

Abstract
Background
Recent advances in genomics methodologies, in particular the availability of next-generation sequencing approaches have made it possible to identify risk loci throughout the genome, in particular the exome. In the current study, we present findings from an exome study conducted in five affected individuals of a multiplex family with cleft palate only (CPO).
Methods
The GEnomeMINIng (GEMINI) pipeline was used to functionally annotate the SNPs, insertions and deletions (In/Del). Filtering methods were applied to identify variants that are clinically relevant and present in affected individuals at minor allele frequencies (≤1%) in the 1000 Genomes Project, Single Nucleotide Polymorphism (dbSNP), Exome Aggregation Consortium (ExAC), and Exome Variant Server (EVS) databases. The bioinformatics tool Systems Tool for Craniofacial Expression-based Gene Discovery (SysFACE) was used to prioritize cleft candidates in our list of variants, and Sanger sequencing was used to validate the presence of identified variants in affected and unaffected relatives.
Results
Our analyses approach narrowed the candidates down to the novel missense variant in ARHGAP29 (GenBank: NM_004815.3, NP_004806.3;c.1654T>C [p.Ser552Pro]. A functional assay in zebrafish embryos showed that the encoded protein lacks the activity possessed by its wild-type (WT) counterpart, and migration assays revealed that keratinocytes transfected with WT ARHGAP29 migrated faster than counterparts transfected with the p.Ser552Pro ARHGAP29 variant or empty vector (control).
Conclusions
These findings reveals ARHGAP29 to be a regulatory protein essential for proper development of the face, identifies an amino-acid that is key for this, and provides a potential new diagnostic tool.
Keywords: Exome, Cleft palate, Missense mutation
Introduction
With the rapid improvement in technology, in particular bioinformatics algorithms and next-generation sequencing approaches, we are witnessing tremendous advances in genomics research. We now have the ability to interrogate the entire exome and genome concurrently in order to identify risk loci – a significant shift from the candidate-gene approach. Since Ng et al. (2010) performed the first the exome study for Miller syndrome, several other studies of this kind have been conducted, proving the usefulness of this approach for clinical molecular diagnosis (Worthey et al.,2011) and identifying the causes of complex traits (Pottier et al., 2012) including cleft lip and palate (Bureau et al., 2014).
Orofacial clefts (OFCs) are the most common birth defects affecting the craniofacial region. They occur in approximately 1 in 700 live births world-wide, with prevalence varying by population (Mossey et al., 2009). OFCs arise during gestation as a result of genetic and environmental factors that interfere with the normal, systematic development and movement of craniofacial processes between the 4th and 8th week of embryonic life. Historically, OFCs have been divided into cleft of the lip only (CL), cleft of the palate only (CPO), and clefts of the lip and palate (CLP) (Leslie and Marazita, 2013). In addition, CL and CLP are often co-classified, as cleft of the lip with or without cleft palate (CL/P). These defects can be repaired, at the levels of both function and aesthetics, through a series of surgeries, orthodontic procedures, and intervention by other healthcare professionals.
OFCs are complex traits whose appearance in any individual is influenced by genes as well as both environmental and stochastic factors (Dixon et al., 2011). The etiology of non-familial OFCs as complex traits has been studied extensively in order to identify risk factors, reduce the risk of clefts, and design strategies for prevention. Complementary approaches have been applied to identifying potential genetic factors, and some successes have been recorded. These approaches include linkage studies of large affected families (Kondo et al., 2002; Zucchero et al., 2004; Marazita et al., 2009); family-based association studies (Moreno et al., 2009); genome-wide association studies using case control and case trio designs (Birnbaum et al., 2009; Grant et al., 2009; Beaty et al., 2010; Ludwig et al., 2012; and recently, whole-exome sequencing of affected second- and third-degree relatives with CL/P in a multiplex family (Bureau et al., 2014). However, gaps in our knowledge base need to be filled in order to improve our understanding of the genetic factors that cause clefts, and to aid in developing strategies toward prevention of these common birth defects.
During the last few years, progress has been made in identifying genes that play roles in isolated CPO using sporadic cases. These include TXB22, (Marcano et al., 2004), SATB2 (FitzPatrick et al., 2003), and Fas-associated factor-1 (FAF1) (Ghassibe et al., 2011). In addition, a common coding variant of GRHL3 was recently shown to be associated with risk for non-syndromic CP in Europeans (Leslie et al., 2016, Mangold et al., 2016). Rare mutations in this gene have also been reported in families with Van der Woude syndrome accompanied by CPO (Peyrard-Janvid et al. 2014).
Evidence from population-based studies suggests that a family with a particular cleft phenotype is more likely to have additional offspring with the same phenotype than is the general population (Grosen et al., 2010). Evidence from Grosen et al (2010) study suggest that distinct cleft phenotypes have genetic underpinnings, and thus studying them in separate cohorts will bridge current knowledge gaps, leading to new and refined strategies for understanding the etiology. To date, the genetic factors leading to an increased risk for CPO in families with multiple affected individuals with CPO have not been studied despite evidence from epidemiology that there is an increased risk within families with a CPO offspring. Previous studies have focused on sporadic cases. Investigating families with multiple affected individuals with CPO will reduce heterogeneity and help identify Mendelian sub-forms that exist (Pengelly et al., 2016). The current study aims to fill this knowledge gap, by identifying additional Mendelian contributions to isolated CPO. Specifically, we conducted exome sequencing in a multiplex family in which isolated CPO was the sole craniofacial phenotype. Our study is the first to implicate a novel loss-of-function variant in ARHGAP29 (MIM:610496) in a multi-generation family with isolated CPO.
Materials and Methods
Ethics Statement
These studies were performed in the Butali, Cornell, Amendt, Kinghorn Centre for Clinical Genomics (KCCG) and Murray laboratories, in compliance with the Institutional Review Board and Office of Animal Resources requirements of the University of Iowa and the Health Research Ethics Committee (HREC) approvals in place at the Garvan institute.
Recruitment and Sample Collection
A multiplex, CPO-affected family of European descent (Figure 1) self-enrolled into our craniofacial genetic study (IRB approval number 199804080). In general the CPO phenotype in this family is variable, with four of the affected individuals having cleft of the soft palate only, and three having cleft of both the hard and soft palates. Informed consent was obtained, and saliva sample collection kits were shipped to the family. In the laboratory, DNA was extracted from the saliva samples using the Oragene protocol (http:www.dnagenotek.com), and quality control steps were taken, including quantification of the DNA (Qubit) and XY genotyping.
Figure 1. Family pedigree and individuals with exome sequencing data.

Affected individuals are indicated by shaded symbols and unaffected family members are shown in white. Red stars indicate members who were selected for exome sequencing because DNA of good quality was available.
Exome Sequencing and Genomic Data Generation
DNA samples from 5 individuals with over 1ug of DNA were selected for exome sequencing (Figure 1). An Agilent Sure Select kit was used for enrichment capture and samples were sequenced using an Ilumina HiSeq 2000 instrument. A 3.48 GB data per sample passed quality control by sequencing. Raw sequences were aligned to the human genome build hg19 using Burrows Wheeler Alignment (BWA) software. Single nucleotide polymorphism and insertion/deletion (In/Del) analyses were performed using Picard tool (https://broadinstitute.github.io/picard/) Sequence Aignment Map (SAM tools,) and Genome Analysis Tool Kit (GATK). Single nucleotide variants and In/Dels were originally called in separate Variant Call File (VCF) files for each individual. Files from each individual were merged, using VCF tools (http://vcftools.sourceforge.net) to present all variant types within a single file. As the GATK UnifiedGenotyper program (McKenna et al., 2010; DePristo et al., 2011) provides missing genotypes (i.e., ‘no calls’), homozygous reference sites were embedded in missing genotype sites. The numbering system for the DNA mutations is based on the cDNA sequence.
Bio-informatics and Genomic Data analysis
The GEneMINIng (GEMINI) platform was used to annotate the variants called using the Ensembl Variant Effect Predictor (VEP) (https://gemini.readthedocs.org/en/latest/). The variants were then filtered using the web-based SEAVE platform, assuming a heterozygous inheritance model. After standard filters for variant quality were applied, candidate variants were prioritised based on rarity (<1%) in the general population using the1000 genome (1KG) database (http://www.1000genomes.org/), the exome variant sequence (EVS) database (http://snp.gs.washington.edu/EVS/), the Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org) database and the The Kinghorn Centre for Clinical Genomics, Sydney, Australia
(KCCG) internal databases of whole exome sequence (WES) and whole genome sequencing (WGS) variants. We also filtered based on the conservation of the affected sites and likely functional impact, i.e. either loss of function (stop or start codon, essential splice site, frameshift insertion or deletion, or missense variant predicted to be pathogenic (CADD, SIFT, Polyphen2 and custom software). The likely pathogenicity of variants and their effects on protein function were predicted using in silico tools such as Polyphen (http://genetics.bwh.harvard.edu/pph2/) (Adzhubei et al., 2010), SIFT (http://sift.jcvi.org/) (Kumar et.al.,2009), v1.3 CADD (http://cadd.gs.washington.edu) and HOPE (http://www.cmbi.ru.nl/hope) (Venselaar et al., 2010). The tolerance of each gene to variation was calculated for all variants using the Residual Variation Intolerance Score (RVIS) (http://genic-intolerance.org/ and conservation). This was combined with the PROVEAN score for missense variants, which incorporates Grantham and conservation and scores (Choi et al., 2015).
The Variant Annotation, Analysis and Search Tool (VAAST) and pedigree VAAST (pVAAST) (Hu et al., 2013) were used to rank the exome-filtered variants according to pathogenicity relative to that of 179 control datasets (http://www.yandell-lab.org/software/vaast.html). The merged multi-sample VCF file was converted to CONDENSER (CDR) file format, based on information about family and phenotype. All references for both FASTA and GFF3 annotations were in human genome hg19 format. pVAAST was run with a parameter file specifying an autosomal dominant inheritance model and complete penetrance, with the input file containing genes from upstream exome filtering analysis. The input filtered gene list was first converted to GFF3 format by creating an interim GTF format file in the UCSC table browser, and then converted to GFF3 format using a Perl script. A pedigree file was required for this, and was generated using Panogram (http://pngu.mgh.harvard.edu/~purcell/plink/data.shtml#ped).
Gene selection using SysFACE (Systems tool for craniofacial expression-based gene discovery)
The bioinformatics tool SysFACE was used to prioritize candidate genes in the list of variants identified using the GEMINI pipeline with respect to their relevance to craniofacial tissue morphogenesis. SysFACE is based on a unique processing protocol of microarray-based genome-level gene expression profiles in FaceBase of craniofacial tissue (Mandible, Palate, Frontonasal, and Maxillary) isolated from mouse embryos (FB00000467.01, FB00000468.01, FB00000474.01, NCBI Gene Expression Omnibus (GEO) - GSE35091, GSE7759, GSE11400, GSE31004. This analysis estimates enrichment scores based on fold-change in these tissues relative to those in microarray datasets generated from mouse whole embryonic body (WB) tissue at E10.5, E11.5, and E12.5 (GSE32334) (http://bioinformatics.udel.edu/Research/SysFACE). Such a comparative analysis with the reference WB dataset has been demonstrated to effectively remove housekeeping genes while enriching for genes that likely function in the tissue of interest, and has resulted in the identification of several new genes linked to developmental defects (Lachke et al., 2012a; Lachke et al., 2012b; Dash et al., 2015). The SysFACE-based CF-tissue enrichment as well CF-tissue expression scores are presented in a heatmap figure (Figure 2). The criteria used for CF-tissue enrichment and expression are ≥1.5 fold enrichment in CF tissues and a ≥100 expression intensity score in more than one dataset. The development of SysFACE will be described in detail elsewhere. However, in brief, facial tissue microarray data in FaceBase and GEO databases were analyzed by a method termed “WB in silico subtraction” as described previously (Anand and Lachke 2016). This method allows for the identification of enriched genes in tissues of interest.
Figure.2. Assessment of absolute expression and enrichment of reported genes in craniofacial tissues datasets.
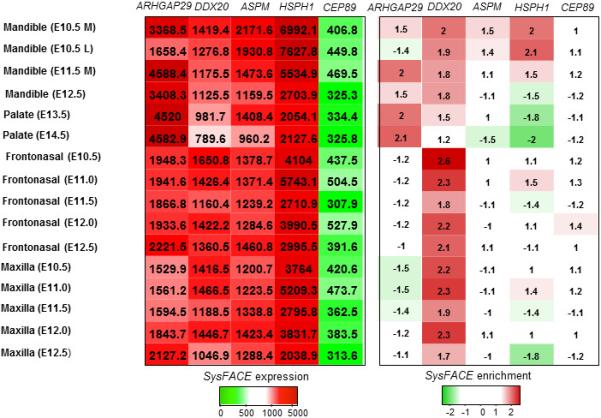
ARHGAP29 and DDX20 exhibit both high expression (>100 absolute expression) and significant enrichment (>1.5) in all four tissues. The enrichment of ASPM and HSPH1 relative to ARHGAP29 is limited to the mandible.
Sanger Sequencing
Sanger sequencing was used to validate identified variants in affected and unaffected relatives. Primers encompassing the genomic region of the variant in the ARHGAP29 (GenBank: NM_004815.3, NP_004806.3;c.1654T>C [p.Ser552Pro] gene were designed using Primer 3 (http://bioinfo.ut.ee/primer3-0.4.0/) and optimized in the Butali laboratory. Gradient PCR was used to determine the annealing temperature for each primer set. A master mix containing 10xNH4 buffer, 5% DMSO, 200μM DNTPs, 50μM MgCl, water, 20μM of forward and reverse primers and the 5u/μl Taq polymerase enzyme was prepared. For each individual of the family from whom a sample was taken, 1μl of DNA was deposited in a 96-well plate, and 9μl of the master mix was added. Two CephHapMap samples and two water samples were used as controls. The size of the amplified DNA products was confirmed using a 2% agarose gel run at 100 Amp and 220V for 20min. The DNA bands were viewed under UV light. DNA samples were sequenced using an ABI 3730XL at Functional Biosciences, Inc. (Madison, WI). Chromatograms generated following sequencing were transferred to a Unix workstation, and bases were called using PHRED (v.0.961028). (www.phrap.org/phredphrapconsed.html) Sequences were assembled using PHRAP (v. 0.960731), scanned using POLYPHRED (v. 0.970312), and viewed using CONSED (v. 4).
To test for co-segregation of the p.Ser552Pro ARHGAP29 variant and isolated CPO in this family, we carried out parametric linkage analysis in MERLIN (Abecasis et al., 2002) using the dominant model in our analysis.
Functional Studies
Zebrafish experiment
We used site-directed mutagenesis for our functional study in zebrafish. A full-length human ARHGAP29 cDNA was purchased as a cDNA clone from Origene (http://www.origene.com/) and sub-cloned into pCS2+Destination plasmid using the Gateway system. Primers were designed to encompass the ARHGAP29 (GenBank: NM_004815.3, NP_004806.3;c.1654T>C [p.Ser552Pro] nucleotide variant and are “available from the Butali laboratory on request”. PCR fragments were subcloned into the x vector using restriction enzyme y, and the variant DNA was extracted for Sanger sequencing to confirm mutagenesis. WT ARHGAP29 and the p.Ser552Pro ARHGAP29 mutant were linearized using Asp718 (Roche), and capped mRNA was synthesized in vitro (mMESSAGE mMACHINE SP6 kit, Ambion). Approximately 1ng of mRNA was injected into WT zebrafish embryos at the 1-cell stage. acZ mRNA was used as a control. When more than 90% of lacZ-injected embryos reached 100% epiboly, embryos in the other groups were counted.
Cell based assays
Immortalized human keratinocytes (iNHKs) were grown in Keratinocyte-SFM (Invitrogen, Carlsbad, CA). For overexpression studies, these cells were co-transfected with WT ARHGAP29 or the p.Ser552Pro ARHGAP29 mutant and CMV-GFP (to measure the transfection efficiency), using lipofectamine 2000 (Life Technologies). For siRNA-mediated knockdown of ARHGAP29, the cells were co-transfected with a validated siRNA (Arhgap29siRNA) or a scrambled control (siCon) (Santa Cruz Biologicals) and a GFP plasmid. Each transfection was estimated to be 20-30% efficient. For assays of cell migration, 8 ~1000um scratches per condition were made in cultures of iNHK cells at 90% confluence, and two locations along the scratch margins were made for identification. Photos were taken of these two locations on each scratch (n=16), at the time of the scratch (t0), and at 12 and 24 hours afterward (t12, t24). For each photo, 5 measurements were made and averaged using ImageJ. These experiments were repeated three times in different batches of cells. For analyses of transcription and translation of the p.Ser552Pro ARHGAP29 variant, cells were transfected with Arhgap29siRNA and siCon. These were done in 6 different replicates for each transfection.
Western Blotting
5-10 ug of cell lysates were run on 10% SDS-PAGE gels, and proteins were tranferred to PVDF membranes and botted with anti-Arhgap29 (Novus Biologicals) or anti-GAPDH (Santa Cruz Biologicals) antibody. Signal was detected using HRP-conjugated secondary antibodies against rabbit or mouse (GE Healthcare).
Results
Identification of the p.Ser552Pro ARHGAP29 variant in a family with isolated CPO
The GEMINI platform was utilized to identify potential pathogenic variants (see Methods). Filtering of the dataset for revealed 19 variants representing 13 genes (Table 1). The 19 variants are heterozygous and represent frameshifts, missense mutations, splice-site, and inframe deletions. Validation using Sanger revealed that the variants in the other genes except for ARHGAP29 have been previsouly identified and reported in control databases (Supp. Table 1). The variant in GOLGA6L2 was not validated in the Sanger Sequencing and thus the variant was excluded. pVAAST ranked the p.Ser552Pro ARHGAP29 highest (Supp. Table S2). The p.Ser552Pro ARHGAP29 variant was thus deposited into the Leiden Open Variation Database with submission number 0000063396.
Table 1.
Analysis showing gene variants identified through filtering with the GEMINI platform
| HGVS Nomenclature* | Genes | NCBI ID | impact | polyphen | sift | cadd_scaled |
|---|---|---|---|---|---|---|
| n.612_615del | LINC00955 | NR_040045.1 | frame_shift | None | None | None |
| n.614_615del | LINC00955 | NR_040045.1 | frame_shift | None | None | None |
| c.1654T>C (p.Ser552Pro) | ARHGAP29 | NM_004815.3 | non_syn_coding | probably_damaging | deleterious | 25.3 |
| c.1210+18del | DDX20 | NM_007204.4 | splice_region | None | None | None |
| c.10332-5_10332-4insT | ASPM | NM_018136.4 | splice_region | None | None | None |
| c.577-5_577-4del | TSPAN8 | NM_004616.2 | splice_region | None | None | None |
| c.1138-3_1138-2del | HSPH1 | NM_006644.3 | splice_region | None | None | None |
| c.1138-2del | HSPH1 | NM_006644.3 | splice_region | None | None | None |
| c.2385_2387del p.Glu795del | GOLGA6L2 | NM_001304388.1 | inframe_codon_loss | None | None | None |
| c.1961C>T (p.Pro654Leu) | FAN1 | NM_014967.4 | non_syn_coding | probably_damaging | deleterious | 25.1 |
| c.455_457del p.Thr152_Cys153delinsSer | KRTAP9-1 | NM_001190460.1 | inframe_codon_loss | None | None | None |
| c.39+179del | CEP89 | NM_032816.4 | splice_region | None | None | None |
| c.404-4_404-3insAA | TASP1 | NM_017714.2 | splice_region | None | None | None |
| c.404-4dup | TASP1 | NM_017714.2 | splice_region | None | None | None |
| c.1963+130_1963+132del | TRAK1 | NM_001042646.2 | inframe_codon_loss | None | None | None |
| c.1963+130_1963+132dup | TRAK1 | NM_001042646.2 | inframe_codon_loss | None | None | None |
| c.2093_2095del p.Glu698del | TRAK1 | NM_001265608.1 | inframe_codon_loss | None | None | None |
| c.2093_2095dup p.Glu698dup | TRAK1 | NM_001265608.1 | inframe_codon_loss | None | None | None |
| n.909-4del | DPY19L2P1 | NR_002833.2 | splice_region | None | None | None |
The DNA mutation numbering system is based on cDNA sequence.
Next, we applied the SysFACE tool to identify both absolute expression and enrichment of the genes based on on publically available craniofacial tissues datasets (Mandible, Palate, Frontonasal, and Maxillary). This analysis of the genes prioritized by the GEMINI pipeline (Table 1) identified ARHGAP29, DDX20, ASPM and HSPS1 as top craniofacial candidates (Figure 2); all the other genes in Table 1 either obtained low expression scores or were completely absent from craniofacial tissues at the stages analyzed (E10.5, E11.5 and E12.5). SysFACE-based comparative analysis of ARHGAP29 and the previously identified human and mouse CP-associated genes Smad4, Bmp4 and Fgfr2 shows that levels of absolute expression are high in all four facial tissues and across several developmental stages, but that they are particularly high for ARHGAP29 and SMAD4 (Supp Figure S1).
Polyphen and SIFT were next applied, and predicted the ARHGAP29 variant affecting exon 15 (p.Ser552Pro) to be damaging and deleterious, respectively. This variant was validated by Sanger sequencing, and 4 of the 5 unaffected individuals (a sibling and 3 who were unrelated), including a CEPH control, did not have the variant. Notably, one of the unaffected siblings did carry the variant (Table 2), indicating incomplete penetrance of phenotype. We were unable to obtain DNA samples from the following members of this family: A9, A10, A11, A15, a16 and 19. Having the exome sequence of these individuals would have allowed for examination of additional genetic variants that are important in clefting, and thus strengthen our understanding of the etiology. We validated all the other variants by Sanger sequencing, and analysis of their segregation suggest that none is associated with CPO. Furthermore, these variants have been previously reported in the ExAC database. Thus, the p.Ser552Pro variant of ARHGAP29 appears to be the only novel variant associated with CPO in this family.
Table 2.
Individuals (affected and unaffected) in the family with or without the ARHGAP29 p.Ser552Pro variant
| Individual identification | Relationship Status | ARHGAP29:NM_004815.3: exon15:c.1654T>C | Status | Types of cleft palate |
|---|---|---|---|---|
| A_2 | Father | TC | Affected | CPO (soft) |
| A_3 | Mother | TT | Unaffected | |
| A_4 | Sibling | TC | Affected | CPO (soft) |
| A_1 | Child | TC | Affected | CPO (hard and soft) |
| A_7 | Paternal grand mother | TT | Unaffected | |
| A_6 | Paternal grand father | TC | Affected | CPO (hard and soft) |
| A_14 | Paternal second cousin | TC | Affected | CPO (hard and soft) |
| A_12 | Paternal grand uncle | TT | Unaffected | |
| CEPH1463_2 unrelated control | Unrelated control | TT | CEPH | |
| A_13 | Grandaunt | TC | Affected | CPO (soft) |
| A_5 | Uncle | TC | Affected | CPO (soft) |
| A_17 | Sibling | TT | Unaffected | |
| A_18 | Sibling | TC | Unaffected |
The parametric analysis testing the model that the p.Ser552Pro ARHGAP29 variant is dominant yielded a maximum LOD score of 1.28. This is insufficient power for detecting significant linkage within the studied family, and is due to its size. Nonetheless, the result supports our initial assumption from the analysis pipeline that the variant is dominant.
The ARHGAP29 variant protein fails to prevent zebrafish epiboly
Based on the statistical analysis of the human family with the p.Ser522Pro variant, we hypothesized that the this amino acid substitution alters a function of ARHGAP29 that is essential for cellular movements underlying morphogenesis of the face. Evidence in the literature has suggested that ARHGAP29 acts in complex with Rap1 and Rasip1 to inhibit Rho, thereby affecting actin re-arrangement, cell-cell adhesion, cell-matrix adhesion, and cell spreading (Post et al., 2013; Post et al., 2015). Because gastrulation depends on actin dynamics, we reasoned that overexpression of WT ARHGAP29 would disrupt gastrulation, and that this effect could be used as an indicator of variants with decreased activity. We engineered a WT human ARHGAP29 cDNA to encode the disease-associated p.Ser522Pro variant, and (separately) two rare coding variants p.Ile513Thr and p.Arg798Gln previously reported in controls in the ExAC database. We synthesized RNA from each variant and injected it (separately) into WT zebrafish embryos at the 1-cell stage. Whereas embryos injected with the lacz mRNA underwent gastrulation on schedule, in those injected with the WT ARHGAP29 RNA this process was significantly delayed. When lacZ-injected embryos had completed epiboly (10 hours post fertilization), those injected with the ARHGAP29 RNA remained at about 50% epiboly (equivalent to about 4 1/3 hours post fertilization). This finding implies that ARHGAP29 interferes with the actin polymerization that underlies cell spreading during epiboly. Embryos injected with mRNAs encoding the variants identified in controls were indistinguishable from those injected with the WT ARHGAP29 mRNA. By contrast those injected with the p.Ser522Pro variant developed on schedule (Figure. 3). These results strongly suggest that the p.Ser522Pro variant has reduced activity relative to the WT form.
Figure 3. The human p.Ser552Pro ARHGAP29 mutant fails to prevent Zebrafish epiboly.

(A-C) Representative lateral views of zebrafish embryos injected with lacZ (control) RNA (A), WT ARHGAP29 (B) or the p.Ser552Pro ARHGAP29 variant RNA. (D) Real-time PCR, validating our injections that they were successful. (E) The number of injected embryos that reach 100% epiboly. Percentage is the average from three separate experiments, each of which included analysis of at least 40 embryos. Error bars represent standard error.
The ARHGAP29 mutant fails to promote keratinocyte migration
Keratinocyte migration is critical to wound healing, and the scratch assay is a well-established model of mammalian cell migration (Biggs et al., 2014). Keratinocytes deficient for Irf6 express less ARHGAP29 than WT controls (Biggs et al., 2014) and, although it has not been clearly established that ARHGAP29 is directly involved in keratinocyte migration, we hypothesize that genes in the IRF6 pathway are involved in cell migration. We therefore used the scratch assay to test the mutation in human ARHGAP29 for an effect on protein function. The transfection of iNHK cells with WT ARHGAP29 accelerated their migration into the scratch as compared to control transfection (no DNA) or transfection with the p.Ser552Pro ARHGAP29 mutant (Figure 4). The cells transfected with WT ARHGAP29 migrated into the scratch faster than either the control or p.Ser552Pro ARHGAP29-transfected cells. We noted no statistically significant differences in migration between the control DNA and the p.Ser552Pro ARHGAP29 mutant (Figure 4a, 4b). Notably, overexpression at the protein level was observed in the case of WT ARHGAP29 (Figure 4c). suggesting that mutant protein is not stable in mammalian cells.
Figure 4. Human p.Ser552Pro ARHGAP29 mutant fails to promote keratinocyte migration.
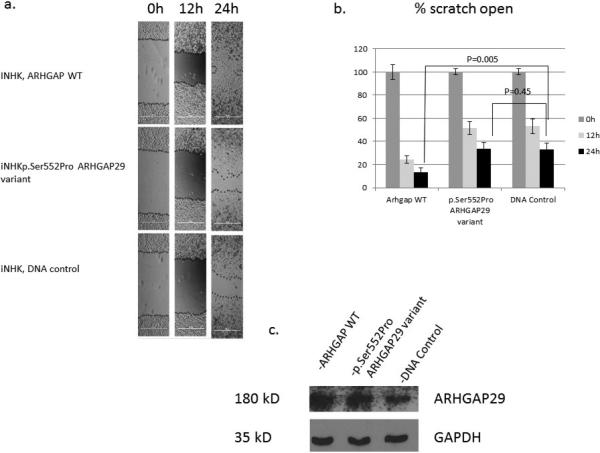
Immortalized human keratinocytes (iNHK) overexpressing WT, p.Ser55Pro, or empty (control) vector were used in a scratch assay as a measure of cell migration. (A) Images of the scratches at T0, T12 and T24, with edges of the migrating cells traced for easier visualization. (B) Percentage of each scratch that remains open (on average) at T0, T12 and T24. Each bar represents data from 8 scratches, at 2 locations per scratch and 5 measurements per location. (C) Western blot analysis of lysates from cells transfected with WT, p.Ser55Pro, or empty vector (control), and probed with ARHGAP29 or GAPDH antibody.
Furthermore, of the 6 cultures of immortalized human keratinocytes (iNHK) cultures transfected with the Arhgap29siRNA, 3 had closed scratches. This is in contrast to cultures transfected with control siRNA (siCon), where 5 out 6 scratches were closed. This observation indicates that the mutant protein was transcribed and translated, consistent with our finding in zebrafish that the p.Ser552Pro ARHGAP29 mutant protein is stably expressed (Supp Figure S2)
Discussion
Here we report, for the first time, on a family with non-syndromic isolated CPO. The variant in question segregates as an autosomal dominant trait caused by an heterozygous missense variant in ARHGAP29 (p.Ser552Pro) that had not previously been identified in a population genomic databases. This variant was predicted to be pathogenic by all in silico tools used for analysis, and was also ranked highest of the genes deemed relevant based on filtering with pVAAST. Analyses of protein structure were also consistent with this mutation disrupting protein function. The pathogenic variant segregated completely in all affected and one unaffected family member, consistent with a low level of non-penetrance. Regardless of the cleft palate type in this family, all affected harbor the p.Ser552Pro ARHGAP29 variant. Genes whose deletion results in autosomal dominant OFC are frequently characterized by variable expression and penetrance. Thus, it is possible that the apparently unaffected carrier of the variant has a subclinical form of clefting that would only have been identified using advance technology such as palatal video recordings for the presence of velopharyngeal insufficiency (Weinberg et al., 2007).
The novel variant in ARHGAP29, p.Ser552Pro, was not reported in any of the available public databases, including 1000 Genomes, the EVS, or the ExAC database. In silico mutation analysis predicted this as a pathogenic variant, with Polyphen classifying it as deleterious and SIFT as probably damaging, and the scaled CADD score of 25.3 indicating a clearly pathogenic level. Analysis of this missense variant using the HOPE mutant analysis server (http://www.cmbi.ru.nl/hope/) confirmed that substitution of a serine at position 522 for a proline could result in a very rigid residue that removes the flexibility required by the protein at this position. The hydrophobic proline was also predicted to lead to loss of a hydrogen bond, thus causing incorrect protein folding. Furthermore, serine is highly conserved at this position in several species, suggesting that the p.Ser522Pro missense variant likely has a deleterious effect on the structure of the ARHGAP29 protein. Our SysFACE analysis identified ARHGAP29 as the top candidate gene based on expression and enrichment in craniofacial tissues. In light of on the bioinformatics analyses that were conducted, ARHGAP29 was considered the best candidate gene for causing cleft palate in the family reported here.
ARHGAP29 gene was identified as a clefting gene following resequencing of the genomic regions surrounding ABCA4 (a genome-wide association candidate gene) (Beaty et al., 2010; Leslie et al., 2012, Letra et al., 2014). ARHGAP29 was found to be expressed in the palatal processes, as assessed by both whole-mount in-situ hybridization and immunofluorescence staining in mice. (Leslie et al.,2012). Together these data strongly suggest that ARHGAP29 is involved in both multifactorial polygenic and Mendelian forms of OFC. Our data now provide evidence that it is one of the causes of CPO in humans, with segregation as a Mendelian trait. Based on the current literature in the field, it is not surprising to find genes that are associated with NSCL/P in CPO. Another example is FOXE1 (Moreno et al., 2009; Ludwig et al., 2015).
ARHGAP29 has GTPase activator activity and regulates small GTP binding proteins including Rho, Rac, and Cdc42 (Saras et al., 1997; Heasman et al., 2008). In endothelial cells ArhGAP29 was recently reported to mediate Rap1-induced inhibition of Rho signaling in the processes of epithelial cell movement and endothelial barrier activity (Post et al., 2013; Post et al., 2015). In the zebrafish experiment performed in this study, we observed that the injection of WT ARHGAP29 significantly delayed epiboly. This is probably due to a requirement for Rho signaling in the context of cell migration during zebrafish gastrulation. In contrast to the normal function of the WT ARHGAP29, the p.Ser552Pro variant had no effect on epiboly, indicating a loss-of-function phenotype.
In the cell-based scratch assay, the cells transfected with WT ARHGAP29 migrated faster than those transfected with the p.Ser552Pro mutant or empty vector (control). This observation is consistent with a report by Biggs et al. (2014), who showed that IRF6 regulates keratinocyte migration through ARHGAP29. Cells deficient for IRF6 had lower ARHGAP29 levels and hyperactive Rho, which led to increases in the levels of stress fibers and cellular area, and to slower migration (Biggs et al., 2014). The ARHGAP29 p.Ser552Pro mutant reported here recapitulates the events described in that study.
In summary, the current study expands our knowledge of biological pathways associated with orofacial clefting. The p.Ser552Pro ARHGAP29 variant was not present in genomic databases and was predicted to be pathogenic by multiple in silico programs. Moreover, the effects of this novel mutant differ from those of the WT protein in a zebrafish development model as well as in a model of human cell migration, providing additional insights into the role of ARHGAP29 in craniofacial development and clefting. Our findings support the inclusion of ARHGAP29 in panels used to diagnose OFC, and suggest that its biological partners should be considered as further candidates in research on clefting.
Supplementary Material
Acknowledgements
We appreciate the support of all members of the family that participated in this research. This work was supported by National Institute of Dental and Craniofacia Research K99/R00 Grant DE022378-04 and Robert Wood Johnson Foundation Grant number 72429 (AB), National Institute of Dental and Craniofacia Research R03 Grant DE024776 (SAL/IS), National Institute of Dental and Craniofacia Research R01 DE023575 (RAC), National Institute of Dental and Craniofacia Research R37 grants DE-08559 and DE-016148 (JCM), National Institute of Dental and Craniofacia Research AR067739 R01(MD) and the Australian National Health and Medica Research Council Grant ID 1045465) (TR).
Footnotes
Author's contributions
AB conceived and designed the project and conducted the exome analysis and wrote the draft of the manuscript. TB, SB, LJJG, AP, and EAAA conducted the mutagenesis and Sanger validation experiments. NN conducted the recruitment, family interview, sample collection and clinical review of the family. HL and RAC performed the zebrafish experiment. DA, IS and SAL conducted the SysFACE analyses. TR also conducted the exome analysis using GEMINI, and YZ developed the bioinformatics pipeline for genomic analysis and performed pVAAST analyses. SE and BA conducted the cell-based experiment. JCM and AA were involved in project design and data interpretation. All authors participated in revising of the manuscript for publication.
REFERENCES
- 1.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 2.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense variants. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anand D, Lachke SA. Systems biology of lens development: A paradigm for disease gene discovery in the eye. Exp Eye Res. 2016;S0014-4835:30040–30049. doi: 10.1016/j.exer.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beaty TH, Murray JC, Marazita ML, Munger RG, Ruczinski I, Hetmanski JB, Liang KY, Wu T, Murray T, Fallin MD, Redett RA, Raymond G, et al. A genome wide association study of cleft lip with / without cleft palate using case-parent trios of European and Asian ancestry identifies MAFB and ABCA4 as novel candidate genes. Nat Genet. 2010;42:525–529. doi: 10.1038/ng.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biggs LC, Naridze RL, DeMali KA, Lusche DF, Kuhl S, Soll DR, Schutte BC, Dunnwald M. Interferon regulatory factor 6 regulates keratinocyte migration. J Cell Sci. 2014;127:2840–2848. doi: 10.1242/jcs.139246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birnbaum S, Ludwig KU, Reutter H, Herms S, Steffens M, Rubini M, Baluardo C, Ferrian M, Almeida de Assis N, Alblas MA, Barth S, Freudenberg J, et al. Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nature Genet. 2009;41:473–477. doi: 10.1038/ng.333. [DOI] [PubMed] [Google Scholar]
- 7.Bureau A, Parker MM, Ruczinski I, Taub MA, Marazita ML, Murray JC, Mangold E, Noethen MM, Ludwig KU, Hetmanski JB, Bailey-Wilson JE, Cropp CD, et al. Whole Exome Sequencing of Distant Relatives in Multiplex Families Implicates Rare Variants in Candidate Genes for Oral Clefts. Genetics. 2014;197(3):1039–1044. doi: 10.1534/genetics.114.165225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–2747. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dash S, Dang CA, Beebe DC, Lachke SA. Deficiency of the RNA binding protein caprin2 causes lens defects and features of peters anomaly. Dev Dyn. 2015;244:1313–1327. doi: 10.1002/dvdy.24303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DePristo M, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. 2011;12:167–178. doi: 10.1038/nrg2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.FitzPatrick DR, Carr IM, McLaren L, Leek JP, Wightman P, Williamson K, Gautier P, McGill N, Hayward C, Firth H, Markham AF, Fantes JA, Bonthron DT. Identification of SATB2 as the cleft palate gene on 2q32-q33. Hum Mol Genet. 2003 Oct 1;12(19):2491–501. doi: 10.1093/hmg/ddg248. Epub 2003 Jul 29. [DOI] [PubMed] [Google Scholar]
- 13.Grant SFA, Wang K, Zhang H, Glaberson W, Annaiah K, Kim CE, Bradfield JP, Glessner JT, Thomas KA, Garris M, Frackelton EC, Otieno FG, et al. A genome -wide association study identifies a locus for non-syndromic cleft lip with or without cleft palate on 8q24. Journal of Pediatr. 2009;155:909–913. doi: 10.1016/j.jpeds.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 14.Grosen D, Chevrier C, Skytthe A, Bille C, Mølsted K, Sivertsen A, Murray JC, Christensen K. A cohort study of recurrence patterns among more than 54,000 relatives of oral cleft cases in Denmark: support for the multifactorial threshold model of inheritance. J Med Genet. 2010;47:162–168. doi: 10.1136/jmg.2009.069385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 16.Hu H, Huff CD, Moore B, Flygare S, Reese MG, Yandell M. VAAST 2.0: improved variant classification and disease-gene identification using a conservation-controlled amino acid substitution matrix. Genet Epidemiol. 2013;37:622–634. doi: 10.1002/gepi.21743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones JL, Canady JW, Brookes JT, Wehby GL, L'Heureux J, Schutte BC, Murray JC, Dunnwald M. Wound complications after cleft repair in children with Van der Woude syndrome. J. Craniofac. Surg. 2010;21:1350–1353. doi: 10.1097/SCS.0b013e3181ec6aad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y, Howard E, de Lima RL, Daack-Hirsch S, Sander A, McDonald-McGinn DM, Zackai EH, et al. Variants in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet. 2002;32:285–289. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 20.Lachke SA, Ho JW, Kryukov GV, O'Connell DJ, Aboukhalil A, Bulyk ML, Park PJ, Maas RL. iSyTE: integrated Systems Tool for Eye gene discovery. Invest Ophthalmol Vis Sci. 2012;53:1617–1627. doi: 10.1167/iovs.11-8839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lachke SA, Higgins AW, Inagaki M, Saadi I, Xi Q, Long M, Quade BJ, Talkowski ME, Gusella JF, Fujimoto A, Robinson ML, Yang Y, Duong QT, et al. The cell adhesion gene PVRL3 is associated with congenital ocular defects. Hum Genet. 2012;131:235–250. doi: 10.1007/s00439-011-1064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leslie EJ, Mansilla MA, Biggs LC, Schuette K, Bullard S, Cooper M, Dunnwald M, Lidral AC, Marazita ML, Beaty TH, Murray JC, et al. Expression and variant analyses implicate ARHGAP29 as the etiologic gene for the cleft lip with or without cleft palate locus identified by genome-wide association on chromosome 1p22. Birth Defects Res A Clin Mol Teratol. 2012;94:934–942. doi: 10.1002/bdra.23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leslie EJ, Marazita ML. Genetics of cleft lip and cleft palate. Am J Med Genet C Semin Med Genet. 2013;163C:246–258. doi: 10.1002/ajmg.c.31381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leslie EJ, Liu H, Carlson JC, Shaffer JR, Feingold E, Wehby G, Laurie CA, Jain D, Laurie CC, Doheny KF, McHenry T, Resick J, Sanchez C, Jacobs J, Emanuele B, Vieira AR, Neiswanger K, Standley J, Czeizel AE, Deleyiannis F, Christensen K, Munger RG, Lie RT, Wilcox A, Romitti PA, Field LL, Padilla CD, Cutiongco-de la Paz EM, Lidral AC, Valencia-Ramirez LC, Lopez-Palacio AM, Valencia DR, Arcos-Burgos M, Castilla EE, Mereb JC, Poletta FA, Orioli IM, Carvalho FM, Hecht JT, Blanton SH, Buxó CJ, Butali A, Mossey PA, Adeyemo WL, James O, Braimah RO, Aregbesola BS, Eshete MA, Deribew M, Koruyucu M, Seymen F, Ma L, de Salamanca JE, Weinberg SM, Moreno L, Cornell RA, Murray JC, Marazita ML. A Genome-wide Association Study of Nonsyndromic Cleft Palate Identifies an Etiologic Missense Variant in GRHL3. Am J Hum Genet. 2016;98:744–754. doi: 10.1016/j.ajhg.2016.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ludwig KU, Mangold E, Herms S, Nowak S, Reutter H, Paul A, Becker J, Herberz R, Al Chawa T, Nasser E, Böhmer AC, Mattheisen M, et al. Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat Genet. 2012;44:968–971. doi: 10.1038/ng.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ludwig KU, Böhmer AC, Rubini M, Mossey PA, Herms S, Nowak S, Reutter H, Alblas MA, Lippke B, Barth S, Paredes-Zenteno M, Muñoz-Jimenez SG, Ortiz-Lopez R, Kreusch T, Hemprich A, Martini M, Braumann B, Jäger A, Pötzsch B, Molloy A, Peterlin B, Hoffmann P, Nöthen MM, Rojas-Martinez A, Knapp M, Steegers-Theunissen RP, Mangold E. Strong association of variants around FOXE1 and orofacial clefting. J Dent Res. 2014;93:376–381. doi: 10.1177/0022034514523987. [DOI] [PubMed] [Google Scholar]
- 27.Mangold E, Böhmer AC, Ishorst N, Hoebel AK, Gültepe P, Schuenke H, Klamt J, Hofmann A, Gölz L, Raff R, Tessmann P, Nowak S, Reutter H, Hemprich A, Kreusch T, Kramer FJ, Braumann B, Reich R, Schmidt G, Jäger A, Reiter R, Brosch S, Stavusis J, Ishida M, Seselgyte R, Moore GE, Nöthen MM, Borck G, Aldhorae KA, Lace B, Stanier P, Knapp M, Ludwig KU. Sequencing the GRHL3 Coding Region Reveals Rare Truncating Mutations and a Common Susceptibility Variant for Nonsyndromic Cleft Palate. Am J Hum Genet. 2016;98:755–762. doi: 10.1016/j.ajhg.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marazita ML, Murray JC, Lidral AC, Arcos-Burgos M, Cooper ME, Govil M, Daack-Hirsch S, Riley B, Jugessur A, Felix T, Morene L, Mansilla MA, et al. Meta-analysis of 13 genome scans reveals multiple cleft lip/palate genes with novel loci on 9q21 and 2q32-35. Am J Hum Genet. 2004;75:161–173. doi: 10.1086/422475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marçano AC, Doudney K, Braybrook C, Squires R, Patton MA, Lees MM, Richieri-Costa A, Lidral AC, Murray JC, Moore GE, Stanier P. TBX22 mutations are a frequent cause of cleft palate. J Med Genet. Jan. 2004;41(1):68–74. doi: 10.1136/jmg.2003.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moreno LM, Mansilla MA, Bullard SA, Cooper ME, Busch TD, Machida J, Johnson MK, Brauer D, Krahn K, Daack-Hirsch S, L'heureux J, Valencia-Ramirez C, et al. FOXE1 association with both isolated cleft lip with or without cleft palate, and isolated cleft palate. Hum Mol Genet. 2009;18:4879–4896. doi: 10.1093/hmg/ddp444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mossey PA, Little J, Munger RG, Dixon M, Shaw WC. Cleft lip and palate. Lancet. 2009;374:1773–1785. doi: 10.1016/S0140-6736(09)60695-4. [DOI] [PubMed] [Google Scholar]
- 33.Pengelly RJ, Arias L, Martínez J, Upstill-Goddard R, Seaby EG, Gibson J, Ennis S, Collins A, Briceño I. Deleterious coding variants in multi-case families with non syndromic cleft lip and/or palate phenotypes. Sci Rep. 2016;26:30457. doi: 10.1038/srep30457. doi: 101038/srep30457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peyrard-Janvid M, Leslie EJ, Kousa YA, Smith TL, Dunnwald M, Magnusson M, Lentz BA, Unneberg P, Fransson I, Koillinen HK, Rautio J, Pegelow M, Karsten A, Basel-Vanagaite L, Gordon W, Andersen B, Svensson T, Murray JC, Cornell RA, Kere J, Schutte BC. Dominant mutations in GRHL3 cause Van der Woude Syndrome and disrupt oral periderm development. Am J Hum Genet. 2014;94:23–32. doi: 10.1016/j.ajhg.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ. Exome sequencing identifies the cause of a Mendelian disorder. Nat Genet0. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Post A, Pannekoek WJ, Ross SH, Verlaan I, Brouwer PM, et al. Rasip1 mediates Rap1 regulation of Rho in endothelial barrier function through ArhGAP29. Proc Natl Acad Sci U S A. 2013;110:11427–11432. doi: 10.1073/pnas.1306595110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Post A, Pannekoek WJ, Ponsioen B, Vliem MJ, Bos JL. Rap1 Spatially Controls ArhGAP29 To Inhibit Rho Signaling during Endothelial Barrier Regulation. Mol Cell Biol. 2015;35:2495–2502. doi: 10.1128/MCB.01453-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pottier C, Hannequin D, Coutant S, Rovelet-Lecrux A, Wallon D, Rousseau S, Legallic S, Paquet C, Bombois S, Pariente J, Thomas-Anterion C, Michon A, et al. High frequency of potentially pathogenic SORL1 variants in autosomal dominant early-onset Alzheimer disease. Mol Psychiatry. 2012;17:875–879. doi: 10.1038/mp.2012.15. [DOI] [PubMed] [Google Scholar]
- 39.Saras J, Franzén P, Aspenström P, Hellman U, Gonez LJ, Heldin CH. A novel GTPase-activating protein for Rho interacts with a PDZ domain of the protein-tyrosine phosphatase PTPL1. J Biol Chem. 1997;272:24333–24338. doi: 10.1074/jbc.272.39.24333. [DOI] [PubMed] [Google Scholar]
- 40.Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G. Protein structure analysis of variants causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics. 2010;11:548. doi: 10.1186/1471-2105-11-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weinberg SM, Neiswanger K, Marazita ML. The use of ultrasound to visualize the upper lips of noncleft and repaired-cleft individuals. Cleft Palate Craniofac J. 2007;44:683–684. doi: 10.1597/07-104.1. [DOI] [PubMed] [Google Scholar]
- 42.Worthey EA, Mayer AN, Syverson GD, Helbling D, Bonacci BB, Decker B, Serpe JM, Dasu T, Tschannen MR, Veith RL, Basehore MJ, Broeckel U, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13:255–262. doi: 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- 43.Zucchero TM, Cooper ME, Maher BS, Daack-Hirsch S, Nepomuceno B, Ribeiro L, Caprau D, Christensen K, Suzuki Y, Machida J, Natsume N, Yoshiura K, et al. Interferon regulatory factor 6 (IRF6) is a modifier for isolated cleft lip and palate. N Engl J Med. 2004;351:769–780. doi: 10.1056/NEJMoa032909. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.