


Abstract
LAGLIDADG homing endonucleases (LHEs) are a class of rare-cleaving nucleases that possess several unique attributes for genome engineering applications. An important approach for advancing LHE technology is the generation of a library of design ‘starting points’ through the discovery and characterization of natural LHEs with diverse specificities. However, while identification of natural LHE proteins by sequence homology from genomic and metagenomic sequence databases is straightforward, prediction of corresponding target sequences from genomic data remains challenging. Here, we describe a general approach that we developed to circumvent this issue that combines two technologies: yeast surface display (YSD) of LHEs and systematic evolution of ligands via exponential enrichment (SELEX). Using LHEs expressed on the surface of yeast, we show that SELEX can yield binding specificity motifs and identify cleavable LHE targets using a combination of bioinformatics and biochemical cleavage assays. This approach, which we term YSD-SELEX, represents a simple and rapid first principles approach to determining the binding and cleavage specificity of novel LHEs that should also be generally applicable to any type of yeast surface expressible DNA-binding protein. In this marriage, SELEX adds DNA specificity determination to the YSD platform, and YSD brings diagnostics and inexpensive, facile protein-matrix generation to SELEX.
INTRODUCTION
LAGLIDADG homing endonucleases (LHEs) are a class of naturally occurring endonucleases that are found within mobile introns and related genetic elements (1). LHEs typically recognize 20–22 base pair target sites and have been developed for applications in demanding genome editing applications due to their high specificity, capacity to generate recombinogenic 3΄ overhangs and compact, non-repetitive structure (2–6). Thus, LHEs excel in applications that require maximal recombination, or that require packaging or propagation (e.g. for gene drive, or generation of retroviral particles) that may preclude the use of CRISPRs, TALENs and ZFNs due to their repetitive elements, easily modified specificities or large sizes. Despite these unique attributes, broad application of LHEs in genome engineering has been limited by difficulties in re-engineering their cleavage specificity due to the high complexity of the protein surface that interacts with their DNA target sites; a typical LHE utilizes between 40 and 50 amino acid side chains to contact bases with negligible predictability.
Homology searches of genomic and metagenomic DNA sequence databases have revealed a substantial number of novel putative LHE open reading frames (ORFs) (7–10). Discovery of new LHEs is an important approach to advancing LHE technology, as the availability of LHEs with diverse recognition sequences provides alternative proximal ‘starting points’ for LHE respecification (7,11,12). Careful examination of genomic sequence adjacent to a mobile genetic element along with comparison to an allele that has not been invaded by the LHE's host mobile genetic element may reveal an LHE's native target site, given the mode of LHE propagation (13,14). However, this method of target site elucidation requires definition of mobile element borders: information that may frequently be unavailable, especially in the case of LHEs identified in partial or metagenomic sequence collections. Although improved bioinformatics methods for target determination have been recently published in an attempt to alleviate this bottleneck (8), the number of enzymes with the information required for their computations is still a minority. Consequently, the description and characterization of novel LHEs for use as scaffolds for target site respecification has been significantly limited.
To lessen our reliance on genome sequence analysis for identifying target sites, and thus better exploit the large number of uncharacterized LHEs identifiable in public sequence databases, we sought to develop a first principles method for determining the DNA binding target specificity of homing endonucleases. Here, we show that standard Systematic Evolution of Ligands by Exponential Enrichment Selection (SELEX) (15,16) protocols can be adapted to use yeast-surface displayed (YSD) LHEs, with the yeast acting as the solid support. We leveraged existing YSD binding and cleavage protocols to optimize a YSD-SELEX protocol and evaluate the selection process in real time, and in a multi-well format. We were then able to generate binding motifs for several novel LHEs identified from genomic sequence databases, and demonstrate that these motifs agree well with cleavage specificities and that YSD-SELEX can be used to identify cleavable target sites.
MATERIALS AND METHODS
Homing endonuclease homolog identification and construct generation
I-OnuI homologs were identified through a basic NCBI online BLAST search (17) of the nucleotide database using the I-OnuI protein sequence as a search query. Homologous proteins were found in the mitochondrial genomes of a wide variety of fungal species. The homing endonucleases were found to exist either as stand-alone ORFs on introns inserted into their host genes or as in-frame fusions to their host proteins. When the homing endonuclease sequences were located on introns with clearly annotated intron/exon boundaries, their target sites were easily predicted by removing the intron and joining the flanking sequence of the host gene. In the case of fusion proteins, a target could be predicted only through alignment to the same gene in a related species with no homing endonuclease present. The six enzymes in our test set include two fusion proteins (I-CpaMIIP: accession AAC24230, I-HjeMII: NP_570153). The remaining enzymes either did not have sufficient intron/exon boundary annotations (I-MveMIP AAW51686, I-OsoMI and I-OsoMII: AB027350) or did not cleave target sites predicted by the annotations provided (I-CpaMVP: AAB84210). Our set of homing endonucleases were named according to the conventions put forth by Roberts et al. (18); notably, a ‘P’ suffix denotes a putative nuclease without verified activity.
Our benchmark and test set homing endonucleases (amino acid sequences shown in Figure 2A) were synthesized by Genscript (Piscataway, NJ, USA) into the parent pETCON yeast surface display vector (Addgene plasmid #41522) between the NdeI and XhoI cloning sites. The pETCON vector allows for expression of a homing endonclease on the surface of yeast (EBY100 S. cerevisiae) as a fusion with N-terminal HA and C-terminal Myc epitope tags.
Figure 2.

SELEX homing endonuclease set. (A) Alignment of I-OnuI with benchmark (I-SmaMI, I-PanMI and I-GzeII) and test (I-HjeMII, I-OsoMI, I-OsoMII, I-CpaMIIP, I-CpaMVP and I-MveMIP, LHEs). Residues making direct contact with target site bases in the I-OnuI crystal structure, and the residues that align with them, are boxed in red to emphasize the diverse recognition sequences and mechanisms represented in the group. (B) The phylogram resulting from a ClustalW2 multiple alignment of the protein sequences of the set of LHEs in the SELEX experiments.
Yeast growth, surface display induction and flow cytometric expression analysis
To induce surface expression, yeast transformed with the vectors above were grown shaking in 1 ml SC media containing 2% glucose at 30°C for one day and seeded into 1 ml SC media containing 2% raffinose + 0.1% glucose and grown shaking at 30°C for one day, before 30 million yeast were washed and transferred to 1 ml SC media containing 2% galactose for 16 h at 25°C without shaking. To measure expression levels, 106 cells were washed in yeast staining buffer (YSB): 180 mM KCl, 10 mM NaCl, 0.2% bovine serum albumin (BSA), 0.1% galactose and 10 mM HEPES, pH 7.5. Cells were then stained for 30 min at 4°C with a 1:100 dilution of αMyc-FITC (ICL Labs) antibody and a 1:250 dilution of biotinylated αHA (Covance) antibody in YSB. The cells were then washed and incubated in a secondary stain of streptavidin-PE (BD Biosciences) diluted to 3.78 nM in YSB for 15 min at 25°C, washed again and run on a BD LSRII™ cytometer (BD Biosciences). The data were analyzed using FloJo software (Tree Star) for the percent fluorescin isothiocyanate (FITC)-positive cells compared to an unstained population.
SELEX library preparation and amplification
We ordered SELEX single stranded hand-mixed randomized oligo pool template with flanking SELEX primer sites (underlined), from Integrated DNA Technologies: CAG GGA TCC ATG CAC TGT ACG TTT (N30) AAA CCA CTT GAC TGC GGA TCC T, along with forward and reverse primers (unlabeled primer for selection experiments, A647 biotin labeled for flow cytometry experiments). The TTT and AAA sequences were included to discourage high affinity base pairing near the constant primer binding regions (which could impair analysis), but should be excluded or altered in cases where that sequence is likely to be part of the binding motif being probed. We used 30 randomized bases to give additional diversity to our library, since each 30-mer contains multiple 20-mer LHE binding sites. We created a dsDNA library from this oligo by running a single round of polymerase chain reaction (PCR) with the reverse primer using Platinum Taq DNA polymerase High Fidelity (Invitrogen): 95°C for 5 min, 59°C for 10 min, 72°C for 10 min. The PCR mix for all amplifications consisted of 1.5 mM MgSO4, 0.2 mM dNTPs, 0.67 μM of each primer and 0.05 u/μl of polymerase in a final volume of 25 μl.
After each round of selection (described below) 8.5 μl of the selected oligo was amplified using 20 PCR cycles: 95°C for 5 min, (95°C for 10 s, 59°C for 15 s, 68°C for 15 s) × 20, 68°C for 3 min. A secondary two-cycle PCR seeded with 6.25 μl of the first PCR product, was then used to ensure that each oligo was double stranded and properly paired for the next round of selection. Fluorescent double stranded oligo was also made for analysis purposes by seeding 0.5 μl of the 20-cycle PCR product into a separate secondary PCR and running six cycles.
SELEX binding selection and analysis
Three million induced yeast per protein per round were washed twice (2000 × g for 1 min) with 200 μl bind and wash buffer (BWB: 0.15 M KCl, 0.002 M CaCl2, 0.01 M NaCl, 0.01 M HEPES, 0.005 M L-Glutamic Acid Potassium Salt Monohydrate, 0.05% BSA, adjusted to pH 7.5 with KOH and filter sterilized), and resuspended to a final concentration of 500 000 yeast/μl. Each round of selection was carried out in a 96-well plate, with each sample containing dsOligo DNA (5 μl 100 μM SELEX0 dsDNA or 2 μl of the previous round's product) brought up to 94 μl with BWB. Six microliters (3 million) yeast expressing each protein were added and the plate was sealed and incubated for 30 min at room temperature with agitation. Each sample was washed 6 times in 150 μl BWB. After the wash steps, the samples were resuspended in 40 μl 10% buffer EB (diluted in H2O), the plate was sealed and the oligo was released by heating the protein past its melting temperature (70°C) for 10 min. Note: for proteins that denature above ∼70°C it may be necessary to release them from the yeast and extract the oligo with phenol chloroform. After release, the yeast were immediately spun down, and using a multichannel pipette, the supernatant was transferred to a new 96-well plate for storage. The oligo was amplified as described above, and 2 μl of the final PCR product was used to seed the next round of selection. The initial 500 pmols of randomized oligo (∼300 × 1012 30mers) was used also because it far exceeds the number of possible 20mer LHE targets (420, or ∼1 × 1012).
For binding analysis, A647-labeled oligo was used in place of unlabeled oligo. Following the 6x washes, the samples were analyzed on a flow cytometer instead of releasing the oligo.
Sequencing and analysis
Once all rounds of selection were complete, the products from each enzyme and round of SELEX to be analyzed were amplified using primers with 5΄ barcodes. The forward primer added a sequence required for next generation sequencing, a 4-base barcode to identify the round and enzyme, and a unique (randomized) 7-base barcode. The reverse primer also added sequence required for next generation sequencing. The PCR products were run on a 3% agarose gel, extracted and sent for sequencing per the provider's instructions (Edge Bio).
The sequences returned from Edge Bio were sorted by each round/enzyme for analysis. Each sequence was parsed for the enzyme/round 4-base barcode and the randomized N30 region (±2 bp), and the N30 information was output to individual FASTA-formatted files, binned by barcode.
Each collection of sequences was then analyzed for a sequence motif by expect maximization using MEME. The following parameters were used with MEME: -dna -mod zoops -noendgaps -minw 19 -maxw 22 -nmotifs 1 -maxsites x -minsites y -revcomp. Here, x was the lesser of 1500 or the total number of sequences, and y was the lesser of x or one third of the total number of sequences.
The motifs found by MEME for uncharacterized LHEs were also used as queries to search their corresponding LHE host genes and close homologs for the original LHE target sites. Close homologs of the host gene, identified by nucleotide BLAST searching, along with the original LHE-inserted host were compiled into a single FASTA file. These sequences were passed to MAST online tool (http://meme.nbcr.net/meme/cgi-bin/mast.cgi) and the default parameters were used to find possible matches to the motifs. We also searched these sequences for half-motifs using MAST since target sites in the host gene are often found split, with each half on either side of the LHE insertion. The MAST search results were used to help identify high quality targets to validate in combination with the MEME results.
Flow cytometric cleavage assay and specificity profiling
The cleavage activity of each putative LHE target was measured using a slightly modified version of the previously described tethered cleavage assay (11,19). Briefly, we tethered Alexa647-fluorescent target DNA to the surface expressed LHE and measured the decrease in fluorescence associated with DNA cleavage. Biotinylated fluorescent DNA was tethered to the HA epitope of the enzyme via an antibody-streptavidin bridge. Approximately 5 × 105 cells were first stained with 1:250 dilution biotinylated αHA (Covance) and 1:100 FITC-conjugated αMyc (ICL Labs) for 30 min at 4°C in the YSB. Pre-conjugated streptavidin-PE:Biotin-DNA-A467 was then tethered to the yeast via the HA-biotin:streptavidin-PE interaction. This secondary stain was performed in the same buffer plus 400 mM KCl to allow biotin-streptavidin conjugation while disallowing alternative LHE-mediated DNA tethering. Cells were washed and resuspended in the cleavage buffer (10 mM NaCl, 113 mM K-Glutamate, 0.05% BSA and 10 mM HEPES, pH 8.2), and split into two wells per sample. The plate was centrifuged and each of the pair of wells was resuspended in cleavage buffer; one with 2 mM MgCl2 (cleavage permissive) and one with CaCl2 (cleavage intolerant). Fluorescence loss due to magnesium-dependent cleavage of the DNA can subsequently be measured by comparing the fluorescence of these otherwise identical samples. After cleaving for 20 min at 37°C, cells were pelleted and resuspended in cold secondary stain buffer plus 4 mM ethylenediaminetetraacetic acid plus 400 mM KCl to aid release of cleaved substrate and mitigate any end-holding effects on DNA–fluorophore release.
Yeast fluorescence was measured on a BD LSRII™ cytometer and the resulting data were analyzed using FlowJo (TreeStar). Relative cleavage efficiencies were calculated by dividing the median DNA-A647 fluorescence value of the Mg++ sample (reduced fluorescence due to cleavage) by the corresponding median fluorescence value of the Ca++ matched pair (no cleavage). A higher Ca++/Mg++ ratio indicates more cleavage.
Cleavage specificity profiles were produced by assaying the cleavage of each of the 66 possible target sequences that differ from the original target by a single base. Each base at each of the 22 positions was substituted with each of the 3 three bases (22 bp target × 3 alternate bases), as in Jarjour's original description of this assay (19), and the targets were tested in parallel using the tethered cleavage assay described above. In these analyses, all Ca++/Mg++ ratios were normalized to the Ca++/Mg++ ratio of the original target site.
In order to simulate the exaggerated preferences that were expected of SELEX, the cleavage position frequency matrices (PFMs) were raised to an arbitrary exponent (5 for I-OnuI and I-OsoMI, 7 for I-HjeMI, 4 for I-OsoMII) before converting to position probability matrices (PPMs) and plotting as sequence logos using Seq2Logo 2.0 (20).
In solution cleavage assay
One million expressing yeast (∼280 pM enzyme; 104 molecules per yeast), were incubated with 50 nM Alexa-647-labeled dsOligo for 30 min at 37°C in cleavage buffer supplemented with 5 mM DTT and 5 mM MgCl2. Oligo was released by heating the protein past its melting temperature (70°C) for 10 min, and the oligo-containing supernatants were collected after centrifugation and run on a 12% non-denaturing polyacrylamide gel. The fluorescent DNA bands were quantified using an Odyssey infrared imaging system (Li-Cor Biosciences).
Central four determination
The target sequences predicted from our SELEX results were located in the original homing endonuclease host gene sequences (both upstream and downstream from the open reading frame for each protein). An extended 60 basepair target site sequence was chosen for each homing endonuclease and cloned into a holding vector with M13 forward and M13 reverse primer binding sequences flanking the inserted target sequence. The vector was then transformed into bacteria and prepped in microgram quantities. To determine the precise center of the target sequence, the vector was linearized by a restriction digest (at a restriction site distant from the cloned target sequence), purified with a PCR purification kit, and used as the substrate in an in vitro cleavage assay.
Five million induced yeast cells with surface-expressed homing endonuclease were incubated in cleavage buffer (components described above) with 10 mM DTT (to release the enzyme from the surface of yeast), 5 mM CaCl2 (no cleavage control) or MgCl2 (to allow cleavage), and 1 μg of purified linear target site substrate plasmid. The mixture was allowed to incubate for at least 1 h at 37°C. The entire digest reaction was loaded into a single large lane of an agarose gel and the cleaved substrate fragments were separated by electrophoresis. The restriction site used for linearization of the target site vector was selected carefully to lead to the formation of distinctly-sized fragments. The two fragments corresponding to successfully cleaved products were purified separately with a gel extraction kit. Finally, run-off sequencing (using either the M13 forward or M13 reverse primer for the respective fragment in the reaction) was performed on each of the purified DNA samples. The resulting sequence chromatogram displayed an abrupt drop-off point that corresponds to the site of cleavage of the substrate. The sequence can be read up to the break, making sure to discount the large additional ‘adenine’ peak at the end added by the Taq polymerase in the BigDye sequencing reaction.
RESULTS
Adaptation of SELEX for Yeast Surface-Displayed LHEs allows target determination
SELEX is traditionally carried out with purified protein coupled to a solid support matrix (e.g. in vitro synthesized, biotinylated protein coupled to streptavidin-conjugated beads) (16,21). The resulting protein-solid support complex allows the putative DNA-binding protein to be incubated with a randomized double-stranded oligonucleotide pool, and then rapidly isolated (e.g. by centrifugation or magnetic separation).
We have previously shown that yeast-surface-displayed homing endonucleases faithfully recapitulate the DNA binding and cleavage properties of purified proteins (11,19). We have also observed that there is a strong relationship between homing endonuclease binding and cleavage. This property is very well exemplified by a plot of binding versus cleavage for the I-OnuI homing endonuclease wherein the relative binding and cleavage is plotted for oligonucleotide targets that differ from the I-OnuI native target sequence by one base pair (Figure 1A). Based on the correlation between LHE binding and target cleavage, we hypothesized that yeast surface displayed LHEs could serve as both the method of expression and as the solid support for SELEX, resulting in a combined method we designated YSD-SELEX (schematized in Figure 1B). Replacing the cofactor required for cleavage—Mg++ with Ca++—allows faithful binding without cleavage and enables our recovery of the intact bound target DNA molecule for amplification and analysis. Compared to traditional SELEX methods, YSD-SELEX has the significant advantage of cutting down on the cost and experimental complexity of generating matrix-bound protein, while simultaneously providing a suite of built-in tools for generation and analysis of libraries of displayed proteins. Importantly, YSD-SELEX allows the use of flow cytometry to evaluate binding of amplified target oligonucleotide pools to the yeast-displayed LHE following each SELEX round, thus providing a means to optimize and directly monitor the evolution of target-ligand binding.
Figure 1.

(A) Plot of binding (Log2(x/WT)) versus cleavage (Ca++/Mg++ normalized to WT) for I-OnuI ‘one-off’ oligonucleotide targets – targets that differ by one base from the native I-OnuI target site at the indicated position. Targets that exhibit high binding (green box) are also typically cleaved efficiently. (B) Schematic of SELEX using yeast surface displayed protein. The homing endonuclease of interest is expressed on the surface of yeast. The yeast is used as a solid support for the protein to facilitate wash steps after binding of randomized oligos. The best-bound sequences are extracted by heating and collecting the supernatant, amplified and subjected to iterative rounds of selection. The initial optimization and later, the success of the experiment are assayed using fluorescently labeled pools and flow cytometry. (C) Flow cytometry plots obtained from staining expressing yeast (green) with labeled target oligonucleotide pools from the indicated SELEX round for I-OnuI and I-AniI. The motif generated by sequencing and analysis of the SELEX5 pool is shown in the far right panel. (D) Representative motif sequence logos for I-OnuI showing that the same profile is obtained by sequence analysis of pools obtained in SELEX oligonucleotide pools 3–5 (top panel), and at very different sequencing breadths (bottom panel). (E) Flow cytometry binding (top) and cleavage analysis (bottom) of WT target oligonucleotides (first column), and targets corresponding to the SELEX motif and the top two most frequent individual sequences obtained from sequencing of the SELEX5 pool. The number in the center of each cleavage plot is the ratio of the mean fluorescence intensity obtained following incubation of the assay with Ca2+ as the divalent ion (non-cleaving conditions) or Mg2+ as the divalent ion (cleaving conditions), and provides a quantitative assessment of the extent of cleavage by the surface expressed enzyme.
To develop a working YSD-SELEX protocol, we initially chose to work with two well-characterized, yeast-displayable homing endonucleases: I-AniI and I-OnuI. I-AniI is representative of a small family of closely related homing endonuclease enzymes whose host gene is apocytochrome B (COB) (22), at least some of which possess RNA maturase activity in addition to DNA endonuclease activity (23,24). In contrast, I-OnuI-family enzymes have invaded a much broader set of host genes (7) and thus cover a wide range of target sites and enzymatic properties. Using these enzymes we optimized the BWB in order to block nonspecific interactions while allowing library binding (see Supplementary Data and Supplementary Data). We also determined the optimal oligo-release and PCR conditions needed to recover and prepare each round's SELEX oligo pool.
Based on these initial analyses, we performed a pilot YSD-SELEX experiment on yeast displayed I-OnuI and I-AniI using 150 mM KCl (Figure 1C–E). As expected, binding of amplified pools of eluted oligos to I-OnuI increased over the five rounds of SELEX (Figure 1C, upper panels). Although these conditions allowed for substantial basal binding of the naïve SELEX oligonucleotide library to yeast-displayed I-AniI, increased target oligonucleotide binding was observed following each round of SELEX for I-AniI as well (Figure 1C, lower panels). As the proportion of bound to unbound enzyme increased with each round of SELEX—especially after round 3—we incorporated increasing stringencies of selection by increasing the salt concentration for later rounds; an approach similar to fixed-stringency SELEX (16). Sequencing of the oligo pools following the 5th round of SELEX (SELEX5) followed by analysis of the resulting sequences (see SELEX sequence analysis, below) resulted in the generation of a binding motif for I-OnuI that closely matched the known I-OnuI target sequence (Figure 1C, top right panel). The I-OnuI motif was robust to both the number of rounds of SELEX, as well as the breadth of sequencing (i.e. the number of unique sequences analyzed) (Figure 1D). In contrast, despite the apparent increase in binding over the five rounds of SELEX, sequencing of the SELEX5 pool was not able to generate any clear motif for I-AniI binding (Figure 1C, bottom right panel), irrespective of round or breadth of sequencing (data not shown). Flow cytometric analysis of the binding and cleavage of target oligonucleotides corresponding to the known WT targets, the SELEX motif and the top two top-ranked sequences from the SELEX5 pools confirmed that SELEX was able to faithfully identify target sequences that were bound and cleaved by I-OnuI (Figure 1E, top panel in each set). Although I-AniI's binding and cleavage of its cognate target was easily detectable using the highly sensitive flow cytometry assay, we were not able to detect any significant cleavage of the putative I-AniI binding motif olignucleotide, or of either of the top two top-ranked sequences from the analysis of the I-AniI SELEX5 oligonucleotide pool. This finding was consistent with the incongruence between the I-AniI cognate target sequence and the weak SELEX binding motif.
Based on the results of the pilot YSD-SELEX experiment, we hypothesized that the conditions we had identified would be generally applicable to identifying cleavable target sequences for homing endonucleases homologous to I-OnuI. To test this hypothesis, we selected a benchmark group of three recently characterized enzymes of the I-OnuI subfamily for whom target sites had been identified using bioinformatics, I-GzeII, I-PanMI and I-SmaMI; and a test group of six surface-expressible putative LHEs for which there is presently insufficient genomic data to predict a target site, I-CpaMIIP, I-CpaMVP, I-HjeMII, I-MveMIP, I-OsoMI and I-OsoMII. These enzymes vary in their level of identity to each other (Figure 2A), and in the host genes within which they are inserted (Figure 2B), though all are found within fungal mitochondrial genomes. These putative I-OnuI homologs represent some of the most distant I-OnuI relatives characterized to date: the closest are about 48% identical at the amino acid level to I-OnuI (I-SmaMI and I-CpaMIIP), and the most distant are about 25% identical (I-MveI and I-CpaMVP). The homologs in the test group also exhibit marked divergence in residues implicated in direct DNA contacts for I-OnuI (Figure 2A, red boxed residues), and are found inserted at new points in the same or different host genes. We thus anticipated that they would likely exhibit significantly different cleavage specificities than known I-OnuI family members. For reference, we have provided the host gene in which the LHE is inserted, the type of insertion within the host gene and the organism name for each homolog in Supplementary Data.
Using the same conditions as the pilot YSD-SELEX experiment, we performed five rounds of YSD-SELEX for yeast-displayed LHEs described in Figure 2. Flow cytometry-based binding patterns over the rounds of SELEX are shown in Figure 3A, with median fluorescent intensities plotted against SELEX round in Figure 3B. SELEX0 pool binding for the displayed LHEs was generally very low, with only one LHE exhibiting a high level of SELEX0 pool binding: I-SmaMI, a benchmark enzyme. Binding increased over each round of SELEX for all enzymes with only one exception: I-CpaMVP, a test enzyme. Notable features of the flow cytometry analysis of SELEX pool binding are that six of the nine enzymes showed approximately two-log increases in bound oligonucleotide in SELEX5 versus SELEX0 pools. I-SmaMI, which exhibited a high level of binding to the initial SELEX0 pool in the KCl concentrations used, achieved only approximately a 1 log increase; I-CpaMIIP achieved only an approximately 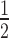 log increase; and the aforementioned SELEX failure, I-CpaMVP, achieved no detectable increase over the course of the five SELEX rounds. Possible explanations for I-CpaMVP's failure include that the initial conditions did not allow any oligo binding at all, or that the predicted ORF encodes a protein which is not a functional DNA binding protein. It is also notable that the surface expression of I-CpaMVP was the lowest of all of the enzymes subjected to SELEX, and this may have been a contributing factor to poor oligonucleotide binding and consequent failure of sequential enrichment.
log increase; and the aforementioned SELEX failure, I-CpaMVP, achieved no detectable increase over the course of the five SELEX rounds. Possible explanations for I-CpaMVP's failure include that the initial conditions did not allow any oligo binding at all, or that the predicted ORF encodes a protein which is not a functional DNA binding protein. It is also notable that the surface expression of I-CpaMVP was the lowest of all of the enzymes subjected to SELEX, and this may have been a contributing factor to poor oligonucleotide binding and consequent failure of sequential enrichment.
Figure 3.

SELEX oligonucleotide pool binding by SELEX round. (A) Flow cytometry binding assays carried out by fluorescently labeling the SELEX pools from each round (SELEX0 is the initial randomized pool) and measuring the amount of fluorescent oligonucleotide bound by the indicated yeast surface displayed enzyme via flow cytometry. Each enzyme's affinity for the targets in the indicated SELEX pool increased with each round with the exception of I-CpaMVP. (B) A summary plot of the binding data, plotting the median fluorescence intensity of bound oligonucleotide by SELEX round. Significant increases in binding occurred over the five rounds of SELEX.
SELEX sequence analysis demonstrates YSD-SELEX robustness
We expected each selected pool to represent a complex family of sequences, so we deep-sequenced each pool in order to obtain information on both the individual sequences and the level of complexity of each pool. The information from each sequenced pool are presented as sequence motifs generated by the Multiple Expect-Maximization for Motif Elicitation (MEME) tool (25) in the left panels of Figure 4, (benchmark enzymes) and Figure 5 (test enzymes). Of the nine enzymes analyzed, only one test enzyme, I-CpaMVP, failed to produce a motif (failure is defined here as not producing a strong motif that was similar across SELEX rounds 3–5), a result that was anticipated based on its similar failure to select an oligunucleotide pool with increased binding affinity over SELEX rounds 3–5 (e.g. see Figure 3). Of the three benchmark enzymes, I-GzeII and I-PanMI produced motifs that closely mirrored the target specificity predicted via bioinformatics, while I-SmaMI's motif was congruent with its predicted target, except that only about half of the target (the N-terminal half) was strongly selected in the motif. Of the five test enzymes for which motifs were generated, each produced strong motifs, consistent across the SELEX rounds SELEX3-5. I-MveI's motif, however, was shorter than the canonical ∼20 bp LHE motif. One feature consistent across many of these motifs was a lower specificity in the central-four region, particularly the central-two. This feature is expected, based on the very few direct base contacts made by LHEs to these bases (7,11,24,26).
Figure 4.

SELEX motif binding and cleavage for benchmark enzymes. The left panel shows benchmark enzyme group motifs found by analyzing the SELEX sequences for each enzyme's SELEX5 oligonucleotide pool using the expect maximization tool, MEME. The wild-type targets are shown below the motifs. The flow cytometry plots represent flow binding analyses (top) and cleavage (bottom) of the indicated target oligonucleotides corresponding to targets previously identified by bioinformatics (first column), the SELEX motif and the top two most frequently represented oligonucleotide targets in the SELEX5 pool.
Figure 5.

SELEX motif binding and cleavage for test enzymes. The left panel shows test enzyme group motifs found by analyzing the SELEX sequences for each enzyme's SELEX5 oligonucleotide pool using the expect maximization tool, MEME. The flow cytometry plots represent flow binding analyses (top) and cleavage (bottom) of the indicated target oligonucleotides corresponding to targets previously identified by bioinformatics (first column), the SELEX motif and the top two most frequently represented oligonucleotide targets in the SELEX5 pool.
Analysis of repeat SELEX experiments run under the same or similar conditions produced very similar results as our initial run, demonstrating the robustness of the method. Motifs for I-OnuI, I-PanMI, I-SmaMI, I-HjeMII, I-OsoMI were recapitulated with nearly identical preferences and stringencies; I-PanMI is shown as a representative example run in identical optimized conditions in Supplementary Data. These results suggest that, at least for enzymes which are within the I-OnuI family, the conditions we have identified provide both sufficient randomization in the initial pool and a sufficient level of initial pool binding to allow isolation of diverse target sequences, as well as a high rate of convergence in the output of the SELEX reaction.
Cleavage of targets predicted by SELEX-generated sequence motifs validates selection
We utilized two approaches to validate the SELEX-generated motifs and identify cleavable target sites for test group LHEs: searching genomic data for a match to the motif in the corresponding host gene, and direct use of the motif consensus in combination with individual sequences from the selected oligo pool. The genomic search tactic is possible because during invasion, LHEs typically leave half of their target site on either side of their insertion. Candidate targets identified by either method were then evaluated biochemically. For the purposes of evaluating the YSD-SELEX method, we biochemically validated motifs and individual SELEX sequences for the benchmark LHE group as well.
For our genomic data searches, we used the Motif Alignment & Search Tool (MAST, part of the MEME suite) (25), which uses a motif's underlying position weight matrix generated by MEME to search genomic sequences for a best-fit match. Generally, divining a target site from genomic data requires the existence of sequence from an insert-less homolog; one can then compare insert-less homologs to homologs with the LHE insertion and reveal the target site (7). Often, however, the homolog is not a close enough relative and the junctions of the insert—and thus the sequence of the target—cannot be determined exactly. SELEX motifs can therefore be greatly helpful by constraining half-site spacing, since exact spacing must be maintained between the putative half-sites to test activity. Furthermore, when no homolog data are available at all, SELEX motifs can be split roughly in half and used to search the sequence of the host gene (with the LHE insert) by itself. Here again, the full SELEX motif is an important aid in constraining the half-site spacing to the required length when reconstructing the original cleavage site from any half-sites that are identified. Using the above motif search tactics, LHE motifs were used to search for genomic sequence matches or partial matches. Although possible genomic matches were found for many of the putative LHEs, three matched to the genomic sequence from an insert-less homolog almost exactly (I-HjeMII, I-OsoMI and I-OsoMII), and were sufficient to identify the corresponding target in the host gene (Figure 5, leftmost flow panels). The remainder of the motifs made only partial matches to genomic sequence from organisms similar to their host, likely due to a lack of highly homologous insert-less host genes.
The YSD-SELEX motifs, sequences from the oligonucleotide pools, and genomic search matches were utilized ensemble to generate potential candidate cleavage targets as follows: Each MEME consensus sequence and two of the top ranking sequences from the expect-maximization were chosen from the SELEX5 pool. The closest possible targets identified in genomic searches (designated as ‘homolog’) were also evaluated. In addition, various partial matched targets, and close matches in related organisms were evaluated (for full lists of evaluated targets, see Supplementary Data). For biochemical assessment, each member of the list of candidate sites was synthesized as an oligonucleotide and used as a template for making fluorescently labeled dsDNA. Individual fluorescent targets were then used as binding and cleavage substrates in flow cytometry-based assays (19). Targets which showed promise in the higher throughput flow assays were further validated in standard solution-based cleavage assays (Supplementary Data). For the three newly discovered enzymes, I-HjeMII, I-OsoMI and I-OsoMII, we also sought to determine the precise site of phosphodiester hydrolysis on each strand, and reveal the location of the center of the target site and the four-base overhangs created by this sub-family of enzymes. To this end we cloned their respective targets into plasmids and used the solution-based cleavage assay followed by gel extraction and Sanger sequencing (Supplementary Data).
The binding and cleavage of the individually selected targets correlated well with the SELEX binding data and quality of the corresponding motifs. Those enzymes that showed the most promise in the SELEX pool binding assay (at least a 1-log increase in fluorescence between the initial randomized pool and rounds 3–5; I-GzeII, I-PanMI, I-HjeMII, I-OsoMI and I-OsoMII in Figure 3) were also the enzymes that produced high-quality motifs of the expected length, and were able to bind and cleave targets predicted by SELEX (I-GzeII and I-PanMI in Figure 4; I-HjeMII, I-OsoMI and I-OsoMII in Figure 5). The cleavage assays for best homolog sequence, the motif sequence, and the two top individual sequences are shown as flow plots – no other class of tested site showed any detectable cleavage (see summary plot in Supplementary Data, summary data tables in Supplementary Data). Those enzymes that showed less than a 1-log increase in fluorescence (I-CpaMIIP, I-MveMIP and I-SmaMI) showed detectable binding to oligonucleotides corresponding to their predicted motifs, but were not able to cleave the motif target (I-SmaMI in Figure 4; I-MveMIP and I-CpaMIIP in Figure 5; also Supplementary Data). The correlation between binding and cleavage was particularly notable in the set of individually selected targets. All targets that demonstrated at least 1.5-log increases in fluorescence above background in the binding assay also demonstrated high levels of cleavage activity (Ca++/Mg++ ratio > 2) against the same targets (Supplementary Data, with summary data tables provided in Supplementary Data). These data support the hypothesis that the best-bound targets have a high probability of yielding cleavable substrates, and thus that binding data obtained during iteration of the YSD-SELEX protocol are a strong predictor of success in identifying a native target.
SELEX binding profiles correlate well with cleavage specificity profiles
Now with cleavable target sequences, we were able to determine the optimal targets and cleavage specificities of the new enzymes, I-HjeMII, I-OsoMI and I-OsoMII. Briefly, we used an established high throughput modification of the yeast surface display cleavage assay (19) to quantify the nucleotide preference of the enzyme at each position of the 22 bp target. At each position the original nucleotide is substituted for each of the other three possible nucleotides while the rest of the target is held constant, generating 66 (3 × 22) singly-substituted targets plus the original target. The cleavage values for each of the 66 targets were normalized to the original target, generating a PFM. I-OnuI's previously published cleavage specificity PFM (7), which was generated in the same fashion, was also included for comparison.
We then used these PFMs to compare the output from SELEX to the actual cleavage specificities. The PFM cleavage values are represented in their standard form in Supplementary Data, with total cleavage on the y-axis. These PFMs were converted to PPMs and plotted as sequence logos (Supplementary Data), which show specificity at each position, rather than total cleavage. Although each of the cleavage sequence logos correlates well with the corresponding SELEX sequence logo (Supplementary Data), many of the bases that have only slight preferences in the cleavage profile appear to have strong preferences in the SELEX profile. This outcome was expected, as the SELEX conditions were optimized to select the most preferred, high affinity targets rather than a diverse population of low- to medium-affinity targets required to generate an accurate binding profile (27). In order to simulate the exaggerated preferences that were expected of SELEX, the cleavage PFMs were raised to an arbitrary exponent (between 4 and 7) before converting to PPMs and plotting as sequence logos (Supplementary Data). The resulting cleavage profiles show even better correlations with, (Supplementary Data) and bear striking resemblances to those generated by SELEX (Figure 6). Importantly, at each position of each profile, the base predicted by SELEX to be the most favored corresponds to the most favored base in the cleavage profile with few exceptions. In the exceptions, the base predicted by SELEX to be the most favored was tolerated, often very well, and was typically located in a region of low enzyme specificity (compare the SELEX profiles in Figure 6 to the raw cleavage profiles in Supplementary Data). A single outlier from this pattern in the cumulative 76 positions assessed was the T predicted by SELEX at the +2 position of I-OsoMII, which is not tolerated; at this position the enzyme showed minimal specificity by SELEX, which predicted a slight preference for T over the correct base, G. Indeed, this mismatch near the active site explains I-OsoMII's inability to cleave the consensus motif shown in Figure 3. Overall, these data demonstrate a strong correlation between homing endonuclease binding and cleavage, and support the hypothesis that YSD-SELEX can be used as a first principles approach for identifying a cleavable substrate.
Figure 6.

Cleavage and SELEX profile comparison. Simulation of the expected outcome of stringent binding selection by transformation of cleavage specificity profiles (top), compared to the SELEX profiles (bottom) for (A) I-HjeMII, (B) I-OsoMII, (C) I-OsoMI and (D) I-OnuI.
DISCUSSION
Here, we have adapted the SELEX method for use with yeast surface display as a means to rapidly generate DNA binding motifs for surface displayed LHEs. The combined method, which we term YSD-SELEX, represents a powerful new tool for characterization of LHEs. Putative LHEs can be assayed for proper folding by surface expression, binding properties can be rapidly determined by SELEX, and the binding and cleavage properties of candidate target sites interrogated at single-base resolution via flow cytometry binding and cleavage assays – all in a multi-well, parallelized fashion over a few days’ time (19,28). The YSD-SELEX method benefits from the ability to test oligo binding in high throughput using yeast surface display and flow cytometry. Selection conditions can be easily tested upfront and as the experiment progresses, and modulated rationally to produce conditions that yield more diverse or narrow target pools depending on whether an investigator desires an accurate binding profile, or simply a few best-bound targets (27). Finally, YSD-SELEX is an improvement upon traditional SELEX insofar as it allows quick, easy and inexpensive expression of matrix-bound protein that does not require further purification or modification. It thus represents a cheap and practical extension of traditional SELEX methods, as well as an addition to the expanding yeast surface display toolbox.
We optimized our SELEX protocol for I-OnuI, and subsequently used a uniform set of binding and amplification conditions for the entire set of enzymes that we characterized. However, the data suggest that the protocol we developed may not be optimal for every LHE, and that optimizing one or more aspects of the protocol for a given individual protein may increase the signal/noise and yield a higher quality binding motif. For example, I-SmaMI and I-MveMIP both showed high levels of binding to the initial randomized pool (Figure 3). This high background binding may explain the poor selection and, at least in part, account for why we obtained incomplete motifs for these enzymes. In contrast, I-AniI's binding affinity is orders of magnitude weaker than I-OnuI for their respective targets (7,19), and it is therefore unlikely that the same stringent binding conditions chosen for I-OnuI and its orthologues would have been permissive to I-AniI binding. Low affinities may have also prevented I-CpaMIIP and I-CpaMVP from binding the oligo pool. Alternatively I-CpaMIIP and I-CpaMVP may be non-functional homologs; we might expect to see a fraction of inactive enzymes in any given set of putative LHEs as they are thought to be susceptible to evolutionary degeneration (29). In either case, suboptimal selection conditions would be predicted to lead to weak or non-existent motifs. Optimization of the number of rounds of SELEX may also have utility for obtaining higher quality selection and output motifs. I-CpaMIIP may have benefitted from additional rounds of selection, given that there was some increase in binding throughout the rounds but not as much as was observed for the other homologs. An attractive feature of integrating YSD into SELEX is that YSD allows easy and rapid identification of these possible issues and lends insight into defining what may be general indicators of successful SELEX reactions for a given class of proteins (e.g. for LHEs: low but detectable initial binding, and a >1 log increase in fluorescence by round three of SELEX).
Although we show that YSD-SELEX can be used to determine target sites for a subset of LHEs, our results also suggest certain limitations of SELEX when applied to LHEs. First, our experiments only included candidate LHEs that could be surface expressed. Although good surface expression correlates well with proper folding, it is still possible that some of the low-expressing homologs that we excluded were functional enzymes, as not all functional proteins will properly transit the yeast secretory pathway. Next, under our specific conditions YSD-SELEX was able to yield only half motifs for I-SmaMI and I-MveMIP, and completely failed to converge on I-AniI's known target site. We speculate that in addition to the insufficiently stringent initial selection conditions, the recently described lack of binding affinity in I-SmaMI's C-terminal half-domain (30)—which corresponds to the weak, 3΄ half of the SELEX motif—was responsible for the asymmetry in binding selection. These observations may also explain the incomplete motif for I-MveMIP, assuming that it is a functional enzyme. Not only is I-AniI's target site DNA-binding energy similarly concentrated in the N-terminal/5΄ half of the enzyme/target interaction (31); I-AniI also has a second nucleic acid binding surface involved in RNA maturase activity (32). The existence of multiple potential nucleic acid interacting surfaces is particularly concerning and should be considered a red flag for any natural protein for which SELEX analysis is contemplated. Ideally, potentially confounding domains should be eliminated or blocked prior to use in SELEX.
Despite the limitations of the YSD-SELEX method, we were able to successfully identify cleavable targets for our benchmark enzymes, and for three of the six (heretofore putative) test enzymes. As one might expect, the motifs generated from our benchmark group closely mirror the target sites that had been previously identified for these enzymes using bioinformatics. One area where the motif is generally weak is around the central-four region, which is to be expected since this is where the enzyme makes contact with the minor groove of the DNA and typically lacks base-specific contacts. For the test enzymes, the target oligonucleotides corresponding to the predicted motif were directly cleavable (I-HjeMII and I-OsoMI), and/or allowed us to identify likely targets in a homologous organism (HjeMII, I-OsoMI and I-OsoMII). These results also highlight an important difference between binding specificity (as determined by SELEX) and cleavage specificity. Because some positions provide cleavage specificity independent of binding specificity (31), and because alignment of the SELEX motif to genomic sequence may not be an option or not fruitful in finding the true genomic target, it may be necessary to combine SELEX motifs with one-off profiling to find true, cleavable targets. Testing closely-related alternative sites by performing YSD one-off cleavage profiling (19) may be able to identify a cleavable target variant from an initially non-cleavable base sequence, especially if performed under highly permissive cleavage conditions (e.g. long duration, high Mg++, high pH). Indeed, a one-off profile using the SELEX motif as a base would have identified any problematic residues and revealed a cleavable substrate for each and every one of our functional enzymes that produced a full motif, without use of genomic reference sequences.
Inspection of the set of SELEX motifs suggests an additional important aspect of LHE biology: di-guanine and di-cytosine bases were specifically recognized and highly selected by SELEX, and therefore play a dominant role in the selection process. This observation likely speaks to the importance of the binding energy created by direct base contacts in the major groove between positively charged amino acid side chains and guanine residues in the target site.
An important issue that we have not explored in depth here is to what extent SELEX could be applied to obtain a binding specificity profile for LHEs. In this regard it is notable that SELEX has been successfully applied for this purpose for a variety of other types of DNA binding proteins (16,27,28), and that LHE SELEX motifs correlate well with the cleavage specificity profiles obtained by using one-off profiling by yeast surface display. It is therefore likely that substituting the high stringency selection conditions used here for those optimized for selection of a diverse set of targets over only a few rounds (as previously described), would allow YSD-SELEX to be used to obtain accurate binding profiles as well. Furthermore, it is likely that YSD-SELEX could also leverage other SELEX modifications such as genomic SELEX (33,34), which could be used to predict LHE binding sites within a genome of interest. This could be of interest to investigators wishing to use a first principles approach to finding their nuclease's off-targets, particularly in cases where the LHE is to be used clinically.
In summary, we have developed SELEX methods that can be used with yeast surface displayed LHEs to identify cleavable target sequences. Our combined method, which we refer to as YSD-SELEX, is a rapid, simple and inexpensive means to determine the binding and cleavage properties of LHEs identified using homology searches of sequence databases. Our method may also complement other purely bioinformatic methods of target prediction (8) that have the potential to produce similar but entirely non-cleavable targets sites if the target halves are joined imprecisely. We have applied the method to six novel LHEs identified from fungal mitochondrial genomes, and determined cleavable target sequences for three of these enzymes. This method offers significant potential to expand the arsenal of homing endonuclease scaffolds available for genome engineering and related biotechnological applications that may benefit from the unique properties of LHEs.
Supplementary Material
Footnotes
Present address: Abigail R. Lambert, Division of Basic Sciences, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Program for Cell and Gene Therapy by Seattle Children's Research Institute; Foundation for the National Institutes of Health grant awarded to Prof. Austin Burt via the Target Malaria project Grand Challenges in Human Health, funded by the Bill and Melinda Gates Foundation; the National Institutes of Health (NIH) [U19AI0966111]; University of Washington Molecular Medicine Training Program through the Howard Hughes Medical Institute. Funding for open access charge: National Institutes of Health (NIH) [U19AI0966111].
Conflict of interest statement. A.M.S. holds equity, consults for, and receives compensation from bluebird bio, a commercial entity that is developing homing endonucleases for therapeutic applications.
REFERENCES
- 1. Jurica M.S., Stoddard B.L.. Homing endonucleases: structure, function and evolution. Cell. Mol. Life Sci. 1999; 55:1304–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Windbichler N., Papathanos P.A., Catteruccia F., Ranson H., Burt A., Crisanti A.. Homing endonuclease mediated gene targeting in Anopheles gambiae cells and embryos. Nucleic Acids Res. 2007; 35:5922–5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gao H., Smith J., Yang M., Jones S., Djukanovic V., Nicholson M.G., West A., Bidney D., Falco S.C., Jantz D. et al. . Heritable targeted mutagenesis in maize using a designed endonuclease. Plant J. Cell Mol. Biol. 2010; 61:176–187. [DOI] [PubMed] [Google Scholar]
- 4. Arnould S., Perez C., Cabaniols J.P., Smith J., Gouble A., Grizot S., Epinat J.C., Duclert A., Duchateau P., Paques F.. Engineered I-CreI derivatives cleaving sequences from the Human XPC gene can induce highly efficient gene correction in mammalian cells. J. Mol. Biol. 2007; 371:49–65. [DOI] [PubMed] [Google Scholar]
- 5. Gouble A., Smith J., Bruneau S., Perez C., Guyot V., Cabaniols J.P., Leduc S., Fiette L., Ave P., Micheau B. et al. . Efficient in toto targeted recombination in mouse liver by meganuclease-induced double-strand break. J. Gene Med. 2006; 8:616–622. [DOI] [PubMed] [Google Scholar]
- 6. Thermes V., Grabher C., Ristoratore F., Bourrat F., Choulika A., Wittbrodt J., Joly J.S.. I-SceI meganuclease mediates highly efficient transgenesis in fish. Mech. Dev. 2002; 118:91–98. [DOI] [PubMed] [Google Scholar]
- 7. Takeuchi R., Lambert A.R., Mak A.N.-S., Jacoby K., Dickson R.J., Gloor G.B., Scharenberg A.M., Edgell D.R., Stoddard B.L.. Tapping natural reservoirs of homing endonucleases for targeted gene modification. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:13077–13082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Szeto M.D., Boissel S.J.S., Baker D., Thyme S.B.. Mining endonuclease cleavage determinants in genomic sequence data. J. Biol. Chem. 2011; 286:32617–32627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thiéry O., Börstler B., Ineichen K., Redecker D.. Evolutionary dynamics of introns and homing endonuclease ORFs in a region of the large subunit of the mitochondrial rRNA in Glomus species (arbuscular mycorrhizal fungi, Glomeromycota). Mol. Phylogenet. Evol. 2010; 55:599–610. [DOI] [PubMed] [Google Scholar]
- 10. Haugen P., Bhattacharya D.. The spread of LAGLIDADG homing endonuclease genes in rDNA. Nucleic Acids Res. 2004; 32:2049–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jacoby K., Metzger M., Shen B.W., Certo M.T., Jarjour J., Stoddard B.L., Scharenberg A.M.. Expanding LAGLIDADG endonuclease scaffold diversity by rapidly surveying evolutionary sequence space. Nucleic Acids Res. 2012; 40:4954–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baxter S., Lambert A.R., Kuhar R., Jarjour J., Kulshina N., Parmeggiani F., Danaher P., Gano J., Baker D., Stoddard B.L. et al. . Engineering domain fusion chimeras from I-OnuI family LAGLIDADG homing endonucleases. Nucleic Acids Res. 2012; 40:7985–8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heath P.J., Stephens K.M., Monnat R.J., Stoddard B.L.. The structure of I-Crel, a group I intron-encoded homing endonuclease. Nat. Struct. Biol. 1997; 4:468–476. [DOI] [PubMed] [Google Scholar]
- 14. Duan X., Gimble F.S., Quiocho F.A.. Crystal structure of PI-SceI, a homing endonuclease with protein splicing activity. Cell. 1997; 89:555–564. [DOI] [PubMed] [Google Scholar]
- 15. Abelson J. Directed evolution of nucleic acids by independent replication and selection. Science. 1990; 249:488–489. [DOI] [PubMed] [Google Scholar]
- 16. Djordjevic M. SELEX experiments: New prospects, applications and data analysis in inferring regulatory pathways. Biomol. Eng. 2007; 24:179–189. [DOI] [PubMed] [Google Scholar]
- 17. Wheeler D.L., Church D.M., Federhen S., Lash A.E., Madden T.L., Pontius J.U., Schuler G.D., Schriml L.M., Sequeira E., Tatusova T.A. et al. . Database resources of the National Center for Biotechnology. Nucleic Acids Res. 2003; 31:28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roberts R.J., Belfort M., Bestor T., Bhagwat A.S., Bickle T.A., Bitinaite J., Blumenthal R.M., Degtyarev Sk., Dryden D.T., Dybvig K. et al. . A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003; 31:1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jarjour J., West-Foyle H., Certo M.T., Hubert C.G., Doyle L., Getz M.M., Stoddard B.L., Scharenberg A.M.. High-resolution profiling of homing endonuclease binding and catalytic specificity using yeast surface display. Nucleic Acids Res. 2009; 37:6871–6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thomsen M.C.F., Nielsen M.. Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res. 2012; 40:W281–W287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Piasecki S.K., Hall B., Ellington A.D.. Nucleic acid pool preparation and characterization. Methods Mol. Biol. 2009; 535:3–18. [DOI] [PubMed] [Google Scholar]
- 22. Scalley-Kim M., McConnell-Smith A., Stoddard B.L.. Coevolution of a homing endonuclease and its host target sequence. J. Mol. Biol. 2007; 372:1305–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takeuchi R., Certo M., Caprara M.G., Scharenberg A.M., Stoddard B.L.. Optimization of in vivo activity of a bifunctional homing endonuclease and maturase reverses evolutionary degradation. Nucleic Acids Res. 2009; 37:877–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bolduc J.M., Spiegel P.C., Chatterjee P., Brady K.L., Downing M.E., Caprara M.G., Waring R.B., Stoddard B.L.. Structural and biochemical analyses of DNA and RNA binding by a bifunctional homing endonuclease and group I intron splicing factor. Genes Dev. 2003; 17:2875–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lambert A.R., Hallinan J.P., Shen B.W., Chik J.K., Bolduc J.M., Kulshina N., Robins L.I., Kaiser B.K., Jarjour J., Havens K. et al. . Indirect DNA sequence recognition and its impact on nuclease cleavage activity. Structure. 2016; 24:862–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roulet E., Busso S., Camargo A.A., Simpson A.J.G., Mermod N., Bucher P.. High-throughput SELEX-SAGE method for quantitative modeling of transcription-factor binding sites. Nat. Biotech. 2002; 20:831–835. [DOI] [PubMed] [Google Scholar]
- 28. Jolma A., Kivioja T., Toivonen J., Cheng L., Wei G., Enge M., Taipale M., Vaquerizas J.M., Yan J., Sillanpää M.J. et al. . Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 2010; 20:861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burt A., Koufopanou V.. Homing endonuclease genes: the rise and fall and rise again of a selfish element. Curr. Opin. Genet. Dev. 2004; 14:609–615. [DOI] [PubMed] [Google Scholar]
- 30. Shen B.W., Lambert A., Walker B.C., Stoddard B.L., Kaiser B.K.. The structural basis of asymmetry in DNA binding and cleavage as exhibited by the I-SmaMI LAGLIDADG meganuclease. J. Mol. Biol. 2016; 428:206–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thyme S.B., Jarjour J., Takeuchi R., Havranek J.J., Ashworth J., Scharenberg A.M., Stoddard B.L., Baker D.. Exploitation of binding energy for catalysis and design. Nature. 2009; 461:1300–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chatterjee P., Brady K.L., Solem A., Ho Y., Caprara M.G.. Functionally distinct nucleic acid binding sites for a group I intron encoded RNA maturase/DNA homing endonuclease. J. Mol. Biol. 2003; 329:239–251. [DOI] [PubMed] [Google Scholar]
- 33. Lorenz C., von Pelchrzim F., Schroeder R.. Genomic systematic evolution of ligands by exponential enrichment (Genomic SELEX) for the identification of protein-binding RNAs independent of their expression levels. Nat. Protoc. 2006; 1:2204–2212. [DOI] [PubMed] [Google Scholar]
- 34. Singer B.S., Shtatland T., Brown D., Gold L.. Libraries for genomic SELEX. Nucleic Acids Res. 1997; 25:781–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.