOI] [PubMed] [Google Scholar]
- 14. Markman B, Tabernero J, Krop I et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol‐3‐kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann Oncol 2012;23:2399–2408. [DOI] [PubMed] [Google Scholar]
- 15. Rodon J, Braña I, Siu LL et al. Phase I dose‐escalation and ‐expansion study of buparlisib (BKM120), an oral pan‐Class I PI3K inhibitor, in patients with advanced solid tumors. Invest New Drugs 2014; 32:670–681. [DOI] [PubMed] [Google Scholar]
- 16. Papadopoulos K, Tabernero J, Markman B et al. Phase I safety, pharmacokinetic and pharmacodynamic study of SAR245409 (XL765), a novel, orally administered PI3K/mTOR inhibitor in patients with advanced solid tumors. Clin Cancer Res 2014;20:2445–2456. [DOI] [PubMed] [Google Scholar]