

Abstract
NMR-spectroscopy enables unique experimental studies on protein dynamics at atomic resolution. In order to obtain a full atom view on protein dynamics, and to study specific local processes like ring-flips, proton-transfer, or tautomerization, one has to perform studies on amino-acid side chains. A key requirement for these studies is site-selective labeling with 13C and/or 1H, which is achieved in the most general way by using site-selectively 13C-enriched glucose (1- and 2-13C) as the carbon source in bacterial expression systems. Using this strategy, multiple sites in side chains, including aromatics, become site-selectively labeled and suitable for relaxation studies. Here we systematically investigate the use of site-selectively 13C-enriched erythrose (1-, 2-, 3- and 4-13C) as a suitable precursor for 13C labeled aromatic side chains. We quantify 13C incorporation in nearly all sites in all 20 amino acids and compare the results to glucose based labeling. In general the erythrose approach results in more selective labeling. While there is only a minor gain for phenylalanine and tyrosine side-chains, the 13C incorporation level for tryptophan is at least doubled. Additionally, the Phe ζ and Trp η2 positions become labeled. In the aliphatic side chains, labeling using erythrose yields isolated 13C labels for certain positions, like Ile β and His β, making these sites suitable for dynamics studies. Using erythrose instead of glucose as a source for site-selective 13C labeling enables unique or superior labeling for certain positions and is thereby expanding the toolbox for customized isotope labeling of amino-acid side-chains.
Electronic supplementary material
The online version of this article (doi:10.1007/s10858-017-0096-7) contains supplementary material, which is available to authorized users.
Keywords: Relaxation, Protein dynamics, Aromatic side chain, Isotope labeling
Introduction
Proteins are dynamic entities. They continuously undergo all kinds of dynamic processes on various time scales, like conformational rearrangements of the backbone, side chains and loops, ring-flips, proton transfers, changing conformations to alternative states, (partially) unfolding, domain reorientation, etc. While it is of fundamental interest to understand intrinsic protein dynamics, many of these processes are also directly linked to function (Mittermaier and Kay 2006). Fast time-scale fluctuations on the ps-ns range are connected to conformational entropy (Akke et al. 1993) and contribute to the free energy of binding or folding (Diehl et al. 2010; Frederick et al. 2007). Slower processes on the µs-ms time-scale are crucial for ligand binding (Malmendal et al. 1999), enzymatic activity (Boehr et al. 2006; Cole and Loria 2002; Eisenmesser et al. 2002) and signal transduction (Volkman et al. 2001).
NMR spectroscopy is a powerful technique to study such dynamic processes on various time-scales at atomic resolution (Palmer 2004). While the majority of studies have focused on the protein backbone using inexpensive and robust 15N labeling (Akke and Palmer 1996; Ishima and Torchia 2003; Jarymowycz and Stone 2006; Loria et al. 1999), more and more methods have been developed to study amino-acid side chains (Hansen and Kay 2011; Hansen et al. 2012; Lundstrom et al. 2009; Millet et al. 2002; Muhandiram et al. 1995; Mulder et al. 2002; Paquin et al. 2008). These approaches complement existing backbone studies and widen the view on certain processes, but also enable unique additional information of structure (Korzhnev et al. 2010; Neudecker et al. 2012), ring-flips (Weininger et al. 2014b), and proton occupancy and transfer reactions (Hansen and Kay 2014; Wallerstein et al. 2015). A key requirement therefore is to site-selectively label the protein, in order to generate isolated 1H-13C spin pairs (for fast dynamics also isolated 2H) that are not affected by coupling with their neighbours.
Aromatic residues are bulky and form a substantial part of protein hydrophobic cores. They are also over-represented in binding sites (Lo Conte et al. 1999). Especially Tyr and Trp contribute significantly to the binding free energy (Bogan and Thorn 1998). His and Tyr play important catalytic residues for enzyme activity (Bartlett et al. 2002). His can exist in three different states, one protonated and two different tautomeric neutral forms. Transient changes between these states affect hydrogen bonding patterns around the histidine. Studying ring flips of the symmetric Tyr and Phe can give insights into their packing and local transient protein breathing motions (Li et al. 1999; Wagner 1980; Wagner et al. 1976). Thus it is of great interest to monitor the dynamics of aromatic residues on both the ps-ns and µs-ms time scales.
Studies of dynamics of aromatic residues have a long history. The finding and quantification of fast ring-flips and their linkage to protein breathing motions have fundamentally changed our view on proteins (Li et al. 1999; Wagner 1980; Wagner et al. 1976). Early studies were based on proton line-shapes, which often limited the application. With easy and robust labeling protocols to achieve site-selective 13C labeling (Lundstrom et al. 2007; Teilum et al. 2006) studies of dynamics on aromatic side chains are undergoing a renaissance. Improved methods of obtaining relaxation rates have been developed (Weininger et al. 2012a) and the first studies of order parameters have been reported (Boyer and Lee 2008; Kasinath et al. 2015, 2013). Additionally, residual dipolar couplings have been obtained (Sathyamoorthy et al. 2013). Experiments designed to characterize dynamics on the ms (Weininger et al. 2012b) and µs (Weininger et al. 2014a) time-scales have been developed. We have recently reinvestigated the ring-flips in BPTI (Weininger et al. 2014b) using these methods, which enabled us to resolve inconsistencies between experiments (Wagner et al. 1987, 1976) and molecular dynamics simulations (Shaw et al. 2010).
While site-selective 13C enriched glucose (1- and 2-13C) has made it possible to routinely perform advanced heteronuclear studies of dynamics in aromatic side chains, its 13C incorporation yields are far from optimal, typically reaching 20–50%. Furthermore, it is controversial whether additional deuteration is needed (Kasinath et al. 2013) or not (Weininger et al. 2012a) in order to obtain artifact free order parameters. For µs (Weininger et al. 2014a) and ms (Weininger et al. 2012b) dynamics studies however deuteration is usually not required, but serves to prevent rather uncommon strong-coupling effects, which complicate recorded relaxation dispersions, but also contain additional information. Based on the dependence of strong-coupling effects on the refocusing frequency, slow ring-flips for degenerate chemical shifts could be identified (Weininger et al. 2013). Further, deuteration is not needed in order to get improved spectra for structural studies (Milbradt et al. 2015) or studies involving residual dipolar couplings (RDCs) (Sathyamoorthy et al. 2013).
Site-selective 13C enrichment using precursors other than glucose (Lundstrom et al. 2007; Teilum et al. 2006) have been developed recently. Pyruvate (Milbradt et al. 2015), 4-13C erythrose in combination with deuterated pyruvate (Kasinath et al. 2013), and more advanced chemically synthesized precursors for labeling of Trp (Schörghuber et al. 2015), Tyr and Phe (Lichtenecker et al. 2013), including perdeuteration of all other hydrogen positions in the aromatic side-chain. All these methods are common in-vivo labeling strategies using E. coli for protein expression. Additionally, advanced in-vitro strategies using the SAIL approach have been developed for Trp (Miyanoiri et al. 2011), Tyr and Phe (Takeda et al. 2010). Again, all non-13C labeled positions are perdeuterated.
Here we present an easy and robust approach using selectively labeled erythrose (1-, 2-, 3- and 4-13C) in combination with unlabeled glucose. This approach is very close to standard 13C labeling using glucose. The only modification is the additional presence of erythrose. Further, we quantify the 13C incorporation in aromatic side-chains and all other positions of the 20 amino acids for the first time and compare it to that achieved with glucose-based labeling. Erythrose labeling leads to a slight enhancement of 13C levels for Phe and Tyr δ, and roughly to a doubling for all proton-bound carbons in the six-ring moiety of Trp. Further the method efficiently labels Phe (and Tyr) ζ and Trp η2 (2-13C erythrose) and thus makes these positions available for studies of dynamics for the first time. Especially Phe ζ is of great potential interest in order to separate the effects of motions around chi-2 and chi-1 dihedral angles. Additionally, His β becomes significantly 13C-labeled, and Ile β, Lys β and β and Arg β become isolated 13C labeled. Finally, we show that the erythrose-based approach for site-selective 13C labeling can be easily combined with the glucose approach, allowing for more custom labeling.
Materials and methods
Selective 13C enriched isotopes
All isotopes were purchased from cortecnet. Typical prices per gram are: 1-13C glucose, 175 €; 2-13C glucose, 200 €; 1-13C erythrose, 450 €; 2-13C erythrose, 1250 €, 3-13C erythrose, 3400 €; 4-13C erythrose, 1100 €. 2 g/l glucose and 1 g/l (in case of Phe and Tyr) or 2 g/l (in case of Trp) erythrose are usually used. Up to now erythrose is only competitive in costs for desired Phe and Tyr ε* labeling (1 g/l 1-13C erythrose to 2 g/l 2-13C glucose) with similar 13C incorporation levels. Labeling of all other positions is more expensive with erythrose but can be justified by significantly higher 13C incorporation (Trp ε3, ζ3, and ζ2) or effectively labeling positions not labeled by 1-13C or 2-13C glucose (Trp η2, Phe and Tyr ζ).
Expression and purification
An optimised coding sequence for human FK506 binding protein 12 (FKBP12; Uniprot: P62942) was synthesised (GenScript, Piscataway, NJ, USA) and sub-cloned into the plasmid pNIC28-Bsa4 (Savitsky et al. 2010).
Recombinant FKBP12 containing an N-terminal 6x His-tag tag was expressed in M9 minimal medium with 1 g/l 15N NH4Cl and 2 g/l glucose (1-13C, or 2-13C labeled, or unlabeled). In the case of erythrose labeling, site-selective 13C enriched erythrose (1-, 2-, 3- or 4-13C) was additionally present at the beginning at a concentration of 2 g/l, unless otherwise indicated. Protein expression was induced by addition of 1 mM IPTG at an OD600 of ~0.8. Protein expression was carried out for 18 h at 25 °C. The protein was purified on a His-trap column. Afterwards the His-tag was cleaved by Tobacco Etch Virus (TEV) protease. The protein was dialysed, and collected as the flow through of another His-trap column. At the end the buffer was exchanged to NMR buffer and the protein was concentrated to ~12 mg/ml.
NMR spectroscopy
All spectra were run on 800 µM samples containing 25 mM sodium phosphate, pH 7.0 and 10% (v/v) D2O at 25 °C and a static magnetic field strength of 14.1 T. For each sample, a 1H–15N plane of an HNCO, non-constant time 1H–13C HSQCs for the aliphatic and aromatic regions, and a 1D spectrum on 13C were recorded for quantification of 13C incorporation. Intensities of different samples (with possible slightly different concentration) were referenced to the averaged intensities of a 1H–15N HSQC. Assignments were checked using standard 3D experiments. Aromatic 13C relaxation studies were performed using L-optimized TROSY detected relaxation experiments (Weininger et al. 2012a). All spectra were processed using NMRPipe (Delaglio et al. 1995) and analysed using NMRView (Johnson 2004).
Data analysis
The analysis was restricted to well resolved signals that only arise from the same kind of atom (residue type and position). For the fully 13C-enriched reference sample, volumes from both peaks split by the 13C–13C 1J coupling were added. All positions of interest described in this article resulting from erythrose labeling (and glucose labeling for comparison) were isolated and showed no signs of any 13C–13C 1J coupling. Intensities were normalized to the fully 13C enriched sample and expressed in %. By analysing multiple signals of the same kind, the relative error in the intensities of 13C covalently bound to 1H could be estimated to 1%. Errors for 13C not bound to 1H were estimated to 3%.
Results and discussion
Erythrose is a precursor that enters the metabolic pathways closer to the amino-acid product than does glucose, which is of great advantage for achieving site-selective 13C labeling of aromatic side chains in proteins (Fig. 1). To make the labeling procedure as generally applicable and simple as possible, 13C-labeled erythrose (1-, 2-, 3- or 4-13C) was added together with unlabeled glucose to the minimal medium, ensuring that the growth rate of E. coli is essentially the same as for standard minimal media conditions. Furthermore, this approach allows for combined 13C labeling by erythrose and glucose. Preliminary tests showed that adding the erythrose at the very beginning does not lead to any scrambling in the aromatic side chains compared to the result obtained when adding it shortly before induction. Since the level of 13C incorporation is slightly higher when erythrose is added at the start this procedure was followed in all experiments. The level of 13C incorporation was monitored for all aromatic side-chains, with exception of Tyr γ, His γ, and Trp δ2 and ε2, as well as for all other carbon sites in the 20 amino acids. All the missing positions do not have any attached proton. The resulting data provides information on background labeling, scrambling, and unexpected selective incorporations, as described below.
Fig. 1.
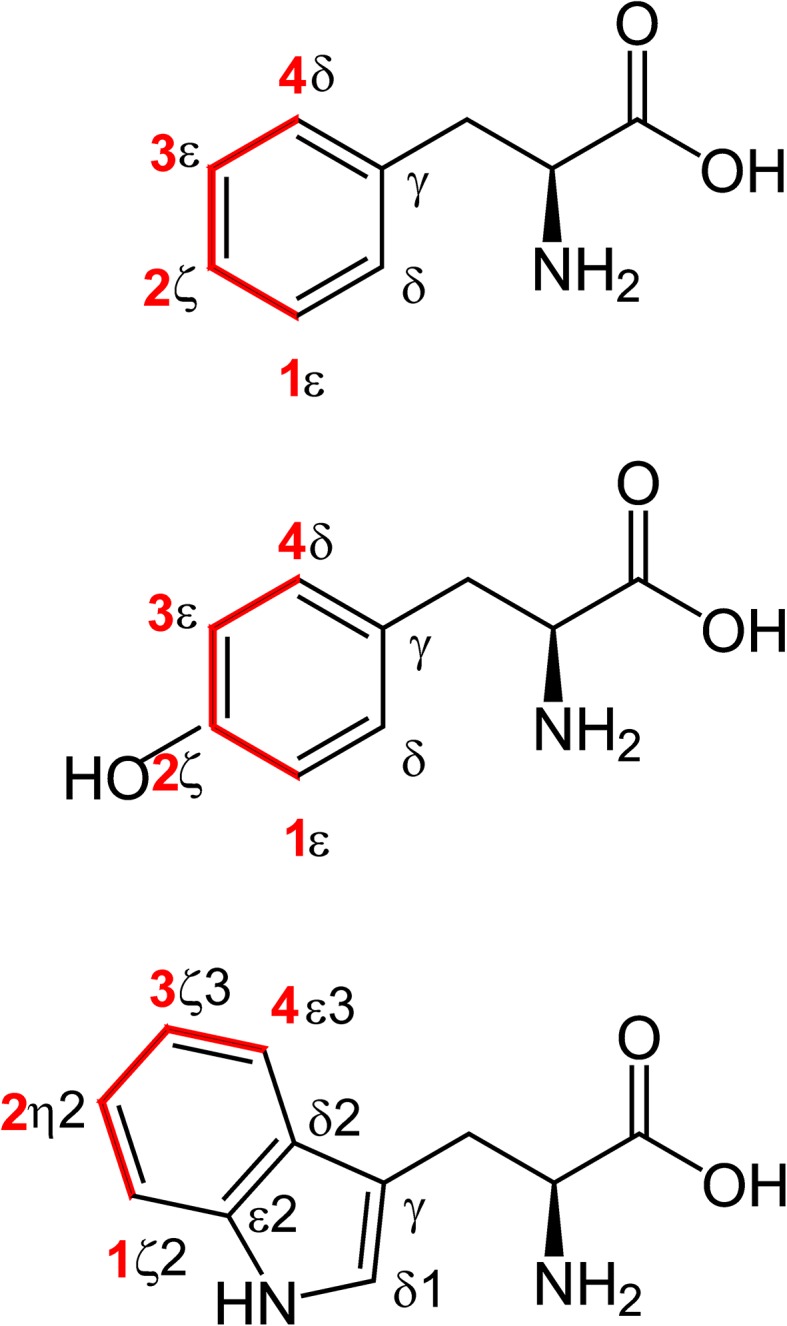
Site-selective 13C incorporation in aromatic side-chains using site-selectively labeled erythrose. Phenylalanine, tyrosine and tryptophan (from top to bottom) are shown with the positions in the aromatic rings labeled. Incorporation of carbons from erythrose is shown in red, with the positions of erythrose (1–4) labeled. Other positions (shown in black) can be labeled if erythrose is scrambling into other pathways
Site-selective 13C labeling of aromatic side-chains
The above mentioned erythrose labeling strategy leads to following general observations. In aromatic side-chains isolated 13C labeling occurs at the expected positions (Fig. 1) and the background labeling of other positions is less than that obtained using glucose as the sole carbon source. Next, the optimal amount of labeled erythrose in the expression medium was tested using different amounts of 1-13C1-erythrose (Fig. 2). Phe and Tyr reach a maximum in 13C incorporation already at 1 g erythrose per liter medium, whereas for Trp the level increases to 2 g/l. Since signals from Trp are weaker in general (in Phe and Tyr two positions normally contribute to the same signal for δ and ε because of fast ring-flips), 2 g/l erythrose were used for the following study. However, if one is only interested in Phe and Tyr, 1 g/l should be enough.
Fig. 2.

13C incorporation level in aromatic side-chains resulting from different amounts of 113C erythrose in the medium. Incorporation in Phe ε* (green), Tyr ε* (blue) and Trp ζ2 red are shown in % relative to fully 13C enriched glucose. Solid lines are single exponential fits. Note that for Phe and Tyr the referencing is for two carbons (1 and 2, represented by asterisk), since the aromatic rings undergo fast ring-flips on the NMR time-scale
13C incorporation levels in Phe, Tyr and Trp using differently labeled erythrose or glucose are summarized in Table 1 (incorporation levels for all positions and amino acids using erythrose labeling are listed in SI Tables 1, 2). All 13C labeled positions do not show any signs of 13C–13C couplings in the spectra (SI Fig. 1) in agreement with the low 13C incorporation for neighbored positions (Table 1). For Phe and Tyr, erythrose (4-13C) labeling leads to a higher incorporation yield in position δ. Additionally position ζ becomes accessible (with 2-13C erythrose), which is potentially very useful to differentiate fluctuations around chi-2 from fluctuations around chi-1, or ring flips from general conformational exchange. For the ε position the 13C incorporation level is very similar for the two carbon sources (1-13C or 3-13C erythrose, or 2-13C glucose). As for Trp, position δ1 is not labeled at all, which is expected. In case of Trp ε3, ζ3 and ζ2, erythrose (4-13C, 3-13C, and 1-13C) yields at least twice as high 13C incorporation. Additionally η2 becomes efficiently labeled by 2-13C erythrose. Since His δ2 is not labeled (analogously to Trp δ1), erythrose (1-13C and 3-13C) labeling allows for studies on Tyr ε without potential disturbance of His δ2, which shares the same spectral region (Fig. 3). This could be of particular interest for studies of ring flips where Tyr ε signals might be broad and are harder to track.
Table 1.
Site-selective 13C incorporation in aromatic side-chains using glucose (G) or erythrose (E)
| 1-13C G | 2-13C G | 1-13C E | 2-13C E | 3-13C E | 4-13C E | |
|---|---|---|---|---|---|---|
| Phe γ | 6 | 55 | 2 | 4 | 24 | 0 |
| Phe δ* | 34 | 4 | 1 | 2 | 1 | 41 |
| Phe ε* | 2 | 22 | 20 | 4 | 23 | 1 |
| Phe ζ | 1 | 1 | 5 | 39 | 1 | 1 |
| Tyr γ | n.d | n.d | n.d | n.d | n.d | n.d |
| Tyr δ* | 32 | 4 | 2 | 2 | 1 | 45 |
| Tyr ε* | 1 | 19 | 17 | 3 | 23 | 1 |
| Tyr ζ | 0 | 0 | 7 | 48 | 11 | 5 |
| Trp γ | 9 | 10 | 1 | 3 | 0 | 1 |
| Trp δ1 | 26 | 49 | 4 | 3 | 2 | 2 |
| Trp δ2 | n.d | n.d | n.d | n.d | n.d | n.d |
| Trp ε2 | n.d | n.d | n.d | n.d | n.d | n.d |
| Trp ε3 | 26 | 2 | 1 | 2 | 1 | 54 |
| Trp ζ3 | 1 | 24 | 1 | 1 | 52 | 1 |
| Trp η2 | 1 | 2 | 6 | 35 | 1 | 0 |
| Trp ζ2 | 2 | 12 | 27 | 5 | 0 | 1 |
Values are in %. Errors are estimated to 1% for 1H bound 13C, 3% for others. 1% for non labeled positions is expected because of natural abundance of 13C
*Represents an averaged signal of position 1 and 2 because of fast exchange of the aromatic rings on the NMR time-scale
Fig. 3.

Tyr ε* His δ2 region of an aromatic 1H13C-TROSY-HSQC of FKBP12. Signals arising from a 2-13C1-glucose labeled sample are shown in black, while signals from a 1-13C1-erythrose labeled sample are shown in red. His δ2 signals are broadened because 15N was not decoupled. Asterisk represents an averaged signal of position 1 and 2 because of fast exchange of the aromatic rings on the NMR time-scale
While the experimental 13C incorporation (Table 1) of 1-13C and 2-13C erythrose is closely following the expected incorporation (Fig. 1, red), 3-13C and 4-13C erythrose show some signs of scrambling and labeling of unexpected positions (Fig. 1, black). For 3-13C erythrose 13C is ending in Phe γ, and for 4-13C erythrose the incorporation of 13C in the δ positions (40% of δ1 and δ2) is higher than for the ε (20% of ε1 and ε2) and z (40% of only one ζ). It is unclear if the additional non expected 13C (Fig. 1, black) end up in the same molecule as the expected (Fig. 1, red), causing possible 2J 13C-13C couplings, or in different molecules. Please note, that this is the default situation in glucose labeling. On the other hand, the absence of effective 13C incorporations in other positions than the expected, in case of 1-13C and 2-13C erythrose clearly indicates the absence of any possible 2J or 3J 13C-13C couplings.
13C relaxation studies
Both erythrose and glucose labeling lead to site-selective 13C labeling in aromatic side-chains. Are they both equally well suited for 13C relaxation studies or are potential long-range 13C-13C couplings affecting the results? Since erythrose labeling leads to less 13C background in the protein and a more distinct labeling of the aromatic side-chains, potential problems are expected to be less. However, comparing R 1 (SI Fig. 2), R 2 (SI Fig.3) and {1H-}13C NOE for identical positions between erythrose- (1-13C, 3-13C, and 4-13C) and glucose- (1-13C and 2-13C) labeled samples, we observe an excellent agreement (Fig. 4). Thus, the two approaches give virtually the same result; small deviations do not follow any trend indicating systematic differences, but appear to be random. Only for poorly 13C labeled positions obtained with glucose labeling (Trp), the relaxation data are slightly different, which can be explained by the higher uncertainty of the glucose-based probe (due to the lower S/N).
Fig. 4.

Comparison of aromatic 13C relaxation experiment using glucose (1-13C and 2-13C) or erythrose (1-13C, 3-13C, and 4-13C) labeled FKBP12. R 1 (a, b), R 2 (c, d) and {1H-}13C NOE (e, f) experiments were conducted using site-selective labeled FKBP12 based on glucose (black) or erythrose (red) labeling. Results from 4 to 13C1-erythrose and corresponding sites from glucose are shown in (a, c, e), results from 1 to 13C1- and 3-13C1-erythrose and corresponding sites from glucose are shown in (b, d, f)
While this does not clarify if additional deuteration is needed for artifact free relaxation data (Kasinath et al. 2013) or not (Weininger et al. 2012a), it shows that remote 13C do not play a role and potentially any method resulting in isolated 13C is equally well suited for relaxation studies. 13C relaxation dispersion experiments both for CPMG (Weininger et al. 2012b) and R 1ρ (Weininger et al. 2014a) were validated for glucose labeled samples previously. These experiments can be directly applied to samples resulting from erythrose labeling, since the relaxation behaviour is identical.
Site-selective 13C labeling of aliphatic side-chains
Labeling with erythrose is more selective then glucose-based labeling, since it is a precursor closer to the aromatic side-chain end products. Therefore it is not surprising that the level of 13C incorporation in aliphatic side-chains is generally lower (SI Tables 1, 2). However, a few positions are worth mentioning, which become efficiently labeled with isolated 13C. First, in histidine the α and β positions are significantly labeled (Table 2) by 3-13C and 2-13C erythrose, indicating a crossover into the pentose-5-phosphate pathway. Indeed erythrose 4-phosphate can be transformed to ribose 5-phosphate via sedoheptulose 7-phosphate by transaldolase and transketolase. (Schwender et al. 2003) In contrast, there is no 13C incorporation in the aromatic moiety of His. Especially His β is of potential interest, where the 13C incorporation is fairly significant at 34%. Since this site is not 13C labeled at all using 1-13C and 2-13C glucose, information in relaxation dispersion studies on the β carbon was missing (Lundstrom et al. 2009). The situation is similar for Ile and Lys β, which both are 13C labeled at 21% (SI Table 2 by 3-13C and 4-13C erythrose. They are efficiently labeled in the glucose (2-13C and 1-13C) approach as well, but not free from 13C–13C couplings. Furthermore, Lys δ and Arg γ are labeled at 22 and 16%, respectively, in an isolated fashion (SI Table 2), by 4-13C erythrose. These might be of interest as additional positions for dynamics studies in long and charged side-chains.
Table 2.
Site-selective 13C incorporation in histidine using glucose (G) or erythrose (E)
| 1-13C G | 2-13C G | 1-13C E | 2-13C E | 3-13C E | 4-13C E | |
|---|---|---|---|---|---|---|
| His CO | 31 | 7 | 1 | 0 | 0 | 3 |
| His α | 3 | 40 | 1 | 1 | 24 | 1 |
| His β | 0 | 0 | 9 | 34 | 1 | 0 |
| His γ | n.d | n.d | n.d | n.d | n.d | n.d |
| His δ2 | 26 | 52 | 4 | 4 | 3 | 2 |
| His ε1 | 37 | 8 | 1 | 1 | 3 | 14 |
Values are in %. Errors are estimated to 1% for 1H bound 13C, 3% for others. 1% for non labeled positions is expected because of natural abundance of 13C
Combined 13C labeling using both erythrose and glucose
Since the general labeling protocol presented here is based on site-selectively 13C-labeled erythrose in addition to unlabeled glucose, it is straightforward to combine site-selective labeling from both sources in order to get more positions per sample labeled or to increase 13C labeling of some sites. This strategy was verified by two approaches.
First, we combined 1-13C1-glucose, which labels Phe and Tyr δ, His ε1 and δ2 and Trp δ1 and ε3 (Fig. 5a, black), with 2-13C1-erythrose, which labels Phe (and Tyr) ζ and Trp η2 (Fig. 5a, blue), positions that are not covered by glucose (1-13C and 2-13C) labeling. The combined approach (Fig. 5a, red) gives the following results. His ε1 becomes as efficiently labeled as with protocols using only glucose and Phe ζ and Trp η2 as efficiently as with erythrose. His δ2 and Trp δ1 are labeled less than in the 1-13C1-glucose-only case. However these positions are better studied with the 2-13C1-glucose approach, which results in much higher 13C. Phe and Tyr δ are also labeled less than with glucose labeling. However, these sites are still labeled at a reasonable level, similar to what glucose labeling achieves for Phe and Tyr ε. Since the δ signals arise from two identical positions (due to fast ring flips), they are of the same signal strength as the Phe ζ signals. The only real drawback is observed for Trp ε3, whose labeling is rather poor in the glucose approach but even worse combined with erythrose. However, the combined approach is ideal to study the δ and ζ positions of Phe and Tyr in a single sample, because the spectral regions are well separated.
Fig. 5.

13C incorporation in aromatic side-chains using a combined glucose erythrose approach. Incorporation from glucose only is shown in black, incorporation from erythrose only in blue, labeling from both erythrose and glucose in red. a Shows results from 1 to 13C1-glucose and 2-13C1-erythrose, b from 2 to 13C1-glucose and 3-13C1-erythrose. All 13C labeled positions are isolated, no signs from 13C-13C couplings could be detected. * represents an averaged signal of position 1 and 2 because of fast exchange of the aromatic rings on the NMR time scale
Second, we combined 2-13C1-glucose, which labels Phe and Tyr ε, His δ2 and Trp δ1, ζ3 and ζ2 (Fig. 5b, black), with 3-13C1-erythrose, which also labels Phe and Tyr ε and Trp ζ3 (Fig. 5b, blue). Replacing 3-13C1-erythrose with 113C1-erythrose gives very similar results, only replacing erythrose based labeling of Trp ζ3 by Trp ζ2. Combining 1-13C1- and 3-13C1-erythrose would label both Trp positions but only half as effective. These approaches were not tested experimentally however. The combined approach of 2-13C1-glucose and 3-13C1-erythrose (Fig. 5b, red) labels His δ2 and Trp δ1 slightly better than what is observed for glucose only. Trp ζ3 and Phe and Tyr ε have improved 13C levels than with either of the two single-label approaches. However, the gain is less than what is expected from theoretical considerations, which suggest levels slightly higher than 60% (Trp ζ3) or 30% (Phe and Tyr ε). As expected the 13C level in Trp ζ2 decreases. This approach leads to results similar to that observed when using 2-13C1-glucose only, but with a moderate increase in 13C levels for Phe and Tyr ε (if sensitivity is crucial) and a large increase for the Trp ζ3 (3-13C1-erythrose) or Trp ζ2 (1-13C1-erythrose).
There are various other possible combinations, as long as glucose and erythrose do not result in covalent 13C-13C neighbors (Fig. 1, 1-13C and 4-13C result in 13C labeling next to positions that can be labeled by glucose according to Table 1). For instance, combining 1-13C1-glucose and 4-13C1-erythrose should result in the highest 13C incorporation for Phe and Tyr δ and Trp ε3.
Based on the 13C incorporation levels achieved with the erythrose-only approach (1-13C to 4-13C) and the combined erythrose–glucose approaches described above, one can estimate to what extent a certain amino acid is built from glucose and erythrose precursors (Fig. 6). The following results are based on experiments using 2 g/l of each carbon source, both present in the expression medium at the beginning. For most amino acids, 60% for the carbon incorporation originates from glucose and 40% from erythrose. While this result is close to the expected 50/50 distribution according to the amount in the medium, it does not agree with the result from studies with varying erythrose concentrations (Fig. 2). The highest amount of glucose based synthesis is 80% or more, which is observed for Arg, the aromatic moiety of His and the aromatic 5-ring moiety of Trp. The lowest amount of glucose based synthesis (and thereby the highest amount of erythrose based) is observed for the aliphatic moiety of His, the aromatic 6-ring moiety of Trp with 40%, and Phe and Tyr ε and ζ with 50%.
Fig. 6.

Amount of incorporation arising from glucose when erythrose is present for all amino acids. Both from individual (1-13C, 2-13C, 3-13C, and 4-13C erythrose, 1-13C glucose, and 2-13C glucose) and combined (1-13C glucose with 2-13C erythrose, 2-13C glucose with 3-13C erythrose) labeling approaches the amount of incorporation from glucose could be determined. The amount arising from erythrose is the complementary fraction. This scenario uses 2 g/l glucose and 2 g/l erythrose. Backbone carbonyl incorporation behaves differently. Whenever results from all other positions of an amino acid are in agreement with each other only one value per amino acid is shown. If certain positions of amino acids show significantly different behaviour than the rest they are shown right from the amino acids and are labeled according to their position (for Met, His, Trp, Phe and Tyr)
Further improvements
13C labeling of aromatic (and other) side-chains based on site-selectively 13C-enriched erythrose together with unlabeled glucose enables similar growth of cells as that resulting from growth on glucose only, and similar or improved 13C incorporation with a higher selectivity. However, labeling yields are far from 100%, which leaves room for further improvement. One way to increase the labeling yield would be to use cells with improved erythrose uptake. This will likely shift the ratio of amino acid biosynthesis more to the erythrose-based side. However, this would most likely come at the price of reduced selectivity. A more straightforward approach would be to use doubly 13C-enriched erythrose, which unfortunately does not appear to be commercially available at present. As long as the two 13C sites are separated in the erythrose they will lead to isolated 13C sites in the aromatic side-chains with the same level of incorporation as that obtained with the singly 13C-labeled erythrose. 1,3-13C2-erythrose would double the 13C incorporation of Phe and Tyr ε and label Trp ζ3 and ζ2 at the same time. 2,4-13C2-erythrose would label Phe and Tyr δ and ζ, and Trp ε3 and η2 at the same time. 1,4-13C2-erythrose would label Phe and Tyr δ and ε, but in this case the 13C sites are not expected to be isolated. Since the 13C incorporation in Phe and Tyr δ for 4-13C erythrose is higher (Table 1) than for ε and ζ (only one carbon), the other δ (Fig. 1, black) must be labeled as well by 4-13C1-erythrose, which will lead to 13C-13C couplings between δ (from 4 to 13C erythrose, Fig. 1, black) and ε (from 1 to 13C erythrose, Fig. 1, red).
Conclusions
We have shown that erythrose as a source for site-selective 13C labeling of amino acids yields more selective incorporation patterns than what is achieved using glucose. Erythrose leads to a slight improvement of the 13C level for Phe and Tyr δ, and a significant improvement (doubling) for proton-bound carbons in the six ring moiety of Trp. Further Phe (and Tyr) ζ and Trp η2 become available for measuring dynamics for the first time. Labeling of Phe ζ make it possible to separate the effects of motions around chi-2 and chi1 dihedral angles. His β becomes significantly 13C labeled via erythrose, and isolated 13C appears in the Ile β, Lys β and δ, and Arg γ sites. Finally, we have shown that the present approach for site-selective 13C labeling can be easily combined with the glucose-based approach, to yield labeling patterns optimized for specific purposes.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Protein expression and purification was performed by the Lund Protein Production Platform (LP3), Lund University, Sweden (http://www.lu.se/lp3). This research was supported by the Royal Physiographic Society of Lund and the Deutsche Forschungsgemeinschaft (WE 5587/1–1). The author thanks Mikael Akke for helpful comments on the manuscript.
References
- Akke M, Palmer AG. Monitoring macromolecular motions on microsecond–millisecond time scales by R1ρ–R1 constant-relaxation-time NMR spectroscopy. J Am Chem Soc. 1996;118:911–912. doi: 10.1021/ja953503r. [DOI] [Google Scholar]
- Akke M, Bruschweiler R, Palmer AG. Nmr order parameters and free-energy—an analytical approach and its application to cooperative Ca2+ binding by calbindin-D(9K) J Am Chem Soc. 1993;115:9832–9833. doi: 10.1021/ja00074a073. [DOI] [Google Scholar]
- Bartlett GJ, Porter CT, Borkakoti N, Thornton JM. Analysis of catalytic residues in enzyme active sites. J Mol Biol. 2002;324:105–121. doi: 10.1016/S0022-2836(02)01036-7. [DOI] [PubMed] [Google Scholar]
- Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- Boyer JA, Lee AL. Monitoring aromatic picosecond to nanosecond dynamics in proteins via C-13 relaxation: expanding perturbation mapping of the rigidifying core mutation, V54A, in Eglin C. BioChemistry. 2008;47:4876–4886. doi: 10.1021/bi702330t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole R, Loria JP. Evidence for flexibility in the function of ribonuclease A. BioChemistry. 2002;41:6072–6081. doi: 10.1021/bi025655m. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. Nmrpipe—a multidimensional spectral processing system based on unix pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Diehl C, et al. Protein flexibility and conformational entropy in ligand design targeting the carbohydrate recognition domain of Galectin-3. J Am Chem Soc. 2010;132:14577–14589. doi: 10.1021/ja105852y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenmesser EZ, Bosco DA, Akke M, Kern D. Enzyme dynamics during catalysis. Science. 2002;295:1520–1523. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- Frederick KK, Marlow MS, Valentine KG, Wand AJ. Conformational entropy in molecular recognition by proteins. Nature. 2007;448:325–329. doi: 10.1038/nature05959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AL, Kay LE. Quantifying millisecond time-scale exchange in proteins by CPMG relaxation dispersion NMR spectroscopy of side-chain carbonyl groups. J Biomol NMR. 2011;50:347–355. doi: 10.1007/s10858-011-9520-6. [DOI] [PubMed] [Google Scholar]
- Hansen AL, Kay LE. Measurement of histidine pK(a) values and tautomer populations in invisible protein states. Proc Natl Acad Sci USA. 2014;111:E1705–E1712. doi: 10.1073/pnas.1400577111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AL, Lundstrom P, Velyvis A, Kay LE. Quantifying millisecond exchange dynamics in proteins by CPMG relaxation dispersion NMR using side-chain H-1 probes. J Am Chem Soc. 2012;134:3178–3189. doi: 10.1021/ja210711v. [DOI] [PubMed] [Google Scholar]
- Ishima R, Torchia DA. Extending the range of amide proton relaxation dispersion experiments in proteins using a constant-time relaxation-compensated CPMG approach. J Biomol NMR. 2003;25:243–248. doi: 10.1023/A:1022851228405. [DOI] [PubMed] [Google Scholar]
- Jarymowycz VA, Stone MJ. Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem Rev. 2006;106:1624–1671. doi: 10.1021/cr040421p. [DOI] [PubMed] [Google Scholar]
- Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules . Methods Mol Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- Kasinath V, Valentine KG, Wand AJ. A C-13 labeling strategy reveals a range of aromatic side chain motion in calmodulin. J Am Chem Soc. 2013;135:9560–9563. doi: 10.1021/ja4001129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasinath V, Fu YN, Sharp KA, Wand AJ. A sharp thermal transition of fast aromatic-ring dynamics in ubiquitin. Angew Chem Int Edit. 2015;54:102107. doi: 10.1002/anie.201408220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzhnev DM, Religa TL, Banachewicz W, Fersht AR, Kay LE. A Transient and low-populated protein-folding intermediate at atomic resolution. Science. 2010;329:1312–1316. doi: 10.1126/science.1191723. [DOI] [PubMed] [Google Scholar]
- Li H, Yamada H, Akasaka K. Effect of pressure on the tertiary structure and dynamics of folded basic pancreatic trypsin inhibitor. Biophys J. 1999;77:2801–2812. doi: 10.1016/S0006-3495(99)77112-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenecker RJ, Weinhaupl K, Schmid W, Konrat R. alpha-Ketoacids as precursors for phenylalanine and tyrosine labelling in cell-based protein overexpression. J Biomol NMR. 2013;57:327–331. doi: 10.1007/s10858-013-9796-9. [DOI] [PubMed] [Google Scholar]
- Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- Loria JP, Rance M, Palmer AG. A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. J Am Chem Soc. 1999;121:2331–2332. doi: 10.1021/ja983961a. [DOI] [Google Scholar]
- Lundstrom P, et al. Fractional C-13 enrichment of isolated carbons using [1-C-13]- or [2-C-13]-glucose facilitates the accurate measurement of dynamics at backbone C-alpha and side-chain methyl positions in proteins. J Biomol NMR. 2007;38:199–212. doi: 10.1007/s10858-007-9158-6. [DOI] [PubMed] [Google Scholar]
- Lundstrom P, Lin H, Kay LE. Measuring (13)C(beta) chemical shifts of invisible excited states in proteins by relaxation dispersion NMR spectroscopy. J Biomol NMR. 2009;44:139–155. doi: 10.1007/s10858-009-9321-3. [DOI] [PubMed] [Google Scholar]
- Malmendal A, Evenas J, Forsen S, Akke M. Structural dynamics in the C-terminal domain of calmodulin at low calcium levels. J Mol Biol. 1999;293:883–899. doi: 10.1006/jmbi.1999.3188. [DOI] [PubMed] [Google Scholar]
- Milbradt AG, Arthanari H, Takeuchi K, Boeszoermenyi A, Hagn F, Wagner G. Increased resolution of aromatic cross peaks using alternate C-13 labeling and TROSY. J Biomol NMR. 2015;62:291–301. doi: 10.1007/s10858-015-9944-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet O, Muhandiram DR, Skrynnikov NR, Kay LE. Deuterium spin probes of side-chain dynamics in proteins. 1. Measurement of five relaxation rates per deuteron in C-13-labeled and fractionally H-2-enriched proteins in solution. J Am Chem Soc. 2002;124:6439–6448. doi: 10.1021/ja012497y. [DOI] [PubMed] [Google Scholar]
- Mittermaier A, Kay LE. Review - New tools provide new insights in NMR studies of protein dynamics. Science. 2006;312:224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- Miyanoiri Y, Takeda M, Jee J, Ono AM, Okuma K, Terauchi T, Kainosho M. Alternative SAIL-Trp for robust aromatic signal assignment and determination of the chi(2) conformation by intra-residue NOEs. J Biomol NMR. 2011;51:425–435. doi: 10.1007/s10858-011-9568-3. [DOI] [PubMed] [Google Scholar]
- Muhandiram DR, Yamazaki T, Sykes BD, Kay LE. Measurement of H-2T-1 and T-1P Relaxation-Times in Uniformly C-13-Labeled and Fractionally H-2-Labeled Proteins in Solution. J Am Chem Soc. 1995;117:11536–11544. doi: 10.1021/ja00151a018. [DOI] [Google Scholar]
- Mulder FAA, Hon B, Mittermaier A, Dahlquist FW, Kay LE. Slow internal dynamics in proteins: Application of NMR relaxation dispersion spectroscopy to methyl groups in a cavity mutant of T4 lysozyme. J Am Chem Soc. 2002;124:1443–1451. doi: 10.1021/ja0119806. [DOI] [PubMed] [Google Scholar]
- Neudecker P, et al. Structure of an intermediate state in protein folding and aggregation. Science. 2012;336:362–366. doi: 10.1126/science.1214203. [DOI] [PubMed] [Google Scholar]
- Palmer AG. NMR characterization of the dynamics of biomacromolecules. Chem Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- Paquin R, Ferrage F, Mulder FAA, Akke M, Bodenhausen G. Multiple-timescale dynamics of side-chain carboxyl and carbonyl groups in proteins by c-13 nuclear spin relaxation. J Am Chem Soc. 2008;130:1580515807. doi: 10.1021/ja803794g. [DOI] [PubMed] [Google Scholar]
- Sathyamoorthy B, Singarapu KK, Garcia AE, Szyperski T. Protein conformational space populated in solution probed with aromatic residual dipolar C-13-H-1 couplings. Chembiochem. 2013;14:684–688. doi: 10.1002/cbic.201300016. [DOI] [PubMed] [Google Scholar]
- Savitsky P, Bray J, Cooper CDO, Marsden BD, Mahajan P, Burgess-Brown NA, Gileadi O. High-throughput production of human proteins for crystallization: The SGC experience. J Struct Biol. 2010;172:3–13. doi: 10.1016/j.jsb.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schörghuber J, Sara T, Bisaccia M, Schmid W, Konrat R, Lichtenecker RJ. Novel approaches in selective tryptophan isotope labeling by using escherichia coli overexpression media. Chembiochem. 2015;16:746–751. doi: 10.1002/cbic.201402677. [DOI] [PubMed] [Google Scholar]
- Schwender J, Ohlrogge JB, Shachar-Hill Y. A flux model of glycolysis and the oxidative pentosephosphate pathway in developing Brassica napus embryos. J Biol Chem. 2003;278:29442–29453. doi: 10.1074/jbc.M303432200. [DOI] [PubMed] [Google Scholar]
- Shaw DE, et al. Atomic-level characterization of the structural dynamics of proteins. Science. 2010;330:341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- Takeda M, Ono AM, Terauchi T, Kainosho M. Application of SAIL phenylalanine and tyrosine with alternative isotope-labeling patterns for protein structure determination. J Biomol NMR. 2010;46:45–49. doi: 10.1007/s10858-009-9360-9. [DOI] [PubMed] [Google Scholar]
- Teilum K, Brath U, Lundstrom P, Akke M. Biosynthetic C-13 labeling of aromatic side chains in proteins for NMR relaxation measurements. J Am Chem Soc. 2006;128:2506–2507. doi: 10.1021/ja055660o. [DOI] [PubMed] [Google Scholar]
- Volkman BF, Lipson D, Wemmer DE, Kern D. Two-state allosteric behavior in a single-domain signaling protein. Science. 2001;291:2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- Wagner G. Activation volumes for the rotational motion of interior aromatic rings in globular-proteins determined by high-resolution h-1-Nmr at variable pressure. FEBS Lett. 1980;112:280–284. doi: 10.1016/0014-5793(80)80198-0. [DOI] [PubMed] [Google Scholar]
- Wagner G, Demarco A, Wuthrich K. Dynamics of aromatic amino-acid residues in globular conformation of basic pancreatic trypsin-inhibitor (Bpti) 0.1. H-1 Nmr-studies. Biophys Struct Mech. 1976;2:139–158. doi: 10.1007/BF00863706. [DOI] [PubMed] [Google Scholar]
- Wagner G, Bruhwiler D, Wuthrich K. Reinvestigation of the aromatic side-chains in the basic pancreatic trypsin-inhibitor by Heteronuclear two-dimensional nuclear-magnetic-resonance. J Mol Biol. 1987;196:227–231. doi: 10.1016/0022-2836(87)90524-9. [DOI] [PubMed] [Google Scholar]
- Wallerstein J, Weininger U, Khan MA, Linse S, Akke M. Site-specific protonation kinetics of acidic side chains in proteins determined by ph-dependent carboxyl (13)c nmr relaxation. J Am Chem Soc. 2015;137:3093–3101. doi: 10.1021/ja513205s. [DOI] [PubMed] [Google Scholar]
- Weininger U, Diehl C, Akke M. C-13 relaxation experiments for aromatic side chains employing longitudinal- and transverse-relaxation optimized NMR spectroscopy. J Biomol NMR. 2012;53:181–190. doi: 10.1007/s10858-012-9650-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weininger U, Respondek M, Akke M. Conformational exchange of aromatic side chains characterized by L-optimized TROSY-selected C-13 CPMG relaxation dispersion. J Biomol NMR. 2012;54:9–14. doi: 10.1007/s10858-012-9656-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weininger U, Respondek M, Low C, Akke M. Slow aromatic ring flips detected despite near-degenerate NMR frequencies of the exchanging nuclei. J Phys Chem B. 2013;117:9241–9247. doi: 10.1021/jp4058065. [DOI] [PubMed] [Google Scholar]
- Weininger U, Brath U, Modig K, Teilum K, Akke M. Off-resonance rotating-frame relaxation dispersion experiment for C-13 in aromatic side chains using L-optimized TROSY-selection. J Biomol NMR. 2014;59:23–29. doi: 10.1007/s10858-014-9826-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weininger U, Modig K, Akke M. Ring flips revisited: C-13 relaxation dispersion measurements of aromatic side chain dynamics and activation barriers in basic pancreatic trypsin inhibitor. BioChemistry. 2014;53:4519–4525. doi: 10.1021/bi500462k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.