


Abstract
Specific targeting of protein–nucleic acid interactions is an area of current interest, for example, in the regulation of gene-expression. Most transcription factor proteins bind in the DNA major groove; however, we are interested in an approach using small molecules to target the minor groove to control expression by an allosteric mechanism. In an effort to broaden sequence recognition of DNA-targeted-small-molecules to include both A·T and G·C base pairs, we recently discovered that the heterocyclic diamidine, DB2277, forms a strong monomer complex with a DNA sequence containing 5΄-AAAGTTT-3΄. Competition mass spectrometry and surface plasmon resonance identified new monomer complexes, as well as unexpected binding of two DB2277 with certain sequences. Inherent microstructural differences within the experimental DNAs were identified through computational analyses to understand the molecular basis for recognition. These findings emphasize the critical nature of the DNA minor groove microstructure for sequence-specific recognition and offer new avenues to design synthetic small molecules for effective regulation of gene-expression.
INTRODUCTION
Regulation of the binding affinity in protein-nucleic acid complexes is an attractive concept for development of novel therapeutics and agents for control of gene expression (1–4). Several innovative approaches have used small molecules to target disease-associated DNA binding transcription factors or TFs (5–15). Most TFs of interest bind in the major groove (16) and an alternative approach to control expression is to use small molecules to modulate TF activities by interacting directly with the minor groove of DNA where most of these agents bind (17–19). There are two possible mechanisms whereby a minor groove binding compound could disrupt protein-nucleic acid interactions in the major groove to modulate TF association. First, when bound to the minor groove, the small molecule could distort DNA so that the structure of the TF no longer complements its target recognition site, such as an allosteric inhibition mechanism (20,21). Alternatively, direct competition is another possible mechanism which may be significant for TFs that position side chains into or near the DNA minor groove. By knowing how small molecule inhibitors recognize DNA, it is possible to preemptively block TF binding to DNA. Our main goal is to understand, in detail, the minor groove binding variations of synthetic small molecules with different DNA sequences and how they vary with sequence-dependent DNA structure.
Small molecules that bind in the minor groove of DNA have been validated for this approach from studies using synthetic polyamides (22–24). However, polyamides have limitations such as aggregation and cell uptake and a wider variety of agents is needed for diverse biological systems (25,26). We are approaching this problem with a class of sequence-specific, DNA-targeted minor groove binders based on a heterocyclic cation design since these compounds have shown good cell uptake and biological properties through human clinical studies (27,28). Few non-polyamide minor groove agents, including heterocyclic diamidines, have been identified to selectively recognize mixed, A·T and G·C base pair-containing DNA sequences (29,30). This constitutes a significant barrier to progress in the area of designed synthetic agents for the disruption of TF-DNA complexes. To interact with the edges of A·T base pairs in the minor groove, compounds must have hydrogen bond donor groups for the thymidine carbonyl and an N3 of adenine acceptor. To recognize a G·C base pair, the compound must have an acceptor to hydrogen bond to the guanine NH2 group. It is also critical that a successful small molecule have the appropriate shape and charge to complement the DNA minor groove (31,32).
A synthetic effort has led to cationic diamidines that strongly and selectively recognize the minor groove in mixed-site DNA sequences (33). The lead compound in this development is DB2277, which contains a nitrogen hydrogen bond acceptor in an aza-benzimidazole (Figure 1). Strong binding of DB2277 requires the 2-amino group of guanine and suggests an aza-N·G-NH2 hydrogen bond. These observations show that DB2277 binds best to mixed-site sequences with a single G·C base pair flanked by A·T base pair sites (34). Key questions in the design effort for new mixed-sequence minor groove compounds that recognize G·C base pairs with flanking A·T base pairs: In addition to monomer binding to recognize a single G·C base pair, can the compound form dimers to recognize two G·C base pair sequences? What is the effect of the flanking A·T base pairs? How could this influence binding affinity?
Figure 1.

(A,B) Structures of DB2277 and (C) DNA sequences used to screen for binding with the DB2277 using ESI-MS.
To address these questions, a systematic set of DNAs were tested with DB2277 and their interactions, affinities and stoichiometries were investigated. The composition of A·T base pairs was maintained (i.e. number of A·T base pairs per binding site) to see how interactions vary due to A·T base pair order with one and two, central G·C base pairs. Electrospray ionization mass spectrometry (ESI-MS) and surface plasmon resonance (SPR) were used to examine stoichiometry and binding behavior for the DNA–DB2277 complexes. Significant variations in affinity and stoichiometry for binding of DB2277 to the different, closely related sequences were observed. To help understand these sequence-dependent variations, extensive molecular dynamics (MD) simulations were conducted to provide specific details regarding the structural properties intrinsic to each DNA sequence that govern small molecule recognition. Large differences in the local DNA structure were observed with these closely related sequences and the differences correlate with observed differences in DB2277 binding affinity and stoichiometry. The results described here provide new and fundamental information in design research for DNA sequence-specific recognition and structural complementarity between a small molecule and its target site.
MATERIALS AND METHODS
Compound and DNAs
Stock solutions of 1.5 mM DB2277 were prepared in ddH2O and stored at 4°C. DNA sequences were from Integrated DNA Technologies and were dissolved in the appropriate experimental buffer. All buffers were filtered and degassed. See Supplementary Data for more details.
Electrospray ionization mass spectrometry
DNAs were combined (10 μM each) with DB2277 in 150 mM NH4OAc buffer. Samples were scanned from m/z 500–3000 in negative ion mode at a rate of 5 μl·min−1 on a Waters Micromass ESI-Q-ToF spectrometer and analyzed with MassLynx 4.1 software. See Supplementary Data for more details.
Surface plasmon resonance
SPR experiments used a Biacore T200 and the Biacore T200 Evaluation Software. Samples in 50 mM Tris–HCl buffer (pH 7.4) were injected over the sensor chip at a rate of 100 μl·min−1 and dissociated with buffer flow, followed by surface regeneration and rinsing with experimental buffer. Additional details regarding fitting and chip preparation can be found in the Supplementary Data.
Molecular dynamics simulations
DNA sequences for simulations were built in AMBER 14. Systems were solvated with TIP3P water and neutralized to reach a salt concentration of 150 mM NaCl. Systems were relaxed and heated with harmonic restraints enforced on heavy atoms of the residues. Restraints were released with a 2 fs time step, totaling 500 ps. Production level simulations were extended to 200 ns with trajectory snapshots saved every 1 ps. Details regarding protocol and analyses can be found in the Supplementary Data.
RESULTS AND DISCUSSION
Competition electrospray ionization mass spectrometry identifies new interactions
Competition ESI-MS can simultaneously identify affinity, stoichiometry and cooperativity in multiple DNA-small molecule interactions (35). The molecular weights of all possible DNA species are controlled through sequence modifications, making each sequence distinguishable (Figure 1C). The target binding sites of the tested DNA sequences are listed as DNA 1–9 in Figure 2. DNA 2, which contains the AAAAGTTTT target site, was used as a reference to compare binding due to the extensive data available for DB2277 with that and similar sequences. Target binding sites were designed to test the interactions of the compound with a range of closely related DNA sequences. Each sequence has two sets of four A·T base pairs as AAAA, AATT or ATAT. Only DNA 1 lacks a central G·C base pair. Two categories of mixed sequences are grouped with either one or two G·C base pairs.
Figure 2.

(A) DNA sequences in the absence of DB2277 with m/z 1580–1,780 signifying -6 charged species. Molar concentration ratio of [DB2277] to [DNA] expressed as: (B) [0.5 to 1], (C) [1 to 1], (D) [1.5 to 1], (E) [2 to 1], (F) [2.5 to 1], and (G) [3 to 1]. Concentrations of DNA were fixed at 5 μM. Unbound DNA, 1 to 1, and 2 to 1 complexes labelled above respective boxes.
Figure 2 illustrates the changes in relative peak intensity for DB2277–DNA complexes. Changes in the relative intensities are based on the binding of DB2277 to DNA and compared to a reference sequence present in the sample. In the presence of DB2277, peak intensities for free DNA disappear while intensities for DB2277–DNA complexes emerge. For instance, in Figure 2B, half of the unbound AAAAGTTTT (DNA 2) is present at a concentration molar ratio of [0.5 to 1] as well as ≈ 50% of 1:1 complex. As the concentration of DB2277 is increased, unbound AAAAGTTTT decreases with a concurrent increase in 1:1 binding. Based on these results, sequences which contain A-tracts (DNA 1–3, 9) prefer 1:1 binding. The −6 charged species were used for illustrative purposes in Figure 2 since they were the most abundant of the multiply charged species. Relative binding affinities were measured using deconvoluted spectra which takes into account all multiply charged species and can be found in the Supplementary Data (Supplementary Figure S1) for a simple comparison of the titration ratios.
Sequences in the AATT subcategory (DNA 5–8) allow us to examine the transition from A-tracts to sites with an ApT base pair step. Surprisingly, for this closely related sequence, the ESI-MS results show that 2:1 binding is strongly preferred over 1:1 complex formation for AATT DNAs. Finding such preference was especially interesting since many minor groove binding compounds cannot differentiate among A·T base pair sites (36,37). Based on the results from AATT sequences, one might expect to find strong 2:1 complexes formed between DB2277 and ATAT sequences since alternating A·T sites have wide minor grooves similar to AATT. In another surprise, the compound preferentially formed a 1:1 complex with ATATGCATAT (Figure 2). DB2277 can bind tightly to sites with a single G base flanked by A-tract sites (31); however, little is known regarding sequences with two G·C base pairs. In summary, the ESI-MS results show that monomer complexes are the preferred systems for AAAA and ATAT base pair sites that flank a core G·C base pair whereas 2:1 complexes are preferred for sequences containing AATT sites.
Surface plasmon resonance confirms Sequence-Specific behavior identified by ESI-MS
Surface plasmon resonance (SPR) is a powerful method to define the thermodynamic and kinetic properties of biomolecular interactions (38,39). In our experiments, increasing concentrations of DB2277 were injected over a set of immobilized DNA sequences. Binding curves for DB2277 are shown in Figure 3. Binding affinities for steady-state equilibrium and kinetics-fitted analyses are compared in Table 1 as well as the binding on and off-rates. The unexpected 2:1 binding of AATTGCAATT was of considerable interest, especially since it contains two central G·C base pairs. To directly compare flanking base pair sequence and its role in small molecule recognition, binding of DB2277 was measured with AATTGCAATT, as well as with AAAAGCTTTT and ATATGCATAT since all three sequences contain the same G·C base pair core. A representative sensorgram of DB2277 binding to AATTGCAATT is shown in Figure 3A. Results of AATTGCAATT binding from SPR are in direct agreement with those obtained from ESI-MS with two binding sites, KD1 and KD2, near 15 and 30 nM, respectively. Also in agreement, a strong 1:1 complex for DB2277 and AAAAGCTTTT was observed (KD ≈ 50 nM). With the ATATGCATAT sequence, a weaker 1:1 complex was formed (KD ≈ 100 nM) and a second, much weaker 2:1 complex was detected at high compound concentrations, which were well above the first KD. These results are in agreement with those obtained by ESI-MS for preferred 1:1 binding.
Figure 3.

(A) SPR sensorgram of AATTGCAATT binding DB2277. Injected concentrations of DB2277 shown are 5, 10, 30, 70 and 200 nM. (B) Steady-state fits for binding with AAAAGCTTTT, AATTGCAATT and ATATGCATAT fit using a two site binding model. (C) Competition SPR steady-state fits of competitor DNA sequences AAAAGTTTT, AATTGAATT and ATATGATAT fit using a one-site binding model.
Table 1. Comparison of kinetics-fitted and steady-state equilibrium binding constants (KD × 10−9 M).
| Kinetic rates | Dissociation constants | |||
|---|---|---|---|---|
| ka (10+6·M−1 s−1) | kd (10−1·s−1) | Kinetics-fit (10−9 M) | Steady-state (10−9 M) | |
| AAAAGCTTTT | 5.3 ± 0.7 | 2.4 ± 0.5 | 44.8 ± 4.7 | 49.6 ± 1.1 |
| ATATGCATAT | 1.6 ± 0.2 | 1.4 ± 0.2 | 87.8 ± 3.8 | 120.3 ± 5.0 |
| AATTGCAATT | ||||
| KD1 | 1.0 ± 0.2 | 0.1 ± 0.01 | 14.1 ± 3.1 | 12.5 ± 1.3 |
| KD2 | 0.5 ± 0.08 | 0.2 ± 0.02 | 35.5 ± 3.0 | 33.3 ± 3.8 |
| AAAAGTTTT | ND | ND | ND | a 4.4 ± 0.7 |
| ATATGATAT | ND | ND | ND | a 50.8 ± 16.1 |
| AATTGAATT | a 41.7 ± 6.1 | |||
| KD12 | 4.4 ± 1.0 | 1.3 ± 0.4 | 28.7 ± 1.6 | b, c 44.6 ± 2.6 |
| KD1 | 3.1 ± 1.2 | 1.6 ± 0.7 | 51.1 ± 2.6 | d 25.0 ± 2.3 |
| KD2 | d 57.0 ± 5.9 | |||
| KD12 |
b, d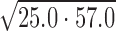 ≈ 39.2 ≈ 39.2 |
|||
NDNot determined.
aDetermined using competition SPR.
bKD12 value determined by 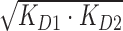 with KD1 and KD2 values obtained through direct-binding SPR.
with KD1 and KD2 values obtained through direct-binding SPR.
cValue determined by direct-binding SPR and fit with one-site binding model.
dValue determined by direct-binding SPR and fit with two-site binding model.
Kinetic rates and fits were determined using direct-binding SPR. Steady-state fits were compared using both direct-binding and competition binding SPR. Sequences with multiple binding constants are listed as KD1 and KD2.
Competition SPR (40) was used to measure the binding of three single G·C base pair sequences against the original immobilized DNA. Similar to direct-binding SPR experiments, the DB2277 was added to the sample solution and the observed response at steady-state is plotted to determine the binding constant. In the competition SPR experiments, the compound was held at a fixed concentration while the competing DNA was added to the sample solution. The observed response, however, decreased as concentrations of competing DNA were increased, which resulted in less available free compound in solution. Calculated dissociation constants of AAAAGTTTT, AATTGAATT and ATATGATAT were determined from Figure 3C and are listed in Table 1. The observed response (RUobs) was plotted as a function of competitor DNA concentration (38,39) and fit to a 1:1 binding model using Equation 2 (see Supplementary Data, Methods and Materials).
Using competition SPR, the strongest 1:1 complex formed within this DNA series was AAAAGTTTT (KD ≈ 4 nM) as expected from ESI-MS and literature (33,34). A 10-fold weaker complex was formed with the compound and AAAAGCTTTT. Results by competition SPR for AATTGAATT show it formed a strong 1:1 complex with a binding constant of 40 nM. Since its two G·C counterpart (i.e. AATTGCAATT) forms both 1:1 and 2:1 complexes, a direct-binding SPR approach was also used to compare the binding affinities and determine if multiple binding modes occur. In this experiment, AATTGAATT forms both 1:1 and 2:1 complexes with KD values near 25 and 60 nM, respectively. Competition SPR results were fit using a one-site binding model; however, when more than one compound binds (e.g. AATTGAATT) the calculated value for two binding constants is KD12 or 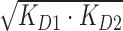 ). Various forms of analyzing DB2277 with AATTGAATT were compared, such as direct-binding SPR, competition SPR, and using one and two-site binding models. Likewise, a comparison of kinetics-fitted and steady-state binding constants, determined by the two SPR methods, are in excellent agreement for AATTGAATT (Table 1).
). Various forms of analyzing DB2277 with AATTGAATT were compared, such as direct-binding SPR, competition SPR, and using one and two-site binding models. Likewise, a comparison of kinetics-fitted and steady-state binding constants, determined by the two SPR methods, are in excellent agreement for AATTGAATT (Table 1).
Interestingly, association rate constants for 1:1 binding of DB2277 to DNA are similar for all sequences (ka ≈ 106 M−1 s−1) whereas association rate constants are comparatively slower for the second DB2277 molecule binding with AATTGCAATT or AATTGAATT. On the other hand, the second off-rate for DB2277 is faster than the first off-rate. The calculated binding constants of AATTGAATT are similar to those for AATTGCAATT (Table 1) and further suggest a binding mechanism for the compound unique to the AATT sequences. Results obtained by ESI-MS and SPR are in excellent agreement and indicate binding of DB2277 differs when the order of flanking A·T base pairs is varied. Clearly, the exact order of the flanking bases influences binding of the test compound since both global and local structure of the DNA are contingent on base pair sequence. In order to probe the basis of these binding differences in molecular detail, we turned to molecular dynamics simulations.
Molecular dynamics identifies microstructural differences in the experimental DNAs
Honig et al. have shown that the local structure within the DNA minor groove can depend on base pair sequence (41–44). Such microstructural variations may explain why binding of DB2277 varies greatly even though base pair composition is maintained. To elucidate how DNA microstructure may influence small molecule binding, extensive molecular dynamics (MD) simulations of a systematic set of closely related experimental DNA sequences with central G·C base pairs flanked by A·T base pairs of different sequence were carried out. Variations of the resulting structures in the MD trajectories were analyzed with Curves+ (45) to predict their roles in DB2277–DNA recognition. We measured helical parameters with specific emphasis on minor groove width and depth (Supplementary Figures S2–S4). Additionally, simulations for select sequences were repeated and analyzed over 100 ns intervals to validate structure and flexibility convergences (Supplementary Figures S5, S6). This is the first highly detailed structural analysis on the effects of systematic changes in DNA sequence focused on one and two core G·C base pairs flanked by varying sequences of A·T-rich sites over long trajectories for 200 ns (46,47).
Varying the A·T flanking sequence around the core G·C produced a surprisingly large deviation in minor groove widths and depths. Comparison of the flanking sequences reveals unique groove width variations. For example, the 2D contour histogram for the minor groove width of AAAAGTTTT (Figure 4A) indicates a high probability of adopting a narrow (4.5 Å) and deep (5.0 Å) minor groove with little variation along the target binding site (see also Supplementary Data for molecular model). This sequence has the highest binding affinity of all the DNA sequences investigated and is explained by the inherently narrow and deep groove pre-formed for energetically favorable binding of the compound. DB2277 and similar compounds thus bind and fit well into A-tract flanking sequences in strong 1:1 complexes.
Figure 4.

2D contour histograms of minor groove width for (A) AAAAGTTTT, (B) AATTGAATT, (C) ATATGATAT, (D) AAAAGCTTTT, (E) AATTGCAATT and (F) ATATGCATAT, respectively. The color gradient indicates increasing probability (navy to red) of distance in Angstroms (in Å) for each base pair.
In contrast, AATTGAATT has a strong preference toward maintaining a narrow and deep groove at the terminal AA·TT regions. The groove width increases to 8.0 Å, becoming much wider than AAAAGTTTT, at the central G·C base pair of the sequence (Figure 4B). The depth of the sequence at the central G·C is also less than the AAAAGTTTT sequence. Due to the change in groove width and depth, the central G·C therefore provides a less favorable binding site for DB2277 monomer binding. The competition SPR results have, thus, revealed sequence-specific variations in both stoichiometry and affinity. We speculate that a single DB2277 binds first with AATTGAATT and because of the wider groove and variable depth, it is possible that two molecules of DB2277 can fit the optimum groove structure by staggered stacking at the central G·C base pair.
Finally, within the single G·C base pair series, the widest and most shallow measurable groove occurs in the ATATGATAT sequence throughout the course of the trajectory. Unlike the previous two sequences, it is energetically unfavorable for ATATGATAT to exist in a deep and narrow groove conformation. Instead, there is a strong preference for the groove to remain wide (8.0 Å) and shallow (4.0 Å). The MD described combination of an intrinsically wide groove and shallow depth would be expected to bind two DB2277 molecules. Our findings, however, suggest the DNA is ill-suited for binding a curved, planar small molecule such as DB2277. Instead, the wide and shallow groove must undergo an induced fit to bind DB2277 with a high deformation energy penalty.
Sequences with two G·C base pairs were next simulated to better understand the structural (dis)similarities among sequences with one or two central G·C base pairs in the target binding site. Altering the core from G to GC increases the overall probability of adopting a wide and shallow groove. For instance, a contour 2D histogram of AAAAGCTTTT in Figure 4D indicates a higher probability of the groove width expanding to 12.0 Å compared to AAAAGTTTT. It is also less probable for AAAAGCTTTT to maintain a deep groove, but rather becomes shallowest with the additional G·C base pair in its core. This observed decrease in probability of a narrow and deep minor groove may help to explain the weaker binding found by DB2277 with AAAAGCTTTT compared to binding with AAAAGTTTT (KD = 49.6 ± 1.1 nM and KD = 4.4 ± 0.7 nM, respectively).
A wide groove also exists at the GC region in AATTGCAATT (Figure 4E), and is more probable than its AATTGAATT counterpart. In addition to the widened groove, a correlated decrease in groove depth also occurs at this region (Supplementary Figure S4). Comparing both histograms, it is evident that the shallowest region occurs at the GC step and is much more pronounced in the AATTGCAATT sequence over AATTGAATT. Interestingly, altering the central G·C base pairs in the sequence to create AATTGGAATT reveals little change in width or depth of the minor groove (Supplementary Figures S3 and S4). Little change in groove width and depth between AATTGCAATT and AATTGGAATT explains why little detectable difference occurred for binding of DB2277 to these two sequences. The MD results therefore provide a rationale for why the AATT sequences are favorable for a 2:1 complex in both ESI-MS and SPR.
Like AATTGCAATT, the ATATGCATAT sequence also has a higher probability of existing in a wider state than its counterpart ATATGATAT (Figure 4F). Changing G to GC in the core of the alternating A·T base pair flanking sequences stabilizes a wider minor groove. This stability is even more evident when looking at the minor groove depth histograms (Supplementary Figure S4). For the sequence ATATGCATAT, there is a clear preference for shallow groove depth throughout the entire sequence and unlike ATATGATAT, does not break at its core G·C region. This would, therefore, indicate that a wide and stable minor groove within the two GC sequence is consistent for ATATGCATAT in having the lowest binding affinity for DB2277. Upon binding a single DB2277 molecule, the flexibility of the ATAT sequence allows it to favorably constrict to a narrow groove, rather than binding two molecules in an unfavorable wider groove conformation. These MD simulations complement ESI-MS and SPR studies and indicate that DB2277 binding should be more favorable where the minor groove is intrinsically narrow and deep and is related to the pre-organized groove width prior to binding the compound. These local structural differences also influence how and where the molecule will bind in the minor groove. It is somewhat surprising that the large diversity of microstructural characteristics, such as groove width, observed for the minor groove are not found in the major groove, which has a much more constant structure (Supplementary Figures S7 and S8).
A comparison of the sequences with matched flanking sites (AAAAGTTTT versus AAAAGCTTTT) shows little variation in local DNA structure (Figure 4 and Supplementary Figure S2). On the other hand, a comparison of unmatched flanking sequences, for example AAAA to AATT, indicates a larger variation in microstructure which in turn governs binding stoichiometry. For sequences with AAAA sites, there are similar distributions of a well-maintained narrow groove (4.5–5.0 Å) along the target site in the region of AAAAGTTTT and AAAAGCTTTT. Likewise, there is a consistently wide groove for both ATATGATAT and ATATGCATAT. With a range of 7.0–8.5 Å, the narrowest regions occur along the ends of the target site while the widest portions are at the T to G/C transitions (i.e. TpG and TpC of the complementary strand). Alternatively, large groove width variations within the target site occur for both AATTGAATT and AATTGCAATT. Specifically in AATT regions at the ApT base steps, there exist large differences in minor groove width (Δwidth ≈ 3.5 Å) compared to variations of Δwidth < 2 Å among AAAA and ATAT flanks. This type of intra-target-site variation is significant to the AATT sequences resulting in two bound DB2277 molecules. Current observations of a monomeric DB2277–AATT system would suggest that a wide minor groove at the core G or G·C base pair region, adjacent to a narrow AATT site, may support staggered stacking of two DB2277 molecules at the G/C core with the unstacked ends of DB2277 in the AATT sites.
The sequence-dependency of other helical parameters for the DNA was also compared. Differences in propeller twist were very informative and the averages for each sequence are shown in Figure 5 (see also Supplementary Figure S9, Tables S1 and S2). For all sequences there is characteristic ‘W’ shape to each of the curves but the range varies. For instance, the degree of propeller twist for AAAAGTTTT is large and quite constant along the target site. On the other hand, AATTGAATT has a much wider range of propeller twist along the target binding site. Interestingly, the AATTGAATT sequence is closely related to the AATTGCAATT sequence, which may partially explain the similar binding measured for both sequences by SPR (Table 1). In general, sequences with consecutive A or T bases (e.g. AAAA) have steric clash due to CH3 groups of thymidine (48). Propeller twists in A·T base pairs form bifurcated hydrogen bonds between the NH2 of adenosine to O4 of the adjacent thymidine on the complementary strand (Figure 5A) which reduces the amount of fluctuation. Likewise, sequences with AATT have two consecutive A·T base pairs and are also likely to form bifurcated hydrogen bonds; however, two fewer possible hydrogen bonds likely increases the structural flexibility as shown by the break at the G or GC core in the minor groove width comparisons. Alternatively, sequences with alternating A·T base pairs do not experience steric clash of the CH3 groups and are therefore more flexible with lower propeller twist.
Figure 5.

(A) Cartoon of bifurcated hydrogen bond network within consecutive AAAA bases. Most probable propeller twist per base pair for (B) one G·C and (C) two G·C base pair sequences.
In addition to propeller twist of our sequences, other base step parameters were evaluated for influence on DNA structure. Comparing the single G-containing sequences, only nominal differences occur in all of the parameters except for roll (Supplementary Figure S10). Roll increases after thymidine-purine steps in AATTGAATT and ATATGATAT. On the other hand, comparison of the two central GC sequences shows interesting results within helical parameters. Significant increases are seen at TpA steps for slide, rise and twist while other sequences are constant. With shift, both up and down changes are seen in ATATGCATAT (Supplementary Figure S10) while no consistent patterns occur within the set of sequences. Increases also occur in slide at TpA steps for ATATGCATAT. Additionally, tilt and roll decrease as AATTGAATT transitions to AATTGCAATT at the CpA step. It is interesting to note how the addition of the second core G·C base pair causes ATATGCATAT to become an outlier compared to the other sequences. Specifically, the marked deviations that occur at the pyrimidine-purine steps exhibit the most dramatic changes in helical parameters. Propeller twists, rolls, and tilts likely compensate for each other in AAAA·TTTT and AATT sites due to the bifurcated hydrogen bonding networks.
The observable changes in ATATGCATAT for every measurable parameter is likely from an inherent flexibility due to the alternating 5΄ to 3΄ purine-pyrimidine steps that can perturb canonical B-DNA conformations (49). The ATATGCATAT sequence is the only consistently alternating purine-pyrimidine sequence within this series and is the sequence with the lowest binding affinity for DB2277. There is a high degree of dynamic helical bending in ATATGCATAT compared to AAAAGTTTT. Early reports by Charney and co-workers demonstrated alternating poly (dA·dT) sequences are nearly twice as flexible as ‘random’ DNA (50). Therefore, the apparent increased flexibility and dynamic bending of our alternating purine-pyrimidine sequences can explain the relatively poor binding of DB2277 with ATATGCATAT and ATATGATAT. These findings suggest that for ATATGCATAT, no single base pair parameter contributes substantially to minor groove width or depth. Instead, minor groove characteristics are a collective contribution of intra and inter-base pair parameters.
Simulations of the 1:1, monomeric complexes were next performed for DB2277 binding with single G·C sequences. Because the DNA sequences are asymmetric about the DB2277 binding site (i.e. 5΄-AAAAGTTTT-3΄ vs. 5΄-AAAACTTTT-3΄) and because of asymmetry in the small molecule, DB2277 was oriented in both the 5΄ to 3΄ and the 3΄ to 5΄ directions, totalling six simulations. For all orientations (six total), two-dimensional contour histograms of the simulated complexes are shown in Figure 6. To our surprise, the minor groove width distributions for the 1:1 complexes changed markedly and were nearly identical for all the simulated orientations. Upon binding DB2277, the preferred sequence, AAAAGTTTT, undergoes very small change in minor groove width. In both AATTGAATT and ATATGATAT simulations, the minor groove becomes constricted at the central G·C base pair, indicative of an induced fit recognition mechanism. This phenomenon is especially prevalent in ATATGATAT, yielding a Δwidth ≈ 3.5 Å. The comparison of effects for AAAAGTTTT and ATATGATAT is interesting since the intrinsic minor groove structure of AAAAGTTTT did not change much upon binding DB2277, in contrast to ATATGATAT. This phenomenon is worth noting since AAAAGTTTT showed the highest affinity for DB2277 while ATATGATAT had the lowest affinity within this series. Regardless of sequence, in the presence of DB2277, minor groove width conforms to the same pattern at its target binding site. Therefore, sequences that the start (free) and end (bound) most similarly have the most favorable binding as a result of lower deformation energy of the DNA. This implies that the sequence with the highest binding affinity for our test compound already has a shape complementary to the small molecule and further suggests that inherent microstructure of the DNA strongly influences binding affinity.
Figure 6.

2D contour histograms of minor groove width for 1:1, DB2277–DNA complexes. (A) AAAAGTTTT, (B) AATTGAATT and (C) ATATGATAT. The color gradient indicates increasing probability (navy to red) of distance in Angstroms (in Å) for each base pair.
CONCLUSIONS
In this study, the molecular basis for sequence-specific binding by a synthetic minor groove binder is explained by inherent differences in the local DNA structure for an investigated set of sequences. This is the first reported use of competition mass spectrometry to identify unique DNA–ligand interactions that are explained by highly detailed, long time-scale molecular dynamics simulations. Our current understanding suggests that planar, synthetic small molecules, such as our test compound, bind best to sequences with a narrow and deep minor groove. Increased flexibility in specific sequences contributes to a wide and shallow groove that is unfavorable for strong 1:1 binding, while unexpected 2:1 binding of the compound for certain sequences further illustrates the sequence-dependent, microstructural variations within DNA. These findings emphasize the need for structural complementarity between the shape of a designed small molecule binder and the local structure of the DNA minor groove, and is therefore critical for understanding small molecule, sequence-specific recognition of DNA. Such site-specific recognition would prove useful for selectively targeting and modulating transcription factor activity and can become a powerful therapeutic tool for treating genetic-related diseases.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Siming Wang for helpful discussions, Prof. David W. Boykin and Dr Yun Chai for helpful discussions and providing DB2277, and to Dr Ananya Paul for validation of competition SPR.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [GM111749 to W.D.W., GM110387 to I.I.]; National Science Foundation CAREER award [MCB-1149521 to I.I.]; National Science Foundation XSEDE allocation [CHE110042 to I.I.] (in part); U.S. Department of Energy Office of Science [DE-AC02-05CH1231 to I.I.]; National Energy Research Scientific Computing Center (NERSC) (to I.I.); Molecular Basis of Disease Area of Focus fellowship (to S.L.T.); Dean's fellowship (to E.K.C.). Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1.Babu M.M., Luscombe N.M., Aravind L., Gerstein M., Teichmann S.A.. Structure and evolution of transcriptional regulatory networks. Curr. Opin. Struct. Biol. 2004; 14:283–291. [DOI] [PubMed] [Google Scholar]
- 2.Bouhlel M.A., Lambert M., David-Cordonnier M.H.. Targeting transcription factor binding to DNA by competing with DNA binders as an approach for controlling gene expression. Curr. Top. Med. Chem. 2015; 15:1323–1358. [DOI] [PubMed] [Google Scholar]
- 3.Chen B.J., Wu Y.L., Tanaka Y., Zhang W.. Small molecules targeting c-Myc oncogene: promising anti-cancer therapeutics. Int. J. Biol. Sci. 2014; 10:1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mann M.J., Dzau V.J.. Therapeutic applications of transcription factor decoy oligonucleotides. J. Clin. Invest. 2000; 106:1071–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arndt H.D. Small molecule modulators of transcription. Angew. Chem. Int. Ed. Engl. 2006; 45:4552–4560. [DOI] [PubMed] [Google Scholar]
- 6.Backus K.M., Correia B.E., Lum K.M., Forli S., Horning B.D., González-Páez G.E., Chatterjee S., Lanning B.R., Teijaro J.R., Olson A.J. et al. . Proteome-wide covalent ligand discovery in native biological systems. Nature. 2016; 534:570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Darnell J.E., Jr Transcription factors as targets for cancer therapy. Nat. Rev. Cancer. 2002; 2:740–749. [DOI] [PubMed] [Google Scholar]
- 8.Koehler A.N. A complex task? Direct modulation of transcription factors with small molecules. Curr. Opin. Chem. Biol. 2010; 14:331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petrovic V., Costa R.H., Lau L.F., Raychaudhuri P., Tyner A.L.. Negative regulation of the oncogenic transcription factor FoxM1 by thiazolidinediones and mithramycin. Cancer Biol. Ther. 2010; 9:1008–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rahim S., Uren A.. Emergence of ETS transcription factors as diagnostic tools and therapeutic targets in prostate cancer. Am. J. Transl. Res. 2013; 5:254–268. [PMC free article] [PubMed] [Google Scholar]
- 11.Raskatov J.A., Meier J.L., Puckett J.W., Yang F., Ramakrishnan P, Dervan P.B.. Modulation of NF-κB-dependent gene transcription using programmable DNA minor groove binders. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:1023–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shoulders M.D., Ryno L.M., Cooley C.B., Kelly J.W., Wiseman R.L.. Broadly applicable methodology for the rapid and dosable small molecule-mediated regulation of transcription factors in human cells. J. Am. Chem. Soc. 2013; 5:8129–8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taniguchi M., Fujiwara K., Nakai Y., Ozaki T., Koshikawa N., Toshio K., Kataba M., Oguni A., Matsuda H., Yoshida Y. et al. . Inhibition of malignant phenotypes of human osteosarcoma cells by a gene silencer, a pyrrole-imidazole polyamide, which targets an Ebox motif. FEBS Open. Biol. 2014; 4:328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tian H., Qian G.W., Li W., Chen F.F., Di J.H., Zhang B.F., Pei D.S., Ma P., Zheng J.N.. A critical role of Sp1 transcription factor in regulating the human Ki-67 gene expression. Tumour Biol. 2011; 32:273–283. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y., Cesena T.I., Ohnishi Y., Burger-Caplan R., Lam V., Kirchhoff P.D., Larsen S.D., Larsen M.J., Nestler E.J., Rudenko G.. Small molecule screening identifies regulators of the transcription factor ΔFosB. ACS Chem. Neurosci. 2012; 18:546–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharrocks A.D. The ETS-domain transcription factor family. Nat. Rev. Mol. Cell Biol. 2001; 2:827–837. [DOI] [PubMed] [Google Scholar]
- 17.Grossman S.A., Ye X., Peereboom D., Rosenfeld M.R., Mikkelsen T., Supko J.G., Desideri S.. Phase I study of terameprocol in patients with recurrent high-grade glioma. Neurol. Oncol. 2012; 14:511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kong D., Park E.J., Stephen A.G., Calvani M., Cardellina J.H., Monks A., Fisher R.J., Shoemaker R.H., Melillo G.. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005; 65:9047–9055. [DOI] [PubMed] [Google Scholar]
- 19.Nickols N.G., Jacobs C.S., Farkas M.E., Dervan P.B.. Modulating hypoxia-inducible transcription by disrupting the HIF-1-DNA interface. ACS Chem. Biol. 2007; 2:561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunt R.A., Munde M., Kumar A., Ismail M.A., Farahat A.A., Arafa R.K., Say M., Batista-Parra A., Tevis D., Boykin D.W. et al. . Induced topological changes in DNA complexes: Influence of DNA sequences and small molecule structures. Nucleic Acids Res. 2011; 39:4265–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang S., Munde M., Wang S., Wilson W.D.. Minor groove to major groove, an unusual DNA sequence-dependent change in bend directionality by a distamycin dimer. Biochemistry. 2011; 50:7674–7683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buchmueller K.L., Taherbhai Z., Howard C.M., Bailey S.L., Nguyen B., O'Hare C., Hochhauser D., Hartley J.A., Wilson W.D., Lee M.. Design of a hairpin polyamide, ZT65B, for targeting the inverted CCAATT box (ICB) site in the multidrug resistant (MDR1) gene. ChemBioChem. 2005; 6:2305–2311. [DOI] [PubMed] [Google Scholar]
- 23.Kiakos K., Pett L., Satam V., Patil P., Hochhauser D., Lee M., Hartley J.A.. Nuclear localization and gene expression modulation by a fluorescent sequence-selective p-anisyl-benzimidazolecarboxamido imidazole-pyrrole polyamide. Chem. Biol. 2015; 22:862–875. [DOI] [PubMed] [Google Scholar]
- 24.Lai Y.M., Fukuda N., Ueno T., Matsuda H., Saito S., Matsumoto K., Ayame H., Bando T., Sugiyama H., Mugishima H. et al. . Synthetic pyrrole-imidazole polyamide inhibits expression of the human transforming growth factor-beta1 gene. Pharmacol. Exp. Ther. 2005; 315:571–575. [DOI] [PubMed] [Google Scholar]
- 25.Crowley K.S., Phillion D.P., Woodard S.S., Schweitzer B.A., Singh M., Shabany H., Burnette B., Hippenmeyer P., Heitmeier M., Bashkin J.K.. Controlling the intracellular localization of fluorescent polyamide analogues in cultured cells. Bioorg. Med. Chem. Lett. 2003; 13:1565. [DOI] [PubMed] [Google Scholar]
- 26.Wang S., Aston K., Koeller K.J., Harris G.D. Jr, Rath N.P., Bashkin J.K., Wilson W.D.. Modulation of DNA-polyamide interaction by β-alanine substitutions: A study of positional effects on binding affinity, kinetics and thermodynamics. Org. Biomol. Chem. 2014; 12:7523–7536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lansiaux A., Dassonneville L., Facompré M., Kumar A., Stephens C.E., Bajic M., Tanious F., Wilson W.D., Boykin D.W., Bailly C.. Distribution of furamidine analogues in tumor cells: Influence of the number of positive charges. Med. Chem. 2002; 45:1994–2002. [DOI] [PubMed] [Google Scholar]
- 28.Nyunt M.M., Hendrix C.W., Bakshi R.P., Kumar N., Shapiro T.A.. Phase I/II evaluation of the prophylactic antimalarial activity of pafuramidine in healthy volunteers challenged with Plasmodium falciparum sporozoites. Am. J. Trop. Med. Hyg. 2009; 80:528–535. [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y., Chai Y., Kumar A., Tidwell R.R., Boykin D.W., Wilson W.D.. Designed compounds for recognition of 10 base pairs of DNA with two AT binding sites. J. Am. Chem. Soc. 2012; 134:5290–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paul A., Nanjunda R., Kumar A., Laughlin S., Nhili R., Depauw S., Deuser S.S., Chai Y., Chaudhary A.S., David-Cordonnier M.H. et al. . Mixed up minor groove binders: Convincing A·T specific compounds to recognize a G·C base pair. Bioorg. Med. Chem. Lett. 2015; 25:4927–4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laughlin S., Wang S., Kumar A., Farahat A.A., Boykin D.W., Wilson W.D.. Resolution of mixed site DNA complexes with dimer-forming minor-groove binders by using electrospray ionization mass spectrometry: compound structure and DNA sequence effects. Chemistry. 2015; 21:5528–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Munde M., Wang S., Kumar A., Stephens C.E., Farahat A.A., Boykin D.W., Wilson W.D., Poon G.M.. Structure-dependent inhibition of the ETS-family transcription factor PU.1 by novel heterocyclic diamidines. Nucleic Acids Res. 2014; 42:1379–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chai Y., Paul A., Rettig M., Wilson W.D., Boykin D.W.. Design and synthesis of heterocyclic cations for specific DNA recognition: From AT-rich to mixed-base pair DNA sequences. J. Org. Chem. 2014; 79:852–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paul A., Chai Y., Boykin D.W., Wilson W.D.. Understanding mixed sequence DNA recognition by novel designed compounds: The kinetic and thermodynamic behavior of azabenzimidazole diamidines. Biochemistry. 2015; 54:577–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laughlin S., Wilson W.D.. May the best molecule win: Competition ESI mass spectrometry. Int. J. Mol. Sci. 2015; 16:24506–24531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abu-Daya A., Brown P.M., Fox K.R.. DNA sequence preferences of several AT-selective minor groove binding ligands. Nucleic Acids Res. 1995; 23:3385–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y., Kumar A., Boykin D.W., Wilson W.D.. Sequence and length dependent thermodynamic differences in heterocyclic diamidine interactions at AT base pairs in the DNA minor groove. Biophys. Chem. 2007; 131:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen B., Tanious F.A., Wilson W.D.. Biosensor-surface plasmon resonance: Quantitative analysis of small molecule-nucleic acid interactions. Methods. 2007; 42:150–161. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y., Wilson W.D.. Quantitative analysis of small molecule-nucleic acid interactions with a biosensor surface and surface plasmon resonance detection. Methods Mol. Biol. 2010; 613:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Mol N.J., Gillies M.B., Fischer M.. Experimental and calculated shift in pK(a) upon binding of phosphotyrosine peptide to the SH2 domain of p56(Ick). J. Bioorg. Med. Chem. 2002; 10:1477–1482. [DOI] [PubMed] [Google Scholar]
- 41.Lu X.J., El Hassan M.A., Hunter C.A.. Structure and conformation of helical nucleic acids: Analysis program (SCHNAaP). J. Mol. Biol. 1997; 273:668–680. [DOI] [PubMed] [Google Scholar]
- 42.Rohs R., West S.M., Sosinsky A., Liu P., Mann R.S., Honig B.. The role of DNA shape in protein-DNA recognition. Nature. 2009; 461:1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rohs R., Jin X., West S.M., Joshi R., Honig B., Mann R.S.. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010; 79:233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou T., Yang L., Lu Y., Dror I., Dantas Machado A.C., Ghane T., Di Felice R., Rohs R.. DNAshape: A method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res. 2013; 41:W56–W62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lavery R., Moakhar M., Maddocks J.H., Petkeviciute D., Zakrzewska K.. Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 2009; 37:5917–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zgarbova M., Luque F.J., Sponer J., Cheatham T.E. 3rd, Otyepka M., Jurečka P.. Toward improved description of DNA backbone: Revisiting epsilon and zeta torsion force field parameters. J. Chem. Theory Comput. 2013; 9:2339–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dans P.D., Danilāne L., Ivani I., Dršata T., Lankaš F., Hospital A., Walther J., Pujagut R.I., Battistini F.1, Gelpí J.L. et al. . Long-timescale dynamics of the Drew-Dickerson dodecamer. Nucleic Acids Res. 2016; 44:4052–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nelson H.C., Finch J.T., Luisi B.F., Klug A.. The structure of an oligo(dA).oligo(dT) tract and its biological implications. Nature. 1987; 330:220–226. [DOI] [PubMed] [Google Scholar]
- 49.Kladde M.P., Kohwi Y., Kohwi-Shigematsu T., Gorski J.. The non-B-DNA structure of d(CA/TG)n differs from that of Z-DNA. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:1898–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen H.H., Rou D.C., Charney E.J.. The flexibility of alternating dA-dT sequences. J. Biomol. Struct. Dyn. 1985; 2:709–719. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.