

Abstract
A male patient, born in 1999, was diagnosed with ornithine transcarbamylase deficiency as neonate and was managed with a strict low-protein diet supplemented with essential amino acids, l-citrulline, and l-arginine as well as sodium benzoate. He had an extensive history of hospitalizations for hyperammonemic crises throughout childhood and early adolescence, which continued after the addition of sodium phenylbutyrate in 2009. In December 2013 he was switched to glycerol phenylbutyrate, and his metabolic stability was greatly improved over the following 7 months prior to liver transplant.
Keywords: Glycerol phenylbutyrate, Hyperammonemia, Ornithine transcarbamylase deficiency, Sodium phenylbutyrate, Urea cycle disorder
Introduction
Urea cycle disorders (UCDs) are inherited enzyme and transporter deficiencies that impair urea cycle function and result in the accumulation of toxic levels of ammonia in the blood and brain (Brusilow and Maestri 1996). Hyperammonemia-related neurological injuries range from lethal cerebral edema to mild or subclinical cognitive impairment, depending on the severity of the defect (Gropman et al. 2008), with the most severely affected patients typically presenting early in life (Brusilow and Maestri 1996; Summar et al. 2008; Tuchman et al. 2008). Ornithine transcarbamylase (OTC) deficiency is the most common UCD, accounting for slightly more than half of all UCD cases (Summar et al. 2008; Tuchman et al. 2008; Batshaw et al. 2014).
Medical management of UCDs is aimed at controlling hyperammonemia by reducing waste nitrogen through the restriction of dietary protein, the use of essential amino acid supplements, as well as the supplementation of l-citrulline and/or l-arginine. Patients whose symptoms cannot be adequately controlled by the abovementioned diet and treatment strategies alone are usually treated with alternative pathway drugs such as sodium benzoate and/or sodium phenylbutyrate (sodium PB) (Häberle et al. 2012). The latter lowers ammonia by enhancing excretion of waste nitrogen in the form of phenylacetylglutamine (Batshaw et al. 1982; Brusilow 1991).
Glycerol phenylbutyrate (GPB; Ravicti®, Horizon Pharmaceuticals, Brisbane, CA) is approved in the USA for the treatment of UCD patients 2 years and older whose ammonia levels cannot be controlled by conventional therapy. GPB has the same mechanism of action as sodium PB, but is a pre-pro drug consisting of phenylbutyric acid (PBA) linked to glycerol. Following hydrolysis by pancreatic lipases, PBA is released and converted to the active moiety, phenylacetic acid (Lee et al. 2010; McGuire et al. 2010; Monteleone et al. 2013). In clinical trials, GPB has been shown to provide effective ammonia control in adult and pediatric UCD patients (Lichter-Konecki et al. 2011; Smith et al. 2013; Diaz et al. 2013; Berry et al. 2014).
Case Report
A 16-year-old male patient was diagnosed with OTC deficiency as a neonate and was hospitalized approximately every 2 months for hyperammonemic crises as an infant. Hospitalizations were less frequent during childhood but still occurred several times a year. This report describes his treatments, hospitalizations, and laboratory results over a 7.5-year period, including the effect of GPB treatment for 7 months prior to liver transplant in 2014.
The period reported here starts in April 2007 (age 8 years and 2 months) when the patient was maintained on a low-protein diet (0.5 g/kg bodyweight (bw)/day) supplemented with essential amino acids (UCD2 and/or E-AM Anamix), l-citrulline (2–3 mmol/kg bw/day), and l-arginine (1.4–2.2 mmol/kg bw/day) and received sodium benzoate (166–311 mg/kg bw/day). In addition, to avoid prolonged fasting, the patient received overnight gastric-tube feeding receiving special protein-free infant formula and essential amino acids. Due to metabolic instability with recurrent hyperammonemic crises and hospitalizations, in June 2009, sodium PB (143–338 mg/kg bw/day) was added, which was replaced with GPB (296 mg/kg bw/day) in December 2013.
The patient was hospitalized for metabolic crises 15 times between April 2007 and May 2009 (7.2 hospitalizations/year, prior to receiving sodium PB) and 39 times between June 2009 and December 2013 (8.7 hospitalizations/year, while receiving sodium PB). The average length of stay was 5.4 days (range 2–27 days). Seven of the crises (four in 2008 and one each in 2007, 2010, and 2012) included 1–3 days in the intensive care unit. Most of the crises were of unknown etiology although six events were associated with infections (two respiratory tract infections, three gastroenteritides, one catheter infection), and three were associated with increased physical activity. Almost all events involved vomiting or nausea, and reduced vigilance was noted in 31 of 54 events (57%). Twenty-six of the hospitalizations occurred during the two calendar years prior to starting GPB (11 in 2012 and 15 in 2013, illustrated in Fig. 1).
Fig. 1.
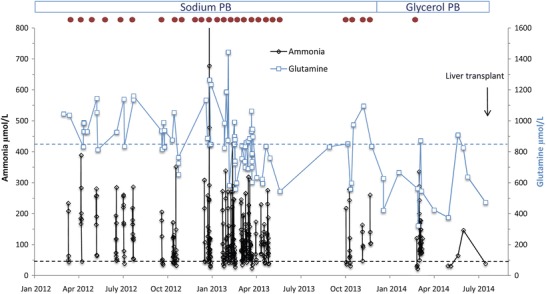
Plasma glutamine and ammonia and hospitalizations in an adolescent with OTC deficiency. Illustration of main parameters of the course in the adolescent OTC deficient patient from January 2012 to July 2014. During this time, the patient received sodium phenylbutyrate (sodium PB) until he was switched to glycerol phenylbutyrate (glycerol PB) in December 2013. Plasma glutamine (in blue) and ammonia (in black) levels obtained during the entire period are shown; upper limits of normal are given as dashed lines in respective colors. Hospitalizations are indicated as red dots at the top of the figure
Peak ammonia levels during metabolic crises for the period from April 2007 to December 2013 ranged from 81 to 1,106 μmol/L (mean, 261.2 μmol/L; reference range 12–48 μmol/L). Glutamine levels for the same period ranged from 346 to 1,445 μmol/L (mean 899 μmol/L; reference range 457–857 μmol/L). For the 2 years prior to starting GPB (2012 and 2013), mean peak ammonia levels were 359 and 257 μmol/L, respectively, and mean glutamine levels were 960 and 899 μmol/L, respectively (Fig. 1).
After starting GPB, during the first 7 months of 2014, the patient had a single hospitalization. This hospitalization (total duration 11 days) was for a metabolic crisis associated with partial respiratory insufficiency due to pleuropneumonia. The patient finally received a successful liver transplant in August 2014.
Discussion
GPB is a recently approved treatment for UCD. It has been shown to significantly lower ammonia levels in pediatric UCD patients compared with sodium PB and to be associated with normal glutamine levels and fewer hyperammonemic crises (Berry et al. 2014). The lower ammonia levels likely reflect the slower absorption of PBA from GPB, presumably because GPB requires digestion by pancreatic lipases, compared with sodium PB, which is a salt and is more rapidly absorbed (Smith et al. 2013). The patient described here was diagnosed with OTC deficiency as a neonate and suffered from frequent hyperammonemic crises, many of unknown etiology, throughout his childhood and early adolescence, even after the introduction of sodium PB at 10 years of age. After switching to GPB short before the patient turned 15 years, his metabolic stability was clearly improved, with fewer and less severe hyperammonemic crises and lower glutamine levels. In fact, Fig. 1 shows that most plasma glutamine levels after introduction of GPB were within the reference range. Under GPB treatment for 7 months, there was only one hospitalization with a severe pleuropneumonia. In both years preceding start of GPB treatment, the patient required monthly hospitalizations. Although it cannot be excluded that the improvement was purely spontaneous, this is unlikely given the following considerations: patients with inherited metabolic disorders tend to become unstable during adolescence and numbers of decompensations often increase (MacDonald et al. 2012). Further, the response to the change in drug treatment was rather prompt rendering a purely age-related effect unlikely. Finally, the relatively mild course during the pleuropneumonia episode is highly suggestive for an actual improved metabolic stability.
Moreover, although not specifically evaluated, it was obvious that the significant reduction of metabolic crises and hospitalizations had a positive impact on the patient’s and his family’s quality of life. It would be of interest in future patients to quantify in more detail the positive effects of GPB treatment, on a biochemical level (plasma ammonia and glutamine) and regarding improvement of quality of life.
We conclude that GPB may be a useful therapeutic option in unstable UCD patients and helped to stabilize our patient until a liver transplant could be performed.
Acknowledgment
Glycerol phenylbutyrate (Ravicti®) was provided free of charge on a compassionate use basis by Hyperion Therapeutics. Medical writing assistance was provided by Jacqueline Wu, PhD, with funding provided by Horizon Pharma (current license holder of Ravicti®), but this had no influence on the content of the report or on interpretation of data. Alexander Laemmle was supported by competitive research grants from the EMDO Foundation Zurich (Grant 851 to JH and AL) and by the Children’s Research Center – University Children’s Hospital Zurich (Grant 10511 to AL) while working on this study. The work on urea cycle disorders is supported by the Swiss National Science Foundation (grant 310030_153196 to JH).
Synopsis
An adolescent with ornithine transcarbamylase deficiency suffering from recurrent hyperammonemic crises showed a significant reduction of plasma ammonia and glutamine levels and a concomitant improvement of metabolic stability when therapy was switched from sodium phenylbutyrate to glycerol phenylbutyrate.
Compliance with Ethics Guidelines
Conflict of Interest
Alexander Laemmle and Tamar Stricker declare that they have no conflict of interest. Johannes Häberle declares the following conflict of interest: at the time of treatment with Ravicti®, he held a “Healthcare Professional Consulting Agreement” with Hyperion Therapeutics, then license holder of Ravicti®. This Consulting Agreement is continued with Horizon Pharma, the current license holder of Ravicti®.
However, all of the authors confirm full independence while treating the patient and while writing this article.
All of the authors participated in planning and performing conception, design, analysis and interpretation of data, drafting, and revising of the article.
Johannes Häberle is the responsible principle investigator and has decided to publish this case report which has not been previously reported elsewhere.
Written informed consent from the patient and the parents to use Ravicti® was obtained. Approval by the responsible ethical committee for using Ravicti® was not required as this was compassionate use.
Footnotes
Competing interests: None declared
Contributor Information
Johannes Häberle, Email: Johannes.Haeberle@kispi.uzh.ch.
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Batshaw ML, et al. Treatment of inborn errors of urea synthesis: activation of alternative pathways of waste nitrogen synthesis and excretion. N Engl J Med. 1982;306(23):1387–1392. doi: 10.1056/NEJM198206103062303. [DOI] [PubMed] [Google Scholar]
- Batshaw ML, et al. A longitudinal study of urea cycle disorders. Mol Genet Metab. 2014;113(1–2):127–130. doi: 10.1016/j.ymgme.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry SA, et al. Glycerol phenylbutyrate treatment in children with urea cycle disorders: pooled analysis of short and long-term ammonia control and outcomes. Mol Genet Metab. 2014;112(1):17–24. doi: 10.1016/j.ymgme.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusilow SW. Phenylacetylglutamine may replace urea as a vehicle for waste nitrogen excretion. Pediatr Res. 1991;29(2):147–150. doi: 10.1203/00006450-199102000-00009. [DOI] [PubMed] [Google Scholar]
- Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology, and therapy. Adv Pediatr. 1996;43:127–170. [PubMed] [Google Scholar]
- Diaz GA, et al. Ammonia control and neurocognitive outcome among urea cycle disorder patients treated with glycerol phenylbutyrate. Hepatology. 2013;57(6):2171–2179. doi: 10.1002/hep.26058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gropman AL, et al. 1H MRS identifies symptomatic and asymptomatic subjects with partial ornithine transcarbamylase deficiency. Mol Genet Metab. 2008;95(1–2):21–30. doi: 10.1016/j.ymgme.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häberle J, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32. doi: 10.1186/1750-1172-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, et al. Phase 2 comparison of a novel ammonia scavenging agent with sodium phenylbutyrate in patients with urea cycle disorders: safety, pharmacokinetics and ammonia control. Mol Genet Metab. 2010;100(3):221–228. doi: 10.1016/j.ymgme.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichter-Konecki U, et al. Ammonia control in children with urea cycle disorders (UCDs); phase 2 comparison of sodium phenylbutyrate and glycerol phenylbutyrate. Mol Genet Metab. 2011;103(4):323–329. doi: 10.1016/j.ymgme.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald A, et al. Adherence issues in inherited metabolic disorders treated by low natural protein diets. Ann Nutr Metab. 2012;61(4):289–295. doi: 10.1159/000342256. [DOI] [PubMed] [Google Scholar]
- McGuire BM, et al. Pharmacology and safety of glycerol phenylbutyrate in healthy adults and adults with cirrhosis. Hepatology. 2010;51(6):2077–2085. doi: 10.1002/hep.23589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteleone JP, et al. Population pharmacokinetic modeling and dosing simulations of nitrogen-scavenging compounds: disposition of glycerol phenylbutyrate and sodium phenylbutyrate in adult and pediatric patients with urea cycle disorders. J Clin Pharmacol. 2013;53(7):699–710. doi: 10.1002/jcph.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith W, et al. Ammonia control in children ages 2 months through 5 years with urea cycle disorders: comparison of sodium phenylbutyrate and glycerol phenylbutyrate. J Pediatr. 2013;162(6):1228–1234. doi: 10.1016/j.jpeds.2012.11.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summar ML, et al. Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year, multicentre study of acute hyperammonaemic episodes. Acta Paediatr. 2008;97(10):1420–1425. doi: 10.1111/j.1651-2227.2008.00952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuchman M, et al. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94(4):397–402. doi: 10.1016/j.ymgme.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]