


Abstract
Aminoacyl-tRNA synthetases (AARSs) are a superfamily of enzymes responsible for the faithful translation of the genetic code and have lately become a prominent target for synthetic biologists. Our large-scale analysis of >2500 prokaryotic genomes reveals the complex evolutionary history of these enzymes and their paralogs, in which horizontal gene transfer played an important role. These results show that a widespread belief in the evolutionary stability of this superfamily is misconceived. Although AlaRS, GlyRS, LeuRS, IleRS, ValRS are the most stable members of the family, GluRS, LysRS and CysRS often have paralogs, whereas AsnRS, GlnRS, PylRS and SepRS are often absent from many genomes. In the course of this analysis, highly conserved protein motifs and domains within each of the AARS loci were identified and used to build a web-based computational tool for the genome-wide detection of AARS coding sequences. This is based on hidden Markov models (HMMs) and is available together with a cognate database that may be used for specific analyses. The bioinformatics tools that we have developed may also help to identify new antibiotic agents and targets using these essential enzymes. These tools also may help to identify organisms with alternative pathways that are involved in maintaining the fidelity of the genetic code.
INTRODUCTION
Aminoacyl-tRNA synthetases (AARSs) are very ancient house-keeping enzymes that are present in all eukaryotes, archaea and bacteria. They mediate the accurate esterification of amino acids (aa) to their cognate tRNAs and thus represent an essential superfamily of enzymes responsible for the faithful translation of the genetic code. Apart from the twenty AARSs that are responsible for the incorporation of the twenty standard proteinogenic aa, two additional AARSs, PylRS and SepRS, are used by some organisms during incorporation of the rare aa pyrolysine and phosphoserine, respectively. AARSs are divided into two non-homologous classes: class I and class II, mainly based on distinct structural folds of their catalytic domains and on which side of the tRNA acceptor-stem will be recognized by the enzyme (1,2). A common misconception is that the genome of almost every organism contains a complete set of 20 AARS, each being individually responsible for coding the enzyme that charges a cognate tRNA with one of the 20 naturally occurring aa. With the ever-increasing availability of complete genome sequences, it is becoming evident that gene duplication, horizontal gene transfer, and gene loss are much more frequent events among the AARSs than originally thought.
The absence of an AARS-encoding gene from a genome is possible because it does not necessarily correlate with the absence of the corresponding essential biochemical function. For example, the absence of glutaminyl-tRNA synthetase (GlnRS) is rescued by a non-discriminating glutamyl-tRNA synthetase (ND-GluRS) that can mis-acylate Glu to a tRNAGln, which is then modified to Gln-tRNAGln by a tRNA-dependent amidotransferase (3). Enzymatic modification of a mischarged aminoacyl-tRNA (aa-tRNA) is documented for Asn, Gln, Cys, selenocysteine and formylmethionine (4–8). Therefore, cataloguing all those cases where classical AARS genes are missing is a necessary first step in identifying known alternative pathways that enable cognate charging of the tRNA species for which the cognate AARS is missing. Genetic code decoding is a much more variable step than originally thought and needs to be quantified (9).
There are numerous reports of genomes with more than one gene for the same AARS enzyme or even paralogous fragments consisting of free-standing domains of AARSs (e.g. catalytic-, anticodon-binding- and editing domains). These paralogs and paralog fragments have been the focus of intense interest since their gene products exhibit diverse functions outside translation. These range from tRNA-dependent aa synthesis, tRNA posttranscriptional modification, editing of misactivated aa and antibiotic resistance in bacteria, to molecular hubs within essential signaling pathways that regulate tumorigenesis in humans (10–16). Evolutionary analyses have highlighted the importance of horizontal gene transfer (HGT) in the evolution of the AARS family (17) and it has been found that this is often linked to antibiotic resistance, especially in microbes (11,18–21). The fact that bacterial AARSs do not often (22) participate in complex protein-protein interactions and that they are frequently compatible with tRNAs from phylogenetically distant organisms suggests that they are frequently functional (and hence selectable) following HGT.
Many microorganisms have evolved low molecular weight toxins that target these essential enzymes in other microorganisms. Such toxins have already been identified for AlaRS, AspRS, AsnRS, IleRS, LeuRS, LysRS, MetRS, ProRS, SerRS, ThrRS, TyrRS, TrpRs, PheRS (11,23). Correspondingly, the microorganisms under attack either acquire low-level resistance via point mutations to the targeted AARS, or they acquire a resistant AARS paralog from other organisms via HGT (24,25). The most prominent example of these toxins is mupirocin (13), a natural antibiotic that has been commercialized. Pseudomonas fluorescens NCIMB 10586 has a large gene cluster that contains several polyketide synthase genes and is responsible for the production of pseudomonic acid (otherwise known as mupirocin), a natural compound that targets/inhibits IleRS. Interestingly, within that cluster, there exists a divergent mupirocin-resistant IleRS paralog (designated as mupA) in addition to the native, mupirocin-sensitive, IleRS. Therefore, both the toxin and the antidote are in the same gene cluster (25,26). The discovery that divergent AARS-paralogs confer resistance to natural AARS-inhibitors has been documented for MetRS, TrpRS, IleRS and SerRS paralogs; for a thorough review see (11,27). Thus, the detection of AARS paralogs may lead to the discovery of new antibiotics that are either encoded as a gene cluster in the vicinity of the paralog, or somewhere else in the genome. Furthermore, potential natural AARS inhibitors may be identified by comparative genomics between closely related strains, where one of the strains has an AARS duplicate (and may also have the inhibitor gene) whereas the other strains lack the AARS duplicate (and the inhibitor).
The manipulation and extension of the genetic code is considered a cornerstone of synthetic biology and biotechnology. In particular, the charging of certain tRNAs with unnatural aa is crucial for both the construction of safe GMOs and the engineering of enzymes with novel properties (28–31). Toward this goal, it is important to understand how the specificity of AARSs for aa and tRNAs is determined. Therefore, the unbiased detection of conserved motifs that characterize each specific AARS enzyme is a pre-requisite for future manipulation and rational site-directed mutagenesis studies.
The goal of our study was 5-fold: (i) to identify, in an unbiased manner, highly conserved motifs and domains for each AARS, (ii) to develop a novel and sensitive web-based computational tool (based on these motifs and domains) that is capable of identifying orthologs and paralogs, or even divergent fragments of AARS enzymes, from all kingdoms of life and on a genome-wide scale, (iii) to analyze more than 2500 prokaryotic genomes in order to provide the first thorough and comprehensive evolutionary analysis of AARS genes and their paralogs, (iv) to organize and store all the above information in a well-organized database to make it publicly accessible for studies of alternative pathways or new antibiotics and (v) to identify potential candidates for AARS inhibitors/antibiotics.
MATERIALS AND METHODS
All annotated eukaryotic, prokaryotic and organellar AARS sequences (11 984) were retrieved from the Uniprot/Swissprot database (32). Redundancy was filtered out at the 95% and 70% sequence similarity level with the BlastClust software, resulting in non-redundant sets of 7517 and 3276 AARS sequences, respectively. 2588 complete prokaryotic proteomes were downloaded from the NCBI ftp server with their sequences in fasta format and their corresponding coordination ptt files for each gene.
Conserved motifs were identified in each AARS enzyme using the MEME software (33). It has been suggested by previous analyses of this superfamily that domain architecture is a good marker for phylogenetic reconstruction (21). In general, domain emergence is very ancient, but domain re-arrangements are a very common phenomenon (34–37). Custom-made HMM generation and scanning was performed with the HMMER software (38). Multiple alignments were performed with the Muscle software (39) whereas distance-based BioNJ phylogenetic analysis was performed with the Seaview software (40). Treedyn was used for phylogenetic tree visualization (41) and integration of phylogenetic trees together with MEME-motif architecture was performed in html pages by using Javascript. All data handling was performed with custom-made Perl scripts. Smith-Waterman pairwise alignment and protein similarity calculation for pairs of homologs within the same genome were estimated with the EMBOSS Water program.
Construction of HMMs of conserved core-catalytic, editing and tRNA-binding domains was based on integration of information from many sources, such as manual inspection of alignments, MEME-motif architecture, expert knowledge from literature (20,21), domain characterization from PDB (42), Astral (43), Interpro (44) and CDD (45).
Data organization and storage was implemented in a MySQL database. A web graphical interface was generated with Java Language and Spring Framework for the back-end and Angular JS Framework for the front-end that is developed in a single-page application format. Front-end and back-end communication is established through an authenticated RESTful API.
RESULTS AND DISCUSSION
Discovery of highly conserved motifs in each AARS class
Initially, 3276 Swissprot annotated AARS protein sequences (redundancy filtered at the 70% sequence similarity level) from each of the two classes (1739 in class I and 1537 in class II) were analyzed with the MEME software, in order to identify shared conserved motifs within each of the two non-homologous classes. It is already well established that class I contains a Rossman fold with the two characteristic HIGH and a KMSKS conserved signatures, whereas class II contains three other conserved motifs (1). Our MEME analysis identified successfully the HIGH and KMSKS motifs in 98% (1708/1739) and 80% (1394/1739) of the Swissprot class I proteins respectively. It also identified motifs 1, 2 and 3 in 59% (901/1537), 73% (1121/1537) and 70% (1069/1537) of the Swissprot class II proteins respectively. The reason is that these three motifs and especially motif 1 are not so strongly conserved in all class II AARSs. Interestingly, for the first time, a fourth motif, designated as motif 2B, due to its C-terminal proximity to motif 2 was also identified in 73% (1124/1537) of the Swissprot annotated class II proteins. Figure 1 and Supplementary file 1 summarizes the findings and the logos of the conserved motifs in each of the AARS classes. From these logos, it is evident that they are very short and dependent on only a few aa. When the MEME algorithm analyzes homologs of the two AARS classes it is capable of identifying these very short motifs that are highly enriched in each class of this super-family. Nevertheless, profile Hidden Markov models based on these very short motifs have very limited detection power, when it comes to proteome-wide scanning of many diverse non-homologous families. Therefore, detection of AARS homologs based solely on these highly conserved, but very short motifs, is problematic. Another approach is needed, where a series of highly conserved motifs are used at the enzyme level (i.e. conserved within that particular AARS enzyme), rather than at the class level.
Figure 1.
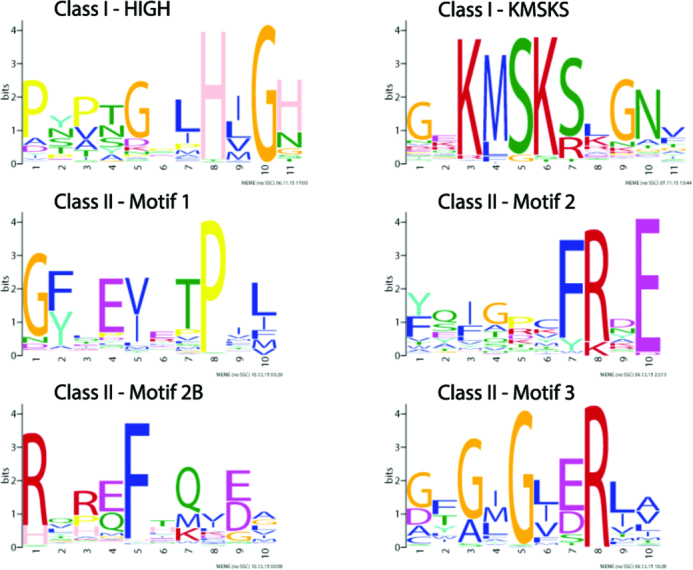
Highly conserved motifs of class I and class II AARSs.
Discovery of conserved motifs in each AARS enzyme and phylogenetic analyses
For each of the 20 AARS enzymes, as well as for PylRS and SepRS, the corresponding Swissprot annotated protein sequences (redundancy filtered at the 95% sequence similarity level) were analyzed with the MEME software in order to identify shared conserved motifs. The identification and architecture of MEME-motifs allowed us to separate each enzyme group to its distinct phylogenetic subgroups. In addition, it provided a better understanding of the degree of diversification of each ortholog and very importantly, of the various paralogs or paralog fragments. Towards this goal, MEME was run with a parameter set that aimed to identify 10 motifs for each AARS enzyme, except Asp–Asn–AARS and Glu–Gln–AARS enzymes, where MEME was set to identify 20 motifs.
Based on the number and sequence-regions of identified motifs that were present in all protein sequences of a certain enzyme (that ranged between 2 and 10 motifs), BioNJ phylogenetic trees were constructed for each enzyme and the trees were visualized with the Treedyn software. More specifically, the sequence regions of the motifs that were common in all sequences were concatenated for each sequence, and then the concatenated motif sequences were used to build a tree. Next, a tree for a certain enzyme was integrated with the MEME-motif architecture of each sequence (in that tree) in html pages, using Javascript. Based on the phylogenetic tree and the motif architecture, whenever applicable, phylogenetic subgroups were identified by manual inspection within a certain enzyme. Each of these subgroups underwent another round of MEME-motif discovery, again using a parameter set of 10 motifs and subsequent phylogenetic analysis using only the shared motifs of the subgroup. The new phylogenetic trees and their motif architectures were visualized again with the Treedyn software and Javascript in html. This iterative approach allowed us to cope efficiently with highly divergent subgroups within one AARS enzyme, or even subgroups that are paraphyletic, like GlyRS (46).
An example of the above analysis is the following. For the ProRS enzyme, there were 500 annotated sequences available in Swissprot. They underwent MEME-motif discovery with a parameter set of 10 motifs. Two motifs (that together comprised 36 aa) were common in all sequences and based on these, the first BioNJ tree was constructed. That tree was integrated with the above MEME motifs for each of the 500 sequences (see Supplementary Figure S1). Based on that tree and motif architecture, it was decided to treat ProRS as two distinct phylogenetic subgroups. Subgroup 1 comprised 386 sequences and subgroup 2 had the remaining 114 sequences. In this new round, the 386 sequences of subgroup 1 again underwent MEME-motif discovery with a parameter set of 10 motifs. This time, eight motifs were identified that were common in all the 386 sequences of this subgroup. Based on these 8 motifs that together comprised 241 amino acids, a second phylogenetic analysis was performed specifically for the 386 sequences. The new tree was integrated with the new motif architecture and visualized with Treedyn and Javascript in HTML (see one of the two ProRS trees in the database). The same pipeline was implemented for the 114 sequences of ProRS subgroup 2. This time, nine motifs were identified that were common in all the 114 sequences of this subgroup. Based on these nine motifs that together comprised 229 amino acids, a third phylogenetic analysis was performed specifically for the 114 sequences. The new tree was integrated with the new motif architecture and visualized with Treedyn and Javascript in HTML (see the other ProRS tree in the database).
For all the Uniprot AARS enzymes that were used for training, the resulting phylogenetic trees together with the corresponding motifs and motif architectures are available in html format in the website (http://bioinf.bio.uth.gr/aars). The above analyses resulted in 33 different phylogenetic trees for 22 aaRS enzymes. This is because an AARS enzyme may need to be broken to two or more sub-groups, so, for each subgroup there exists a new tree and meme motif architecture. The members of each kingdom (bacteria, archaea, eukaryotes) are distinguished by different colors.
For each of the 10 motifs of every phylogenetic group or subgroup, we extracted the motif-sequences from all members of that subgroup and performed multiple sequence alignment with the MUSCLE algorithm (39). Next, each of these alignments was used to build a HMM with the HMMER software (38). In total, 330 HMMs were constructed for their corresponding MEME-motifs. All annotated AARS from Swissprot were scanned with these 330 HMMs using the HMMER software. The output file was filtered with a custom-made perl script that assigned a query protein to a certain AARS enzyme, based on majority rule. More specifically, if two hmms from two different enzymes were hitting a certain protein region and their coordinates were overlapping, the hmm with the best bitscore would be kept. In this way, hmms from different enzymes may compete for the same protein region. Finally, the enzyme that had the most hmms found in the protein would be retained as the best assignment. By applying a minimum cut-off of two or five motifs, the resulting hmms and the majority rule filters applied resulted in 99.9% and 99.8% correct locus assignment respectively (see supplementary file 1: Swissprot evaluation). This protocol had high precision. Therefore, although a cut-off of five motifs is stringent, its performance is very satisfactory. This modular approach of individual MEME-motifs is very robust to domain re-arrangements or large insertions that may hinder detection with other methods. The principle applied in this analysis is similar to the PRINTS database (47).
Despite all this, a small number of distant AARS paralogs, such as HisZ could not be detected by the MEME-motif approach. To overcome this problem, an additional set of highly sensitive HMMs was developed, that was based on the whole catalytic domain (sometimes including an insertion domain). Again, all non-redundant annotated AARS sequences were used for the construction of catalytic HMMs for each AARS enzyme, taking into account the evolutionary sub-groups within each AARS. In addition, HMMs for editing and tRNA binding domains were created. Again, this set of catalytic domain HMMs for each locus could correctly identify 99.9% of Swissprot annotated AARS proteins. Furthermore, distant AARS paralogs that could not be detected by the MEME-motif approach were now detected. Only 9/883 Swissprot annotated GluRS were identified by our HMM domains as Gln-GltxRS. Therefore, the two sets of HMMs, based on short and specific MEME-motifs as well as longer catalytic domains were integrated in our computational tool and the subsequent bioinformatic analyses.
Development of a database and web-based detection tool
In order to investigate the AARS evolutionary profile of prokaryotes, the newly developed computational tool/pipeline was used to analyze ∼8 million proteins from 2588 prokaryotic proteomes, that were downloaded from NCBI. Proteins were first scanned by the highly sensitive, but not so highly specific, catalytic domain HMMs, resulting in 56 469 protein hits. Next, these hits were further scanned by the highly specific meme-motifs, applying a less stringent cutoff of two motifs and thus resulting in 52 595 protein hits. Application of an even more stringent cutoff of five motifs resulted in 49788 protein hits (see supplementary file 1: NCBI_protein_info).
The results from the large-scale NCBI prokaryotic proteome-wide scanning have been organized and saved in a MySQL database (bioinf.bio.uth.gr/aars). The user may query, in the database, only those proteins (NCBI protein accessions) whose number of AARS-specific motifs exceeds a threshold (set by the user). Other types of filters may also be applied in order to narrow the query results - such as organism name, evolutionary group and subgroup, or search by AARS type. In addition, a master table is generated, where the number of genes/paralogs for each AARS enzyme are displayed for each genome or a filtered/selected set of genomes. User selection of a certain protein also displays graphically the domain and motif architecture as well as the neighboring genes and their NCBI annotation. To our knowledge, the only other database that has been developed specifically for AARSs was constructed in 2001 (48).
A web-based AARS motif/domain detection tool is also integrated into the database, where the user may upload sequences in FASTA format for scanning. Such a tool is useful not only for annotating the AARS superfamily in a new genome (prokaryotic or even eukaryotic), but also for detecting paralog fragments or identifying genomes with alternative pathways, by observing the absence of a particular type of AARS gene. Recently, there has been intense interest in identifying human AARS fragments, even those that are catalytic nulls, because they seem to be involved in diverse functions. Paralog fragments of AARSs represented by appended domains and exhibiting various biological functions have been identified in recent years in almost all organisms. The new roles and diverse functions of the AARS fragments or, in some cases, their gene duplicates are mostly related to pathways outside translation, ranging from editing and antibiotic resistance in bacteria to molecular hubs within essential signaling pathways that regulate tumorigenesis in humans (15,28,49,50). The domain architecture of AARSs seems to be critical for conferring additional functions; however, the exact repertoire of these domains when they act in trans still remains elusive to a great extent. Interestingly, it was recently shown that the manipulation of such domains revealed their direct involvement in many metabolic networks that regulate essential cellular processes (51).
A global overview of the evolutionary profile of prokaryotic AARS
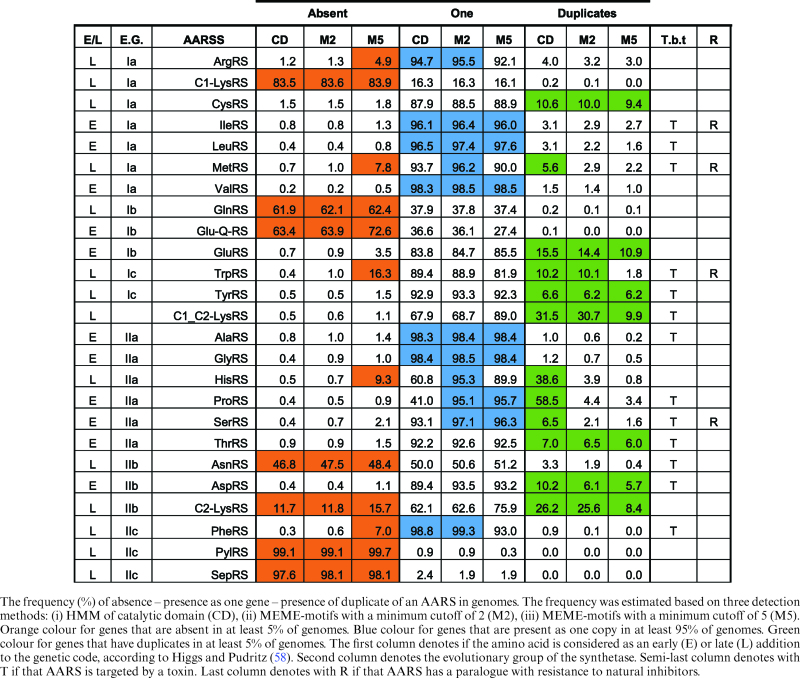
By analyzing 2588 prokaryotic proteomes, it was possible to obtain a very comprehensive evolutionary profile of this complex family that plays a central role in ensuring faithful translation of the genetic code and also in natural toxin resistance. For many of the subsequent analyses, we applied different criteria. In the first and more stringent criterion, a gene was considered to be a member of an AARS enzyme if it displayed at least 5 out of 10 motifs. Our previous evaluation analysis, based on Swissport annotated AARSs, showed that this cut-off was capable of capturing and correctly annotating 99.8% of the proteins. In a second (and more relaxed) criterion, a gene was considered to be a member of an AARS locus if it had at least 2 out of 10 motifs. In an even more relaxed criterion, due to the failure of MEME-motifs to capture some very distant homologs, such as HisZ, a gene was considered to be a member of an AARS enzyme if it was detected by the HMM of a catalytic domain. In this way, the more distant paralogs could be captured as well. The performance of the MEME-motifs (with cut-off two and five) and the catalytic domain HMMs is shown in supplementary file 1.
The catalytic domains have a very similar performance to that of the MEME-motifs with cut-off two, except for ProRS and HisRS. The same conclusions regarding the reduced detection power of MEME-motifs, but only for ProRS and HisRS distant homologs are also drawn from the analysis of the NCBI proteomes (see Table 1). In the case of HisRS, the catalytic domain HMMs are capable of capturing the HisZ paralogs that are known to lack aminoacylation activity (16) whereas the MEME-motifs could not capture the vast majority of such paralogs. NCBI has annotated the majority of HisZ paralogs as ATP phosphoribosyltransferases. In the case of ProRS, the catalytic domain HMMs are capable of capturing the ybaK/ebsC/ProX proteins that constitute an editing domain of one ProRS sub-group that may function in trans, whereas the MEME-motifs could not capture the vast majority of the members of this sub-group, probably because the motifs were situated in the catalytic core. Visual inspection of the NCBI annotation of detected proteins by the three criteria (catalytic domain HMM only, MEME-motif cutoff, two and MEME-motif cutoff five) revealed that 93.2% (52662/56531), 98.7% (51975/52653) and 99.1% (49380/49846) of detected proteins respectively were clearly annotated as tRNA synthetases or as a known paralog. This is a very strong indication that the MEME-motif detection methods are not only very sensitive, but also very specific.
Table 1. Evolutionary volatility profile of AARSs.
|
Distribution of AARS genes within genomes reflects the evolution of the genetic code
Although it is a common belief that most prokaryotic genomes have 20 AARS genes, our large-scale analysis clearly showed that this is not the case. Rather, the most frequent number of AARS genes in a prokaryotic genome is 19, although there also exist many genomes with 20 AARS, albeit less frequently. A distribution of the number of AARS genes per genome is shown in Figure 2. This reduced number is mostly due to the frequent absence of GlnRS (62% of scanned genomes; see Table 1). This finding is in accordance with an evolutionary scenario where GlnRS emerged later in evolution in the eukaryotic lineage via duplication of GluRS and moved to bacteria via HGT (17,52,53). In all prokaryotes missing GlnRS, Gln incorporation is mediated via a transamidation pathway (3,54). Gamma-Proteobacteria were considered as the GlnRS entry-point in the bacterial world. By analyzing the present distribution of GlnRS in the sequenced genomes and comparing it to an expected by-chance scenario, the most enriched phylogenetic lineage for GlnRs is indeed Proteobacterial (hypergeometric test: 1.5e–169). In addition, no Gln-GltxRS was detected in the analyzed Archaeal genomes.
Figure 2.

Distribution of total AARSs per genome (including SepRS and PylRS). Blue bars: detection based on the catalytic domain: orange bars: detection based on a cut-off of two motifs; gray bars: detection based on a cut-off of five motifs.
Along the same line of reasoning, it is very probable that AsnRS followed an evolutionary pathway similar to that of GlnRs, since it is missing in 47–48% of prokaryotic genomes studied (missing in 69–74% of analyzed archaeal genomes) (see Table 1 and supplementary file 1: Genomes_tables). In this case, by analyzing the present distribution of AsnRS in the 2588 sequenced genomes and comparing it to an expected by-chance scenario, the most enriched phylogenetic lineage for AsnRs originates among the Firmicutes (hypergeometric test: 2.7e-116).
Concerning TrpRS and TyrRS, it has been suggested (based on structural alignments) that the more ancient gene is that for TyrRS whereas that for TrpRS emerged by duplication in the archaeal lineage and later moved to bacteria via HGT (55). Indeed, this scenario is also supported by our evolutionary profile, but only for a cut-off of five motifs, where 16% of prokaryotes are missing a TrpRS whereas only 1.5% seem to be missing a TyrRS. Interestingly, only for a cut-off of five motifs, HisRS, is missing from 9% of studied genomes. Histidine is also considered a rather ‘young’ aa. The order that an aa entered the genetic code, as suggested by the co-evolution code theory, is in very good agreement with our evolutionary profile of AARS enzymes, but only for a cut-off of five motifs, as shown in Table 1 (56,57).
Intriguingly, there seem to be some broad correlations between the evolutionary age and the reactivity of aa and the deletion/duplication frequency of their corresponding AARSs. Amino acids such as Arg, Cys, Met, Gln, Tyr, Trp, His, Asn, Phe have been proposed to be relatively later additions to the genetic code (58). For a strict cut-off of five MEME-motifs, the AARSs that esterify these ‘younger’ aa (with the exception of Cys and Tyr) also tend to be completely absent from prokaryotic genomes more frequently compared to the group of AARSs that esterify the older aa. It is also very interesting that many of the AARSs that charge older and hydrophobic/non-reactive aa (Leu, Ile, Val, Ala, Gly) to tRNAs seem to be very stable (only one copy of the gene per genome) in their evolutionary profile. Taken together, our findings support the notions that amino acid emergence mirrors genetic code structure, with tRNA and AARS assignments evolving consequently.
The presence of paralogs is very frequent
To ensure a conservative approach, the results of this section are based on a cut-off of five motifs, unless stated otherwise. This analysis revealed that 40–61% (for five motifs and for two motifs) of scanned genomes had at least one AARS paralog detected.
The highest number of AARS genes detected in a genome is 29 in Ketasatospora setae and 28 in Bacillus cereus. In general, the Bacillus genus had several genomes with a high number of AARS genes/paralogs. Twenty-two percent (581/2588) and 3% (85/2588) of scanned NCBI genomes had ≥21 and ≥23 AARSs, respectively. Moreover, the group of genomes with ≥23 AARSs was enriched for Firmicutes (64% instead of 21% background; hypergeometric test: 5e–17). On the other hand, Nasuia deltocephalinicola str. NAS-ALF was the genome with the lowest number (only one LeuRS for a cutoff of five motifs and five genes for a cutoff of two motifs) of AARS genes detected. This is probably due to the endosymbiotic lifestyle of the smallest prokaryotic genome yet sequenced (169 genes in total) (59). If one considers that 20 is the expected number of AARS genes that should be found in a genome, our analysis shows that 59% (1531/2588) of the genomes scanned had less than that number, whereas 22% had more than 20 AARS genes. In terms of paralogs, the most extreme case was of Kitasatospora setae with four alleles for the SerRS gene. Each of the four genes had seven or more motifs and were all annotated as SerRS by NCBI. Similarly, there were three alleles of the same gene for AspRS, CysRS, GluRS, LeuRS, LysRS, ThrRS and ValRS in various species.
LysRS is an interesting case, because this AARS can either be from class I or class II (60). A total of 2143 genomes had only class II LysRS, whereas 377 genomes had only class I LysRS. Thirty nine genomes (mostly Bacillus thuringiensis and Bacillus cereus strains and members of the Streptomyces genus) had genes from both classes. It is more common to have a paralogous gene of the same class (218 genomes with class-II Lys paralogs; only one case of a paralog in class I) instead of having genes from the two classes (39 cases).
One crucial question for understanding the molecular mechanisms of evolution of AARS paralogs found within the same genome, is whether they result from either gene duplication or HGT. Toward this goal, a basic assumption was made that if two homologs of the same enzyme have very high protein sequence similarity, then they are most probably the result of a recent gene duplication. On the other hand, if two homologs have rather low similarity, then either they are the result of HGT or represent survivors from a very old gene duplication event that was soon followed by rapid divergence. By using 1751 pairs of homologs, the distribution of the percentage of protein similarity was calculated (shown in supplementary file 1: Paralog_similarities). From the graph, it is evident that (based on the above assumptions) <10% of homologous pairs are the result of a recent gene duplication within the genome. This calculation is in accordance with previous large-scale analyses on other protein families, where HGT instead of intra-chromosomal gene duplication is considered the main driving force behind protein family expansion in prokaryotes with an estimated contribution of 88–98% (61). Furthermore, a previous analysis with much less genomic data available at that time (1999) also supported HGT as a driving force in the early evolution of this family (21). On average, two homologs of the same AARS enzyme in a genome had 57% sequence similarity. The 60% of analyzed genomes had no evidence of paralog presence in any AARS at all. Moreover, the vast majority of AARSs were detected in bacterial chromosomes, whereas only 4% of AARSs were found in plasmids (see supplementary file 1: NCBI_protein_info). Apparently, HGT plays a significant role in the expansion of AARS paralogs, but complex evolutionary scenarios including gene duplications followed by rapid divergence, domain shuffling and gene loss should also be considered, as in the case of the bona fide AARSs (2,19,62,63).
It is tempting to speculate that the extended presence of paralogs is mainly driven by resistance to inhibitors, as for example in the case of bacterial MprF proteins that are fused to LysRSs (64). Nevertheless, the integration of available knowledge about known AARS paralogs with such resistance, together with the extent of paralog presence in prokaryotic genomes, as shown in Table 1, does not provide strong support for such a claim. An alternative explanation is that these paralogs are involved in other biochemical functions (65), many of them as yet unknown, in accordance with various observations where AARSs form complexes with other types of proteins, thus participating in other functions beyond translation (22,66). It is also intriguing that atypical AARS paralogs have been identified as participating in peptide formation via the non-ribosomal peptide synthesis mechanism (67). In principle, manual inspection of domain and MEME-motif architecture by our computational tools should provide better insight on which is the typical and which is the diverse AARS allele.
Computational detection of potential natural AARS inhibitors
Recently, there has been a growing concern that misuse of antibiotics is triggering the emergence of resistant strains that could eventually lead to a post-antibiotic world (68). Therefore, the discovery of new antibiotics, although neglected for some time, is now back in focus. AARSs have drawn considerable attention during recent years as targets of new antibacterial inhibitors.
Mupirocin is the product of a cluster of multimodular polyketide synthetases in Pseudomonas fluorescens and is an inhibitor of the cognate IleRS, acting as an antibiotic against methicillin-resistant Staphylococcus aureus. The modified IleRS paralog mupA that lies within the mupirocin biosynthetic cluster (in P. fluorescens) confers resistance to mupirocin by complementing the function of the mupirocin-inhibited cognate IleRS, thus acting as an antidote (26,69). To nullify the mupirocin inhibitor, S. aureus has evolved another paralog, mupB (69). In order to identify other potential antibiotic clusters, we looked for AARS paralogs/xenologs that have within their vicinity (±10 gene neighbors) NCBI-annotated polyketide synthetase genes. The results of this in silico genomic search are summarized in Table 2 and need to be verified experimentally in the future. The mupirocin cluster was not detected because the whole-genome sequence of P. fluorescens strain was not available for scanning in NCBI. With the cost of sequencing being reduced, very soon, metagenomic approaches will be used to scan and identify environmental contigs that harbor both PKS and AARS genes in the same sequence contig.
Table 2. Potential AARS-inhibiting antibiotic clusters.
| Organism | Phylogenetic group | AARS paralogue/ xenologue | Organismal characteristics | Number of PKS within vicinity |
|---|---|---|---|---|
| Paenibacillus polymyxa E681 | Firmicutes | AspRS | Produces compounds with antifungal or antibacterial activity. | 7 |
| Azospirillum sp. B510 | Proteobacteria | PheRS | Plant growth promoting organism. | 2 |
| Mycobacterium abscessus | Actinobacteria | CysRS | Causes a chronic lung infection, similar to tuberculosis, in patients with cystic fibrosis. | 6 |
| Very resistant to many commonly used antibiotics. | ||||
| Mycobacterium smegmatis JS623 | Actinobacteria | CysRS | Associated with soft tissue lesions following trauma or surgery.A possible factor in penile carcinogenesis. | 4 |
CONCLUSIONS
One of the major goals of this study was to develop computational tools for the rapid and sensitive detection of AARS proteins encoded in genome sequences. This was accomplished by using automatic and unbiased detection of short and conserved motifs within each of the 22 AARS enzymes, using the MEME algorithm in combination with phylogenetic analyses. Next, the detected motifs were used to build HMMs. To ensure detection of distant homologs, HMMs of the whole catalytic domain of each AARS were developed as well. This approach generated a web-based tool that uses all of the above HMMs to rapidly scan for AARS coding sequences on a genome-wide scale.
The second major goal of this study was to analyze more than 2500 prokaryotic genomes for the presence and absence of AARS-coding sequences, with the results organized and stored in a publicly available database. This large-scale and comprehensive evolutionary profile quantified, for the first time, the large variability that exists within a specific, but essential component of the apparatus required for the translation of the genetic code—the acylation of tRNAs with their cognate amino acids. Furthermore, it appears that the presence of more than one AARS homolog of the same enzyme is usually the result of horizontal gene transfer or gene duplication followed by rapid divergence, most probably linked to alternative biochemical functions, although resistance towards AARS-targeting toxins from other bacteria cannot be excluded.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Prof. Stephen G. Oliver, Department of Biochemistry, University of Cambridge, UK for critical reading of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
‘ARISTEIA’ and ‘ARISTEIA II’ Action of the ‘OPERATIONAL PROGRAMME EDUCATION AND LIFELONG LEARNING’ and is co-funded by the European Social Fund (ESF) and National Resources [MIS 1225, No. D608 to C.S., 4288 to G.D.A.]; Postgraduate Program ‘Applications of Molecular Biology-Genetics, Diagnostic Biomarkers’ [code 3817 to G.D.A.] of the University of Thessaly, School of Health Sciences, Department of Biochemistry & Biotechnology; French National Program ‘Investissement d'Avenir’ administered by the ‘Agence National de la Recherche’ (ANR), ‘MitoCross’ Laboratory of Excellence (Labex) [ANR-10-IDEX-0002-02] and the University of Strasbourg (to H.B.); National Science Foundation [MCB 1412611 to M.I.]. This work was also supported in part by ‘Fondation Sante’ Grants 2016 (E515 to CS). The open access publication charge for this paper has been waived by Oxford University Press - NAR.
Conflict of interest statement. None declared.
REFERENCES
- 1.Eriani G., Delarue M., Poch O., Gangloff J., Moras D.. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature. 1990; 347:203–206. [DOI] [PubMed] [Google Scholar]
- 2.Ribas de Pouplana L, Schimmel P.. Two classes of tRNA synthetases suggested by sterically compatible dockings on tRNA acceptor stem. Cell. 2001; 104:191–193. [DOI] [PubMed] [Google Scholar]
- 3.Curnow A.W., Hong K.W., Yuan R., Kim S.I., Martins O., Winkler W., Henkin T.M., Söll D.. Glu-tRNAGln amidotransferase: a novel heterotrimeric enzyme required for correct decoding of glutamine codons during translation. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:11819–11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becker H.D., Kern D.. Thermus thermophilus: a link in evolution of the tRNA-dependent amino acid amidation pathways. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:12832–12837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leinfelder W., Zehelein E., Mandrand-Berthelot M.A., Böck A.. Gene for a novel tRNA species that accepts L-serine and cotranslationally inserts selenocysteine. Nature. 1988; 331:723–725. [DOI] [PubMed] [Google Scholar]
- 6.Ibba M., Curnow A.W., Söll D.. Aminoacyl-tRNA synthesis: divergent routes to a common goal. Trends Biochem. Sci. 1997; 22:39–42. [DOI] [PubMed] [Google Scholar]
- 7.Sheppard K., Yuan J., Hohn M.J., Jester B., Devine K.M., Söll D.. From one amino acid to another: tRNA-dependent amino acid biosynthesis. Nucleic Acids Res. 2008; 36:1813–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sauerwald A., Zhu W., Major T.A., Roy H., Palioura S., Jahn D., Whitman W.B., Yates J.R., Ibba M., Söll D.. RNA-dependent cysteine biosynthesis in archaea. Science. 2005; 307:1969–1972. [DOI] [PubMed] [Google Scholar]
- 9.Ling J., O'Donoghue P., Söll D.. Genetic code flexibility in microorganisms: novel mechanisms and impact on physiology. Nat. Rev. Microbiol. 2015; 13:707–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahel I., Korencic D., Ibba M., Söll D.. Trans-editing of mischarged tRNAs. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:15422–15427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andam C.P., Fournier G.P., Gogarten J.P.. Multilevel populations and the evolution of antibiotic resistance through horizontal gene transfer. FEMS Microbiol. Rev. 2011; 35:756–767. [DOI] [PubMed] [Google Scholar]
- 12.Blaise M., Becker H.D., Keith G., Cambillau C., Lapointe J., Giegé R., Kern D.. A minimalist glutamyl-tRNA synthetase dedicated to aminoacylation of the tRNAAsp QUC anticodon. Nucleic Acids Res. 2004; 32:2768–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilbart J., Perry C.R., Slocombe B.. High-level mupirocin resistance in Staphylococcus aureus: evidence for two distinct isoleucyl-tRNA synthetases. Antimicrob. Agents Chemother. 1993; 37:32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim S., You S., Hwang D.. Aminoacyl-tRNA synthetases and tumorigenesis: more than housekeeping. Nat. Rev. Cancer. 2011; 11:708–718. [DOI] [PubMed] [Google Scholar]
- 15.Lo W.-S., Gardiner E., Xu Z., Lau C.-F., Wang F., Zhou J.J., Mendlein J.D., Nangle L.A., Chiang K.P., Yang X.-L. et al. . Human tRNA synthetase catalytic nulls with diverse functions. Science. 2014; 345:328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sissler M., Delorme C., Bond J., Ehrlich S.D., Renault P., Francklyn C.. An aminoacyl-tRNA synthetase paralog with a catalytic role in histidine biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:8985–8990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamour V., Quevillon S., Diriong S., N'Guyen V.C., Lipinski M., Mirande M.. Evolution of the Glx-tRNA synthetase family: the glutaminyl enzyme as a case of horizontal gene transfer. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:8670–8674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fournier G.P., Andam C.P., Gogarten J.P.. Ancient horizontal gene transfer and the last common ancestors. BMC Evol. Biol. 2015; 15:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Donoghue P., Luthey-Schulten Z.. On the evolution of structure in aminoacyl-tRNA synthetases. Microbiol. Mol. Biol. Rev. MMBR. 2003; 67:550–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woese C.R., Olsen G.J., Ibba M., Söll D.. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol. Mol. Biol. Rev. MMBR. 2000; 64:202–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolf Y.I., Aravind L., Grishin N.V., Koonin E.V.. Evolution of aminoacyl-tRNA synthetases–analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999; 9:689–710. [PubMed] [Google Scholar]
- 22.Laporte D., Huot J.L., Bader G., Enkler L., Senger B., Becker H.D.. Exploring the evolutionary diversity and assembly modes of multi-aminoacyl-tRNA synthetase complexes: lessons from unicellular organisms. FEBS Lett. 2014; 588:4268–4278. [DOI] [PubMed] [Google Scholar]
- 23.Pham J.S., Dawson K.L., Jackson K.E., Lim E.E., Pasaje C.F.A., Turner K.E.C., Ralph S.A.. Aminoacyl-tRNA synthetases as drug targets in eukaryotic parasites. Int. J. Parasitol. Drugs Drug Resist. 2014; 4:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antonio M., McFerran N., Pallen M.J.. Mutations affecting the Rossman fold of isoleucyl-tRNA synthetase are correlated with low-level mupirocin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 2002; 46:438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yanagisawa T., Kawakami M.. How does Pseudomonas fluorescens avoid suicide from its antibiotic pseudomonic acid?: Evidence for two evolutionarily distinct isoleucyl-tRNA synthetases conferring self-defense. J. Biol. Chem. 2003; 278:25887–25894. [DOI] [PubMed] [Google Scholar]
- 26.El-Sayed A.K., Hothersall J., Cooper S.M., Stephens E., Simpson T.J., Thomas C.M.. Characterization of the mupirocin biosynthesis gene cluster from Pseudomonas fluorescens NCIMB 10586. Chem. Biol. 2003; 10:419–430. [DOI] [PubMed] [Google Scholar]
- 27.Ochsner U.A., Sun X., Jarvis T., Critchley I., Janjic N.. Aminoacyl-tRNA synthetases: essential and still promising targets for new anti-infective agents. Expert Opin. Investig. Drugs. 2007; 16:573–593. [DOI] [PubMed] [Google Scholar]
- 28.Guo L.-T., Wang Y.-S., Nakamura A., Eiler D., Kavran J.M., Wong M., Kiessling L.L., Steitz T.A., O'Donoghue P., Söll D.. Polyspecific pyrrolysyl-tRNA synthetases from directed evolution. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:16724–16729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hadd A., Perona J.J.. Recoding aminoacyl-tRNA synthetases for synthetic biology by rational protein-RNA engineering. ACS Chem. Biol. 2014; 9:2761–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Passioura T., Suga H.. Reprogramming the genetic code in vitro. Trends Biochem. Sci. 2014; 39:400–408. [DOI] [PubMed] [Google Scholar]
- 31.Wang L., Xie J., Schultz P.G.. Expanding the genetic code. Annu. Rev. Biophys. Biomol. Struct. 2006; 35:225–249. [DOI] [PubMed] [Google Scholar]
- 32.UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2015; 43:D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bailey T.L., Johnson J., Grant C.E., Noble W.S.. The MEME Suite. Nucleic Acids Res. 2015; doi:10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chothia C., Gough J., Vogel C., Teichmann S.A.. Evolution of the protein repertoire. Science. 2003; 300:1701–1703. [DOI] [PubMed] [Google Scholar]
- 35.Marsh J.A., Teichmann S.A.. How do proteins gain new domains. Genome Biol. 2010; 11:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Teichmann S.A., Rison S.C., Thornton J.M., Riley M., Gough J., Chothia C.. The evolution and structural anatomy of the small molecule metabolic pathways in Escherichia coli. J. Mol. Biol. 2001; 311:693–708. [DOI] [PubMed] [Google Scholar]
- 37.Weiner J., Moore A.D., Bornberg-Bauer E.. Just how versatile are domains. BMC Evol. Biol. 2008; 8:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eddy S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011; 7:e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004; 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gouy M., Guindon S., Gascuel O.. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010; 27:221–224. [DOI] [PubMed] [Google Scholar]
- 41.Chevenet F., Brun C., Bañuls A.-L., Jacq B., Christen R.. TreeDyn: towards dynamic graphics and annotations for analyses of trees. BMC Bioinformatics. 2006; 7:439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rose P.W., Prlić A., Bi C., Bluhm W.F., Christie C.H., Dutta S., Green R.K., Goodsell D.S., Westbrook J.D., Woo J. et al. . The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015; 43:D345–D356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fox N.K., Brenner S.E., Chandonia J.-M.. SCOPe: Structural Classification of Proteins—extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 2014; 42:D304–D309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mitchell A., Chang H.-Y., Daugherty L., Fraser M., Hunter S., Lopez R., McAnulla C., McMenamin C., Nuka G., Pesseat S. et al. . The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 2015; 43:D213–D221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marchler-Bauer A., Derbyshire M.K., Gonzales N.R., Lu S., Chitsaz F., Geer L.Y., Geer R.C., He J., Gwadz M., Hurwitz D.I. et al. . CDD: NCBI's conserved domain database. Nucleic Acids Res. 2015; 43:D222–D226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valencia-Sánchez M.I., Rodríguez-Hernández A., Ferreira R., Santamaria-Suárez H.A., Arciniega M., Dock-Bregeon A.-C., Moras D., Beinsteiner B., Mertens H., Svergun D. et al. . Structural insights into the polyphyletic origins of glycyl tRNA synthetases. J. Biol. Chem. 2016; doi:10.1074/jbc.M116.730382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Attwood T.K., Coletta A., Muirhead G., Pavlopoulou A., Philippou P.B., Popov I., Romá-Mateo C., Theodosiou A., Mitchell A.L.. The PRINTS database: a fine-grained protein sequence annotation and analysis resource–its status in 2012. Database J. Biol. Databases Curation. 2012; bas019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szymanski M., Deniziak M.A., Barciszewski J.. Aminoacyl-tRNA synthetases database. Nucleic Acids Res. 2001; 29:288–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo M., Chong Y.E., Shapiro R., Beebe K., Yang X.-L., Schimmel P.. Paradox of mistranslation of serine for alanine caused by AlaRS recognition dilemma. Nature. 2009; 462:808–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han J.M., Jeong S.J., Park M.C., Kim G., Kwon N.H., Kim H.K., Ha S.H., Ryu S.H., Kim S.. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell. 2012; 149:410–424. [DOI] [PubMed] [Google Scholar]
- 51.Guo M., Schimmel P., Yang X.-L.. Functional expansion of human tRNA synthetases achieved by structural inventions. FEBS Lett. 2010; 584:434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brown J.R., Doolittle W.F.. Gene descent, duplication, and horizontal transfer in the evolution of glutamyl- and glutaminyl-tRNA synthetases. J. Mol. Evol. 1999; 49:485–495. [DOI] [PubMed] [Google Scholar]
- 53.Koonin E.V., Makarova K.S., Aravind L.. Horizontal gene transfer in prokaryotes: quantification and classification. Annu. Rev. Microbiol. 2001; 55:709–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tumbula D.L., Becker H.D., Chang W.Z., Söll D.. Domain-specific recruitment of amide amino acids for protein synthesis. Nature. 2000; 407:106–110. [DOI] [PubMed] [Google Scholar]
- 55.Dong X., Zhou M., Zhong C., Yang B., Shen N., Ding J.. Crystal structure of Pyrococcus horikoshii tryptophanyl-tRNA synthetase and structure-based phylogenetic analysis suggest an archaeal origin of tryptophanyl-tRNA synthetase. Nucleic Acids Res. 2010; 38:1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Francklyn C. tRNA synthetase paralogs: evolutionary links in the transition from tRNA-dependent amino acid biosynthesis to de novo biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:9650–9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wong J.T. A co-evolution theory of the genetic code. Proc. Natl. Acad. Sci. U.S.A. 1975; 72:1909–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Higgs P.G., Pudritz R.E.. A thermodynamic basis for prebiotic amino acid synthesis and the nature of the first genetic code. Astrobiology. 2009; 9:483–490. [DOI] [PubMed] [Google Scholar]
- 59.Bennett G.M., Moran N.A.. Small, smaller, smallest: the origins and evolution of ancient dual symbioses in a Phloem-feeding insect. Genome Biol. Evol. 2013; 5:1675–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ibba M., Morgan S., Curnow A.W., Pridmore D.R., Vothknecht U.C., Gardner W., Lin W., Woese C.R., Söll D.. A euryarchaeal lysyl-tRNA synthetase: resemblance to class I synthetases. Science. 1997; 278:1119–1122. [DOI] [PubMed] [Google Scholar]
- 61.Treangen T.J., Rocha E.P.C.. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet. 2011; 7:e1001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kunin V., Ouzounis C.A.. The balance of driving forces during genome evolution in prokaryotes. Genome Res. 2003; 13:1589–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kyrpides N., Overbeek R., Ouzounis C.. Universal protein families and the functional content of the last universal common ancestor. J. Mol. Evol. 1999; 49:413–423. [DOI] [PubMed] [Google Scholar]
- 64.Roy H., Ibba M.. RNA-dependent lipid remodeling by bacterial multiple peptide resistance factors. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:4667–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Giegé R., Springer M.. Aminoacyl-tRNA Synthetases in the Bacterial World. EcoSal Plus. 2016; 7, doi:10.1128/ecosalplus.ESP-0002-2016. [DOI] [PubMed] [Google Scholar]
- 66.Rubio M.Á., Napolitano M., Ochoa de Alda J.A.G., Santamaría-Gómez J., Patterson C.J., Foster A.W., Bru-Martínez R., Robinson N.J., Luque I.. Trans-oligomerization of duplicated aminoacyl-tRNA synthetases maintains genetic code fidelity under stress. Nucleic Acids Res. 2015; 43:9905–9917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mocibob M., Ivic N., Bilokapic S., Maier T., Luic M., Ban N., Weygand-Durasevic I.. Homologs of aminoacyl-tRNA synthetases acylate carrier proteins and provide a link between ribosomal and nonribosomal peptide synthesis. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:14585–14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu Y.-Y., Wang Y., Walsh T.R., Yi L.-X., Zhang R., Spencer J., Doi Y., Tian G., Dong B., Huang X. et al. . Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect. Dis. 2015; doi:10.1016/S1473-3099(15)00424-7. [DOI] [PubMed] [Google Scholar]
- 69.Seah C., Alexander D.C., Louie L., Simor A., Low D.E., Longtin J., Melano R.G.. MupB, a new high-level mupirocin resistance mechanism in Staphylococcus aureus. Antimicrob. Agents Chemother. 2012; 56:1916–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.