


Abstract
RIG-I is an innate immune receptor that detects and responds to infection by deadly RNA viruses such as influenza, and Hepatitis C. In the cytoplasm, RIG-I is faced with a difficult challenge: it must sensitively detect viral RNA while ignoring the abundance of host RNA. It has been suggested that RIG-I has a ‘proof-reading’ mechanism for rejecting host RNA targets, and that disruptions of this selectivity filter give rise to autoimmune diseases. Here, we directly monitor RNA proof-reading by RIG-I and we show that it is controlled by a set of conserved amino acids that couple RNA and ATP binding to the protein (Motif III). Mutations of this motif directly modulate proof-reading by eliminating or enhancing selectivity for viral RNA, with major implications for autoimmune disease and cancer. More broadly, the results provide a physical explanation for the ATP-gated behavior of SF2 RNA helicases and receptor proteins.
INTRODUCTION
Pattern recognition receptors (PRRs) protect against host invasion by detecting pathogen associated molecular patterns and initiating an immune response (1–4). RIG-I-like receptors (RLRs), which include Retinoic Acid-Inducible Gene I (RIG-I), Melanoma Differentiation-Associated Gene 5 (MDA5) and Laboratory of Genetics and Physiology 2, are a structurally related group of cytosolic PRRs that recognize structural variations among viral RNA molecules and play a critical role in the vertebrate antiviral response (5–8). RLRs contain a central DExD/H-box ATPase core comprised of two RecA-like domains, HEL1 and HEL2, and a conserved insertion domain, HEL2i, which promotes recognition of duplex RNA (9–11). To facilitate the detection of a broad range of pathogenic RNAs, each RLR contains a related C-terminal domain (CTD) that mediates specific interactions with bound nucleic acids or neighboring protein partners (8,12–13). In addition to these specialized target recognition domains, RIG-I and MDA5 contain a pair of tandem caspase activation and recruitment domains (CARDs) that initiate immune signaling by activating the downstream adaptor protein MAVS (14,15).
To prevent an unwanted immune response, these receptors must discriminate between endogenous and pathogenic RNAs. In RIG-I, the CTD confers viral RNA specificity by engaging in a high affinity interaction with 5΄ triphosphate or 5΄ diphosphate moieties located at the terminus of RNA duplexes on viral genomic RNA or replicative intermediates (13,16–18). While surveying the cytoplasm, RIG-I maintains an autorepressed conformation that is incapable of signaling. When viral RNA is detected, the CTD caps the duplex terminus, making extensive contact with the 5΄ triphosphate group and terminal base pairs, while the helicase core wraps around the RNA duplex (9–10,19–20). The RNA-bound RIG-I is now competent to bind ATP, which is involved in presenting the CARDs for a productive interaction with MAVS, thereby stimulating subsequent downstream signaling (11,15,21).
In addition to the RNA selectivity governed by the CTD, RIG-I employs its ATP binding and hydrolysis activities to specifically recognize and respond to viral RNA during the initiation of immune signaling. We have shown that ATP binding, but not hydrolysis, contributes to activation of RIG-I signaling in the presence of an optimized RNA ligand (22). When RIG-I is bound to triphosphorylated, pathogenic RNA, ATP binding causes a pronounced conformational change in the complex (10,19,23–25), constricting the structure in a manner that promotes domain rearrangement (9–10,23,25–26), potentially promoting CARDs expulsion and RIG-I activation (9–10,23). When RIG-I is bound to internal duplex sites and inappropriate targets, such as RNAs within the host, the high-affinity CTD is disengaged and interactions with the RNA occur exclusively through contacts involving the helicase domain (13). In these cases, ATP binding serves to weaken the RIG-I:RNA interaction, resulting in RNA dissociation (22). When RIG-I is confronted with an abundance of non-pathogenic RNAs, this may help to recycle the protein, enabling it to dissociate without signaling and search for an appropriate viral target RNA.
Some form of proofreading mechanism is essential for proper RIG-I function because it prevents the protein from inducing an aberrant immune response (22,27). Several groups have suggested that RIG-I mutations within the active-site for ATP hydrolysis disrupt the ability of RIG-I to differentiate host from viral RNA molecules, thereby causing dysregulated signaling and disease. For example, an ATPase active-site mutation that causes constitutive signal activation has been linked with the autoimmune disease Singleton–Merton syndrome (SMS, associated with mutation E373A) (28). It has been suggested that SMS mutants are defective in proofreading (29), however, the physical basis for this apparent loss of RIG-I specificity and the mechanism of activation by off-target RNAs is not known (30). In these types of mutants, the linkage between RNA and ATP binding, and the actual act of recycling was not directly investigated, so the basis of dysregulated ‘proofreading’ has not been established. The Walker A and B motifs in HEL1 (Motif I and II) are necessary for ATP binding and hydrolysis in RIG-I and all other Superfamily 2 Helicase (SF2) proteins (6,31–33). It is therefore unsurprising that mutations within these motifs can alter RIG-I function. Furthermore, given the key role of ATPase site mutants in multiple aspects of RIG-I signaling (11,22), mutations can induce pleiotropic effects that defy simple mechanistic characterization without direct, quantitative biochemical analysis of their behavior. We therefore set out to directly study the role of ATP in recycling by RIG-I active-site mutants and to determine whether certain mutations can specifically abrogate RIG-I target fidelity through a bona-fide proof-reading mechanism.
Here, we employ a set of enzymological and cell-based approaches to investigate the role of ATP in the behavior of RIG-I variants that are defective in proof-reading. We identified a mutation within the conserved Motif III region (S411A) that causes RIG-I to signal constantly within the cell, and which is aberrantly activated by non-pathogenic RNA. We then characterized the ability of this mutant to couple ATP binding with differential RNA discrimination. We find that ‘constitutively active’ RIG-I variants such as S411A have lost the ability to effectively sample between RNA molecules in the cell. In these mutants, ATP binding no longer promotes the rapid dissociation of RIG-I from inappropriate targets, resulting in apparently constitutive signaling. In this way, we reveal the molecular basis for an important type of dysregulated RIG-I signaling, potentially providing a mechanistic framework for certain classes of autoimmunity.
MATERIAL AND METHODS
RNA hairpin preparation
Oligoribonucleotides (OH10L: 5΄ - GGACGUACGUUUCGACGUACGUCC – 3΄) were synthesized on an automated MerMade synthesizer (BioAutomation) using standard phosphoramidite chemistry. The hydroxylated oligonucleotides were deprotected and gel purified as previously described (34). Triphosphorylated oligonucleotides (5΄ ppp10L: 5΄ – pppGGACGUACGUUUCGACGUACGUCC – 3΄) were synthesized as described (35,36), and deprotected and gel purified. The triphosphorylation state and purity were confirmed using mass spectrometry (Novatia, Newtown, PA, USA). The oligonucleotides were resuspended in RNA storage buffer (10 mM MOPS pH 6, 1 mM EDTA for OH10L and 10 mM MOPS pH 7, 1 mM EDTA for 5΄ppp10L), and snap cooled to ensure hairpin formation. To confirm the monomeric state of the hairpins, the pure RNA hairpins were run on a 15% native polyacrylamide gel alongside a 22 base pair duplex, which would mimic the migration of a hairpin dimer.
Fluorophore-labeled RNA hairpin used in stopped flow fluorescence spectroscopy and equilibrium RNA binding experiments was generated using amino-modified RNA (on a U in the UUCG tetraloop), which is synthesized using a 3΄ amino modifier with a C3 linker (Glen Research). Cy3 Mono NHS ester (GE Healthcare) or 6-FAM (GE Healthcare) was conjugated to the modified oligonucleotides as per the manufacturer's instructions. Fluorophore-labeled and unlabeled RNA were separated on a 20% denaturing polyacrylamide gel, and purified by gel extraction.
HEK 293T cell culture and IFN-β induction assays
Vector pUNO-hRIG-I for constitutive WT RIG-I expression in mammalian cells was purchased from Invivogen. Mutations were introduced into the parent plasmid using primers S411A forward: 5΄- GTCATTGGGCTGACTGCCGCGGTTGGTGTTGGGGATGCC -3΄ and S411A reverse: 5΄- GGCATCCCCAACACCAACCGCGGCAGTCAGCCCAATGAC -3΄ or S411L forward: 5΄- CATTGGGCTGACTGCCTTGGTTGGTGTTGGG -3΄ and S411L reverse: 5΄- CCCAACACCAACCAAGGCAGTCAGCCCAATG -3΄ and PfuUltra Hotstart PCR Master Mix (Agilent) per the manufacturer's protocol. Mutagenesis was validated by sequencing.
Cell based experiments were conducted in HEK 293T cells, which lack endogenous RIG-I expression (proteinatlas.org). Cells were grown and maintained in 15 cm dishes containing Dulbecco's Modified Eagle Medium (Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum (Hyclone) and Non-Essential Amino Acids (Life Technologies). IFN-β induction assays were conducted in 24-well format. Briefly, 0.5 ml of cells at 100 000 cells/ml were seeded in each well of a tissue culture treated 24-well plate. After 24 h, each well of cells was transfected with the indicated amount of WT or mutant pUNO-hRIG-I, 6 ng pRL-TK constitutive Renilla luciferase reporter plasmid (Promega), and 150 ng of an IFN-β/FireflyLuc reporter plasmid using the Lipofectamine 2000 transfection reagent (Life Technologies) per the manufacturer's protocol. Protein expression was allowed to proceed for 24 h, at which point the cells were challenged by transfection of 1 μg of the indicated dsRNA, also using the Lipofectamine 2000 reagent. After 12 h, cells were harvested for luminescence analysis.
To assess IFN-β induction using a dual luciferase assay, cells were harvested and lysed as follows: Growth media was aspirated from each well, and 100 μl of passive lysis buffer (Promega) was added. Lysis proceeded for 15 min at room temperature. The lysates were transferred to a 96-well PCR plate (Eppendorf) and clarified by centrifugation. Next, 20 μl samples of the supernatant were transferred to a 96-well assay plate for analysis using the Dual-Luciferase Reporter Assay System (Promega). Luminescence was measured using a Biotek Synergy H1 plate reader. The resulting Firefly luciferase activity (i.e. the induction of IFN-β) was normalized to the activity of the constitutively expressed Renilla luciferase to account for differences in confluency, viability and transfection efficiency across sample wells.
Protein purification
Wild-type hRIG-I was cloned into the pET-SUMO vector (Life Technologies) using the manufacturer's protocols. Mutations were introduced using the same primers and technique described for pUNO-hRIG-I mutation above.
For expression, plasmids were transformed into Rosetta II (DE3) Escherichia coli cells (Novagen) and grown at 37°C to an Abs.600 of 0.6. Expression was induced at 16°C for 24 h by the addition of 0.5 mM isopropyl-β-D-thiogalactopyranoside. RIG-I was then purified as previously described (22). Briefly, after lysis, RIG-I was purified using nickel affinity chromatography, followed by cationic exchange and size exclusion chromatography using Heparin Sepharose and HiLoad Superdex 200 (16/60) columns (GE Healthcare). RIG-I was concentrated in storage buffer (25 mM MOPS pH 7.4, 300 mM NaCl, 5% glycerol, 5 mM βME) and concentrations were determined spectrophotometrically using an extinction coefficient of ε = 99 700 M−1 cm−1 at λ = 280 nm. Protein preparations were aliquoted, flash frozen using liquid nitrogen and stored at −80°C.
NADH coupled ATPase activity assay
RIG-I ATPase activity was measured using an established absorbance-based coupled assay system. The RIG-I protein of interest was diluted into ATPase assay buffer (25 mM MOPS pH 7.4, 150 mM NaCl, 5 mM DTT) to a final concentration of 10 nM for KM, RNA experiments in the presence of a coupled assay mix (1 mM NADH, 100 U/ml lactic dehydrogenase, 500 U/ml pyruvate kinase, 2.5 mM phosphoenol pyruvic acid).
For KM, RNA experiments, the RNA of interest was diluted into ME buffer (25 mM MES pH 6.0, 0.5 mM EDTA) over a 12-pt concentration series and added to the protein/NADH sample mix resulting in RNA concentrations varying from approximately 0.5–500 nM. Samples were incubated for at least 2 h at room temperature. The reaction was initiated by the addition of 5 mM ATP/5 mM MgCl2 to all wells.
The rate of ATP hydrolysis was determined indirectly by monitoring the conversion of NADH to NAD+ which results in a loss of sample absorbance at 340 nM. The assay was performed in 96-well format and absorbance was measured over a 10 min time course using a Biotek Synergy H1 Plate Reader. Mean velocities were extrapolated for each time course and plotted as a function of either ATP or RNA concentration. These data were then fit to the quadratic solution of the Briggs–Haldane equation:
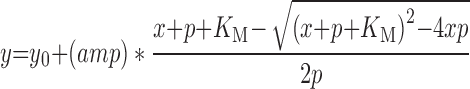 |
(1) |
Where y0 = basal activity, defined as background catalytic velocity observed in the absence of RIG-I, amp = vmax – y0 = kcat, x = total ATP or RNA concentration, P = total protein concentration and KM is the Michaelis constant for the variable substrate.
Equilibrium fluorescent RNA binding assays
Binding assays were carried out in 384-well plate format. Briefly, fluorophore-labeled hairpin RNA was diluted into binding buffer (25 mM MOPS pH 7.4, 150 mM NaCl, 5 mM DTT, 2 mM MgCl, 0.01% Triton X-100) to a concentration of 2 nM. The RIG-I protein construct of interest was then diluted into binding buffer over a 12-pt series of concentrations and mixed 1:1 with RNA samples (final RNA concentration of 2 nM) to a volume of 20 μl. Final RIG-I concentrations varied from 1.5 to 1500 nM. Samples were equilibrated at room temperature for 2 h. Fluorescence polarization was measured using a Biotek Synergy H1 plate reader. Samples were excited through a bandpass filter at 485/20 nM and fluorescence emission was measured through a bandpass filter at 528/20 nM. Polarization was calculated using the following Equation (2):
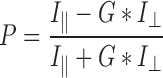 |
(2) |
Where I∥ is the intensity of the fluorescent light parallel to the plane of excitation, I⊥ is the intensity of fluorescent light perpendicular to the plane of excitation and G is an empirically determined correction factor accounting for instrumental bias toward the detection of horizontally polarized light; in this case G = 0.87.
An individual experiment consisted of two replicates of each protein concentration for which polarization measurements were taken 3 times, yielding 6 values for each condition. The mean polarization values were then plotted against protein concentration and fit to a one-site total binding Equation (3):
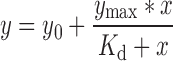 |
(3) |
Where y0 represents the polarization value when [enzyme] = 0 nM, ymax represents the polarization achieved at a saturating enzyme concentration, and Kd is the dissociation constant. Three experiments were performed for each RIG-I construct, with each reported Kd value representing the mean and standard deviation across these experiments.
RNA dissociation rate constant measurements
To obtain RNA dissociation rate constants (koffRNA) for RIG-I, stopped-flow fluorescence spectroscopy was employed. Stopped-flow experiments were performed in stopped flow buffer (25 mM HEPES pH 7.4, 150 mM NaCl, 2 mM DTT, 0.1 mg/ml BSA) at 24°C using a Kintek Auto-SF stopped-flow instrument supplied with a 150 W xenon arc lamp. For detection, the Cy3-labeled hairpin RNA was excited at 515 nm and the fluorescence emission was monitored at ≥570 nm using a 570 bandpass filter (Newport Corporation). Briefly, RIG-I was pre-incubated with Cy3-labeled hairpin RNA in equimolar amounts at room temperature for 2–6 h to form a protein–RNA complex. The protein-Cy3 RNA complex was then rapidly mixed with a 100-fold excess of unlabeled hairpin RNA for a specified period of time in which 2000 points were collected. For experiments with nucleotide analog, adenosine 5΄-[γ-thio]triphosphate (ATPγS) (Sigma-Aldrich) and MgCl2 were included with the trap RNA and rapidly mixed with the protein–Cy3 RNA complex. The average fluorescence measurements (4–6 traces) for each condition were then used in data analysis.
Data were fit using non-linear regression to a double exponential Equation (4) using GraFit 5,
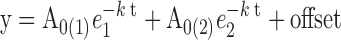 |
(4) |
where A is the amplitude, k is the rate constant, t is the reaction time (s) and the offset is the fluorescence value (V) of free Cy3-RNA.
MANT-ATP binding
Using stopped flow fluorescence spectroscopy, RIG-I binding to nucleotide was measured via Förster resonance energy transfer from RIG-I (λex = 290 nm) to the MANT-ATP (Invitrogen Life Technologies) (λem > 400 nm). RIG-I (1 μM) bound to the specified RNA hairpin (1 μM) in buffer A (see above) was mixed with varying concentrations of mant-nucleotide under pseudo-first order conditions (>4x [RIG-I]) to measure the observed association rate constant. Upon binding (λex = 290 nm), the change in fluorescence was monitored through a 400 nm long-pass filter using the Kintek Auto-SF stopped flow, and the resulting traces were fit to a double exponential Equation (4, above). The kobs (k1) corresponding to the initial binding event (obtained in triplicate) is then plotted versus [mant-nucleotide] and fit to a linear Equation (5) using GraFit 5,
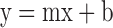 |
(5) |
where the intercept y is koff and the slope m is kon. A calculated Kd can then be obtained (koff/kon) using the experimentally derived koff and kon values.
RESULTS
Mutations within the RIG-I ATPase active-site influence efficiency and fidelity of signaling
To screen for RIG-I mutants that display aberrant signaling behavior, we used a cell-based assay to evaluate the activity of RIG-I variants that contain mutations within conserved helicase motifs (Figure 1) (9,22). To identify highly active mutants that stimulate signaling in the absence of added viral RNA (which might represent a form of constitutive activity, or an acquired ability to signal from host RNA), we transfected HEK 293T cells with a set of mutant RIG-I constructs and an IFN-β/firefly luciferase reporter gene, then measured luciferase activity in the absence of added ‘target’ RNA (Figure 2A). In parallel, a second set of transfected cells was challenged with the RNA target ligand poly(I:C) (Figure 2B). The RIG-I variants tested contain mutations within a diverse set of conserved motifs that contribute to ATP binding and hydrolysis, specifically ATPase Motif I, II, III and the Q motif. These include the recently reported mutation E373Q, which is located in motif II (29). In addition, we mutated an amino acid that is conserved only within the RLR family, and which places a phenylalanine close to the site of ATP binding (Phe 241). A few of these mutants display slightly elevated levels of RIG-I signaling in the absence of transfected, activating RNA (Figure 2A). However, one particular mutant (S411A) displayed high levels of IFN induction under these conditions, suggesting robust constitutive signaling activity or activity that is stimulated by endogenous cellular RNA (Figure 2A). Intriguingly, many of the mutants tested also displayed enhanced signaling in the presence of poly(I:C), suggesting greater RNA-stimulated activity than is observed in WT RIG-I (Figure 2B).
Figure 1.

Domain architecture of RIG-I: Schematic representation of the RIG-I domains. RIG-I is comprised of four major domain groups including the CARDs (black), and RNA-stimulated ATPase core (green), a helical regulatory and binding domain inserted into the second lobe of the ATPase called HEL2i (cyan), and a triphosphate recognition and RNA binding domain at the c-terminus annotated the CTD (orange). Hel1 of the ATPase core contains four conserved motifs, motifs I, II, III and the Q-motif. Residues in these motifs are necessary for proper RIG-I function, and the sites of disease related mutants, E373 and S411, are indicated (*).
Figure 2.

Signaling activities of RIG-I variants containing mutations within conserved ATPase motifs. (A) IFNβ induction in HEK 293T cells without the transfection of exogenous RNA. (B) IFNβ induction in HEK 293T cells that are stimulated with Poly(I:C).
The S411A variant contains a mutation within helicase Motif III, which is a highly conserved alcohol–alanine–alcohol motif that is located within the ATPase core of all SF2 helicase proteins. Mutations in Motif III influence both RNA binding and ATPase activity of DEAD-box proteins and related enzymes (37,38). Specifically, Motif III is thought to mediate communication between the nucleic acid binding and ATPase core domains (39,40), thereby coordinating the functions of RNA and ATP ligands within SF2 proteins. While biochemical studies are generally consistent with this model, until now, there has been no direct physical or structural evidence that Motif III couples RNA binding with ATP binding and hydrolysis. Consistent with a key role for this highly conserved region, many Motif III mutants have pathogenic phenotypes in SF2 helicase subfamilies, including RIG-I (vide infra).
Motif III is critical for proper enzymatic and signaling activities of RIG-I
Given the striking behavior of the S411A mutant and the fact that Motif III mutants are implicated in coordination between RNA and ATP binding, we reasoned that enzymological studies on the S411A mutant might reveal a physical basis for dysregulated RIG-I signaling and its contribution to autoimmune disorders.
As a first step in understanding the interplay between S411A and ATP, we measured the steady state ATP hydrolysis activities of WT RIG-I and S411A when stimulated by 5΄ppp10L, which has been previously shown to optimally activate RIG-I ATPase and signaling activity (22,41). The RNA-stimulated ATPase activity of S411A is reduced relative to WT RIG-I, with kcat values of 5.2 and 18.2 s−1, respectively (Figure 3A, Table 1), suggesting that defects in ATPase activity of S411A are on the rate of catalysis. The ATPase activity of S411A is similarly reduced when stimulated by OH10L (data not shown), which was previously shown to activate WT RIG-I ATPase, but not signaling activity (22,41). To determine whether the reduction in kcat is attributable to impaired RNA binding by S411A, we measured the equilibrium dissociation constant of S411A for a FAM-labeled 10 base pair RNA hairpin (OH10L) using fluorescence anisotropy. We find that S411A and WT RIG-I have similar affinities for OH10L (23 and 17 nM, respectively, Table 1), indicating that the reduction in S411A ATPase activity is attributable to a defect within the ATPase active site and that it is not the result of an inability to bind RNA.
Figure 3.

S411A enzymatic and signaling activities differ from WT RIG-I. (A) Steady state ATP hydrolysis by wild type and S411A RIG-I proteins stimulated with varying concentrations of the RNA hairpin 5΄ppp10L. (B) IFN-β induction in HEK 293T cells transfected with the indicated amount (ng) of the constitutive expression plasmid pUNO-hRIG-I containing either wild type or mutant RIG-I constructs. Cells expressing the indicated construct were challenged by transfection of 5΄ppp10L. (C) IFN-β induction of WT RIG-I and S411A in HEK-293T cells challenged with 5΄OH10L, 5΄ppp10L and mock. (D) IFN-β induction in HEK 293T cells transfected with the indicated amount (ng) of the constitutive expression plasmid pUNO-hRIG-I containing either wild type or mutant RIG-I constructs. Cells expressing the indicated construct were challenged by transfection of OH10L.
Table 1. Table of Measured Biophysical Constants.
| Construct | WT RIG-I | S411A | S411L |
|---|---|---|---|
| Steady state ATPase activity | |||
| 5΄ppp10L: | |||
| k cat (ATP/RIG-I•s) | 18.2 ± 0.3 | 5.1 ± 0.4 | Inactive |
| K M,RNA (nM) | 10.0 ± 1.0 | 5.0 ± 1 | Inactive |
| Maximum IFN-β Production | |||
| 5΄ppp10L (Firefly Luc/Renilla Luc) | 20.1 ± 1.7 | 56.6 ± 10.8 | 7.8 ± 1.5 |
| 5΄OH10L (Firefly Luc/Renilla Luc) | 3.8 ± 0.3 | 19.5 ± 3.8 | 0.2 ± 0..2 |
| PIC (Firefly Luc/Renilla Luc) | 59.2 ± 15 | 209.8 ± 17 | 1.8 ± 0.4 |
| Mock (Firefly Luc/Renilla Luc) | 0.99 ± 0.4 | 3.5 ± 0.5 | 0.2 ± 0.01 |
| MANT-ATP binding | |||
| 5΄ppp10L (μM) | 72 ± 3 | 82 ± 6 | 110 ± 8 |
| OH10L (μM) | 136 ± 6 | 75 ± 5 | – |
| Equilibrium RNA binding | |||
| OH10L (nM) | 17 ± 1.0 | 23 ± 1.8 | 41 ± 1.8 |
Biophysical constants for binding, catalysis and cell-based signaling assays performed for wild type, S411A and S411L constructs of RIG-I. Values represent mean ± SD (n = 3). See Materials and Methods for details of the experimental setups and data analysis used in each assay.
Several concentrations of each protein expression plasmid were transfected prior to dsRNA challenge in order to compare the dose-dependence of signaling by the wild type and S411A RIG-I constructs. We observe that S411A elicited a higher level of RNA-stimulated IFN-β promotor response at each of the RIG-I plasmid concentrations tested relative to WT (Figure 3B, Table 1). Interestingly, when the cells were challenged with a vehicle control and a 5΄-hydroxyl 10 base pair hairpin (OH10L), we also observed enhanced IFN- β promoter activation by S411A (Figure 3C, Table 1), albeit at lower levels than for 5΄ppp10L. As expected, wild-type RIG-I is not activated by vehicle control or OH10L (Figure 3C). Increasing the concentration of S411A in cells that are challenged with OH10L resulted in a concomitant increase in signaling activity (Figure 3D). Unlike WT RIG-I, high concentrations of the S411A mutant induce robust signaling in the presence of the suboptimal OH10L RNA (Figure 3D). This suggests that S411A is activated by different types of RNA, and specificity is reduced for the 5΄ triphosphorylated, virus-like RNA constructs that activate WT RIG-I. In contrast, WT RIG-I exhibits minimal IFN-β promoter activation, even at higher levels of protein expression (Figure 3D). Importantly, these values do not increase with increasing RIG-I concentration, and they are much lower than those observed with S411A, indicating that OH10L does not readily activate WT RIG-I signaling activity. Taken together, these data indicate that S411A signaling activity is improperly regulated, that S411A is activated by diverse RNA molecules, and that it is no longer selectively activated by viral RNA determinants.
Enhanced signaling by S411A on 5΄ppp10L is not caused by enhanced ATP binding
Previous studies have shown that ATP binding affinity, rather than ATP hydrolysis activity, is correlated with RIG-I signaling (22). We therefore hypothesized that the higher level of IFN-β promoter activation observed in S411A transfected cells might be due to tighter ATP binding by this mutant. Using a stopped flow fluorescence binding assay (see Materials and Methods), we measured the S411A binding affinity for ATP. To evaluate ATP binding, we monitored the association of S411A with a fluorescent ATP analog, MANT-ATP, in the presence of the 5΄ppp10L ligand using stopped flow fluorescence spectroscopy (Figure 4A). Using the observed rate constants, we extrapolated the apparent dissociation constant (Kd) for MANT-ATP for S411A (Figure 4A, Table 2). We observed that S411A binds MANT-ATP similarly to WT RIG-I, with Kd values of 82 and 72 μM for S411A and WT RIG-I (22), respectively. These results indicate that enhanced signaling by S411A is not attributable to an increase in ATP binding activity.
Figure 4.

RNA and ATP binding activities of S411A. (A) Average linear fit of [mant-ATP] versus kobs for WT RIG-I bound to 5΄ppp10L (black) and S411A bound to 5΄ppp10L (red), showing the [mant-ATP] dependence of the observed rate constants. Plotted kobs values are mean ± SD (n = 3). The kon (slope) and koff (intercept) values were derived from three independent trials and averaged. Using these values (koff/kon), a Kd was calculated for each protein/RNA combination. WT RIG-I/5΄ppp10L: koff = 110.0 ± 5, kon = 1.5 ± 0.1, Kd(calc.) = 72 ± 4 μM. S411A/5΄ppp10L: koff = 137.4 ± 10., kon = 1.7 ± 0.2, Kd(calc.) = 82 ± 6 μM. (B) koffRNA traces for S411A. Four traces monitoring displacement of S411A from 5΄ppp10L (black) were averaged and fit to a double exponential equation. Also shown are koff traces in the presence of 3 mM ATPγS (red) and 3 mM ADP (blue). (C) Average linear fit of [mant-ATP] versus kobs for WT RIG-I bound to OH10L (black) and S411A bound to OH10L (red), showing the [mant-ATP] dependence of the observed rate constants. Plotted kobs values are mean ± SD (n = 3). Kd values were calculated as in (A). WT RIG-I/OH10L: koff = 162.8 ± 7, kon = 1.2 ± 0.2, Kd(calc.) = 136 ± 6 μM. S411A/OH10L: koff = 135.7 ± 10, kon = 1.8 ± 0.2, Kd(calc.) = 75 ± 5 μM. (D) Bar plot of extrapolated dissociation constants for MANT-ATP binding by wild type and S411A RIG-I proteins bound to 5΄ppp10L and OH10L.
Table 2. RNA dissociation rate constants.
| Protein | Nucleotide | k 1 (× 10−3 s−1) | Amplitude | k 2 (× 10−3 s−1) | Amplitude |
|---|---|---|---|---|---|
| S411A | None | 45 ± 0.2 | 52% | 4 ± 0.1 | 48% |
| ATPγS | 68 ± 0.9 | 42% | 2 ± 0.04 | 58% | |
| ADP | 70 ± 0.9 | 62% | 6 ± 0.1 | 38% | |
| S411L | None | 27 ± 0.2 | 52% | 2 ± 0.04 | 48% |
| ATPγS | 210 ± 1 | 81% | 10 ± 0.1 | 19% |
Rate constants for the dissociation of S411A and S411L measured using stopped-flow fluorescence spectroscopy as described in Materials and Methods. Dissociation was measured from a 5΄ppp10L hairpin RNA in all cases. The data were fit to a biphasic exponential decay equation. Values represent mean ± SD (n = 3).
RNA dissociation from S411A is not stimulated by nucleotide binding
We have previously shown that ATP binding facilitates RIG-I turnover by stimulating RNA dissociation (22). The ATP-dependent release of RIG-I from RNA may contribute to a proofreading mechanism by reducing the ability of RIG-I to bind and signal from non-pathogenic RNAs. Because S411A exhibits enhanced signaling activity on both 5΄ triphosphorylated and hydroxylated RNAs, it is possible that this proofreading mechanism has been disrupted. To investigate the effects of nucleotide binding on RNA dissociation, microscopic rate constants for the dissociation of S411A (koffRNA) from 5΄ppp10L were determined either alone or in the presence of various nucleotides and nucleotide analogs, as previously determined for WT RIG-I (22). Briefly, koffRNA values were measured using stopped flow fluorescence spectroscopy, with a pre-bound S411A:Cy3-RNA complex that was rapidly mixed with an excess of unlabeled RNA, resulting in the release of S411A from Cy3-RNA and a concomitant decrease in fluorescence intensity (see Materials and Methods, Figure 4B).
The koffRNA for S411A was first measured in the absence of nucleotide to establish a baseline rate constant for dissociation. The observed time courses exhibited biphasic behavior and were best fit to a double exponential equation (Figure 4B). S411A dissociates slowly from 5΄ppp10L, with koff values of 45 ± 0.2 × 10−3 s−1 and 4.0 ± 0.1 × 10−3 s−1, respectively (Figure 4B, Table 2). These rate constants are similar to those observed for WT RIG-I (22), further validating the similarity in RNA binding behavior between wild type and mutant.
Next, we measured the koffRNA in the presence of different nucleotides to evaluate the effects of ATP binding. As above, we rapidly mixed pre-formed RIG-I:Cy3-RNA complex with an excess of trap RNA with saturating amounts of nucleotide. We observe that the presence of nucleotide did not significantly change the rate of dissociation from 5΄ppp10L by S411A (Figure 4B, Table 2). By contrast, the presence of either ATPγS or ADP causes a 5-fold increase in the rate of dissociation from 5΄ppp10L by WT RIG-I (22). These data demonstrate that ATP binding does not influence S411A dissociation from RNA. We therefore conclude that ATP binding does not act to challenge the S411A interaction with RNA in order to promote dissociation from off-target RNAs, as is the case for WT RIG-I. Thus, S411A effectively lacks the ATP-dependent proofreading mechanism that enables RIG-I to sample between different RNA molecules, thereby enabling S411A to signal from low affinity off-target sites, such as internal duplexes or non-triphosphorylated ends, while also promoting increased signaling on target, triphosphorylated RNAs.
S411A binds ATP more tightly in the presence of suboptimal ligands
In the presence of the 5΄ppp10L ligand, S411A binds ATP with an affinity that is similar to WT RIG-I (Figure 4A), but it does not exhibit WT levels of ATPase activity (Figure 3A). Additionally, we observe that ATP binding does not stimulate RNA dissociation by S411A (Figure 4B), suggesting the loss of a proof-reading function. To test whether S411A displays deviations in ATP binding when it is bound to suboptimal RNA ligands, we tested ATP binding to S411A in complex with OH10L. Using the stopped flow fluorescence assay described above, we measured the association of S411A and WT RIG-I with MANT-ATP when bound to OH10L (Figure 4C). The extrapolated Kd values reveal 2-fold greater affinity of S411A for MANT-ATP relative to WT (Figure 4D, Table 1). Furthermore, the affinities for MANT-ATP are similar when S411A is bound to OH10L and 5΄ppp10L, with Kd values of 82 and 75 μM, respectively (Figure 4D, Table 1). WT RIG-I, however, has a weaker affinity for MANT-ATP when bound to OH10L relative to 5΄ppp10L, with Kd values of 110 and 72 μM (Figure 4D, Table 1). We therefore conclude that ATP binding is no longer coupled to RNA binding in RIG-I constructs that contain mutations within motif III, as observed for related DEAD-box proteins. This indicates that S411A is unable to distinguish between pathogenic and endogenous RNA at the coupled ATP binding step. Furthermore, increased binding of ATP by S411A on non-triphosphorylated RNA, taken together with the lack of ATP-induced RNA dissociation, should readily promote signaling on non-pathogenic RNAs in vivo, as evidenced by the increased signaling behavior of S411A in mock and OH10L treated cells.
Motif III mutant S411L is annotated in tumor samples and exhibits impaired enzymatic and signaling activity
Given our results with S411A, we set out to determine whether RIG-I motif III mutants are associated with disease. Using the UCSC Genome Browser (42), we found two observations of a serine 411 mutation to leucine (S411L) in large intestine and malignant melanoma tumor samples, annotated in the COSMIC and NCBI ClinVar databases (43,44). We were therefore interested in examining whether S411L behaves similarly to S411A and whether its behavior can provide a mechanistic link between RIG-I dysregulation and cancer.
We first measured the ATP hydrolysis activity of S411L when activated by 5΄ppp10L using the coupled ATPase assay, and were unable to detect any measurable ATPase activity (Figure 5A, Table 1). We then repeated this experiment using RNA duplexes of varying lengths to ensure that the length preference was not different from wild type, and again found that all RNAs were unable to stimulate S411L ATPase activity (data not shown). Fluorescence anisotropy RNA binding assays show that RNA binding by S411L is not impaired. Indeed, S411L binds the 10-mer hairpin with an affinity similar to WT (Table 1). Therefore, while S411L is capable of binding RNA, this activity does not stimulate ATP hydrolysis.
Figure 5.

S411L displays impaired signaling activity due to enhanced ATP-induced RNA dissociation. (A) Steady state ATP hydrolysis by wild type and S411A RIG-I proteins stimulated with varying concentrations of the RNA hairpin 5΄ppp10L. (B) IFN-β induction of WT RIG-I and S411L in HEK-293T cells challenged with 5΄ppp10L, low molecular weight poly(I:C) (PIC) and mock. (C) Average linear fit of [mant-ATP] versus kobs for WT RIG-I bound to 5΄ppp10L (black, as in Figure 4A) and S411L bound to 5΄ppp10L (blue), showing the [mant-ATP] dependence of the observed rate constants. Plotted kobs values are mean ± SD (n = 3). The kon (slope) and koff (intercept) values were derived from three independent trials and averaged. Using these values (koff/kon), a Kd was calculated. S411L/5΄ppp10L: koff = 164.4 ± 12, kon = 1.5 ± 0.3, Kd(calc.) = 110 ± 8 μM. (D) koffRNA traces for S411L. Four traces monitoring displacement of S411L from 5΄ppp10L (black) were averaged and fit to a double exponential equation. Also shown are koff traces in the presence of 3 mM ATPγS (red).
We next investigated the ability of S411L to induce an RNA-stimulated IFN-β response in cells. When S411L is challenged by 5΄ppp10L, a 3-fold loss in IFN-β activation is observed relative to WT (Figure 5B). As in the case of WT, S411L signaling is not significantly activated by OH10L (data not shown). Intriguingly, we observe a striking loss (31-fold) in IFN-β activation when S411L is challenged by poly I:C (Figure 5B). This contrasts starkly with the signaling behavior of S411A, which is hyper-activated by all RNAs tested. The behavior of S411L is reminiscent of Motif I K270X mutations, which lack ATP hydrolysis activity and are nonetheless capable of signaling on short, idealized ligands such as 5΄ppp10L, but incapable of signaling on longer RNAs (22). One possible explanation for the reduction in cellular S411L activity might be that the protein misfolds or is inappropriately expressed. However, Western blots show that the reduction in activity is not due to any loss in S411L protein expression level (Supplementary Methods, Figure S1). The contrasting behaviors of S411A and S411L indicate that motif III mutations can disrupt regulated signaling activation in different ways, underscoring the centrality of this motif in coordinating functional ATP and RNA binding by SF2 proteins.
ATP binding by S411L enhances RNA dissociation
We next sought to understand the role of ATP binding in the mechanism of RNA recognition and signaling by S411L. It has been shown that ATP binding, rather than hydrolysis, is most strongly correlated with IFN-β activation, particularly in cells that are challenged by short, triphosphorylated RNA duplexes (22). We therefore hypothesized that severely impaired ATP binding by S411L might explain its reduction in signaling capacity (Figure 5B). However, S411L in complex with 5΄ppp10L readily binds ATP (Figure 5C). The extrapolated Kd value is 109 μM, which is only slightly weaker than the WT Kd of 73 μM, and is tighter than the Kd measured for the signaling competent, 5΄ppp30L-bound RIG-I (22). Thus, the slight decrease in ATP binding affinity cannot solely account for the significant reduction in IFN-β promoter activation in cell culture. Like S411A, aberrant signaling by S411L cannot be fully explained by atypical ATP binding activity.
Because the enhanced signaling activity of S411A is attributable to an increased lifetime on RNA due to a lack of ATP-induced RNA dissociation, we hypothesized that the observed reduction in S411L signaling might also be linked to aberrant dissociation from RNA. First, we measured the koffRNA for S411L in the absence of nucleotide and observed that S411L, like WT RIG-I, dissociates from 5΄ppp10L slowly, with a rate of 27 ± 0.2 × 10−3 s−1 (Figure 5D, Table 2). We next measured the koffRNA in the presence of nucleotide to evaluate the effects of ATP binding, and determined that ATPγS stimulates koffRNA by 7.8-fold (Figure 5D, Table 2). This effect is significantly greater than that observed for WT, and demonstrates a hyper-stimulatory effect of ATP on RNA dissociation by S411L. We therefore conclude that ATP binding weakens the S411L interaction with all RNAs, promoting rapid dissociation and resulting in a highly stringent proofreading mechanism that leads to a severe reduction of S411L signaling, even on pathogenic RNAs. This is in stark contrast to S411A, where ATP binding does not stimulate dissociation from any RNA, and thus promotes enhanced signaling even on non-pathogenic RNAs. Thus, the proofreading mechanisms of S411A and S411L are dysregulated in opposing ways: S411A is too promiscuous (signaling all the time) and S411L is too discriminating (rarely signaling).
Motif III mutations disrupt conserved residue contacts
In order to understand how mutations in Motif III influence the behavior of RIG-I, we examined available RIG-I crystal structures and analyzed the molecular interaction networks that involve Ser411. Structural analysis was carried out with PyMol (45). In many of the structures, including the apo-form (PDB ID: 4A2W) (10), the RNA bound form (PDB ID: 2YKG, 3ZD6, 4BPB, 5F9F, 5F9H) (9,41,46) and the RNA–ADP bound form (PDB ID: 4AY2) (20), Ser411 interacts directly with Met761 (762 in duck), which is a residue within helix 1 of the Pincer domain. Ser411 and Met761 form a conserved hydrogen bond that ranges from 2.9 to 3.4 Å in length (Figure 6A). The location of the Ser411-Met761 interaction and its proximity to larger networks of hydrogen bonding suggest that this interaction facilitates the proper orientation of pincer helix 1.
Figure 6.

The molecular interaction network of S411. (A) Hydrogen bonding between Ser411 and Met761 (PDB ID 5F9H). Hydrogen bonds formed by conserved residues in Motif II (colored in wheat) and Motif III (colored in light blue) result in a rigid unit. Motif Va (marine), Motif I (yellow) are shown to indicate the relative position of the motifs around ADP (cyan, placed according to the ADP location in PDB ID 5E3H). (B) Gln726 of Helix721-730 (colored in lemon) mediates the hydrogen bond network that coordinates Motif II (wheat), Motif III (light blue) and Motif Va (marine), which are essential for the ATPase active site. In helix 721–730, residue Arg728 contacts Glu758 in pincer helix I. A hydrophobic cluster (Val721, Ile722, Ile725 and Gln726) on the surface of Helix721-730 is adjacent to Ser411. As in Figure 6A, ADP is placed according to PDB ID 5E3H.
In the hyperactive S411A mutant, the Ser411-Met761 interaction is eliminated, which disrupts the structural linkage between RNA and ATP binding. As a result, RNA dissociates from S411A slowly, in a process that is insensitive to the binding of ATP. Consequently, S411A has a long residence time on all RNA targets and, lacking a proof-reading mechanism, can signal from many targets that it binds.
By contrast, the S411L cancer mutation would reorganize the hydrophobic core adjacent to the ATPase active site. The leucine residue of S411L is likely to pack against the hydrophobic pocket formed by Val721, Ile722 and Ile725 (Figure 6B). As a result of this rearrangement, the RNA complex is hypersensitive to ATP binding, and RNA is rapidly released. Analysis of recent crystal structures also explains why ATP hydrolysis activity is inhibited by S411L. These structures (exemplified by 5F9H (46)) reveal the presence of a short alpha-helix (Helix 721–730), which bridges the conserved ATPase motifs with Motif III, and with the Pincer 1 helix. Specifically, helix 721–730 makes extensive polar contacts with Pincer helix 1, ATPase motifs V and Va, the DECH motif and even with the backbone of bound RNA (Figure 6B, Table 3). When it is well-ordered, helix 721–730 plays a key role in organizing the ATPase active site and it orients the HEL2-HEL2i domains closer to RNA, resulting in a more compact conformation. The interaction network involving helix 721–730 also enables Arg664 to interact with the γ-phosphate of 5΄-triphosphorylated RNA duplexes, suggesting a direct role for these residues in differentiating between self and non-self RNA ligands. S411L is expected to draw helix 721–730 toward residue 411 via non-polar interactions, thereby inducing a rearrangement of the ATPase active site, resulting in a protein that can bind ATP, but is incapable of hydrolysis.
Table 3. Polar interaction involving residues in helix 721–730 (PDB ID: 5F9H).
| Helix 721–730 | Residues interacting with Helix 721–730 |
|---|---|
| G719 | N754 (N-terminal pincer helix) |
| R728 | E758 (N-terminal pincer helix) |
| N720 | RNA backbone |
| K723 | V699 (motif V) |
| Q726 | D701 (motif Va) |
| E373 (DECH motif) | |
| R730 | Q708 |
| R732 | Q708 |
Although Ser411 does not contact RNA or nucleotide directly, structural analysis reveals that it is embedded within an interaction network that includes residues within the pincer domain, helix 721–730 and even the ATPase core. Mutations within motif III, such as S411A and S411L, disrupt these conserved networks and decouple RNA and ATP binding, resulting in atypical enzymatic and signaling activities.
DISCUSSION
Through a series of mutational, enzymological and in-cellulo signaling studies, we have demonstrated a pathway for RIG-I proof-reading, revealing one mechanism by which RIG-I can differentiate pathogenic RNA molecules from the vast assortment of host RNA molecules that are present in the mammalian cytoplasm. Many studies have suggested that RIG-I has a selection mechanism for avidly binding viral RNA while avoiding activation by host RNA molecules and they have shown that this process is somehow linked with the function of ATP (30,47). However, the physical basis for this pathway had not been demonstrated or measured until now. From a screen of RIG-I motif variants, we identified a RIG-I mutation (S411A) that displays strikingly high levels of unregulated signaling activity, and we determined the molecular basis for its loss of RNA discrimination.
Through analysis of S411A and other mutants, we show that ATP binding serves to release RNA molecules from WT RIG-I (22), and that this allows the receptor to sample among RNA species until it identifies a high affinity target that efficiently stimulates signaling (ideally a blunt, 5΄-triphosphorylated RNA duplex). When the coordination between the RNA and ATP binding sites is broken by mutations such as S411A, RIG-I becomes stuck to inappropriate ligands (such as cellular RNA or transfected non-triphosphorylated RNA), triggering constitutive, aberrant interferon induction. In addition, we demonstrate that proof-reading can be specifically defined and directly measured by monitoring the release of RNA from RIG-I. This is important because mutations within the conserved ATPase active site can produce a variety of pleiotropic functional defects, and these are not necessarily linked to the ability of RIG-I to discriminate among RNAs in the cell.
Mutational studies on S411 demonstrate that defects in ATP-coupled RNA binding can dysregulate signaling in several ways. In mutant S411A, the physical connection between the RNA and ATP binding sites has been severed, and the normal anticooperativity that is provided by ATP binding is lost. As a result, S411A signals promiscuously and efficiently even without the exogenous introduction of high-affinity RNA ligands. RIG-I mutants with this type of behavior are expected to induce autoimmunity, as suggested for the E373A mutant that is linked with SMS (28). Patients with the S411A mutation have not been identified yet, but given the extraordinary loss of proof-reading by S411A, it may in fact be a lethal mutation.
The opposite behavior is seen for S411L, which has undergone slight rearrangements in its hydrophobic core that enhance the normal anticooperativity provided by ATP and result in hyperactive proof-reading. Nucleotide binding causes unusually rapid release of RNA from S411L, thereby eliminating signaling from off-target RNAs and reducing signaling from optimal targets such as triphosphorylated duplexes. This causes an overall reduction in S411L signaling, and it is therefore not surprising that this mutation is found in tumors, as it would help maintain the characteristic immunosuppressive tumor microenvironment (48).
These results have implications for a new generation of chemotherapies in which RNA ligands are used to up-regulate RIG-I signaling in tumors. Immunostimulatory RNA molecules such as polyI:C (49) and triphosphorylated RNA duplexes (29) have potent antitumor effects that are believed to be mediated by RIG-I-dependent IFN induction. However, this approach assumes a tumor microenvironment in which RIG-I has not undergone mutation. Tumors containing cells with the S411L mutation, or similar changes, will not respond effectively to RNA adjuvants, and they may be the product of resistance to cellular RNA ligands that become massively upregulated by standard cancer therapies. Only the strongest RIG-I activators (such as short, triphosphorylated hairpins as described here), are expected to promote an antitumor response in these cells.
It is possible that only mild defects in RIG-I signaling will be sufficiently functional to arise in human populations. An example is the E373Q RIG-I variant, which was shown to undergo activation by cellular ribosomal RNA, suggesting a proof-reading defect that leads to autoimmunity (28,29). This finding was significant due to similarities with the E373A mutant, which was identified in SMS patients. However, in our hands, E373Q (29) is only slightly more promiscuous than WT RIG-I, and its host-stimulated activity differs little from that of other RIG-I mutants. The most prominent defect in E373Q is a striking loss in overall RNA-stimulated RIG-I signaling, which is unsurprising given its central location within the enzymatic active-site. Given that proof-reading by E373A/Q was not directly monitored in previous studies, and that it is not unusually promiscuous relative to other mutants (particularly S411A), it is possible that SMS is caused by other aspects of RIG-I dysregulation in patients. This would not be without precedent, as ‘gain of function’ mutations in both MDA5 and TREX1, which were previously thought to be pathogenic and have a causal link with autoimmunity, are now known to play a peripheral role. For example, mutant variants of MDA5 are associated with Aicardi–Goutières syndrome (AGS), which has an upregulated interferon signature (50,51). However, other genetic and environmental factors contribute to the onset of AGS, as multicopy MDA5 transgenic mouse lines with chronically elevated levels of type I interferon do not initiate autoimmunity, although they exacerbate an ongoing autoimmune pathology (52). Similar conclusions have now been reached for mutants of the exonuclease TREX1, which is also associated with AGS (53).
Proof-reading in RIG-I, and probably other SF2 proteins, is controlled by a specific region of the protein known as Motif III, which has long been implicated in the coordination of RNA and ATP ligands by this protein family (37,38). Motif III contains a highly conserved set of three amino acids (usually SAT or TAS) that are found in all SF2 RNA-dependent ATPases. Although this region is frequently referred to as a ‘coupling motif’, a physical explanation for its ability to coordinate ligand binding has been lacking until now. Recent crystal structures of RIG-I show that Motif III is specifically sandwiched between Pincer domain 1, the ATP binding and hydrolysis motifs I, II, V and Va, and a novel helix (721–730) that interacts with all of these core motifs (and with RNA) (23,46). These structures show that mutation of Motif III residues will disrupt an intricate network of interactions that connect RNA and ATP binding sites, thereby eliminating the regulated control of RNA binding and disrupting downstream functions of SF2 motor proteins.
Taken together, the functional information gleaned from Ser411 mutations, combined with recent high resolution structures of RIG-I, provide a framework for understanding the physical basis of autoimmunity, and for the induction of certain cancers. More broadly, our findings provide a molecular explanation for the role of Motif III in coordinating RNA and ligand binding, explaining the basis for regulated, mechanical function of all SF2 family members.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank the members of the Pyle lab, especially Dr Srinivas Somarowthu and Dr Thayne Dickey, for discussion and insightful suggestions. A.M.P. would also like to thank Dr Paul DaSilva-Jardin for helpful discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Howard Hughes Medical Institute and the National Institutes of Health; M.F. and X.R. are Research Associates and A.M.P. is an Investigator with the Howard Hughes Medical Institute; NIH [RO1 AI089826 to A.K. and D.R.]. Funding for open access charge: Howard Hughes Medical Institute (Yale University).
Conflict of interest statement. None declared.
REFERENCES
- 1. Chen G., Shaw M.H., Kim Y.G., Nunez G.. NOD-like receptors: Role in innate immunity and inflammatory disease. Annu. Rev. Pathol. 2009; 4:365–398. [DOI] [PubMed] [Google Scholar]
- 2. Kawai T., Akira S.. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011; 34:637–650. [DOI] [PubMed] [Google Scholar]
- 3. Schlee M. Master sensors of pathogenic RNA - RIG-I like receptors. Immunobiology. 2013; 218:1322–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sun L., Wu J., Du F., Chen X., Chen Z.J.. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013; 339:786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoneyama M., Kikuchi M., Matsumoto K., Imaizumi T., Miyagishi M., Taira K., Foy E., Loo Y.M., Gale M. Jr, Akira S. et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005; 175:2851–2858. [DOI] [PubMed] [Google Scholar]
- 6. Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M., Taira K., Akira S., Fujita T.. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004; 5:730–737. [DOI] [PubMed] [Google Scholar]
- 7. Kang D.C., Gopalkrishnan R.V., Wu Q., Jankowsky E., Pyle A.M., Fisher P.B.. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu B., Peisley A., Richards C., Yao H., Zeng X., Lin C., Chu F., Walz T., Hur S.. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. 2013; 152:276–289. [DOI] [PubMed] [Google Scholar]
- 9. Luo D., Ding S.C., Vela A., Kohlway A., Lindenbach B.D., Pyle A.M.. Structural insights into RNA recognition by RIG-I. Cell. 2011; 147:409–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kowalinski E., Lunardi T., McCarthy A.A., Louber J., Brunel J., Grigorov B., Gerlier D., Cusack S.. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011; 147:423–435. [DOI] [PubMed] [Google Scholar]
- 11. Rawling D.C., Pyle A.M.. Parts, assembly and operation of the RIG-I family of motors. Curr. Opin. Struct. Biol. 2014; 25:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Luo D., Kohlway A., Pyle A.M.. Duplex RNA activated ATPases (DRAs): Platforms for RNA sensing, signaling and processing. RNA Biol. 2013; 10:111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vela A., Fedorova O., Ding S.C., Pyle A.M.. The thermodynamic basis for viral RNA detection by the RIG-I innate immune sensor. J. Biol. Chem. 2012; 287:42564–42573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seth R.B., Sun L., Ea C.K., Chen Z.J.. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005; 122:669–682. [DOI] [PubMed] [Google Scholar]
- 15. Peisley A., Wu B., Xu H., Chen Z.J., Hur S.. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature. 2014; 509:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schlee M., Roth A., Hornung V., Hagmann C.A., Wimmenauer V., Barchet W., Coch C., Janke M., Mihailovic A., Wardle G. et al. Recognition of 5΄ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity. 2009; 31:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weber M., Gawanbacht A., Habjan M., Rang A., Borner C., Schmidt A.M., Veitinger S., Jacob R., Devignot S., Kochs G. et al. Incoming RNA virus nucleocapsids containing a 5΄-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe. 2013; 13:336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baum A., Sachidanandam R., Garcia-Sastre A.. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:16303–16308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hornung V., Ellegast J., Kim S., Brzozka K., Jung A., Kato H., Poeck H., Akira S., Conzelmann K.K., Schlee M. et al. 5΄-Triphosphate RNA is the ligand for RIG-I. Science. 2006; 314:994–997. [DOI] [PubMed] [Google Scholar]
- 20. Luo D., Kohlway A., Vela A., Pyle A.M.. Visualizing the determinants of viral RNA recognition by innate immune sensor RIG-I. Structure. 2012; 20:1983–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hou F., Sun L., Zheng H., Skaug B., Jiang Q.X., Chen Z.J.. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011; 146:448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rawling D.C., Fitzgerald M.E., Pyle A.M.. Establishing the role of ATP for the function of the RIG-I innate immune sensor. Elife. 2015; 4:09391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jiang F., Ramanathan A., Miller M.T., Tang G.Q., Gale M. Jr, Patel S.S., Marcotrigiano J.. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011; 479:423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takahasi K., Yoneyama M., Nishihori T., Hirai R., Kumeta H., Narita R., Gale M. Jr, Inagaki F., Fujita T.. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell. 2008; 29:428–440. [DOI] [PubMed] [Google Scholar]
- 25. Civril F., Bennett M., Moldt M., Deimling T., Witte G., Schiesser S., Carell T., Hopfner K.P.. The RIG-I ATPase domain structure reveals insights into ATP-dependent antiviral signalling. EMBO Rep. 2011; 12:1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saito T., Hirai R., Loo Y.M., Owen D., Johnson C.L., Sinha S.C., Akira S., Fujita T., Gale M. Jr. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Loo Y.M., Gale M. Jr. Immune signaling by RIG-I-like receptors. Immunity. 2011; 34:680–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jang M.A., Kim E.K., Now H., Nguyen N.T., Kim W.J., Yoo J.Y., Lee J., Jeong Y.M., Kim C.H., Kim O.H. et al. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am. J. Hum. Genet. 2015; 96:266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lassig C., Matheisl S., Sparrer K.M., de Oliveira Mann C.C., Moldt M., Patel J.R., Goldeck M., Hartmann G., Garcia-Sastre A., Hornung V. et al. ATP hydrolysis by the viral RNA sensor RIG-I prevents unintentional recognition of self-RNA. Elife. 2015; 4:10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Louber J., Brunel J., Uchikawa E., Cusack S., Gerlier D.. Kinetic discrimination of self/non-self RNA by the ATPase activity of RIG-I and MDA5. BMC Biol. 2015; 13:54–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cordin O., Banroques J., Tanner N.K., Linder P.. The DEAD-box protein family of RNA helicases. Gene. 2006; 367:17–37. [DOI] [PubMed] [Google Scholar]
- 32. Cordin O., Tanner N.K., Doere M., Linder P., Banroques J.. The newly discovered Q motif of DEAD-box RNA helicases regulates RNA-binding and helicase activity. EMBO J. 2004; 23:2478–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Walker J.E., Saraste M., Runswick M.J., Gay N.J.. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982; 1:945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wincott F., DiRenzo A., Shaffer C., Grimm S., Tracz D., Workman C., Sweedler D., Gonzalez C., Scaringe S., Usman N.. Synthesis, deprotection, analysis and purification of RNA and ribozymes. Nucleic Acids Res. 1995; 23:2677–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zlatev I., Lackey J.G., Zhang L., Dell A., McRae K., Shaikh S., Duncan R.G., Rajeev K.G., Manoharan M.. Automated parallel synthesis of 5΄-triphosphate oligonucleotides and preparation of chemically modified 5΄-triphosphate small interfering RNA. Bioorg. Med. Chem. 2013; 21:722–732. [DOI] [PubMed] [Google Scholar]
- 36. Zlatev I., Manoharan M., Vasseur J.J., Morvan F.. Solid-phase chemical synthesis of 5΄-triphosphate DNA, RNA, and chemically modified oligonucleotides. Curr. Protoc. Nucleic Acid Chem. 2012; 50, doi:10.1002/0471142700.nc0128s50. [DOI] [PubMed] [Google Scholar]
- 37. Banroques J., Doere M., Dreyfus M., Linder P., Tanner N.K.. Motif III in superfamily 2 ‘helicases’ helps convert the binding energy of ATP into a high-affinity RNA binding site in the yeast DEAD-box protein Ded1. J. Mol. Biol. 2010; 396:949–966. [DOI] [PubMed] [Google Scholar]
- 38. Bamming D., Horvath C.M.. Regulation of signal transduction by enzymatically inactive antiviral RNA helicase proteins MDA5, RIG-I, and LGP2. J. Biol. Chem. 2009; 284:9700–9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Caruthers J.M., McKay D.B.. Helicase structure and mechanism. Curr. Opin. Struct. Biol. 2002; 12:123–133. [DOI] [PubMed] [Google Scholar]
- 40. Pause A., Sonenberg N.. Mutational analysis of a DEAD box RNA helicase: the mammalian translation initiation factor eIF-4A. EMBO J. 1992; 11:2643–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kohlway A., Luo D., Rawling D.C., Ding S.C., Pyle A.M.. Defining the functional determinants for RNA surveillance by RIG-I. EMBO Rep. 2013; 14:772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Forbes S.A., Bindal N., Bamford S., Cole C., Kok C.Y., Beare D., Jia M., Shepherd R., Leung K., Menzies A. et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011; 39:D945–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Landrum M.J., Lee J.M., Benson M., Brown G., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Hoover J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016; 44:D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schrödinger L. The PyMOL Molecular Graphics System. 2015; Version 1.8, Schrödinger, LLC.
- 46. Devarkar S.C., Wang C., Miller M.T., Ramanathan A., Jiang F., Khan A.G., Patel S.S., Marcotrigiano J.. Structural basis for m7G recognition and 2΄-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kato H., Takahasi K., Fujita T.. RIG-I-like receptors: cytoplasmic sensors for non-self RNA. Immunol. Rev. 2011; 243:91–98. [DOI] [PubMed] [Google Scholar]
- 48. van den Boorn J.G., Hartmann G.. Turning tumors into vaccines: co-opting the innate immune system. Immunity. 2013; 39:27–37. [DOI] [PubMed] [Google Scholar]
- 49. Palchetti S., Starace D., De Cesaris P., Filippini A., Ziparo E., Riccioli A.. Transfected poly(I:C) activates different dsRNA receptors, leading to apoptosis or immunoadjuvant response in androgen-independent prostate cancer cells. J. Biol. Chem. 2015; 290:5470–5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rice G.I., del Toro Duany Y., Jenkinson E.M., Forte G.M., Anderson B.H., Ariaudo G., Bader-Meunier B., Baildam E.M., Battini R., Beresford M.W. et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet. 2014; 46:503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Oda H., Nakagawa K., Abe J., Awaya T., Funabiki M., Hijikata A., Nishikomori R., Funatsuka M., Ohshima Y., Sugawara Y. et al. Aicardi-Goutieres syndrome is caused by IFIH1 mutations. Am. J. Hum. Genet. 2014; 95:121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Crampton S.P., Deane J.A., Feigenbaum L., Bolland S.. Ifih1 gene dose effect reveals MDA5-mediated chronic type I IFN gene signature, viral resistance, and accelerated autoimmunity. J. Immunol. 2012; 188:1451–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wolf C., Rapp A., Berndt N., Staroske W., Schuster M., Dobrick-Mattheuer M., Kretschmer S., Konig N., Kurth T., Wieczorek D. et al. RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA. Nat. Commun. 2016; 7:11752–11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.