

Abstract
Glycine N-methyltransferase deficiency is an inherited disorder of methionine metabolism, reported so far in only four patients and characterised by permanent hypermethioninemia. This disorder has been considered as probably benign because moderate hepatomegaly in two patients was the only obvious symptom and mild to moderate elevation of aminotransferases the only laboratory abnormality. Our experience with the current novel patient points out that this disease, due to very high hypermethioninemia, is not harmless and that there may be diagnostic pitfalls in interpretation of biochemical hallmarks of the disease. Since the first description of glycine N-methyltransferase deficiency, other disorders of this metabolic pathway affecting the liver have been reported pointing to dysmethylation as the common pathogenetic mechanism. Therefore, we suggest the whole group to be named dysmethylating liver diseases.
Electronic supplementary material
The online version of this chapter (doi:10.1007/8904_2016_543) contains supplementary material, which is available to authorized users.
Keywords: Glycine-N-methyltransferase deficiency, Hypermethioninemia, Inherited liver disease, Methylation disorders
Introduction
Glycine N-methyltransferase (GNMT) deficiency (OMIM 606664; E.C. 2.1.1.20) is an autosomal recessive inherited disorder of methionine metabolism (Fig. 1), which has been reported in extenso so far in only three patients from two families (Mudd et al. 2001; Augoustides-Savvopoulou et al. 2003). In 2015, the fourth patient was described in an abstract (Rakic et al. 2015). This disorder has been considered to be probably benign because moderate hepatomegaly in two patients was the only obvious symptom and mild to moderate elevation of aminotransferases (up to 5× normal) was the only routine laboratory abnormality. Liver biopsy, done in two patients, showed only mild centrilobular fibrosis and presence of some eosinophils in one patient and virtually normal result with only a few hepatic cells with mild hydropic degeneration in the other one. During follow-up the three in extenso reported patients have been asymptomatic (R. Cerone, P. Augoustides-Savvopoulou – personal communications). Biochemical hallmarks of the disease are remarkable hypermethioninemia, highly elevated plasma S-adenosylmethionine (AdoMet) with normal S-adenosylhomocysteine (AdoHcy) and total homocysteine (tHcy). Here we report the fifth patient with this disease whose data indicate that the disease may not be a benign one.
Fig. 1.
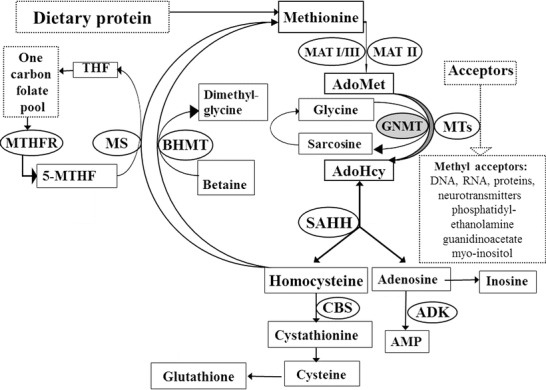
Methionine metabolism. AdoMet S-adenosylmethionine, AdoHcy S-adenosylhomocysteine, THF tetrahydrofolate, 5-MTHF 5-methyltetrahydrofolate, AMP adenosine monophosphate. The following enzymes are in circles: MAT methionine adenosyltransferase (E.C.2.5.1.6), GNMT glycine N-methyltransferase (E.C.2.1.1.20), MTs a variety of AdoMet-dependent methyltransferases, SAHH AdoHcy hydrolase (E.C.3.3.1.1), CBS cystathionine β-synthase (E.C.4.2.1.22), MS methionine synthase (5-MTHF-homocysteine methyltransferase) (E.C.2.1.1.13), BHMT betaine-homocysteine methyltransferase (E.C.2.1.1.5), MTHFR methylenetetrahydrofolate reductase (E.C.1.5.1.20), ADK adenosine kinase (E.C.2.7.1.20)
Since the first description of GNMT deficiency, other disorders of this metabolic pathway affecting the liver have been reported pointing to the common pathogenetic mechanism of this group of the diseases, which we therefore suggest may be named dysmethylating liver diseases.
Case Report
The subject of this report is a Turkish boy at the age of 5 and half years. He is the second child of consanguineous healthy parents. His brother had etiologically unexplained muscle weakness and epilepsy from the age of 4 years and died at the age of 7 years. The mother’s brother and sister had muscle weakness and could not walk since the age of 11 years and had epilepsy since the age of 15 years. Neither their samples nor the tissue from the patient’s brother is available for additional analyses. Our patient was born after unremarkable pregnancy and delivery. He was considered healthy until the age of 2 years and 8 months, when he experienced a single episode of febrile convulsions. At that time his aminotransferases were checked for the first time and found to be permanently elevated, up to 6× normal (AST) and 3× normal (ALT). Physical findings have always been normal. Diagnostic work-up showed highly elevated plasma methionine, which turned out to be a permanent abnormality (815, 1,018, 1,246, and 1,247 μmol/L, reference range 7–47 μmol/L). tHcy was mildly elevated at 21.4 and 28.3 μmol/L (reference range 5–12 μmol/L). Other amino acids were either normal or showed mild unspecific changes. Creatine kinase, albumin, gamma-glutamyl transpeptidase, bilirubin, alkaline phosphatase, alpha-fetoprotein, coagulation tests, folic acid, vitamin B12, and liver ultrasound were normal. Liver biopsy was denied. Because of unexplained isolated hypermethioninemia, AdoMet and AdoHcy were measured. In whole blood, AdoMet was 1,790 nmol/L (reference range 1,000–1,800 nmol/L) and AdoHcy 30 nmol/L (reference range 20–170). In plasma, which became available after the diagnosis has been confirmed by the gene analysis, AdoMet was highly elevated – 3,348 nmol/L (reference range 71–118 nmol/L), whilst AdoHcy was 63 nmol/L (reference range 9.3–14.1 nmol/L). Increased whole blood ratio of AdoMet to AdoHcy, in combination with clinical presentation, raised suspicion on GNMT deficiency which was confirmed by GNMT gene analysis showing the previously unreported homozygous c.296 G>A (R99H) mutation. Parents were heterozygotes for this mutation. Expression studies confirmed pathogenicity of the mutation (see details in supplementary material). During the follow-up period, the patient has been well, without any treatment.
Materials and Methods
Amino Acid Measurement
Plasma amino acids were measured with a kit for quantitative LC-MS/MS analysis of amino acids in biological fluids (Zivak Technologies, Holland) using HPLC-MS-MS technology according to manufacturer’s protocol.
Total Homocysteine Measurement
Plasma tHcy was measured by chemiluminescence immunoassay (CLIA) on IMMULITE 2000 (Siemens Healthcare Diagnostics) according to manufacturer’s instructions.
S-Adenosylmethionine and S-Adenosylhomocysteine Measurement
In whole blood, AdoMet and AdoHcy were measured in perchloride acid-treated whole blood according to Fux and coworkers (Fux et al. 2005) and modified as described in supplementary material. In plasma, AdoMet and AdoHcy were measured as previously described (Gellekink et al. 2005).
Gene Expression Studies and Enzymatic Activity
Heterologous expression of recombinant GNMT was performed in prokaryotic (E. coli) and eukaryotic (HEK293) hosts. Major differences in protein solubility in between recombinant wild-type and mutant R99H GNMT were observed. Namely, wild-type protein showed high solubility, whereas only a minor fraction of mutant protein was remained soluble during extraction indicating significant structural changes leading to high amounts of misfolded protein.
Enzymatic activity was determined using a modified coupled assay including two major steps based on the protocol from R&D systems (https://www.rndsystems.com). Firstly, recombinant GNMT is incubated with glycine to generate sarcosine and AdoHcy. Addition of recombinant adenosine deaminase and S-adenosylhomocysteine hydrolase leads to hydrolysis of AdoHcy into homocysteine and inosine. Subsequently, free homocysteine is reacted with thiol reagent (5,5′-dithiobis-(2-nitrobenzoic acid)) (Belužić et al. 2006), and the amount of thiols produced is measured spectrophotometrically at a wavelength of 405 nm. The retrieved soluble fraction of mutated GNMT lacked measurable enzymatic activity, whereas wild-type protein exhibits specific activity of >200 pmol/min/μg.
To overcome known limitations of prokaryotic expression, human HEK293 cells were transfected with either wild-type or R99H GNMT, cloned as a N-terminal GFP fusion in order to be able to distinguish between endogenous and foreign mRNA levels. Subsequently, mRNA and protein levels were determined using real-time PCR and Western blotting, respectively. Whilst we could not detect any differences in transcript levels of wild-type and mutant GNMT, again the GFP-R99H protein was barely detectable. Taken together, these findings strongly support the adversive effect of R98H mutation on GNMT translation or stability and thereby its function. Detailed experimental procedures are available in supplementary material.
Discussion
Data from our patient point to an important aspect of GNMT deficiency which has not been discussed as yet in the literature. Although his clinical presentation and routine liver tests support the thesis on a benign disorder, his plasma methionine was on three occasions above 1,000 μmol/L, which was estimated as approximate borderline for increased risk of various neurological complications related to hypermethioninemia regardless of the cause (Barić and Fowler 2014; Mudd 2011). Plasma methionine levels above 1,000 μmol/L were observed also in the third reported patient (Augoustides-Savvopoulou et al. 2003). Very recent data from patients with methionine adenosyltransferase I/III deficiency (MAT I/III; OMIM 250850; E.C. 2.5.1.6) suggest that patients with mean values above 800 μmol/L almost always have central nervous system abnormalities, whereas those with means less than 800 μmol/L usually do not (Chien et al. 2015). The most characteristic brain imaging changes related to high hypermethioninemia are demyelination and oedema of white matter, more pronounced in the dorsal brain stem, resulting in separation of myelin layers – the so-called vacuolating myopathy (Braverman et al. 2005; Chamberlin et al. 1996; Devlin et al. 2004). Accordingly, treatment recommendations could be regular monitoring and low-methionine diet if symptoms appear or if methionine is very high.
Experience with this patient revealed interesting possible diagnostic pitfalls of GNMT deficiency. Plasma AdoHcy was unexpectedly more than four times above the reference range. If clinical data and plasma AdoMet are neglected, this could suggest other diseases, for instance, adenosine kinase deficiency (OMIM 614300, E.C. 2.7.1.20). A plausible explanation for increased AdoHcy in this patient could be extremely high plasma AdoMet which secondarily caused an increase of AdoHcy. Actually, AdoMet is an unstable compound and can be converted in part due to inappropriate sampling, shipment or storage to AdoHcy, particularly if the sample is not properly acidified. Whether plasma AdoHcy in our patient was normal or decreased or even slightly increased, we cannot be sure. The instability of AdoMet could be the reason also for the AdoMet levels in the upper normal range when measured in the whole blood, whilst in the same sample, AdoHcy was in the lower normal range. This indicates that, when measured in the whole blood, AdoMet/AdoHcy ratio could be more accurate diagnostic key than isolated AdoMet value and, even more importantly, that plasma is the sample of choice for assaying AdoMet and AdoHcy. The plasma tHcy elevation in our patient (not reported in other patients) may seem confusing because hypermethioninemia and hyperhomocysteinemia characterise cystathionine beta-synthase deficiency (classic homocystinuria; OMIM 263200, E.C. 4.2.1.22), although in the latter tHcy is usually higher. The elevation of homocysteine may be explained by inhibition of betaine-homocysteine methyltransferase or methylenetetrahydrofolate reductase by methionine or AdoMet. Finally, the GNMT activity assay does not seem to be feasible in peripheral white blood cells, lymphocytes or cultured skin fibroblasts, thus making the way to diagnosis more difficult, i.e. requiring either liver biopsy or enzyme assay in a specialised laboratory or gene analysis. Even then, the pathogenicity of mutations may not always be clear.
In our patient, there are several factors indicating that the R99H mutation is pathogenic. First of all, the mutated residue is in the conserved region of the gene. Secondly, the mutant protein in E. coli is almost insoluble and completely inactive. Thirdly, mutant GNMT protein levels in eukaryotic HEK293 cells were at the threshold of detection, whereas wild-type protein was overexpressed as expected. We excluded mechanisms of transcript control/degradation, e.g. ‘no-go decay’, which would lower the mRNA levels. Taken together, the most plausible explanation is that this single amino acid change results in drastically lowered stability of GNMT protein, thereby leading to rapid degradation and/or proteolysis by cellular mechanisms.
Concerning pathogenesis of the GNMT deficiency, it seems that at the moment more can be learned from conditions associated with similar biochemical pattern, i.e. elevated AdoMet and disturbed AdoMet/AdoHcy ratio than from just monitoring patients with GNMT deficiency. These conditions, i.e. AdoHcy hydrolase deficiency and adenosine kinase deficiency, are two disorders sharing the same metabolic pathway with GNMT deficiency. In all three, elevated AdoMet and disturbed AdoMet/AdoHcy ratio possibly lead to complex changes in numerous methylation processes in the body, some of them affecting the liver. Remarkably elevated plasma AdoMet, with much less elevated AdoHcy, was found in all patients with adenosine kinase deficiency, reported first time in 2011 (Bjursell et al. 2011). In that report all six patients had signs of liver disease. In a recent review of 11 patients, nine of them had liver disease, and liver biopsies from four out of five patients showed fibrosis (Staufner et al. 2015). Liver disease is a frequent finding also in AdoHcy hydrolase deficiency (OMIM 613752; E.C. 3.3.1.1), described for the first time by our group (Barić et al. 2004), where AdoMet elevation is a constant finding in untreated patients. In this disorder, the methylation ratio is decreased due to highly elevated AdoHcy. Interestingly, high AdoMet and high methylation index were recently found also in hepatic mtDNA depletion syndromes with poor outcome (Mudd et al. 2012).
All these examples indicate that changes in AdoMet concentration and methylation index are harmful for the liver and that dysmethylation has to be considered as one of pathogenetic mechanisms of liver diseases or, in other words, that this group of disorders affecting the liver may be pathogenetically considered as ‘dysmethylating liver diseases’. GNMT deficiency seems to be an underdiagnosed disorder, but a valuable natural model for studying effects of methylation changes on the liver, which in the long run may cause, not only in GNMT deficiency, serious liver pathology, as shown in animal and in vitro models. In GNMT knock-out mouse, Gnmt−/− mice developed liver steatosis, fibrosis, cirrhosis, chronic hepatitis and hepatocellular carcinoma (Luka et al. 2006; Liu et al. 2007; Martínez-Chantar et al. 2008; for review see also Luka et al. 2009). This is in accordance with previous reports that GNMT is down-regulated in the liver of patients with hepatitis C virus-induced and alcohol-induced cirrhosis who are at risk for hepatocellular carcinoma (Avila et al. 2000) and is not expressed in hepatocellular carcinoma (Chen et al. 1998). Therefore, it seems recommendable to regularly check the liver status in GNMT-deficient patients, particularly for development of hepatocellular carcinoma. An important, very recently reported similarity to GNMT-deficiency in mice is the occurrence of hepatocellular carcinoma in an adult with AdoHcy hydrolase deficiency (Stender et al. 2015). This points to the possible common, liver-related pathogenetic mechanism of these two disorders.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Acknowledgements
The study was supported by the project “European Network and Registry for Homocystinurias and Methylation Defects (E-HOD)” which is co-funded by the European Union in the framework of the Health Program (No.2012_12_02). OV and RB were supported by FP7-REGPOT-2012-2013-1, Grant Agreement Number 316289-InnoMol.
Take-Home Message
Glycine-N-methyltransferase deficiency does not seem a harmless disease and interpretation of diagnostic biochemical abnormalities requires caution.
Description of Authors’ Contributions to the Study
Ivo Barić- study design and supervision, writing of the manuscript; he contributed pertinent aspects of the planning, conducting and reporting of the work described in the article.
Sahin Erdol and Halil Saglam- diagnosis and follow-up of the patient, providing medical data of the patient for the manuscript and critical revision of the manuscript.
Mila Lovrić- biochemical analyses, contributing to the manuscript writing and critical revision of the manuscript.
Robert Belužić- gene analysis and expression studies, contributing to the manuscript writing and critical revision of the manuscript.
Oliver Vugrek- gene analysis and expression studies, contributing to the manuscript writing and critical revision of the manuscript.
Henk J. Blom- biochemical analyses, contributing to the manuscript writing and critical revision of the manuscript.
Ksenija Fumić- biochemical analyses, contributing to the manuscript writing and critical revision of the manuscript.
Guarantor for the Article
Ivo Barić
Conflict of Interest
Ivo Barić, Sahin Erdol, Halil Saglam, Mila Lovrić, Robert Belužić, Oliver Vugrek, Henk J. Blom, and Ksenija Fumić declare that they have no conflict of interest.
Details of Funding
The study was supported by the project “European Network and Registry for Homocystinurias and Methylation Defects (E-HOD)” which is co-funded by the European Union in the framework of the Health Program (No.2012_12_02). OV and RB were supported by FP7-REGPOT-2012-2013-1, Grant Agreement Number 316289-InnoMol. The author(s) confirm(s) independence from the sponsors; the content of the article has not been influenced by the sponsors.
Compliance with Ethics Guidelines
Ethics approval was not required for all research studies.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from the patient’s parents for investigations reported in this study.
Animal Rights
This article does not contain any studies with animal subjects performed by any of the authors.
Footnotes
Competing interests: None declared
Contributor Information
Ivo Barić, Email: ibaric@kbc-zagreb.hr.
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Augoustides-Savvopoulou P, Luka Z, Karyda S, et al. Glycine N-methyltransferase deficiency: a new patient with a novel mutation. J Inherit Metab Dis. 2003;26:745–759. doi: 10.1023/B:BOLI.0000009978.17777.33. [DOI] [PubMed] [Google Scholar]
- Avila MA, Berasain C, Torres L, et al. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J Hepatol. 2000;33:907–914. doi: 10.1016/S0168-8278(00)80122-1. [DOI] [PubMed] [Google Scholar]
- Barić I, Fowler B. Sulphur amino acids. In: Blau N, Duran M, Gibson KM, Dionisi-Vici C, editors. Physicians guide to the diagnosis, treatment and follow-up of inherited metabolic diseases. Berlin: Springer; 2014. pp. 33–46. [Google Scholar]
- Barić I, Fumić K, Glenn B, et al. S-adenosylhomocysteine hydrolase deficiency in a human: a genetic disorder of methionine metabolism. Proc Natl Acad Sci U S A. 2004;101:4234–4239. doi: 10.1073/pnas.0400658101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belužić R, Ćuk M, Pavkov T, et al. A single mutation at Tyr143 of human S-adenosylhomocysteine hydrolase renders the enzyme thermosensitive and affects the oxidation state of bound cofactor nicotinamide-adenine dinucleotide. Biochem J. 2006;400:245–253. doi: 10.1042/BJ20060749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjursell MK, Blom HJ, Cayuela JA, et al. Adenosine kinase deficiency disrupts the methionine cycle and causes hypermethioninemia, encephalopathy, and abnormal liver function. Am J Hum Genet. 2011;89:507–515. doi: 10.1016/j.ajhg.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braverman NE, Mudd SH, Barker PB, Pomper MG. Characteristic MR imaging changes in severe hypermethioninemic states. Am J Neuroradiol. 2005;26:2705–2706. [PMC free article] [PubMed] [Google Scholar]
- Chamberlin ME, Ubagai T, Mudd SH, Wilson WG, Leonard JV, Chou JY. Demyelination of the brain is associated with methionine adenosyltransferase I/III deficiency. J Clin Invest. 1996;98:1021–1027. doi: 10.1172/JCI118862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Shiu JY, Tzeng SJ, et al. Characterization of glycine N-methyltransferase gene expression in human hepatocellular carcinoma. Int J Cancer. 1998;75:787–793. doi: 10.1002/(SICI)1097-0215(19980302)75:5<787::AID-IJC20>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Chien YH, Abdenur JE, Baronio F et al (2015) Mudd’s disease (MAT I/III deficiency): a survey of data for MAT1A homozygotes and compound heterozygotes. Orphanet J Rare Dis 10:99. doi:10.1186/s13023-015-0321-y [DOI] [PMC free article] [PubMed]
- Devlin AM, Hajipour L, Gholkar A, Fernandes H, Ramesh V, Morris AA. Cerebral edema associated with betaine treatment in classical homocystinuria. J Pediatr. 2004;144:545–548. doi: 10.1016/j.jpeds.2003.12.041. [DOI] [PubMed] [Google Scholar]
- Fux R, Kloor D, Hermes M, et al. Effect of acute hyperhomocysteinemia on methylation potential of erythrocytes and on DNA methylation of lymphocytes in healthy male volunteers. Am J Physiol Renal Physiol. 2005;289:F786–F792. doi: 10.1152/ajprenal.00465.2004. [DOI] [PubMed] [Google Scholar]
- Gellekink H, van Oppenraaij-Emmerzaal D, van Rooij A, Struys EA, den Heijer M, Blom HJ. Stable-isotope dilution liquid chromatography-electrospray injection tandem mass spectrometry method for fast, selective measurement of S-adenosylmethionine and S-adenosylhomocysteine in plasma. Clin Chem. 2005;51:1487–1492. doi: 10.1373/clinchem.2004.046995. [DOI] [PubMed] [Google Scholar]
- Liu SP, Li YS, Chen YJ, et al. Glycine N-methyltransferase −/− mice develop chronic hepatitis and glycogen storage disease in the liver. Hepatology. 2007;46:1413–1425. doi: 10.1002/hep.21863. [DOI] [PubMed] [Google Scholar]
- Luka Z, Capdevila A, Mato JM, Wagner C. A glycine N-methyltransferase knockout mouse model for humans with deficiency of this enzyme. Transgenic Res. 2006;15:393–397. doi: 10.1007/s11248-006-0008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luka Z, Mudd SH, Wagner C. Glycine N-methyltransferase and regulation of S-adenosylmethionine levels. J Biol Chem. 2009;284:22507–22511. doi: 10.1074/jbc.R109.019273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Chantar ML, Vázquez-Chantada M, Ariz U, et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 2008;47:1191–1199. doi: 10.1002/hep.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudd SH. Hypermethioninemias of genetic and non-genetic origin: a review. Am J Med Genet C Semin Med Genet. 2011;157C:3–32. doi: 10.1002/ajmg.c.30293. [DOI] [PubMed] [Google Scholar]
- Mudd SH, Cerone R, Schiaffino MC, et al. Glycine N-methyltransferase deficiency: a novel inborn error causing persistent isolated hypermethioninemia. J Inherit Metab Dis. 2001;24:448–464. doi: 10.1023/A:1010577512912. [DOI] [PubMed] [Google Scholar]
- Mudd SH, Wagner C, Luka Z, et al. Two patients with hepatic mtDNA depletion syndromes and marked elevations of S-adenosylmethionine and methionine. Mol Genet Metab. 2012;105:228–236. doi: 10.1016/j.ymgme.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic B, Sinclair G, Stockler S, Vallance H (2015) Is glycine N-methyltransferase (GNMT) deficiency underdiagnosed? In: Garrod symposium “metabolic medicine in motion”, book of abstracts, Vancouver, 114
- Staufner C, Lindner M, Dionisi-Vici C, et al. Adenosine kinase deficiency: expanding the clinical spectrum and evaluating therapeutic options. J Inherit Metab Dis. 2015 doi: 10.1007/s10545-015-9904-y. [DOI] [PubMed] [Google Scholar]
- Stender S, Chakrabarti RS, Xing C, Gotway G, Cohen JC, Hobbs HH. Adult-onset liver disease and hepatocellular carcinoma in S-adenosylhomocysteine hydrolase deficiency. Mol Genet Metab. 2015;116:269–274. doi: 10.1016/j.ymgme.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.