

Abstract
Deficiency of the mitochondrial trifunctional protein (TFP) and long-chain 3-Hydroxy Acyl-CoA dehydrogenase (LCHAD) impairs long-chain fatty acid oxidation and presents with hypoglycemia, cardiac, liver, eye, and muscle involvement. Without treatment, both conditions can be life-threatening. These diseases are identified by newborn screening (NBS), but the impact of early treatment on long-term clinical outcome is unknown. Moreover, there is lack of consensus on treatment, particularly on the use of carnitine supplementation. Here, we report clinical and biochemical data in five patients with TFP/LCHAD deficiency, three of whom were diagnosed by newborn screening. All patients had signs and symptoms related to their metabolic disorder, including hypoglycemia, elevated creatine kinase (CK), and rhabdomyolysis, and experienced episodes of metabolic decompensation triggered by illness. Treatment was started shortly after diagnosis in all patients and consisted of a diet low in long-chain fats supplemented with medium chain triglycerides (MCT), essential fatty acids, and low-dose carnitine (25 mg/kg/day). Patients had growth restriction early in life that resolved after 2 years of age. All patients but the youngest (2 years old) developed pigmentary retinopathy. Long-chain hydroxylated acylcarnitines did not change significantly with age, but increased during acute illnesses. Free carnitine levels were maintained within the normal range and did not correlate with long-chain hydroxylated acylcarnitines. These results show that patients with LCHAD deficiency can have normal growth and development with appropriate treatment. Low-dose carnitine supplements prevented carnitine deficiency and did not result in increased long-chain hydroxylated acylcarnitines or any specific toxicity.
Keywords: Acylcarnitine, Carnitine, Fatty acid oxidation, Long-chain 3-Hydroxy Acyl-CoA dehydrogenase (LCHAD) deficiency, Newborn screening
Introduction
Long-chain 3-Hydroxy Acyl-CoA dehydrogenase (LCHAD) deficiency (OMIM # 609016) is a disorder of fatty acid oxidation caused by specific mutations in the HADHA gene (Wanders et al. 1990; IJlst et al. 1994). This gene encodes for the alpha subunit of the trifunctional protein (TFP), which is composed of 4 alpha and 4 beta subunits and catalyzes three activities (hydratase, dehydrogenase, and thiolase) in mitochondrial long-chain fatty acids oxidation. In LCHAD deficiency, the second of the three reactions is more severely impaired, whereas in TFP deficiency (OMIM # 609015), there is decreased activity of all three enzymes. TFP and LCHAD deficiency cannot be differentiated biochemically: both are characterized by increased long-chain 3-hydroxyacylcarnitines in plasma and excessive excretion of 3-hydroxy-dicarboxylic acids in urine, the latter only at the time of acute decompensation (den Boer et al. 2002). The diagnosis is confirmed by mutation analysis of the two genes (HADHA and HADHB) encoding for the alpha and beta subunits of the enzyme. The most common mutation causing LCHAD deficiency is c.1528 G>C (p. E510Q) in HADHA (IJlst et al. 1996).
Clinically, patients with TFP and LCHAD deficiency present with hypoketotic hypoglycemia after fasting or illness, with or without cardiomyopathy, liver dysfunction, and rhabdomyolysis (den Boer et al. 2002). Left untreated, morbidity and mortality are high prompting the inclusion of LCHAD and TFP deficiency in expanded newborn screening (NBS) programs (American College of Medical Genetics Newborn Screening Expert 2006). Therapy includes fasting avoidance, a diet restricted in long-chain fatty acids and supplemented with both medium chain triglycerides (MCT) and essential fatty acids. Successful treatment reduces long-chain 3-hydroxyacylcarnitines in plasma and normalizes the dicarboxylic aciduria (Spiekerkoetter et al. 2009). Fasting avoidance and supplementation with MCT and essential fatty acids are part of the clinical management of all patients with TFP/LCHAD deficiency, but the use of l-carnitine is controversial (Spiekerkoetter et al. 2009; Potter et al. 2012) since in animal models of very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency carnitine supplements might increase long-chain acyl carnitines with potential arrhythmogenic effect (Primassin et al. 2008; Tucci et al. 2014).
This manuscript reports clinical outcome and biochemical parameters in five patients with TFP/LCHAD deficiency. All patients were regularly followed in the Metabolic Clinic with an average follow-up time of nearly 10 years (9.2 ± 5.9 years).
Patients and Methods
Diagnosis
This study was reviewed and approved by the Institutional Review Board at the University of Utah. All five patients (three males and two females) were followed in the Metabolic Clinic at the University of Utah from the time of diagnosis (since birth for patients 3, 4, and 5) (Table 1). Patient 2 was last evaluated at 15 years of age. Demographic, clinical, and laboratory data were obtained by retrospective chart review. Patients 1 and 2 were born before the introduction of the expanded newborn screening and presented clinically: patient 1 at 4 months of age with lethargy and patient 2 at 6 months of age with severe hypoglycemia followed by cardiorespiratory arrest. The other three patients were identified by newborn screening, even though patients 4 and 5 already had hypoglycemia at birth (or shortly after). In all patients, confirmatory biochemical testing was consistent with a diagnosis of TFP or LCHAD deficiency, showing increase in plasma long-chain 3-hydroxyacylcarnitines. DNA sequencing confirmed homozygosity for the common c.1528G>C (p.E510Q) mutation in the HADHA gene in all patients except patient 5 who is compound heterozygote for the previously reported pathogenic mutation c.1025T>C (p.L342P) associated with LCHAD deficiency (IJlst et al. 1997) and for a novel change c.1493A>G (p.H498R) in the HADHA gene. This change has been identified in 1/121,392 alleles in the normal population, and it is predicted to be probably damaging by Polyphen and deleterious by SIFT (exac.broadinstitute.org).
Table 1.
Clinical characteristics of patients with LCHAD/TFP deficiency
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | |
|---|---|---|---|---|---|
| Gender | M | F | M | F | M |
| Age at diagnosis | 4 months | 6 months | Birth (NBS) | Birth (NBS) | Birth (NBS) |
| Current age | 22 years | 19 years | 10 years | 5 years | 24 months |
| Follow-up years | 17 years | 12 years | 10 years | 5 years | 2 years |
| Genotype | c.1528 G>C [p.E510Q] | c.1528 G>C [p.E510Q] | c.1528 G>C [p.E510Q] | c.1528 G>C [p.E510Q] | c.1025 T>C [p.L342P]a |
| c.1528 G>C [p.E510Q] | c.1528 G>C [p.E510Q] | c.1528 G>C [p.E510Q] | c.1528 G>C [p.E510Q] | c.1493 A>G [p.H498R]b | |
| Family history | One sister died at 9 months of age; one healthy sister | No siblings | Two healthy brothers | One healthy brother, one healthy sister | One healthy brother |
| Pregnancy | HELLP syndrome | Unremarkable | HELLP syndrome | Preeclampsia | Placenta previa |
| Gestational age | Preterm | Term | 37 0/7 | 35 0/7 | 35 0/7 |
| Weight at birth (Z-score) | N/A | N/A | 2,330 g (0.64) | 1,685 g (−1.61) | 2,275 g (−0.74) |
| Presentation at diagnosis | Lethargy | Hypoglycemia | Asymptomatic | Hypoglycemia | Hypoglycemia |
| Medical history | Muscle pain; depression; cardiac episode with ventricular tachycardia | Exercise-induced muscle pain; multiple hospitalizations (hypoglycemia) | Exercise-induced muscle pain | Failure to thrive; muscle pain | Failure to thrive; hypospadias |
| Retinitis pigmentosa | Yes | Yes | Yes | Yes | No |
N/A not available
aFound in LCHAD deficiency compound heterozygotes (IJlst et al. 1997)
bPreviously unreported
Patient 1 had a family history of LCHAD deficiency, as his older sister died at 9 months of age with hypoglycemia, liver failure, and cardiac arrest with LCHAD deficiency diagnosed post-mortem. All other patients were the only affected individuals in their extended family.
Clinical Descriptions (Table 1)
Patient 1 (LCHAD deficiency) had intermittent muscle pain involving arms, shoulders, and legs, exacerbated by activity and cold; depression treated with psychotherapy and medications; one episode of myoglobinuria followed by renal failure and ventricular tachycardia requiring defibrillation (at 20 years of age). This episode occurred while he was in college, away from home, and he had history of poor compliance with therapy. Administration of intravenous glucose resolved both the renal failure and the ventricular tachycardia and stabilized cardiac function. He developed retinitis pigmentosa with night vision loss as a child. Therapy, followed with variable compliance, included a low-fat diet (10–20% calories from proteins; 45–65% from carbohydrates; 30% calories from fat of which 10% were from natural fat, and 20% from medium chain triglycerides), essential fatty acids (walnut oil, part of the 10% calories from natural fat), docosahexaenoic acid (DHA), CoQ10 and carnitine (25 mg/kg/day).
Patient 2 (LCHAD deficiency) had intermittent muscle pain (lower extremities) exacerbated by activity, hypoglycemia, and multiple hospitalizations for dehydration. Mild retinal degeneration was noted at 15 years of age, with modest loss of the visual field and no significant night blindness. Treatment consisted of a low-fat diet (10–20% calories from proteins; 45–65% from carbohydrates; 30% calories from fat of which 10% were from natural fat, and 20% from medium chain triglycerides such as Portagen), essential fatty acids (walnut oil, part of the 10% calories from natural fat), cornstarch at bedtime, and carnitine (25 mg/kg/day).
Patient 3 (LCHAD deficiency) was identified at birth by newborn screening. His growth and development are appropriate and he is very physically active. However, he experienced several episodes of exercise-induced muscle pain with prolonged exertion. At 3 years of age, changes consistent with retinitis pigmentosa were noted. He followed a low-fat diet (10–20% calories from proteins; 45–65% from carbohydrates; 35% calories from fat of which 10% were from natural fat, and 25% from medium chain triglycerides such as Portagen), essential fatty acids (flaxseed oil, part of the 10–15% calories from natural fat), cornstarch at bedtime, and carnitine (25 mg/kg/day). Creatine (4 g/day) was added at 5 years of age to improve exercise tolerance.
Patient 4 (LCHAD deficiency) was born prematurely at 35 weeks gestation for maternal preeclampsia. She developed severe hypoglycemia at birth that required IV glucose and was further complicated by sepsis. LCHAD deficiency was detected by newborn screening. Her medical history includes several episodes of muscle pain after illness. Mild retinal pigmentary changes were noted in the retina at 2 years of age. Therapy consisted of low-fat diet (10–20% calories from proteins; 45–65% from carbohydrates; 35% calories from fat of which 10% were from natural fat, and 25% from medium chain triglycerides such as Lipistart), essential fatty acids (flaxseed and walnut oil, part of the 10–15% calories from natural fat), cornstarch (at bedtime), and carnitine (25 mg/kg/day).
Patient 5 (LCHAD/TFP deficiency) had severe hypoglycemia at birth and difficulty regulating his temperature, but quickly recovered after IV glucose. LCHAD/TFP deficiency was detected by newborn screening. He had several preventive hospitalizations for fever and gastroenteritis. No signs of pigmentary changes have been detected yet (at 2 years of age). He has been treated with a low-fat diet (10–20% calories from proteins; 45–65% from carbohydrates; 35% calories from fat of which 10% were from natural fat, and 25% from medium chain triglycerides), DHA, cornstarch (at bedtime), and carnitine (25 mg/kg/day). This child subsequently started therapy with triheptanoin that substituted MCT oil.
Laboratory Studies
Metabolic and routine testing was performed by the hospital laboratory or by a reference laboratory (ARUP Laboratories, Salt Lake City, UT, USA, aruplab.com) according to standard procedures. Occasional metabolic decompensation episodes, varying in number and severity among patients, required additional testing, including creatine kinase (CK) activity and transaminases (aspartate aminotransferase, AST; alanine aminotransferase, ALT) levels, and clinical evaluation. Growth parameters were assessed during routine visits. Weight, length/height, BMI percentiles, and Z-scores were assessed using the standard charts of the Centers for Disease Control and Prevention (CDC). Specifically, the World Health Organization (WHO) growth standards (Borghi et al. 2006) were used for data collected from birth to 2 years of age (correcting for gestational age up to 1 year of age), and the CDC growth standards (Kuczmarski et al. 2000) were used for data collected after 2 years of age. BMI percentiles were only assessed for patients >2 years of age.
Statistics
Values for descriptive statistics are presented as means ± SD. Comparison of means was performed using the paired or unpaired t-test with Welch’s correction (not assuming equal variances). The correlation between parameters (patients’ age and biochemical measurements) was assessed by linear regression. Results were considered statistically significant with p < 0.05. GraphPad Prism® software (Version 5.04 (2010), GraphPad Software Inc) was used for data analysis.
Results
Growth and Development
Table 1 summarizes the clinical characteristics of patients included in this study. Four out of five pregnancies were complicated: HELLP syndrome (patients 1 and 3), preeclampsia (patient 4), and placenta previa requiring emergency C-section (patient 5) resulting in preterm delivery. Patients 4 and 5 had low birth weight even after correction for prematurity (Z-scores −1.61 and −0.74, respectively), and experienced significant failure to thrive early in life. All patients experienced a slower growth rate in the first few years after diagnosis (Fig. 1). Cumulatively, the average Z-scores were significantly lower than normal (p < 0.0001) in the first 2 years of life for both weight (−1.74 ± 0.75, age 0–2 years, N = 53) and length/height (−1.44 ± 1.16, age 0–2 years, N = 53). After 2 years of age, all patients with TFP/LCHAD deficiency reached normal percentiles for weight, height, and BMI (Fig. 1). When compared to the normal population, there was no difference for weight (−0.068 ± 1.15, age 2–20 years, N = 47, p = 0.69) and a modest difference with height (0.56 ± 1.66, age 2–20 years, N = 47, p = 0.025), with the LCHAD/TFP deficiency patients being taller. We did not observe overweight/obesity issues after the initial slow growth in our LCHAD/TFP deficiency patients as previously reported (Haglind et al. 2013). The average Z-score for the BMI was −0.31 ± 0.90 SD (age 2–20 years, N = 47), mildly reduced when compared to the normal population (p = 0.025). All five patients had normal development and attended normal school.
Fig. 1.
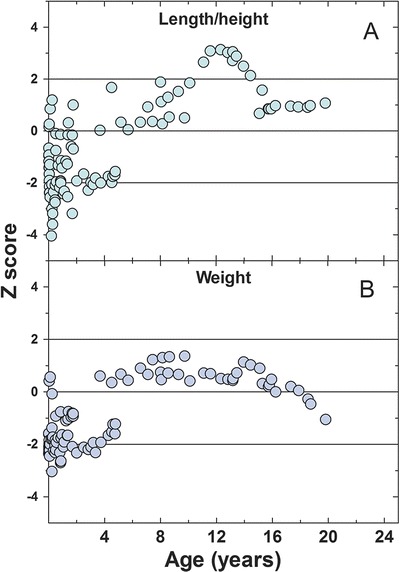
Z-scores for length/height (a) and weight (b) normalize (Z-score = 0) over time (age in years) in mitochondrial trifunctional protein disorders patients. Length measurements were used for patients younger than 2 years of age, while height measurements were used for patients >2 years of age
Long-Term Follow-Up with Laboratory Testing
Laboratory testing was routinely ordered in all five patients to monitor their disease. For patients 3, 4, and 5 data are available from birth to current age (respectively, 10, 5, and 2 years of age); while for patients 1 and 2 early records from the first few years were not available. Values for long-chain hydroxyacylcarnitines (C14:0-OH, C14:1-OH, C16:0-OH, C18:0-OH, and C18:1-OH) were increased in all patients, both as individual species and cumulatively. No changes with age were observed, with nonsignificant correlation between age and levels of individual acylcarnitines or their sum (age range 0–22 years, N = 75, p = 0.58 for C14:0-OH, p = 0.79 for C14:1-OH, p = 0.33 for C16:0-OH, p = 0.69 for C18:0-OH, p = 0.84 for C18:1-OH, and p = 0.83 for the long-chain hydroxyacylcarnitines sum). Similar results have been reported by others (Karall et al. 2015).
All patients in our cohort experienced several illnesses, some of whom required hospitalization and administration of intravenous glucose. During illnesses, CK activity, a measure of muscle involvement, increased dramatically, normalizing with treatment. There was considerable clinical heterogeneity in levels of CK and clinical course despite the fact that four out of five patients were homozygous for the common mutation p.E510Q. Most critical values of CK were recorded for patients 4 and 5 (80%) during episodes of metabolic decompensation and patient 3 never presented with a CK > 1,000 UI/L. Transaminases (AST and ALT) levels and the AST/ALT ratio were also increased, and correlated with CK (Fig. 2), suggesting that most of the increase in transaminases was reflecting release from the muscle rather than liver involvement.
Fig. 2.
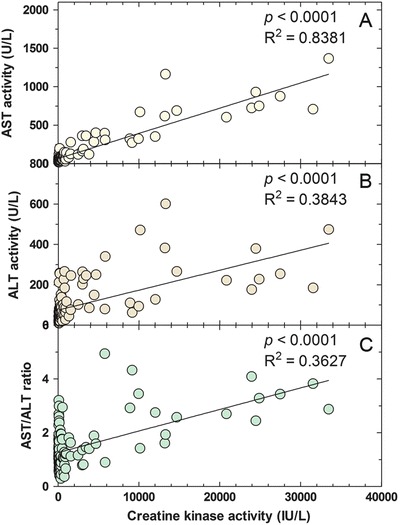
Correlation between creatine kinase activity (IU/L) and (a) aspartate aminotransferase activity (U/L), (b) alanine aminotransferase activity (U/L), and (c) the AST/ALT ratio in mitochondrial trifunctional protein disorders patients. Solid line indicates the best-fit linear regression line
Notably, levels of 3-OH-acylcarnitines significantly correlated with CK levels (Fig. 3a, p < 0.0001), but not with transaminase levels (Fig. 3b, c). The significance of the correlation was lost if values of CK during crises (>5,000 U) were omitted from the analysis (N = 72; R 2 = 0.000; p = 0.98).
Fig. 3.
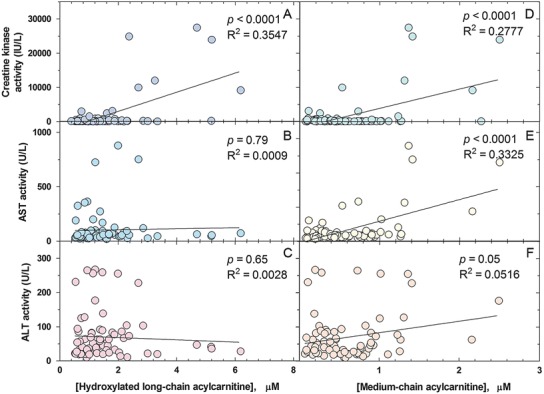
Correlation between cumulative long-chain hydroxyacylcarnitines concentration (μM) and creatine kinase activity (a), aspartate aminotransferase activity (b), and alanine aminotransferase activity (c); and between cumulative medium chain acylcarnitines (μM) and creatine kinase activity (d), aspartate aminotransferase activity (e), and alanine aminotransferase activity (f). Solid line indicates the best-fit linear regression line
Treatment and Clinical Outcome
Dietary intervention and medications were started shortly after diagnosis in all patients. All patients were supplemented with essential fatty acids (walnut or flaxseed oil, and/or DHA), MCT oil and/or medical food containing MCT, and carnitine (25 mg/kg/day). Free carnitine in plasma was 47.5 ± 14.5 μmol/L, ranging from 17 to 77 μmol/L (n = 75, normal range: 22–63 μmol/L), with all values except one (obtained at diagnosis) being within or mildly above the normal range, showing that in our cohort low-dose supplementation prevented carnitine deficiency.
Levels of medium chain acylcarnitines were mildly increased reflecting MCT oil supplements (C6 = 0.16 ± 0.10 μmol/L, normal 0–0.16 μmol/L; C8 = 0.19 ± 0.18 μmol/L, normal 0–0.21 μmol/L; and C10 = 0.28 ± 0.25 μmol/L, normal 0–0.26 μmol/L; N = 82). MCT oil supplements were increased during metabolic crises, probably explaining the positive correlation between the medium chain acylcarnitine cumulative concentration (C6 + C8 + C10) and CK (p < 0.0001; R 2 = 0.278; Fig. 3d) and transaminases (AST p < 0.0001; R 2 = 0.333; ALT p < 0.05; R 2 = 0.052; respectively, Fig. 3e, f). The significant relationship between medium chain acylcarnitine and CK disappeared when the outliers (CK > 5,000 IU/L) were removed (N = 72; R 2 = 0.0198; p = 0.23).
A mild inverse correlation was noted between free carnitine levels and medium chain acylcarnitine (Fig. 4a), possibly reflecting improved utilization or excretion of medium chain fatty acids with carnitine supplements. By contrast, there was no correlation between free carnitine levels and long-chain hydroxyacylcarnitines (Fig. 4b).
Fig. 4.
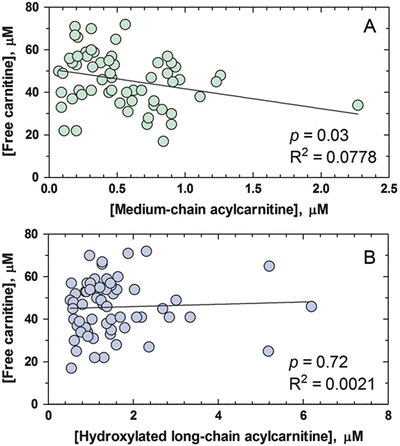
Correlation between free carnitine concentration (μM) and cumulative medium chain acylcarnitines (a) and long-chain hydroxyacylcarnitines (b). Solid line indicates the best-fit linear regression line
Measurement of plasma essential fatty acids indicated no evidence of deficiency (not shown).
Discussion
This study evaluated the longitudinal history of five patients with mitochondrial trifunctional protein disorders (one with TFP deficiency and four with LCHAD deficiency) followed for 9.2 ± 5.7 years in the Metabolic Clinic. Three of these patients were diagnosed at birth by newborn screening. In our five patients, a diet low in long-chain fat and supplemented with MCT, essential fatty acids, and carnitine at low dose (25 mg/kg/day) was started shortly after diagnosis. Some patients also received cornstarch, DHA, and/or creatine. Our retrospective chart review found that, even after correcting for gestational age, most patients had a low birth weight and experienced failure to thrive early in life (Fig. 1). Eventually all patients were able to achieve normal growth, and did not experience overweight issues previously reported in LCHAD deficiency patients (Haglind et al. 2013). Due to the small size of our cohort, it is difficult to determine if growth normalization was due to fewer illness-triggered hospitalizations in older patients, as suggested by Karall et al. (2015). Nevertheless, these findings indicate that normal growth and development can be achieved in TFP/LCHAD deficiency patients with appropriate treatment and management of acute episodes.
It is too early to make any assessment on the effectiveness of newborn screening in preventing the complications of LCHAD/TFP deficiency. The early detection by newborn screening for TFP and LCHAD deficiencies does not prevent metabolic decompensation, which still occurs in patients treated early, before the onset of symptoms (Spiekerkoetter et al. 2009; Karall et al. 2015). In our cohort, patients identified by newborn screening still had illnesses resulting in elevation of CK, AST, and ALT (Fig. 2). There was a strict correlation between these markers suggesting that the elevation of AST and ALT was more likely secondary to muscle involvement (Fig. 2). Patients identified by newborn screening had a similar number of hospitalizations to patients identified clinically, before the onset of newborn screening. However, these were all preventative hospitalizations and not the results of complications (rhabdomyolysis and cardiac arrest) of the disease. Hospital admissions were usually shorter. Finally, one of the patients born before newborn screening developed myopathy and neuropathy after puberty. We have to wait that our cohort reaches that age to see if these functional defects are prevented by early diagnosis and therapy. Biochemical monitoring indicated a correlation between long-chain hydroxyacylcarnitines and creatine kinase levels, with most of the correlation being driven by elevated CK levels during illnesses (Fig. 3a, p < 0.0001), as previously suggested (Gillingham et al. 2005; Karall et al. 2015).
There is general consensus among physicians in recommending fasting avoidance and supplementation with MCT and essential fatty acids for the treatment of TFP/LCHAD deficiencies (Spiekerkoetter et al. 2009; Potter et al. 2012). There is no consensus on the use of carnitine, in part driven by the uncertain effects on long-chain acylcarnitine species. Animal studies indicated an increase in long-chain acyl carnitines with carnitine supplementation in a mouse model of VLCAD deficiency (Primassin et al. 2008; Tucci et al. 2014). On the other hand, carnitine supplementation attenuated myocardial lipid accumulation in long-chain acyl-CoA dehydrogenase knockout mice (Bakermans et al. 2013). Carnitine supplementation is not widely used in the clinical management of patients with TFP/LCHAD deficiency (Potter et al. 2012). Acute administration of carnitine in a single patient with a biochemical diagnosis of LCHAD deficiency was ineffective in preventing death after prolonged fasting (Rocchiccioli et al. 1990). Karall et al. (2015) reported long-term follow-up data on 14 Austrian patients with LCHAD deficiency (ages ranging from 0.9 to 15.3 years), none of whom was supplemented with carnitine. In their cohort, growth and development were normal; however, long-term complications were described: cardiomyopathy (seven patients), hepatopathy (five patients), retinopathy (eight patients), and polyneuropathy (one patient).
We used low-dose oral carnitine supplementation in all five patients (age ranging from 2 to 22 years) with the goal of preventing carnitine deficiency. Free carnitine levels in treated patients were within or mildly above the normal range. There was a mild inverse correlation of plasma carnitine levels with medium chain acylcarnitines (Fig. 4a), which may suggest a better utilization or elimination of the MCT oil with carnitine supplementation. Notably, however, there was no correlation between free carnitine concentration and long-chain hydroxyacylcarnitines (Fig. 4b). None of our patients developed cardiomyopathy. Patient 1 experienced an episode of ventricular tachycardia following myoglobinuria and renal failure (at 20 years of age) triggered by an acute illness and fasting. His cardiac function returned to normal after the resolution of the acute episode. Therapeutic intervention did not prevent the onset of retinopathy in all of our patients, except possibly the youngest (still too early to determine), as previously observed by others (Gillingham et al. 2003, 2005; Fahnehjelm et al. 2008). Longer follow-up studies and a larger number of patients are necessary to determine whether carnitine has any effect on the outcome of patients with mitochondrial trifunctional protein disorders.
Acknowledgements
This work was supported by the ARUP Institute for Clinical and Experimental Pathology®.
Synopsis
Patients with mitochondrial trifunctional protein or long-chain 3-Hydroxy Acyl-CoA dehydrogenase deficiency can achieve normal growth and development with proper treatment, which includes diet and low-dose carnitine supplements.
Individual Authors’ Contributions
IDB, MP, and NL designed the study; NL, KSV, and LB collected the data; IDB, AL, TY, and MP contributed with data collection and interpretation; IDB, KSV, and NL wrote the manuscript. All authors were involved in revising the manuscript critically for content.
Funding
This work was supported by the ARUP Institute for Clinical and Experimental Pathology®. The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors.
Conflict of Interests
Irene De Biase declares that she has no conflict of interest.
Krista S. Viau declares that she has no conflict of interest.
Aiping Liu declares that she has no conflict of interest.
Tatiana Yuzyuk declares that she has no conflict of interest.
Lorenzo D. Botto declares that he has no conflict of interest.
Marzia Pasquali declares that she has no conflict of interest.
Nicola Longo declares that he has no conflict of interest.
Compliance with Ethics Guidelines
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was waived by our Institutional Review Board in view of the retrospective nature of the study and the minimal risks to patients.
Animal Rights
This article does not contain any studies with animal subjects performed by any of the authors.
Contributor Information
Irene De Biase, Email: irene.de-biase@aruplab.com.
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- American College of Medical Genetics Newborn Screening Expert Group Newborn screening: toward a uniform screening panel and system--executive summary. Pediatrics. 2006;117:S296–S307. doi: 10.1542/peds.2005-2633I. [DOI] [PubMed] [Google Scholar]
- Bakermans AJ, van Weeghel M, Denis S, Nicolay K, Prompers JJ, Houten SM. Carnitine supplementation attenuates myocardial lipid accumulation in long-chain acyl-CoA dehydrogenase knockout mice. J Inherit Metab Dis. 2013;36:973–981. doi: 10.1007/s10545-013-9604-4. [DOI] [PubMed] [Google Scholar]
- Borghi E, de Onis M, Garza C, et al. Construction of the World Health Organization child growth standards: selection of methods for attained growth curves. Stat Med. 2006;25:247–265. doi: 10.1002/sim.2227. [DOI] [PubMed] [Google Scholar]
- den Boer ME, Wanders RJ, IJlst L, Morris AA, Heymans HS, Wijburg FA. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: clinical presentation and follow-up of 50 patients. Pediatrics. 2002;109:99–104. doi: 10.1542/peds.109.1.99. [DOI] [PubMed] [Google Scholar]
- Fahnehjelm KT, Holmstrom G, Ying L, et al. Ocular characteristics in 10 children with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: a cross-sectional study with long-term follow-up. Acta Ophthalmol. 2008;86:329–337. doi: 10.1111/j.1600-0420.2007.01121.x. [DOI] [PubMed] [Google Scholar]
- Gillingham MB, Connor WE, Matern D, et al. Optimal dietary therapy of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Mol Genet Metab. 2003;79:114–123. doi: 10.1016/S1096-7192(03)00073-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingham MB, Weleber RG, Neuringer M, et al. Effect of optimal dietary therapy upon visual function in children with long-chain 3-hydroxyacyl CoA dehydrogenase and trifunctional protein deficiency. Mol Genet Metab. 2005;86:124–133. doi: 10.1016/j.ymgme.2005.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglind CB, Stenlid MH, Ask S, et al. Growth in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. JIMD Rep. 2013;8:81–90. doi: 10.1007/8904_2012_164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IJlst L, Wanders RJ, Ushikubo S, Kamijo T, Hashimoto T. Molecular basis of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: identification of the major disease-causing mutation in the alpha-subunit of the mitochondrial trifunctional protein. Biochim Biophys Acta. 1994;1215:347–350. doi: 10.1016/0005-2760(94)90064-7. [DOI] [PubMed] [Google Scholar]
- IJlst L, Ruiter JP, Vreijling J, Wanders RJ. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: a new method to identify the G1528C mutation in genomic DNA showing its high frequency (approximately 90%) and identification of a new mutation (T2198C) J Inherit Metab Dis. 1996;19:165–168. doi: 10.1007/BF01799420. [DOI] [PubMed] [Google Scholar]
- IJlst L, Oostheim W, Ruiter JP, Wanders RJ. Molecular basis of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: identification of two new mutations. J Inherit Metab Dis. 1997;20:420–422. doi: 10.1023/A:1005310903004. [DOI] [PubMed] [Google Scholar]
- Karall D, Brunner-Krainz M, Kogelnig K, et al. Clinical outcome, biochemical and therapeutic follow-up in 14 Austrian patients with Long-Chain 3-Hydroxy Acyl CoA Dehydrogenase Deficiency (LCHADD) Orphanet J Rare Dis. 2015;10:21. doi: 10.1186/s13023-015-0236-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczmarski RJ, Ogden CL, Grummer-Strawn LM, et al. CDC growth charts: United States. Adv Data. 2000;314:1–27. [PubMed] [Google Scholar]
- Potter BK, Little J, Chakraborty P, et al. Variability in the clinical management of fatty acid oxidation disorders: results of a survey of Canadian metabolic physicians. J Inherit Metab Dis. 2012;35:115–123. doi: 10.1007/s10545-011-9352-2. [DOI] [PubMed] [Google Scholar]
- Primassin S, Ter Veld F, Mayatepek E, Spiekerkoetter U. Carnitine supplementation induces acylcarnitine production in tissues of very long-chain acyl-CoA dehydrogenase deficient mice, without replenishing low free carnitine. Pediatr Res. 2008;63:632–637. doi: 10.1203/PDR.0b013e31816ff6f0. [DOI] [PubMed] [Google Scholar]
- Rocchiccioli F, Wanders RJ, Aubourg P, et al. Deficiency of long-chain 3-hydroxyacyl-CoA dehydrogenase: a cause of lethal myopathy and cardiomyopathy in early childhood. Pediatr Res. 1990;28(6):657–662. doi: 10.1203/00006450-199012000-00023. [DOI] [PubMed] [Google Scholar]
- Spiekerkoetter U, Lindner M, Santer R, et al. Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis. 2009;32:488–497. doi: 10.1007/s10545-009-1125-9. [DOI] [PubMed] [Google Scholar]
- Tucci S, Flögel U, Hermann S, Sturm M, Schäfers M, Spiekerkoetter U. Development and pathomechanisms of cardiomyopathy in very long-chain acyl-CoA dehydrogenase deficient (VLCAD−/−) mice. Biochim Biophys Acta. 2014;1842:677–685. doi: 10.1016/j.bbadis.2014.02.001. [DOI] [PubMed] [Google Scholar]
- Wanders RJ, IJ L, van Gennip AH, et al. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: identification of a new inborn error of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 1990;13:311–314. doi: 10.1007/BF01799383. [DOI] [PubMed] [Google Scholar]