

Abstract
Elucidating signaling pathways that regulate cellular metabolism is essential for a better understanding of normal development and tumorigenesis. Recent studies have shown that mitochondrial pyruvate carrier 1 (MPC1), a crucial player in pyruvate metabolism, is downregulated in colon adenocarcinomas. Utilizing zebrafish to examine the genetic relationship between MPC1 and Adenomatous polyposis coli (APC), a key tumor suppressor in colorectal cancer, we found that apc controls the levels of mpc1 and that knock down of mpc1 recapitulates phenotypes of impaired apc function including failed intestinal differentiation. Exogenous human MPC1 RNA rescued failed intestinal differentiation in zebrafish models of apc deficiency. Our data demonstrate a novel role for apc in pyruvate metabolism and that pyruvate metabolism dictates intestinal cell fate and differentiation decisions downstream of apc.
DOI: http://dx.doi.org/10.7554/eLife.22706.001
Research Organism: Zebrafish
eLife digest
Colon cancer remains an important problem in healthcare. Cancer researchers are looking for new ways to detect the disease earlier and treat it more effectively. This is challenging because many of the genetic and molecular causes of colon cancer are still poorly understood. Mutations in the gene that encodes a protein called APC are one of the major causes of the disease. The APC protein normally keeps cells from growing and dividing too fast or in an uncontrolled way and is hence referred to as a tumor suppressor. For example, APC induces stem cells in the intestine to develop into specialized cells that keep the gut working normally. Mutations in tumor suppressor genes are common in many cancers.
Other research has shown that cancer cells must reprogram their own metabolism – in other words, all the chemical processes that keep the cell alive – to meet the demands of proliferating rapidly. In particular, recent studies reveal that colon cancer cells produce less of a protein called mpc1, which is involved in metabolism. These discoveries raised the following questions: does APC have an additional role in maintaining normal metabolism in cells by controlling how much mpc1 is produced? Do mutations in the gene for APC lead to colon cancer because they alter the cell’s metabolism?
Sandoval et al. have now discovered a connection between APC and changes in cancer cells that help them to adapt to a new metabolic program. Experiments with zebrafish – a model animal that is now commonly used in the field of cancer biology – showed that APC acts via mpc1 to regulate how the cell uses energy. This regulation goes awry in colon cells that have abnormal APC activity; however, restoring the cell’s metabolism back to normal was enough to induce cells in the intestine to develop properly.
Together, these findings suggest that restoring the normal balance of energy production in colon cancer cells may be an effective way to make the cells behave normally. This hypothesis remains to be tested and, if confirmed, further studies will be needed to determine whether it will lead to new treatments for colon cancer in humans.
Introduction
Mutations in the adenomatous polyposis coli (APC) gene are responsible for Familial Adenomatous Polyposis (FAP), a genetic predisposition to colorectal cancer, and are also found in the majority of sporadic colonic tumors (Fearnhead et al., 2001). Critical roles for APC in colon carcinogenesis are attributed to its ability to negatively regulate the proliferative consequences of Wnt signaling through degradation of β-catenin, and maintain normal intestinal differentiation by controlling the biosynthesis of retinoic acid (RA) (Jette et al., 2004; Nadauld et al., 2006a, 2004, 2005; Rai et al., 2010; Schneikert et al., 2007; Shelton et al., 2006). Although tremendous progress has been made in understanding the role of APC, its full battery of functions continue to expand.
Altered energy metabolism is an emerging hallmark in cancer (Hanahan and Weinberg, 2011). The observation that cancer cells produce energy to support cell growth and proliferation differently than normal cells is known as the Warburg effect, and refers to neoplastic cells favoring aerobic glycolysis, even in the presence of ample oxygen (Vander Heiden et al., 2009; Warburg, 1956). One of the major molecular mechanisms contributing to Warburg effect is mitochondrial dysfunction through impaired pyruvate metabolism (Diaz-Ruiz et al., 2011).
Pyruvate lies at the junction of glycolysis and the tricarboxylic acid (TCA) cycle. Contingent on the metabolic needs of the cell, pyruvate can be transported into the mitochondria, and through the action mainly of pyruvate dehydrogenase (PDH), it can be used to drive ATP production and generate building blocks for macromolecule biosynthesis through oxidative phosphorylation. Alternatively, pyruvate can be converted to lactate via lactate dehydrogenase (LDH) and exported out of the cell. Aberrations in genes involved in pyruvate metabolism and transport have been reported in human diseases, particularly in cancer (Gray et al., 2014). For example, monocarboxylate transporter 4 (MCT4) and LDHA are overexpressed in cancer (Kim et al., 2013; Rong et al., 2013). An isoform of pyruvate kinase 2 (PKM2) is preferentially expressed in numerous cancer types including pancreatic, colon and lung, and has been shown to promote aerobic glycolysis in HeLa cells by functioning as a transcriptional coactivator for HIF-1 (Cerwenka et al., 1999; Christofk et al., 2008; Luo and Semenza, 2011; Schneider et al., 2002; Yeh et al., 2008). Restoration of the pyruvate dehydrogenase complex activity through inhibition of pyruvate dehydrogenase kinase 1 (PDK1) in head and neck squamous cell carcinoma cell lines led to reduced HIF-1a expression and tumor growth (McFate et al., 2008).
The recently identified mitochondrial pyruvate carrier subunit MPC1 is part of the MPC complex that is responsible for the uptake of pyruvate into the inner mitochondrial matrix (Bricker et al., 2012; Herzig et al., 2012). Recent work has revealed that MPC1 is downregulated in various human cancers and that this correlates with poor survival (Schell et al., 2014). Consistent with a causative role in tumorigenesis, re-expression of MPC1 repressed the Warburg effect in colon cancer cell lines (Schell et al., 2014). It is not clear how MPC1 is regulated or how its activities relate to the known genetic events that contribute to colon cancer development. Given the potential role for MPC1 in colorectal cancer and the importance of APC mutation, we investigated the mechanistic relationship between the mutational status of apc and mpc1. Herein, we report that apc regulates pyruvate metabolism by controlling the levels of mpc1 via RA. Further, mpc1 is required and sufficient for initiating normal intestinal differentiation downstream of apc. Our findings strongly suggest that changes in metabolic profile can drive cell fate and differentiation decisions.
Results
mpc1 and mpc2 are downregulated in apc-deficient zebrafish
To investigate the relationship between apc and mpc, we utilized the apcmcr zebrafish, which is homozygous for a truncating mutation in the Mutation Cluster Region (MCR) of apc and similar to what is found in human colon tumors (Hurlstone et al., 2003; Miyoshi et al., 1992a, 1992b). In parallel, we also knocked down the expression of apc in wild type (WT) embryos using antisense morpholino (apc mo) (Figure 1—figure supplement 1). Evaluating gene expression of mpc1 and mpc2 by qRT-PCR, we found that both genes were significantly downregulated in apcmcr and apc mo embryos compared to WT/het siblings and control mo, respectively (Figure 1A,B). This was confirmed by whole mount in situ hybridization for mpc1 and mpc2 (Figure 1C,D). Additional in situ analyses for mpc1 and mpc2 in WT embryos revealed staining in the head, eyes, vasculature and somites at 24–48 hr post-fertilization (hpf) (Figure 1E). At later time points, expression in the pectoral fin buds, liver and gut emerged (Figure 1E). Cross sections of 72 hpf WT embryos previously probed with mpc1 and mpc2 confirmed gut expression for both genes (black arrows) (Figure 1F).
Figure 1. mpc1 and mpc2 are downregulated in apcmcr and apc morphant embryos.
(A,B) Quantitative RT-PCR analysis of mpc1 and mpc2 gene expression in apcmcr (A) and apc mo (B) embryos. Values represent mean ± SD. Graph shown above is representative of at least three independent experiments. Statistical significance was analyzed using unpaired t-test. (C,D) Whole mount in situ hybridization for mpc1 and mpc2 in 72 hpf apcmcr (C) and apc mo (D) embryos. (E) Whole mount in situ hybridization for mpc1 and mpc2 in wild type (WT) embryos. head (h), eyes (e), somite (som), vasculature (vas), gut (g), liver (l). (F) Cross sections from 96 hpf WT embryos probed with either mpc1 or mpc2 confirmed gut-specific expression of both genes. See also Figure 1—figure supplement 1.
DOI: http://dx.doi.org/10.7554/eLife.22706.003

Figure 1—figure supplement 1. PCR analysis confirming apc knockdown.
Knock down of mpc1 phenocopies apc knock down
Previous studies have established phenotypes associated with impaired apc function in the developing zebrafish including malformation of the gut, eyes, pancreas and jaw, arrested fin buds and failed heart looping (Nadauld et al., 2004; Hurlstone et al., 2003; Nadauld et al., 2006b). To determine whether loss of mpc1 would recapitulate morphological defects related to apc deficiency, we knocked down the expression of mpc1 in WT embryos with a splice-blocking morpholino which we confirmed by PCR (Figure 2—figure supplement 1A). Microinjection of 0.75 mM mpc1 morpholino into WT embryos at the one- to two-cell stage resulted in about 87% of injected embryos appearing morphant (n = 228) (Figure 2—figure supplement 1B). Consistent with downregulation of mpc1 in apcmcr, mpc1 morphants (mpc1 mo) exhibited a range of phenotypes consisting of smaller head and eyes, enlarged hindbrain vesicle (black arrows), pericardial edema, body curvature, and loss of pectoral fins (blue arrows) (Figure 2A).
Figure 2. Knock down of mpc1 expression phenocopies loss of apc.
(A) Gross phenotype associated with mpc1 knock down. (B) Whole mount in situ hybridization analysis for organ-specific markers in mpc1 mo. Alcian blue staining revealed improper cartilage development (*) and confirmed loss of pectoral fins in mpc1 mo. pectoral fin bud (pfb), heart (h), gut (g), pancreas (p). (C) Cross section of the eye and gut from control or mpc1 mo. Prior to sectioning, embryos were previously stained with eye and gut-specific markers, irbp (red arrow) and fabp2 (black arrow), respectively. (D) Co-injection with human MPC1 RNA led to increased percentage of mpc1 mo with normal pectoral fins as determined by in situ staining for idi1. Statistical significance was analyzed using Fisher’s exact test. See also Figure 2—figure supplements 1, 2.

Figure 2—figure supplement 1. mpc1 morphant phenotype is rescued by human MPC1 RNA.

Figure 2—figure supplement 2. mpc2 morphants phenocopy loss of mpc1.

In situ hybridization with gata6 revealed that the primordial gut formed (93%, n = 55) in mpc1 mo but developed abnormally, as shown by reduced staining for fabp2 (100%, n = 36), which marks the differentiated gut (Figure 2B). Histological analyses on mpc1 mo gut confirmed these findings, there were fewer cells comprising the gut tube and they appeared cuboidal and non-polarized (Figure 2C). Additionally, intestinal folds were visibly lacking in the mpc1 mo gut (black arrow, Figure 2C). In contrast, cross-section of the gut from control mo showed polarized columnar intestinal cells, with the nuclei lined up clearly against the basal membrane (Figure 2C).
Since APC has also been reported to play a crucial role in congenital hypertrophy of retinal pigment epithelium (CHRPE) in humans and normal ocular development in the zebrafish embryo, we examined the eyes of mpc1 mo and found that irbp, a marker for photoreceptor and retinal pigmented epithelial cells, was severely reduced in mpc1 mo (95%, n = 40) (Figure 2B) (Nadauld et al., 2006b; Chapman et al., 1989). Cross-section of mpc1 mo eye revealed small lens and disorganized cell layers (Figure 2C). The retinal cells appeared to be undifferentiated as supported by the loss of irbp expression (red arrow, Figure 2C).
Further phenotypic analyses of mpc1 mo by in situ hybridization using tissue-specific markers exposed diminished maturation for brain (95%, n = 22) and fin buds (100%, n = 28) as indicated by ascl1a and id1 expression, respectively (Figure 2B). Also, mpc1 mo hearts failed to loop as determined by myl7 staining (100%, n = 39) (Figure 2B). As with the gut, terminal differentiation of the pancreas in mpc1 mo was severely reduced as assessed by trypsin expression, a marker for exocrine pancreas (100%, n = 28) (Figure 2B). Insulin, denoting the endocrine pancreas, remained normal (100%, n = 25) (Figure 2B). Cartilage staining with alcian blue confirmed the absence of pectoral fins and revealed improper jaw formation in mpc1 mo (100%, n = 152) (Figure 2B).
We verified that the morphological defects we observed in mpc1 mo were specifically due to knock down of mpc1 by co-injecting with 0.5 ng of full length human MPC1 mRNA and analyzing the embryos by in situ hybridization for id1 and otx2, a marker for both the midbrain and eyes. Overexpression of MPC1 mRNA alone resulted mostly in normal-appearing embryos, a small percentage exhibited cyclopia, severe body curvature and truncated tail (29%, n = 187) (Figure 2—figure supplement 1C). However, in co-injected embryos (mpc1 mo + MPC1 RNA), we found that MPC1 mRNA restored fin development as indicated by id1 staining (50%, n = 49) (Figure 2D, Figure 2—figure supplement 1D). We obtained similar results with otx2, MPC1 mRNA was able to rescue normal midbrain and eye development in mpc1 mo (Figure 2—figure supplement 1D and E). The presence of MPC1 transcript was confirmed by PCR (Figure 2—figure supplement 1F).
mpc1 and mpc2 form a heterodimer complex that is responsible for transporting pyruvate from the inner mitochondrial space into the inner mitochondrial matrix (Bricker et al., 2012; Herzig et al., 2012). To examine whether loss of mpc2 would result in similar phenotypes as mpc1 mo, we knocked down its expression in WT embryos using antisense morpholino (Figure 2—figure supplement 2A and B). We found that mpc2 mo exhibited similar developmental defects as mpc1 mo, such as smaller head and eyes, enlarged hindbrain vesicle (black arrows), body curvature, and absence of pectoral fins (blue arrows) (Figure 2—figure supplement 2B). In contrast to mpc1 mo, only a third of mpc2 mo-injected embryos appeared morphant, the majority of which exhibited a mild phenotype (Figure 2—figure supplement 2C, Figure 2—figure supplement 1A, data not shown). MPC2 expression is inconsistently altered in cancer and variably correlated with survival (Schell et al., 2014). We therefore focused further studies on mpc1.
MPC1 rescues intestinal differentiation in apc-deficient embryos
In light of our data relating reduced mpc1 levels to failed intestinal differentiation, we sought to determine if re-expression of MPC1 would rescue intestinal defects in apc-deficient zebrafish embryos. We injected 0.75 mM apc mo with or without 0.1 ng MPC1 mRNA into WT embryos and evaluated intestinal differentiation by in situ hybridization for fabp2. Compared to apc mo (6%, n = 265), there was a significant increase in embryos with differentiated gut in the morpholino plus mRNA group (29%, n = 312) (Figure 3A,C). Control embryos all displayed normal fabp2 expression (data not shown).
Figure 3. MPC1 rescues gut phenotype of apc mo and apcmcr.

(A,B) In situ hybridization for fabp2, in 72 hpf WT embryos injected with cont mo, apc mo or both apc mo and human MPC1 RNA (apc mo + MPC1 RNA) (A). In situ hybridization for fabp2 in 72 hpf apc WT, apcmcr or apcmcr injected with human MPC1 mRNA (apcmcr + MPC1 RNA) (B). (C,D) Quantification of injected embryos with differentiated gut as determined by fabp2 staining. Statistical significance was analyzed using Fisher’s exact test.
We confirmed this finding by injecting 0.5 ng MPC1 mRNA into apcmcr and observed similar results, 40% of injected embryos re-expressed fabp2 (n = 52) (Figure 3B,D). However, only 3% of un-injected apcmcr showed fabp2 staining (n = 29), while WT/het siblings were all positive (n = 80, data not shown). MPC1 was also able to rescue cardiac defects in apcmcr, as we saw improved blood circulation in injected mutants as well (data not shown). These results suggest that re-introduction of mpc1 can drive intestinal differentiation.
Knock down of mpc1 or apc alters mitochondrial function
Because of the integral role of MPC1 in pyruvate metabolism, we next investigated if mpc1 mo harbor metabolic defects as a consequence of diminished mpc1 function. We assessed mitochondrial respiration by measuring oxidative consumption rates (OCR) in 72 hpf embryos and there was a significant reduction in OCR in mpc1 mo compared to control (Figure 4A). We also looked at triglyceride (TG) levels as an indicator of disturbance in normal energy utilization and observed a similar trend (Figure 4B). Moreover, there was an extensive dysregulation of pyruvate metabolism upon loss of mpc1, as we discovered a profound upregulation of pyruvate metabolic genes in mpc1 mo, suggesting a compensatory mechanism to account for reduced pyruvate transport across the inner mitochondrial membrane (Figure 4—figure supplement 1A and B). Knock down of mpc2 did not affect mpc1 transcript level (Figure 4—figure supplement 1C).
Figure 4. Knock down of mpc1 or apc leads to altered mitochondrial respiration and pyruvate metabolism.
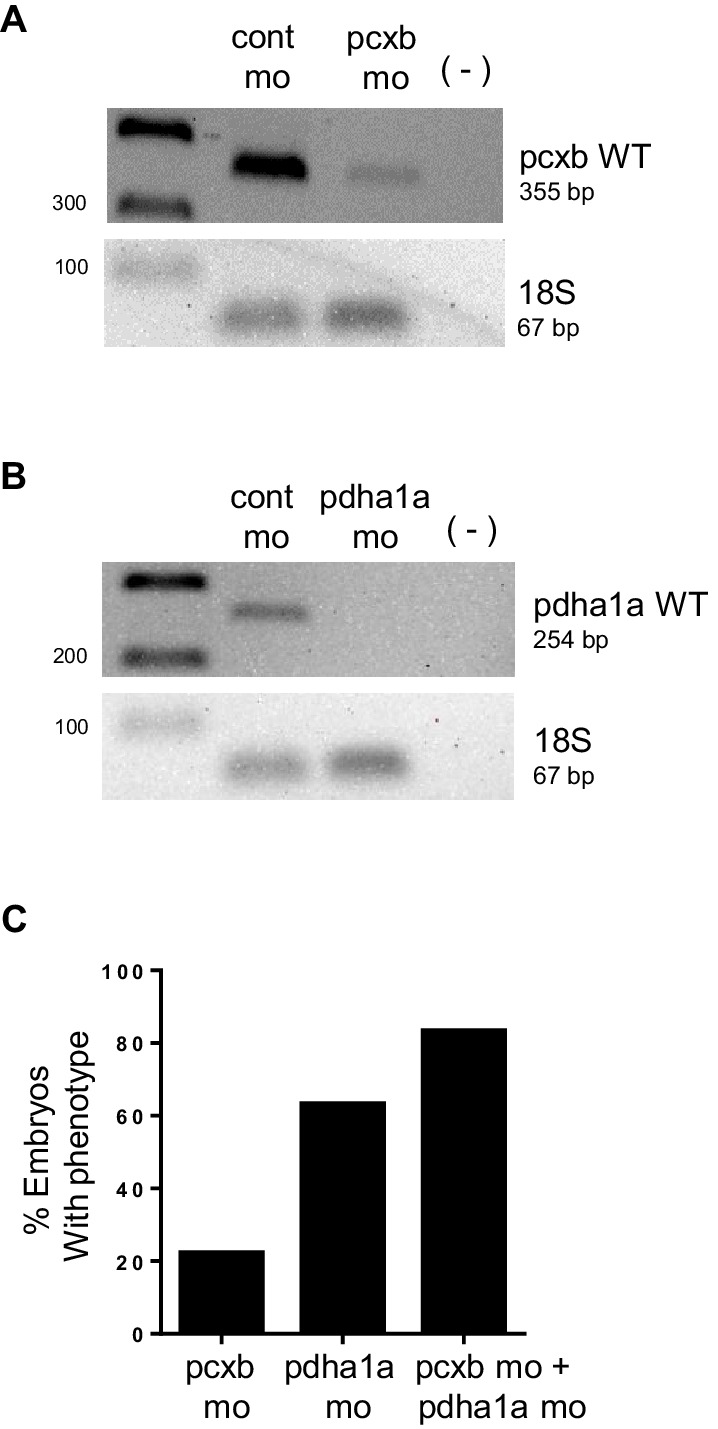
(A) Mitochondrial respiration was evaluated by measuring oxygen consumption rates (OCR) in 72 hpf embryos. (B) Triglyceride (TG) levels were determined in lysates prepared from 72 hpf embryos using a colorimetric assay. (C,D) Quantitative RT-PCR analysis of enzymes involved in pyruvate metabolism in apc mo and (C) apcmcr (D) embryos. pyruvate dehydrogenase alpha 1a (pdha1a); pyruvate dehydrogenase kinase, isozyme 1 (pdk1); pyruvate kinase, muscle, a (pkma); citrate synthase (cs). (E) Lactate levels in apc wild type (WT), un-injected apcmcr (UI) or apcmcr embryos injected with human MPC1 mRNA (MPC1 RNA). For figures A–E, values represent mean ± SD. Graph shown above is representative of at least three independent experiments. Statistical significance was analyzed using unpaired t-test. (F,G,H) Gross phenotype (F), alcian blue staining (G) and in situ hybridization for fabp2 (H) in pdha1a, pcxb, and pcxb + pdha1a mo. pcxb (pyruvate carboxylase b). See also Figure 4—figure supplements 1, 2.
DOI: http://dx.doi.org/10.7554/eLife.22706.010

Figure 4—figure supplement 1. Knockdown of mpc1 leads to dysregulated pyruvate metabolism.

Figure 4—figure supplement 2. PCR analysis confirming knockdown of pdha1a, pcxb.
Earlier studies have reported that MPC1 is downregulated in colon cancer and its expression positively correlates with APC (Schell et al., 2014). Together with our previous data showing that mpc1 is downregulated in apc-deficient zebrafish and that mpc1 mo exhibit impaired oxidative respiration, we hypothesized that apc regulates mpc1 and therefore, pyruvate metabolism overall. To test this, we evaluated mitochondrial respiration and TG levels in apcmcr and apc mo and compared to appropriate controls, we observed significant defects in mitochondrial function upon loss of apc (Figure 4A,B). Several enzymes in the pyruvate pathway were also differentially regulated in apcmcr or apc mo (Figure 4C,D).
To further validate that impaired mitochondrial function in apcmcr is facilitated by reduced mpc1 expression, we injected MPC1 mRNA into apc mutant embryos and looked at lactate levels as indicator of improved mitochondrial function. Compared to un-injected apcmcr, mutant embryos overexpressing MPC1 showed a significant reduction in lactate (Figure 4E). apc WT/het sibs (WT) represent basal lactate levels in normal embryos (Figure 4E).
Pyruvate, after passing through the inner mitochondrial membrane, is converted to oxaloacetate and acetyl-CoA by pyruvate carboxylase (PC) and pyruvate dehydrogenase (PDH), respectively. To ascertain if the knock down of enzymes downstream of mpc1 would result in a phenotype similar to mpc1 mo, we targeted pcxb and pdha1a, separately and in tandem, with antisense morpholinos (Figure 4—figure supplement 2A and B). Loss of either metabolic gene or both, resulted in morphant embryos that lacked pectoral fins (blue arrows), jaw (red arrows) and appeared identical to mpc1 mo (Figures 4F–H, 2A). Interestingly, knock down of pcxb resulted in only 22% of embryos with phenotype (n = 27) while targeting pdha1a gave a higher percentage of morphant embryos (63%, n = 19) (Figure 4—figure supplement 2C). A synergistic effect was observed when both enzymes were diminished (83%, n = 18) (Figure 4—figure supplement 2C). fabp2 in situ staining (black arrows) also revealed intestinal developmental defects in the pdha1a mo, pcxb mo and pdha1a + pcxb morphant embryos (Figure 4H). Knock down of pcxb expression, however, not only resulted in low penetrance but mild phenotype as well (Figure 4F–H). This could be due to the activation of an alternative pathway where oxaloacetate can be derived from glutamine instead of pyruvate (DeBerardinis et al., 2007).
Decreased RA levels lead to aberrant pyruvate metabolism
To further elucidate how apc is controlling mpc1, we initially looked at Wnt signaling as one of the major roles of APC is to regulate degradation of β-catenin (Fearnhead et al., 2001). Perturbation of the Wnt pathway by treatment of apc mo with 10 uM NS-398, a COX-2-specific inhibitor that has been shown to impair β-catenin activity in an apc-deficient background, did not affect mpc1 or mpc2 levels (Figure 5A) (Eisinger et al., 2007). We did see a dramatic reduction in expression of known β-catenin target gene, mmp9, implying that apc regulation of mpc1 is independent of Wnt (Figure 5A).
Figure 5. RA deficiency results in dysregulated pyruvate metabolism that is independent of Wnt pathway.

(A) apc mo treated either with DMSO control or 10 uM NS-398 were analyzed by qRT-PCR to determine expression levels of mpc1, mpc2 and mmp9. (B,C) WT embryos treated with either DMSO control or 5 uM DEAB were analyzed by qRT-PCR to determine expression levels of enzymes involved in pyruvate metabolism. (D) Lactate levels in 72 hpf WT embryos treated with either DMSO or 5 uM DEAB. For figures A–D, values represent mean ± SD. Graph shown above is representative of 3 independent experiments. Statistical significance was analyzed using unpaired t-test. matrix metallopeptidase 9 (mmp9); solute carrier family 16, member 1 (slc16a1)..
DOI: http://dx.doi.org/10.7554/eLife.22706.015
In addition to regulating Wnt signaling, apc controls the program of intestinal differentiation through regulation of RA levels, as we have previously shown (Jette et al., 2004; Nadauld et al., 2006a, 2004, 2005; Rai et al., 2010; Shelton et al., 2006). To interrogate the involvement of RA in pyruvate metabolism downstream of apc, we treated WT embryos with DMSO or 5 uM DEAB, a known inhibitor of RALDH which catalyzes the second step in the conversion of Vitamin A to the active metabolite, RA (Marill et al., 2003; Russo et al., 1988). By qRT-PCR, we found that mpc1 and mpc2 transcript levels went down significantly with inhibition of RA (Figure 5B). Expanding our analyses to include other components of pyruvate metabolism, there were five other genes that were either up- or downregulated upon DEAB treatment, supporting the notion that the pyruvate metabolic program is altered at various points when RA levels are perturbed (Figure 5C). The DEAB-treated embryos also had low lactate levels, further establishing that RA inhibition results in mitochondrial dysfunction (Figure 5D).
Pyruvate metabolism is dysregulated in colon cancer
To further understand the implication of our findings in zebrafish, we employed publicly available curated databases to investigate mutations and gene expression alterations of pyruvate metabolism genes in human cancers. Using samples deposited at The Cancer Genome Atlas (TCGA), we selected for colon adenocarcinomas with mutations upstream of codon 1600 of APC, a region encompassing the MCR, and resulting in a truncated protein similar to those found in a majority of patients with FAP (Fearnhead et al., 2001). Consistent with our previous data, we found that mpc1 expression is significantly downregulated in colon adenocarcinomas with APC deletions (n = 91) compared to normal colon (n = 19) (Figure 6A). We also looked at other genes involved in pyruvate transport and metabolism and interestingly, MPC1, MPC2, PDHA1 and PC showed consistent downregulation in a specific subset of colon adenocarcinomas known as colon mucinous adenocarcinomas (AC) (n = 22) (Figure 6—figure supplement 1A). In addition to altered gene expression, we also found colorectal cancer samples in COSMIC that had mutations both in APC and pyruvate metabolism enzymes. There were five samples with somatic APC deletions that had mutations in multiple pyruvate metabolism enzymes as well, most of which are predicted to be probably (**) or possibly (*) damaging, further supporting a genetic link between APC mutation and dysregulation of pyruvate metabolism (Figure 6—figure supplement 1B).
Figure 6. In silico analyses of APC and pyruvate metabolism gene alterations in cancer.
(A) MPC1 expression levels in TCGA normal colon and colon adenocarcinoma samples harboring truncating mutations in APC upstream of codon 1600. Statistical significance was analyzed using Mann Whitney test. (B) ONCOMINE database was analyzed for gene expression alterations in pyruvate metabolism genes in all cancer types. A control group composed of Uniprot random genes was used for comparison. Graph shows percentage of up- and downregulated genes with respect to the total unique analyses for each gene tested for all cancer groups. Statistical significance was analyzed using Mann Whitney test. (C) cBioportal analysis to estimate Kaplan-Meier overall survival of TCGA colorectal adenocarcinoma and kidney chromophobe patients with or without alterations in pyruvate metabolism (PM) genes. For B–C, pyruvate metabolism genes used in meta-analyses: MPC1, MPC2, CS, PDK1, PDHA1, PC, PKLR, LDHA, SLC16A1, GYS1. See also Figure 6—figure supplement 1, Supplementary files 1, 2.

Figure 6—figure supplement 1. Pyruvate metabolism genes are mutated in human colon carcinomas.

Several studies have shown that individual genes in the pyruvate metabolism pathway are altered in various cancer types (Gray et al., 2014; Schell et al., 2014). Using Oncomine, we extended these studies by treating the genes involved in pyruvate transport and metabolism as a group (n = 10). We discovered that this pathway is significantly dysregulated in cancer compared to a group of randomly-generated Uniprot genes (n = 55) (Figure 6B, Supplementary file 1). We then looked at overall survival for patients, with or without mutations and/or gene expression changes in the pyruvate metabolism gene set using TCGA samples in cBioportal. Out of 21 cancer types that we analyzed, only colorectal adenocarcinoma and kidney chromophobe carcinoma showed a significant difference in overall survival between the two groups (Figure 6C, Supplementary file 2).
Discussion
Recent identification of MPC1 and MPC2, genes responsible for pyruvate uptake into the mitochondrial matrix, has added a new complexity to targeting pyruvate metabolism in human disorders, including cancer (Gray et al., 2014; Bricker et al., 2012; Herzig et al., 2012). How dysregulation of metabolism relates to the accumulation of genetic hits that cause tumor suppression has largely been unstudied. Here, we demonstrate a direct relationship between loss of a key tumor suppressor gene, APC, and dysregulation of MPC1. Our finding that mpc1 expression is downregulated in embryos harboring a genetic mutation (apcmcr) or knocked down expression (apc mo) of apc is reflected in human tumors as well, where we not only discovered a significant downregulation of MPC1 expression in colon adenocarcinomas with APC deletions, but also samples possessing mutations in both APC and several pyruvate metabolism enzymes (Figure 1, Figure 6, Figure 6—figure supplement 1). Our meta-analyses of human cancer data sets also revealed extensive dysregulation of pyruvate metabolism, at multiple points in the pathway, and this incidence of altered gene expression of pyruvate metabolism genes in cancer is significantly more prevalent compared to a group of randomly selected genes (Figure 6, Figure 6—figure supplement 1, Supplementary file 1). It is remarkable how MPC1 and MPC2, PDHA1 and PC—genes that are involved in the transport and conversion of pyruvate in the inner mitochondrial matrix, respectively—are all significantly downregulated in a subset of colon adenocarcinomas categorized as colon mucinous adenocarcinomas, as loss of these genes essentially shuts down oxidative phosphorylation in the mitochondria (Figure 6—figure supplement 1). Interestingly, additional in silico analyses suggest a potential use of pyruvate metabolism genes as prognostic markers for colorectal adenocarcinoma and kidney chromophobe carcinoma, as we found a strong correlation between aberrations in pyruvate metabolism genes with poor overall survival in these cancer types (Figure 6).
It is interesting to note that there are differences in the dysregulation of pyruvate metabolism genes in apcmcr/apc mo and mpc1 mo (Figure 4, Figure 4—figure supplement 1). APC is a multifunctional protein that has critical roles in various cellular processes (Fodde, 2003). In addition, regulation of other metabolic genes by APC could occur in parallel with regulation of mpc1. mpc1 knock down alone, therefore, would not alter the expression of these genes in the same way as knock down of apc. Confirming a functional epistatic relationship between apc and mpc1, knock down of mpc1 in WT embryos resulted in phenotypes that have been previously reported for impaired apc function (Figure 2) (Nadauld et al., 2004; Hurlstone et al., 2003). APC has been shown to positively regulate glycogen synthase kinase-3 (GSK-3) activity, an enzyme that inhibits glycogen synthase (GS) which is involved in converting glucose into glycogen for storage (Bouskila et al., 2010; Valvezan et al., 2012). Taken together, these findings suggest a major role for APC in controlling cellular bioenergetics and homeostasis, as it can affect glycogen synthesis and oxidative phosphorylation through GSK-3 and MPC1, respectively.
The exact role of metabolism in cancer as a driver versus passenger process has been unclear. Indeed, roles for metabolism in directing cell fate and differentiation decisions are only now being considered (Schell et al., 2014; Bracha et al., 2010; Sperber et al., 2015; Yanes et al., 2010; Zhou et al., 2012). The rescue of intestinal differentiation defects in embryos with impaired apc function by exogenous MPC1 mRNA establishes a clear role for metabolic programming as a switch that can control cell fate decisions (Figure 3). Mechanistic insights into regulatory pathways that control metabolism and how perturbations in cellular bioenergetics effect cell differentiation and proliferation can lead to a better understanding of normal development and tumorigenesis. In this respect, the role of retinoic acid in promoting cell fate and differentiation remains undefined. Our studies suggest that RA may control a program of metabolism that is permissive for intestinal differentiation. The actions of RA are complex, and it is likely that the effects of RA on metabolism are indirect. Consistent with this, treatment of either WT or apcmcr embryos with RA did not result in an immediate induction of mpc1 (data not shown).
To conclude, we present a novel role for apc in controlling a metabolic program driving intestinal differentiation through regulation of mpc1. Our data strongly support the notion that metabolic changes are a major part of the decision process in determining cell fate and provide a better understanding of how cancer genetics is linked with biochemical metabolic pathways.
Materials and methods
Zebrafish maintenance
Wild-type (WT) TU (RRID:ZIRC_ZL57) and apcWT/mcr (RRID:ZFIN_ZDB-ALT-050914-2) Danio rerio (zebrafish) were maintained as previously described (Westerfield, 1995). Fertilized embryos were collected following natural spawnings in 1 × E3 medium (286 mg/L NaCl, 13 mg/L KCl, 48 mg/L CaCl2·2H2O, 40 mg/L MgSO4, 0.01% methylene blue) and allowed to develop at 28.5°C.
Morpholino and RNA microinjections
Morpholino oligonucleotides were obtained from Gene Tools LLC (Philomath, OR) and solubilized to 1 mM or 3 mM stock solutions in 1x Danieau buffer. For microinjections, 1 nl of morpholino was injected into WT embryos at the 1- to 2-cell stages (Draper et al., 2001). Knock down of gene expression was assessed by PCR. Primers were designed according to guidelines recommended by Gene Tools (www.gene-tools.com) to amplify WT and splice-blocked morphant bands. z18s was amplified as a control gene.
For RNA rescue experiments, full length human RNA transcripts were transcribed from linearized DNA using mMESSAGE mMACHINE transcription kit (Ambion - Waltham, MA). For microinjections, 1–2 nl of RNA was injected into embryos at the 1- to 2-cell stages. Overexpression of mRNA transcript was assessed by PCR. Statistical analyses were performed using Fisher’s exact test (GraphPad Prism v 6.04, RRID:SCR_002798).
A complete list of morpholinos, PCR primers and working concentrations used are provided in Supplementary files 3–4.
Zebrafish drug treatments
Wild type embryos were given DEAB (VWR International - Radnor, PA) at 5 µM. apc morphants were treated with 10 µM NS398 (Cayman Chemical - Ann Arbour, MI). Embryos were harvested at 72 hpf in RNAlater (Ambion) for RNA/cDNA prep.
In situ hybridization
In situ hybridizations were performed as previously described using digoxigenin-labeled riboprobes for ascl1a (achaete-scute family bHLH transcription factor 1a), fabp2 (fatty acid binding protein 2, intestinal), gata6 (GATA binding protein 6), id1 (inhibitor of DNA binding 1), insulin, irbp (interphotoreceptor retinoid-binding protein), mpc1 (mitochondrial pyruvate carrier 1), mpc2 (mitochondrial pyruvate carrier 2), myl7 (myosin, light chain 7, regulatory), otx2 (orthodenticle homeobox 2) and trypsin (Thisse and Thisse, 2008). Embryos were cleared in 2:1 benzyl benzoate/benzyl alcohol solution and documented using an Olympus SZX12/DP71 imaging system (Olympus Corporation - Japan). RNA Reference Sequences deposited in ZFIN (zfin.org, RRID:SCR_002560) were used in designing the riboprobes.
Quantitative RT-PCR
RNA from zebrafish embryo lysates was isolated using the RNeasy kit (Qiagen - Germany). cDNA was synthesized from 1 µg of total RNA using iScript (Bio-Rad - Hercules, CA). Intron-spanning primers, when possible, were designed using the Universal ProbeLibrary Assay Design Center (Roche Applied Science). A complete list of primer sets is provided in Supplementary file 5.
PCR master mix was prepared with the FastStart Essential DNA Probe Master kit and Universal ProbeLibrary probes according to the manufacturer’s protocols (Roche Applied Science - Germany). PCR was performed in triplicate using the LightCycler 96 System (Roche Applied Science) with 45 cycles of amplification and annealing temperature of 60°C. Fold change in gene expression was measured by normalizing against 18S rRNA and comparing test group with control.
Alcian blue assay
Cartilage of 96 hpf embryos was stained with alcian blue as previously described (Neuhauss et al., 1996). Briefly, embryos were fixed in 4% sucrose-buffered paraformaldehyde, bleached with 30% hydrogen peroxide for 2 hr and stained with alcian blue overnight. The embryos were then cleared in acidic ethanol for 4 hr, dehydrated stepwise in ethanol and stored either in glycerol or 2:1 benzyl benzoate/benzyl alcohol solution. Stained embryos were examined using an Olympus SZX12/DP71 imaging system (Olympus Corporation).
Seahorse bioscience XF assay
Metabolic respiration in 72 hpf embryos, expressed as oxygen consumption rate (OCR), was measured using XF24 Extracellular Flux Analyzer (Seahorse Bioscience - North Billerica, MA) as previously described (Stackley et al., 2011). As a minor modification, mixing step was omitted during measurement cycle. Statistical analyses were performed using unpaired t-test (GraphPad Prism v 7.02, RRID:SCR_002798).
Triglyceride (TG) assay
Embryos were harvested at 72 hpf and homogenized in 0.05% PBST +1X protease inhibitor. TG levels were determined using the Infinity Triglycerides Liquid Stable Reagent (Thermo Scientific - Waltham, MA) by measuring absorbance at 540 nm. Total protein concentration was determined using the DC Protein Assay (Bio-Rad) to normalize TG levels. Statistical analyses were performed using unpaired t-test (GraphPad Prism v 7.02, RRID:SCR_002798).
Lactate assay
Lactate levels were measured in 72 hpf embryos using the EnzyChrom L-Lactate Assay kit (BioAssay Systems - Hayward, CA) as previously described (Bestman et al., 2015). Groups of 25–50 embryos were used in the assay. Statistical analyses were performed using unpaired t-test (GraphPad Prism v 7.02, RRID:SCR_002798).
Histological analyses
Embryos were fixed in 10% neutral buffered formalin, dehydrated in 70% ethanol and embedded in paraffin. Five-micron sections were cut using a Shandon Finesse E Microtome (Thermo Scientific) and stained with hematoxylin and eosin (H and E). Sections were analyzed using a Nikon Eclipse 80i/DS-Fi1 imaging system (Nikon Instruments Inc - Japan).
Bioinformatic analyses
Publicly available curated databases and analysis software were utilized to examine mutations and gene expression alterations in APC and pyruvate metabolism enzymes (MPC1, MPC2, CS, PDK1, PDHA1, PC, PKLR, LDHA, SLC16A1,GYS1). COSMIC (http://cancer.sanger.ac.uk/cosmic, RRID:SCR_002260) was mined for mutations that are found in human cancers (Forbes et al., 2015). Polyphen2 (http://genetics.bwh.harvard.edu/pph2/index.shtml, RRID:SCR_008584) was used to predict functional and structural consequences of amino acid substitutions in proteins mentioned above (Adzhubei et al., 2010).
For MPC1 expression analysis, Oncomine (www.oncomine.org, RRID:SCR_010949) was utilized to determine the colon adenocarcinoma subset (n = 101) in the TCGA (http://cancergenome.nih.gov, RRID:SCR_003193) colorectal carcinoma sample set (n = 237), which was further selected, using COSMIC, for truncating mutations in APC upstream of codon 1600, encompassing the MCR region (n = 91). MPC1 expression level in these samples was compared with normal colon (n = 19). Colon mucinous adenocarcinomas (n = 22) within the same TCGA colorectal carcinoma sample set were analyzed for gene expression of pyruvate metabolism genes, with normal colon as control. Statistical analysis was performed using Mann Whitney test (GraphPad Prism v 6.04, RRID:SCR_002798).
Oncomine also allowed for gene expression analysis of individual pyruvate metabolism genes (n = 10) and randomly selected Uniprot genes (n = 55) (http://www.uniprot.org/uniprot/?query=reviewed:yes+AND+organism:9606&random=yes, RRID:SCR_002380) in multiple cancer types employing these thresholds: p value = 0.001; fold-change = 1.5; gene rank = top 10%; data type = all. A plot was generated to show percentage of datasets meeting set thresholds with respect to total unique analyses for each gene tested in both groups. Statistical analysis was performed using unpaired t-test (GraphPad Prism v 6.04, RRID:SCR_002798).
Overall survival in 21 different TCGA cancer types, segregated by presence of mutations in pyruvate metabolism genes, was analyzed with cBioportal (http://www.cbioportal.org/index.do, RRID:SCR_014555) (Cerami et al., 2012). To verify the specificity of our pyruvate metabolism gene set as predictor of overall survival, four groups of ten random genes from Uniprot were utilized as a negative control gene set.
Statistical analyses
Unpaired t-test was used to compare two unmatched, independent groups. Fisher’s exact test was used to determine if outcome is related to a categorical condition by more than chance. Mann-Whitney test was used to compare distribution of two unmatched groups. For fold change data, statistical significance was determined from t-test analyses of relative gene expression. For sample size calculations, the minimum number of samples per group (95% power) was determined by assuming the probability of the defect in the control group is 5% or lower and 80% in the experimental group.
Acknowledgements
We wish to thank the following: S Lee and HY Lim for assistance with the TG assay, A Vara for technical support, and OMRF Core Labs. This work was supported by NCI/NIH (RO1 CA116468NIH, [DAJ]), Samuel Waxman Cancer Research Foundation, Oklahoma Center for Adult Stem Cell Research (OCASCR) and Oklahoma Medical Research Foundation (OMRF).
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Funding Information
This paper was supported by the following grants:
National Cancer Institute RO1 CA116468NIH to David A Jones.
Samuel Waxman Cancer Research Foundation to David A Jones.
Oklahoma Medical Research Foundation to David A Jones.
Oklahoma Center for Adult Stem Cell Research to David A Jones.
Additional information
Competing interests
The authors declare that no competing interests exist.
Author contributions
ITS, Conceptualization, Formal analysis, Investigation, Visualization, Writing—original draft, Project administration, Writing—review and editing.
RGCD, Formal analysis, Investigation, Visualization, Writing—original draft, Writing—review and editing.
BNM, Investigation, Writing—review and editing.
SH, Investigation.
KAO, Resources.
AEG, Formal analysis, Investigation, Writing—review and editing.
KB, Formal analysis, Investigation, Writing—review and editing.
CS, Investigation, Writing—review and editing.
HVR, Resources.
JR, Resources, Writing—review and editing.
DAJ, Conceptualization, Supervision, Funding acquisition, Writing—original draft, Writing—review and editing.
Ethics
Animal experimentation: This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All of the animals were handled according to approved Institutional Animal Care And Use Committee (IACUC) protocol 14-08 of the Oklahoma Medical Research Foundation.
Additional files
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestman JE, Stackley KD, Rahn JJ, Williamson TJ, Chan SS. The cellular and molecular progression of mitochondrial dysfunction induced by 2,4-dinitrophenol in developing zebrafish embryos. Differentiation. 2015;89:51–69. doi: 10.1016/j.diff.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouskila M, Hunter RW, Ibrahim AF, Delattre L, Peggie M, van Diepen JA, Voshol PJ, Jensen J, Sakamoto K. Allosteric regulation of glycogen synthase controls glycogen synthesis in muscle. Cell Metabolism. 2010;12:456–466. doi: 10.1016/j.cmet.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Bracha AL, Ramanathan A, Huang S, Ingber DE, Schreiber SL. Carbon metabolism-mediated myogenic differentiation. Nature Chemical Biology. 2010;6:202–204. doi: 10.1038/nchembio.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, Redin C, Boudina S, Gygi SP, Brivet M, Thummel CS, Rutter J. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;337:96–100. doi: 10.1126/science.1218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerwenka H, Aigner R, Bacher H, Werkgartner G, el-Shabrawi A, Quehenberger F, Mischinger HJ. TUM2-PK (pyruvate kinase type tumor M2), CA19-9 and CEA in patients with benign, malignant and metastasizing pancreatic lesions. Anticancer Research. 1999;19:849–851. [PubMed] [Google Scholar]
- Chapman PD, Church W, Burn J, Gunn A. Congenital hypertrophy of retinal pigment epithelium: a sign of familial adenomatous polyposis. British Medical Journal. 1989;298:353–354. doi: 10.1136/bmj.298.6670.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. PNAS. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Ruiz R, Rigoulet M, Devin A. The warburg and Crabtree effects: on the origin of cancer cell energy metabolism and of yeast glucose repression. Biochimica Et Biophysica Acta (BBA) - Bioenergetics. 2011;1807:568–576. doi: 10.1016/j.bbabio.2010.08.010. [DOI] [PubMed] [Google Scholar]
- Draper BW, Morcos PA, Kimmel CB. Inhibition of zebrafish fgf8 pre-mRNA splicing with morpholino oligos: a quantifiable method for gene knockdown. Genesis. 2001;30:154–156. doi: 10.1002/gene.1053. [DOI] [PubMed] [Google Scholar]
- Eisinger AL, Nadauld LD, Shelton DN, Prescott SM, Stafforini DM, Jones DA. Retinoic acid inhibits beta-catenin through suppression of Cox-2: a role for truncated adenomatous polyposis coli. Journal of Biological Chemistry. 2007;282:29394–29400. doi: 10.1074/jbc.M609768200. [DOI] [PubMed] [Google Scholar]
- Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Human Molecular Genetics. 2001;10:721–733. doi: 10.1093/hmg/10.7.721. [DOI] [PubMed] [Google Scholar]
- Fodde R. The multiple functions of tumour suppressors: it's all in APC. Nature Cell Biology. 2003;5:190–192. doi: 10.1038/ncb0303-190. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, Teague JW, Stratton MR, McDermott U, Campbell PJ. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Research. 2015;43:D805–D811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray LR, Tompkins SC, Taylor EB. Regulation of pyruvate metabolism and human disease. Cellular and Molecular Life Sciences. 2014;71:2577–2604. doi: 10.1007/s00018-013-1539-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ER, Martinou JC. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;337:93–96. doi: 10.1126/science.1218530. [DOI] [PubMed] [Google Scholar]
- Hurlstone AF, Haramis AP, Wienholds E, Begthel H, Korving J, Van Eeden F, Cuppen E, Zivkovic D, Plasterk RH, Clevers H. The wnt/beta-catenin pathway regulates cardiac valve formation. Nature. 2003;425:633–637. doi: 10.1038/nature02028. [DOI] [PubMed] [Google Scholar]
- Jette C, Peterson PW, Sandoval IT, Manos EJ, Hadley E, Ireland CM, Jones DA. The tumor suppressor adenomatous polyposis coli and caudal related homeodomain protein regulate expression of retinol dehydrogenase L. Journal of Biological Chemistry. 2004;279:34397–34405. doi: 10.1074/jbc.M314021200. [DOI] [PubMed] [Google Scholar]
- Kim S, Jung WH, Koo JS. The expression of Glut-1, CAIX, and MCT4 in mucinous carcinoma. Journal of Breast Cancer. 2013;16:146–151. doi: 10.4048/jbc.2013.16.2.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Semenza GL. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget. 2011;2:551–556. doi: 10.18632/oncotarget.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marill J, Idres N, Capron CC, Nguyen E, Chabot GG. Retinoic acid metabolism and mechanism of action: a review. Current Drug Metabolism. 2003;4:1–10. doi: 10.2174/1389200033336900. [DOI] [PubMed] [Google Scholar]
- McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, Zhou S, Califano JA, Jeoung NH, Harris RA, Verma A. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. Journal of Biological Chemistry. 2008;283:22700–22708. doi: 10.1074/jbc.M801765200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi Y, Ando H, Nagase H, Nishisho I, Horii A, Miki Y, Mori T, Utsunomiya J, Baba S, Petersen G. Germ-line mutations of the APC gene in 53 familial adenomatous polyposis patients. PNAS. 1992a;89:4452–4456. doi: 10.1073/pnas.89.10.4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Human Molecular Genetics. 1992b;1:229–233. doi: 10.1093/hmg/1.4.229. [DOI] [PubMed] [Google Scholar]
- Nadauld LD, Sandoval IT, Chidester S, Yost HJ, Jones DA. Adenomatous polyposis coli control of retinoic acid biosynthesis is critical for zebrafish intestinal development and differentiation. Journal of Biological Chemistry. 2004;279:51581–51589. doi: 10.1074/jbc.M408830200. [DOI] [PubMed] [Google Scholar]
- Nadauld LD, Shelton DN, Chidester S, Yost HJ, Jones DA. The zebrafish retinol dehydrogenase, rdh1l, is essential for intestinal development and is regulated by the tumor suppressor adenomatous polyposis coli. Journal of Biological Chemistry. 2005;280:30490–30495. doi: 10.1074/jbc.M504973200. [DOI] [PubMed] [Google Scholar]
- Nadauld LD, Phelps R, Moore BC, Eisinger A, Sandoval IT, Chidester S, Peterson PW, Manos EJ, Sklow B, Burt RW, Jones DA. Adenomatous polyposis coli control of C-terminal binding protein-1 stability regulates expression of intestinal retinol dehydrogenases. Journal of Biological Chemistry. 2006a;281:37828–37835. doi: 10.1074/jbc.M602119200. [DOI] [PubMed] [Google Scholar]
- Nadauld LD, Chidester S, Shelton DN, Rai K, Broadbent T, Sandoval IT, Peterson PW, Manos EJ, Ireland CM, Yost HJ, Jones DA. Dual roles for adenomatous polyposis coli in regulating retinoic acid biosynthesis and wnt during ocular development. PNAS. 2006b;103:13409–13414. doi: 10.1073/pnas.0601634103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhauss SC, Solnica-Krezel L, Schier AF, Zwartkruis F, Stemple DL, Malicki J, Abdelilah S, Stainier DY, Driever W. Mutations affecting craniofacial development in zebrafish. Development. 1996;123:357–367. doi: 10.1242/dev.123.1.357. [DOI] [PubMed] [Google Scholar]
- Rai K, Sarkar S, Broadbent TJ, Voas M, Grossmann KF, Nadauld LD, Dehghanizadeh S, Hagos FT, Li Y, Toth RK, Chidester S, Bahr TM, Johnson WE, Sklow B, Burt R, Cairns BR, Jones DA. DNA demethylase activity maintains intestinal cells in an undifferentiated state following loss of APC. Cell. 2010;142:930–942. doi: 10.1016/j.cell.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Y, Wu W, Ni X, Kuang T, Jin D, Wang D, Lou W. Lactate dehydrogenase A is overexpressed in pancreatic cancer and promotes the growth of pancreatic cancer cells. Tumor Biology. 2013;34:1523–1530. doi: 10.1007/s13277-013-0679-1. [DOI] [PubMed] [Google Scholar]
- Russo JE, Hauguitz D, Hilton J. Inhibition of mouse cytosolic aldehyde dehydrogenase by 4-(diethylamino)benzaldehyde. Biochemical Pharmacology. 1988;37:1639–1642. doi: 10.1016/0006-2952(88)90030-5. [DOI] [PubMed] [Google Scholar]
- Schell JC, Rutter J. The long and winding road to the mitochondrial pyruvate carrier. Cancer & Metabolism. 2013;1:6. doi: 10.1186/2049-3002-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell JC, Olson KA, Jiang L, Hawkins AJ, Van Vranken JG, Xie J, Egnatchik RA, Earl EG, DeBerardinis RJ, Rutter J. A role for the mitochondrial pyruvate carrier as a repressor of the warburg effect and colon cancer cell growth. Molecular Cell. 2014;56:400–413. doi: 10.1016/j.molcel.2014.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider J, Neu K, Grimm H, Velcovsky HG, Weisse G, Eigenbrodt E. Tumor M2-pyruvate kinase in lung cancer patients: immunohistochemical detection and disease monitoring. Anticancer Research. 2002;22:311–318. [PubMed] [Google Scholar]
- Schneikert J, Behrens J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut. 2007;56:417–425. doi: 10.1136/gut.2006.093310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton DN, Sandoval IT, Eisinger A, Chidester S, Ratnayake A, Ireland CM, Jones DA. Up-regulation of CYP26A1 in adenomatous polyposis coli-deficient vertebrates via a WNT-dependent mechanism: implications for intestinal cell differentiation and colon tumor development. Cancer Research. 2006;66:7571–7577. doi: 10.1158/0008-5472.CAN-06-1067. [DOI] [PubMed] [Google Scholar]
- Sperber H, Mathieu J, Wang Y, Ferreccio A, Hesson J, Xu Z, Fischer KA, Devi A, Detraux D, Gu H, Battle SL, Showalter M, Valensisi C, Bielas JH, Ericson NG, Margaretha L, Robitaille AM, Margineantu D, Fiehn O, Hockenbery D, Blau CA, Raftery D, Margolin AA, Hawkins RD, Moon RT, Ware CB, Ruohola-Baker H. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nature Cell Biology. 2015;17:1523–1535. doi: 10.1038/ncb3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stackley KD, Beeson CC, Rahn JJ, Chan SS. Bioenergetic profiling of zebrafish embryonic development. PLoS One. 2011;6:e25652. doi: 10.1371/journal.pone.0025652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nature Protocols. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- Valvezan AJ, Zhang F, Diehl JA, Klein PS, Coli AP. Adenomatous polyposis coli (APC) regulates multiple signaling pathways by enhancing glycogen synthase kinase-3 (GSK-3) activity. Journal of Biological Chemistry. 2012;287:3823–3832. doi: 10.1074/jbc.M111.323337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Westerfield M. A Guide for the Laboratory Use of Zebrafish (Danio Rerio) 3rd Edn. Eugene, Oregon: University of Oregon Press; 1995. The Zebrafish Book; p. 385. [Google Scholar]
- Yanes O, Clark J, Wong DM, Patti GJ, Sánchez-Ruiz A, Benton HP, Trauger SA, Desponts C, Ding S, Siuzdak G. Metabolic oxidation regulates embryonic stem cell differentiation. Nature Chemical Biology. 2010;6:411–417. doi: 10.1038/nchembio.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh CS, Wang JY, Chung FY, Lee SC, Huang MY, Kuo CW, Yang MJ, Lin SR. Significance of the glycolytic pathway and glycolysis related-genes in tumorigenesis of human colorectal cancers. Oncology Reports. 2008;19:81–91. doi: 10.3892/or.19.1.81. [DOI] [PubMed] [Google Scholar]
- Zhou W, Choi M, Margineantu D, Margaretha L, Hesson J, Cavanaugh C, Blau CA, Horwitz MS, Hockenbery D, Ware C, Ruohola-Baker H. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. The EMBO Journal. 2012;31:2103–2116. doi: 10.1038/emboj.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]