

Abstract
Age is the most important risk factor for the development of infectious diseases, cancer and chronic inflammatory diseases including rheumatoid arthritis (RA). The very act of living causes damage to cells. A network of molecular, cellular and physiological maintenance and repair systems creates a buffering capacity against these damages. Aging leads to progressive shrinkage of the buffering capacity and increases vulnerability. In order to better understand the complex mammalian aging processes, nine hallmarks of aging and their interrelatedness were recently put forward.
RA is a chronic autoimmune disease affecting the joints. Although RA may develop at a young age, the incidence of RA increases with age. It has been suggested that RA may develop as a consequence of premature aging (immunosenescence) of the immune system. Alternatively, premature aging may be the consequence of the inflammatory state in RA. In an effort to answer this chicken and egg conundrum, we here outline and discuss the nine hallmarks of aging, their contribution to the pre-aged phenotype and the effects of treatment on the reversibility of immunosenescence in RA.
Keywords: Aging, immunosenescence, inflammation, rheumatoid arthritis, T-cells
1. Introduction
Age is the most important risk factor for development of chronic inflammatory diseases including rheumatoid arthritis (RA). Aging is a complex process, which is not yet fully understood. In order to better understand the complex mammalian aging processes, nine hallmarks of aging and their interrelatedness were recently put forward. In essence, primary hallmarks involve acquisition of cell biological forms of damage such as genomic instability, telomere attrition, epigenetic alterations and loss of proteostasis. Secondary hallmarks involve antagonistic, compensatory responses to these damages such as deregulated nutrient-sensing, mitochondrial dysfunction and cellular senescence. Lastly, the integrative hallmarks represent the result of the primary and secondary events such as stem cell exhaustion and inflammaging, leading to deterioration of cellular and organismal function as seen with aging [1].
Age-associated changes in immune function termed immunosenescence are thought to be responsible for the increased morbidity with age. Both the innate and the adaptive arm of the immune system undergo marked changes with age and contribute to the process of immunosenescence [2]. With age, innate immune mechanisms generally become more active, whereas functioning of the adaptive immune system generally declines.
Immunosenescence is characterized by i) thymic involution leading to a steady decline in the production of naïve T-cells, ii) shrinkage of the T-cell repertoire through continuous antigen stimulation favoring the development of functionally altered, oligoclonal, senescent T-cells identified by CD28 loss and iii) a chronic low degree of inflammation termed inflammaging as evidenced by increased serum levels of inflammatory cytokines such as TNF-α, IL-6 and acute phase proteins [3]. These changes in cellular composition and cellular functions create a pro-inflammatory environment which might accelerate development of RA.
RA is a chronic auto-inflammatory disorder targeting the joints. Although RA can develop in individuals of any age, its incidence continues to increase with age into the seventh decade [4]. When compared to healthy individuals of the same age, RA patients show significantly more prominent features of immune system aging. Despite the growing experimental data, the question whether accelerated immunosenescence is a primary cause of RA or an event secondary to the chronic inflammatory process remains to be answered.
In this review we summarize the experimental evidence for the pre-aged phenotype of different immune cells in RA and their relevance to disease pathogenesis. We also compare features of senescence in peripheral versus local tissues (bone marrow, peripheral blood and the inflamed joint). Furthermore, we outline the contribution of the hallmarks of aging to the pre-aged phenotype in RA. Also, the effects of treatment on the reversibility of immunosenescence in RA are discussed. Lastly, novel insights in the molecular pathogenesis of RA relevant to the development of the pre-aged phenotype may guide adequate design of conclusive patient-based research to solve this issue.
2. Evidence for a pre-aged phenotype in RA
2.1. Immunosenescence and T Lymphocytes in RA
Although aging affects all cells of the immune system, T-cells appear to be most sensitive. A prominent feature of T-cell aging is the oligoclonal expansion of CD4+ and especially CD8+ T-cells lacking expression of the co-stimulatory molecule CD28. CD28 co-stimulation, required for efficient T-cell activation and proliferation, is progressively lost with age [3]. Consequences of CD28 loss differ between CD4+ and CD8+ T-cells; in CD8+ T-cells loss of CD28 can lead to dysfunction or towards a regulatory phenotype, whereas in CD4+ T-cells CD28 deficiency is associated with acquisition of novel NK-like functionalities. Loss of CD28 in CD4+ T-cells is associated with an increased production of pro-inflammatory cytokines, increased cytotoxicity, via expression of perforin and granzyme B and a propensity to migrate into tissues. Interestingly, CD4+CD28- T-cells were found expanded in patients with several chronic autoimmune conditions, including RA. Expansions were seen in both early and late RA patients and were more prominent in carriers of the RA-susceptibility HLA-DRB1*04 alleles [5]. This suggested that accelerated immunosenescence is a genetically-driven phenomenon that might be causal to RA development rather than the consequence of disease.
Novel functional features of senescent CD28- T-cells have been associated with phenotypical changes, such as de novo expression of NK receptors: Immunoglobulin (Ig)-like (i.e. killer cell activating receptors [KAR], killer cell inhibitory receptors [KIR]) [6-8] and C-type lectin-like superfamily (NKG2D) receptors [9, 10]. KIRs and KARs are specific for classical MHC class I molecules (HLA-A, HLA-B, HLA-C) and the non-classical MHC molecule HLA-G [11]. Ligands for NKG2D include the stress-induced MHC class I polypeptide-related sequence (MIC) proteins. MIC proteins were found upregulated by RA synoviocytes [10]. Also, CD28- T-cells in RA patients demonstrated increased expression of NK cell-associated receptors CD56 [9, 12, 13] and CD57 [14].
Upregulation of NK receptors likely serves a co-stimulatory role in CD28- T-cells. Cross-linking of KAR in the presence of anti-CD3 induced proliferation of CD4+CD28- T-cells [6]. Engagement of NKG2D, in the presence of anti-CD3, induced proliferation, IFN-γ and TNF-α secretion [10]. Triggering of CD56 alone led to the production of IL-2, TNF-α and MIP-1β [12]. Furthermore, several studies showed upregulation of CD70, a member of the TNF superfamily, by CD28- T-cells [15-17]. CD70 expression lowered the activation threshold of CD28- T-cells [15]. In line with their pro-inflammatory potential, RA patient-derived peripheral blood (PB) CD4+CD28- T-cells [14, 18, 19] and CD4+CD28- T-cell clones [14] produced significantly higher levels of TNF-α and IFN-γ than their CD28+ counterparts.
The exact role of senescent T-cells in the clinical course of RA is still unclear. Numbers of circulating CD4+CD28- T-cells did not correlate with RA clinical parameters such as C-reactive protein (CRP), level of anti-cyclic citrullinated protein antibodies (ACPA), swollen and tender joint counts or disease duration [14, 20, 21]. Some studies demonstrated a correlation between CD4+CD28- T-cells and erosions in RA [22, 23] but this was not confirmed by others [21, 24]. Expansions of CD4+CD28- T-cells in RA have been implicated in the development of atherosclerotic disease [24] and in extra-articular manifestations [21, 24].
The role of senescent T-cells at the level of the joint in RA also remains unclear. Several studies demonstrated that CD4+CD28- T-cells are less frequent in RA synovial fluid (SF) or synovium than in PB [14,18, 20, 25]. This seems to contradict the tissue tracking propensity of CD4+CD28- cells suggested based on the expression of adhesion molecules facilitating tissue infiltration (CD11a, CD49d) [9, 18, 26] and receptors (NKG2D, CX3CR1) which ligands (MIC, fractalkine) are readily expressed in RA tissue [9, 10]. A possible explanation may involve the restoration of CD28 expression by IL-12 [27] which is abundantly expressed in RA joints [28]. Consistent with this explanation, several similarities between PB CD28- and SF CD28+CD4+ T-cells have been demonstrated. These include the methylation status of IFN-γ promoter, expression of CXCR3, CCR6, CCR7 [19] and clonotypic composition [25].
Several studies highlighted the importance of CX3CR1, a chemokine receptor exclusively expressed by CD4+CD28- T-cells [9, 29, 30] and its ligand fractalkine (FKN) expressed by fibroblast-like synoviocytes (FLS), as co-stimulatory pair relevant to the local inflammatory process. The CX3CR1-FKN interaction between CD4+CD28- T-cells and FLS induced expression of pro-inflammatory cytokines and relayed survival signals. Furthermore, the CX3CR1-FKN interaction facilitated proliferation of FLS [29, 30].
It has been suggested that CD4+CD28- T-cells in RA are autoreactive. So far, conclusive evidence for this notion is lacking. RA-derived CD4+CD28- T-cell clones were shown to proliferate in response to autologous adherent cells in vitro [25], while CD4+CD28-NKG2D+ T-cell clones secreted IFN-γ when cultured with autologous MIC+ synoviocytes [10]. CD4+CD28- T-cells did not respond to collagen type II [18], a putative RA-associated autoantigen. In contrast, the reactivity of CD4+CD28- T-cells towards CMV has been clearly demonstrated, and did not differ between RA- and healthy control (HC)-derived or RA- and multiple sclerosis-derived senescent T-cells [18, 31, 32]. Thus, clonally expanded CD4+CD28- T-cells in RA are likely CMV-specific but may also include selfreactive clones [31]. Senescence features reported in various stages of T-cell development in RA are depicted in the Fig. (1).
Fig. (1).
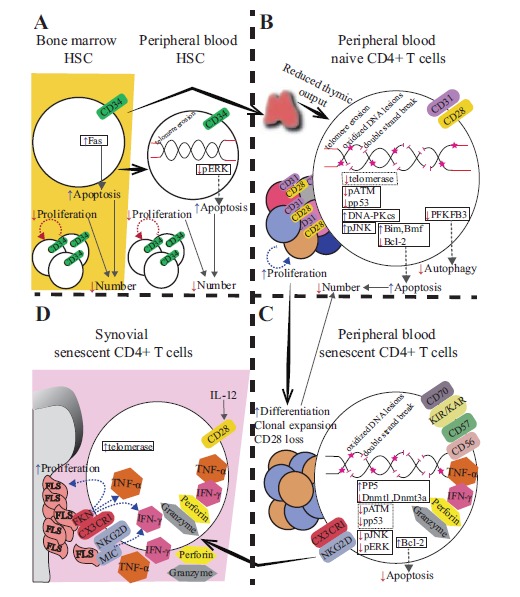
Depiction of senescence-associated alterations reported within A) hematopoietic stem cells (HSC) in bone marrow and peripheral blood, B) naive CD4+ T-cells in peripheral blood, C) senescent (CD28-) CD4+ T-cells in peripheral blood and D) senescent (CD28-) CD4+ T-cells in the inflamed joint of RA patients. A) In RA, bone marrow-derived hematopoietic stem cells (defined as CD34+) show impaired proliferative capacity and increased expression of Fas, rendering them apoptosis-sensitive. Consequently, RA patients show reduced HSC numbers at the level of the bone marrow. Peripheral blood-derived CD34+ HSC show similar defects in proliferation and increased susceptibility to apoptosis. Increased apoptosis sensitivity is via impaired ERK pathway signaling and/or age-inappropriate telomere erosion of CD34+ HSC in peripheral blood. B) Reduced thymic output in RA was evidenced by reductions of circulating recent thymic emigrants (defined as CD31+ or T-cell receptor excision circle (TREC)+ T-cells). This is explained by inadequate supply of HSC from the bone marrow and/or age-inappropriate enhanced thymic atrophy. Peripheral blood naive T-cells (defined as CD31+CD28+) are characterized by telomere shortening, increased levels of oxidized lesions and double-strand DNA breaks, decreased telomerase activity, deficiency of proteins involved in the DNA damage response (phosphorylated forms of ATM and downstream p53), overactivation of DNA-PKCs-JNK pathway, overexpression of proapoptotic proteins Bim, Bmf and downregulation of antiapoptotic protein Bcl-2. Defects of naive CD4+ T-cells may facilitate their accelerated differentiation towards the senescent state, when subjected to proliferative stress and enhance apoptosis. C) Compensatory hyperproliferation of the naive CD4+ T-cell pool leads to the expansion of senescent cells (characterized by the loss of CD28, de novo expression of NK receptors [KIR/KAR, CD56, NKG2D], upregulation of CD57, CD70, CX3CR1, expression of IFN-γ, TNF-α and cytotoxic molecules). Senescent CD4+ T-cells show further increase of DNA damage (double-strand breaks, oxidized DNA lesions and diminished levels of ATM and p53 phosphorylation) relative to naive CD4+ T-cells. Diminished activity of JNK and ERK pathways is associated with the increased expression of anti-apoptotic protein Bcl-2 and concomitant resistance to apoptosis of senescent CD4+ T-cells. D) Expression of CX3CR1 facilitates migration of senescent CD4+ T-cells towards soluble Fractalkine (FKN) abundant at the level of synovium. Interaction with FKN expressed on the surface of fibroblast-like synoviocytes (FLS) augments expression of IFN-γ, TNF-α and stimulates release of cytotoxic granules by senescent T-cells and enhances proliferation and FKN expression by FLS. IFN-γ production is also promoted by the interaction of NKG2D with FLS-expressed MIC. IL-12 increased at the level of inflamed joints, may restore CD28 expression by senescent T-cells.
2.2. CMV as Accelerator of T-cell Immunosenescence
CMV infection has been demonstrated to drive immunosenescence directly, by imposing chronic replicative stress and indirectly, by induction of IFN-α expression by plasmacytoid dendritic cells (PDC). IFN-α has been shown to accelerate differentiation and telomere shortening through inhibition of telomerase activity [33].
Latent CMV infection has been linked to a pro-inflammatory state as evidenced by increased systemic levels of IL-6 and TNF-α [34]. TNF-α is known to downregulate CD28 expression [35]. Latent CMV infection was found to aggravate the clinical course of RA [36]. CMV-specific CD4+CD28- T-cells were found increased in CMV+ but not CMV- RA patients [20]. These cells represented potent producers of IFN-γ [20, 36, 37].
Despite its role in the development of an immunosenescent phenotype, numerous studies undermine the notion of CMV involvement in RA pathogenesis. The most obvious, epidemiological evidence shows a worldwide CMV prevalence of 40-99%, while only ~1% of all individuals develop RA. A study by Pierer et al. which included large cohorts (>200 subjects each) of RA patients and HC, showed similar prevalence of CMV infection as well as comparable titers of anti-CMV antibodies in these groups [36].
Presence of CMV-specific T-cells, CMV DNA or CMV early antigen protein has been demonstrated in RA synovial tissue or synovial fluid but also at inflammatory sites of other autoimmune conditions [38-43].
In conclusion, CMV-driven T-cell immunosenescence is well established in both HC and in RA patients. The data on CD28- T-cells imply that these cells contribute to the inflammatory milieu in several chronic inflammatory conditions including RA. Also, CD28- cells may be involved in (extraarticular) tissue injury. Their contribution to joint pathology is hard to establish, partly because cells may regain CD28 expression via an IL-12 dependent mechanism at the level of the joint.
2.3. Immunosenescence and B Lymphocytes in RA
B-cells play an important role in RA pathogenesis based on their ability to produce and secrete autoantibodies and their role as antigen presenting cells [44-47]. Aging has pronounced effects on both numbers and functions of B-cells. Available evidence shows a decline of PB B-cells with age [48]. Yet, peripheral B-cell numbers were not further decreased in newly-diagnosed RA patients when compared to age-matched controls [49].
To the best of our knowledge, no studies have elucidated whether B-cells from RA patients show features of premature senescence. Recently, Rubtsov et al. identified a novel population of age-associated B-cells (ABCs), defined as CD19+CD11b+CD11c+, in aged female mice. ABCs were significantly increased in autoimmune-prone mouse strains at the onset of autoimmune disease and were found to be the main source of autoantibody production. The human equivalent of mouse ABCs, defined as CD19+CD11c+CD21- B-cells, were identified in the PB of some elderly female patients with RA [50]. Further studies are required to confirm the accumulation and functional role of ABCs in RA pathogenesis. Besides the increase of potentially pathogenic B-cells, a decrease of IL-10 producing B-cells may contribute to RA development. Both the number and function of immature transitional B-cells (with CD19+CD24highCD38high phenotype) regarded as the main IL-10 producers among B-cells, were found to decrease with age. The frequency of IL-10-expressing cells among CD24highCD38high B-cells was negatively correlated with rheumatoid factor (RF) titers [51]. In conclusion, although it is evident that aging and inflammation affect B-cell numbers and functional subsets, there is no sound evidence to support a role of senescent B-cells in RA pathogenesis.
2.4. Immunosenescence and NK-cells in RA
Aging-associated numerical, phenotypical and functional changes in NK-cells have been comprehensively reviewed [52-55]. Healthy aging is associated with an increase in the number of NK-cells, mainly attributed to the expansion of the differentiated, mature CD56dim subset that develops from the immature CD56bright subset [56]. CD56bright NK-cells were reported to either decrease [57, 58] or remain stable with age [56]. While NK-cell cytotoxicity is well-preserved with age [57], the cytokine and chemokine expression pattern of NK-cells, their proliferative capacity and their responsiveness to cytokines were impaired upon aging [56, 57, 59].
In contrast to healthy elderly, RA patients are characterized by a decline of peripheral NK-cell numbers [60-62]. A decline of these cells may accelerate senescence in RA patients. Support for this notion comes from studies demonstrating that NK-cells clear senescent cells in tumor lesions [63] and eliminate senescent cells involved in tissue damage [64]. These data indicate an immunosurveillant role of NK-cells in RA senescence rather than a pro-inflammatory role.
2.5. Immunosenescence and Monocytes in RA
Three different monocyte subsets can be distinguished based on surface expression of CD14 and CD16 [65, 66], namely the classical monocytes (CD14bright CD16-), the intermediate monocytes (CD14bright CD16+) and the non-classical monocytes (CD14dimCD16bright). The latter subset has been suggested to represent senescent monocytes, due to a shorter telomere length compared to classical monocytes and expression of the senescence-associated β-galactosidase (SA-βgal) [67, 68]. The CD14dimCD16bright non-classical monocytes are also more pro-inflammatory and express chemokine receptors facilitating migration to tissues at significantly higher levels than classical monocytes [67, 69]. CD14dimCD16bright monocytes were found to be similarly increased in elderly individuals with atherosclerosis and RA patients when compared to young subjects [67, 70].
Another population of monocytes that was increased with age is the CD56 NK receptor-expressing monocyte subset. These CD14brightCD56+ monocytes were found to be potent producers of cytokines and reactive oxygen species (ROS). Interestingly, only young RA patients (<40 years) showed CD14brightCD56+ monocyte expansion when compared to HC. As the number of CD14brightCD56+ monocytes did not correlate with disease duration, medication or C-reactive protein levels, the cause of the reported increase in these young RA patients remains unclear [71]. More studies are needed to elucidate the nature of senescence-associated changes of monocyte populations and to establish whether these alterations are more profound in RA patients than in aging per se.
In conclusion, ample evidence supports a pre-aged phenotype in RA patients within the T-cell compartment, whereas for other PB cells this is less evident. The culprit phenotype is represented by late stage, pro-inflammatory T-cells that have lost CD28 expression. Senescent cells may thrive because NK-cell surveillance is significantly reduced in RA.
3. The nine hallmarks of aging and RA
Nine common denominators of aging have recently been proposed [1]. The aim of this paragraph is to summarize the available evidence for the presence of these senescence hallmarks in the context of RA.
3.1. Genomic Instability
Cellular senescence or stable cell cycle arrest can be either telomere-dependent (discussed in “3.2. Telomere shortening”) or telomere-independent. The latter develops as a consequence of DNA damage accumulation due to replication errors, ROS, genotoxic drugs or UV light. Excessive DNA damage (including telomere erosion) evokes a persistent DNA damage response (DDR) which induces senescence or programmed cell death, both representing means to prevent malignant transformation. Factors involved in cell fate determination are not known but may include the extent of DNA damage, the strength and duration of DDR signaling and the type of the affected cell [72-74].
Elevated levels of DNA double-strand breaks were demonstrated in PB mononuclear cells (PBMC) [75, 76], or isolated naïve and memory CD4+ T lymphocytes [77, 78] but not neutrophils [79] from RA patients compared to HC. Similarly increased levels of DNA double-strand breaks were seen in naïve CD4+ T-cells from treatment-naïve recently diagnosed RA patients [77]. Also, increased levels of mutagenic DNA adducts such as 8-oxo-guanine (8-oxo-7-hydrodeoxyguanosine or 8-hydroxyguanine) or Heptanone-Etheno-2’-Deoxycytidine (HεdC) were found. 8-oxo-guanine and HεdC represent markers of oxidative DNA damage. In RA, levels of 8-oxo-dG and HεdC were found significantly increased within DNA derived from urine [80], whole blood cells [81], PBMC [75], CD4+ T-cells [77] and naïve CD4+ T-cells [78].
Accumulation of DNA damage in T-cells of RA patients has been associated with defects in DNA repair mechanisms. First evidence came from studies demonstrating an impaired ability of RA patient-derived PBMC to repair the mutagenic base lesion O6-methylguanine induced by the methylating carcinogen N-methyl-N-nitrosourea [82-84]. Also, a decreased rate of repairing double-strand DNA breaks by RA-derived PBMC has been demonstrated using the DNA unwinding in alkaline solution assay [76]. Studies by Shao et al. demonstrated accumulated DNA damage in both naïve and memory T-cells from newly-diagnosed RA patients and related this to decreased levels of the DNA repair kinase ataxia telangiectasia mutated (ATM). ATM deficiency was shown to recapitulate the pre-aged phenotype as seen in RA T-cells whereas overexpression of ATM reconstituted DNA repair capabilities [77, 78]. Thus, both the increase in DNA damage-inducing events such as replicative and oxidative stress as well as the impaired levels of proteins involved in DNA repair are likely the cause of DNA damage accumulation and permanent growth arrest of RA T-cells.
3.2. Telomere Shortening
Telomere erosion is recognized by the cell as a persistent DNA damage [85-87]. Telomere-dependent cell cycle arrest is mediated by upregulation of the p53-dependent DNA damage pathway [88]. Telomeres shorten 50-100 bp after each replication cycle, due to the end-replication problem of DNA polymerase [85, 88]. Activation of telomerase prevents replicative senescence by maintaining telomere function. Telomerase is a ribonucleoprotein complex consisting of the catalytic unit called the telomerase reverse transcriptase (hTERT) and telomerase RNA (TERC). Elongation of telomeres occurs through de novo reverse transcription. Expression of hTERT is a limiting factor for telomerase activity. In healthy conditions, telomerase is active only in stem cells and activated T and B lymphocytes [89].
Compared to age-matched HC, RA patients showed enhanced telomere shortening in granulocytes [5], PBMC [90] and CD4+ T-cells [5]. The latter was attributed to telomere erosion of naive, but not memory CD4+ T-cells by Koetz et al. [91]. Similarly, telomerase activity following TCR-dependent stimulation was found to be significantly decreased in naive, but not memory, CD4+ T-cells. hTERT deficiency was associated with a reduced proliferative capacity and a higher apoptosis rate of naïve T-cells. In contrast, lymphocytes infiltrating the synovium were characterized by high telomerase activity, indicative of their activated status. The telomerase activity levels of infiltrating cells were correlated with intensity of synovial lining hyperplasia, suggesting active lymphocyte involvement in joint destruction [92]. Interestingly, telomerase activity of anti-CD3 stimulated PBMC was similarly decreased in patients with early RA, multiple sclerosis and patients with flu-like symptoms [93], indicating lack of RA-specificity of telomerase insufficiency.
3.3. Changes in Gene Regulation
Aging is associated with alterations of epigenetic processes. These include mechanisms involved in gene regulation at the transcriptional level, i.e. histone modifications (acetylation and methylation), DNA methylation, chromatin remodeling and mechanisms involved in post-transcriptional gene regulation, i.e. non-coding (nc)RNAs expression [1]. A growing body of data demonstrates a substantial role of environmental factors in modulation of the epigenome [94]. Low RA concordance rates between monozygotic twins (~15%) [95-98], suggest a high relevance of the interplay between genetics and environmentally-influenced epigenetic alterations in RA pathogenesis.
Epigenetic changes are characteristic of RA synoviocytes. Reported epigenetic alterations include global hypomethylation [99, 100], local hypomethylation of LINE-1 and DR-3 promoters [101, 102], increased HDAC activity [103, 104], Sirt1 overexpression [105] and hyperacetylation [104], local H4 acetylation within MMP-1 promoter [106], sumoylation [107], and distinct pattern of microRNA expression [108].
3.3.1. Histone Modifications
One of the age-associated post-transcriptional (epigenetic) modifications include histone acetylation [1]. Histone acetylation at lysine residues, mediated by histone acetyltransferases (HAT), leads to decreased levels of chromatin condensation, allowing recruitment of the transcriptional machinery and ensuing gene transciption. Inhibition of gene expression, by removal of acetyl groups, is mediated by histone deacetylases (HDAC) [109]. Aging has been associated with a general increase of the histone acetylation status, as evidenced by increased H4K16 acetylation and decreased expression of the class III HDAC (NAD-dependent protein deacetylases sirtuins [Sirt]) [1].
An in vitro study employing RA synovial fibroblasts demonstrated a TNF-α-induced increase in HDAC and Sirt1 activity, suggesting that altered histone acetylation may be a feature of inflammation [110, 111]. Moreover, RA patient derived PBMC demonstrated increased HDAC activity [112], but no specific change of Sirt1 activity [105]. The current available data suggest that histone acetylation is differently modulated in aging and RA.
3.3.2. DNA Methylation
Chromatin methylation changes the chromatin structure to a more compact and less easily accessible state for transcription factors. Depending on the gene and cell type, both an age-associated decrease or increase in methylation status may occur. Decreased methylation status leads to enhanced mRNA and protein expression [113, 114]. Methylation occurs at deoxycytosine, primarily within CpG islands (repeated CpG sequences) which are associated with ~70% of the promotors in the vertebrate genome. Age-associated methylation changes have also been found to affect genes lacking CpG islands, i.e. CD11a (LFA-1) [115]. Methylation is mediated by DNA methyltransferase Dnmt1, involved in the preservation of the methylation pattern after each cell division, and methyltransferases Dnmt3a and Dnmt3b, which mediate methylation of previously unmethylated DNA [116]. Both Dnmt1 and Dnmt3a expression generally decrease with age [114].
Decreased methylation status has been reported in aging cells of various types, including fibroblasts [117] and stem cells [118]. Hypomethylation of a single CpG in the IL-6 promoter region was found more frequent in RA PBMC when compared to HC PBMC. High levels of IL-6 mRNA in LPS-stimulated macrophages were associated with this single CpG hypomethylation [119]. The most apparent link between hypomethylation and autoimmunity, however, is constituted by the demonstration that functional alterations in senescent CD4+CD28- T-cells are brought about by changes in DNA methylation. Relative to the CD4+CD28+ T-cell population, Dnmt1 and Dnmt3a levels and the DNA methylation status were significantly decreased in senescent CD4+CD28- T-cells. Genes overexpressed in response to hypomethylation included those associated with effector functions of senescent cells, i.e. IFN-γ [19] CD70, KIR2DL4 and perforin [16, 17]. Decreased levels of Dnmt1 and Dnmt3 in RA CD4+CD28- T-cells are the consequence of impaired signaling of the ERK and JNK pathways. Similar defects have been noted in CD4+CD28- T-cells generated in vitro by repeated stimulation as wells as in CD4+CD28- T-cells from elderly subjects [17]. Interestingly, SF-derived CD4+ T-cells showed prominent hypomethylation of the IFN-γ promoter (IFNG), irrespective of CD28 expression [19].
3.3.3. Chromatin Remodeling
Chromatin remodeling is mediated by enzymes involved in DNA and histone post-transcriptional modifications as well as Heterochromatin Protein 1α (HP1α), Polycomb Group (PcG) or the Nucleosome Remodeling and Deacetylase (NuRD) protein complexes. Their expression levels decrease during aging [1]. RA-derived synovial fibroblasts showed altered levels of the PcG protein EZH2 [120].
3.3.4. ncRNAs
The best studied group of ncRNAs in both health and RA are the microRNAs (miRNAs). These miRNAs are small (~22 nucleotides) single-stranded RNAs that have emerged as important post -transcriptional regulators of gene expression based on limited sequence complementarity.
The aging-associated epigenetic changes influence expression of miRNAs. The role of specific miRNAs in RA has recently been reviewed [121]. Here we will review the miRNAs that link changes in gene regulation, aging and RA. Twenty-four miRNAs have been reported to be involved in gene regulation with aging [122]. MiRNA’s may cause aberrant DNA methylation. For example, the miR-29 gene family, miR-143, miR-148a and miR-152 all target Dnmt3a and Dnmt3b. MiRNAs that are implicated in both gene regulation with aging and in RA are miR-16, miR-124a and miR-125a. The expression level of miR-16 was elevated in PBMC of RA patients with active disease and correlated with the erythrocyte sedimentation rate (ESR), CRP and disease activity score 28 (DAS28) [123]. HDAC1, HDAC2 and HDAC3 transcripts are all proven targets of miR-16. It is currently not known if higher miR-16 levels also correspond with lower levels of HDAC transcripts and thus increased histone acetylation in RA patient-derived PBMC. MiR-124a was down regulated in RA synovial fibroblasts [124]. MiR-124a targets the 3` UTR of mRNAs encoding MCP-1 and CDK-2. MiR-124a down regulation thus promotes the production of MCP-1 and CDK-2 proteins by RA synovial fibroblasts. Also, miR-124 (among others) is thought to regulate EZH2, a component of the polycomb repressive complex, involved in chromatin remodeling in various cell types.
Pathways most affected by ageing include genes involved in post transcriptional events such as mRNA splicing [125] Hu antigen R (HuR) is one of the splicing control proteins. HuR is an RNA binding protein which stabilizes mRNA, thereby regulating gene expression [126]. Increased miR-125a levels correlate inversely with HuR in various tumor cells and miR-125a targets the ELAV gene transcript that encodes the HuR protein levels, thereby regulating HuR expression [126]. MiR-125a levels were elevated in plasma from RA patients. These increased plasma levels may reflect the down modulation of HuR expression in PBMC from RA patients [127].
3.4. Loss of Protein Homeostasis
Exogenous or endogenous stressors can lead to unfolding of proteins. Heat shock proteins (hsps) are stress-induced chaperones that refold these proteins or target them for destruction in autophagosomes. The age-associated decline of hsps is associated with reduced longevity [1].
Interestingly, an increased expression of several hsps in RA patient-derived PBMC [128] as well as in RA synovial tissue and fluid has been reported [129-132]. Hsps are upregulated in arthritic joints, likely in response to stress-induced endoplasmic reticulum (ER) hyperreactivity and increased protein turnover. Substantial data support a role for chaperones as autoantigens for T- and B-cells in RA; including detection of hsp-specific autoantibodies [129, 130, 133-135], presence of autoreactivity-inducing citrulline groups within hsps [136], ability to interact with RA-associated HLA-DRB1*0401 allele [137] and stimulation of T-cell responses [129, 130, 138, 139]. In RA patients, hsps serve as targets of the pathological immune response and their levels do not decline as demonstrated in aging.
Damaged, unfolded or incorrectly folded proteins which cannot be re-folded by hsps, are degraded by cellular proteolytic systems such as the ubiquitin-proteasome and lysosomal systems. This process is impaired with age and leads, in tandem with the decrease of chaperone functions, to accumulation and aggregation of erroneous proteins. Together, this results in an age-associated loss of tissue function [1].
Autophagy is a mechanism that involves degradation of cellular components through the actions of lysosomes. The breakdown of cellular components promotes cellular survival during stress by maintaining cellular energy levels. Autophagy is thus an important process relevant to protein homeostasis, energy metabolism and cell death [140]. Normal aging generally reduces autophagy [1].
The joint environment imposes ER stress to synovial cells due to a high protein turnover [141-143]. Consequently, autophagy is significantly increased in RA synovial fibroblasts. Enhanced autophagy correlated with reduced apoptosis. This led to the assumption that autophagy protects synovial cells from cell death. However, in a recent study using ex vivo cultured synovial fibroblasts the authors showed that autophagy may also promote death of RA synovial fibroblasts [144]. Both apoptosis-resistance and apoptosis-induction by autophagy represent means of responding to increased levels of stressors within the inflamed RA joints. Modulation of autophagy in synovial fibroblasts in RA joints was shown to depend on TNF-α [144-146].
Similar to the autophagy-lysosomal system, also elements of the ubiquitin-proteasome system have been found altered in RA synovial fibroblasts [144, 145, 147]. A single nucleotide polymorphism (SNP) mapping to the E3 ubiquitin ligase - cullin1 (CUL1) gene locus, has been associated with RA susceptibility in the Japanese [148] and north Indian populations [149]. E3 ubiquitin-protein ligase synoviolin, was increased not only in synovial fibroblasts in the joint [150-152], but also within PBMC and serum of RA patients [153]. Synoviolin has been suggested to render synovial cells resistant to apoptosis induced by ER stress [152]. Upregulation of synoviolin in vitro by fibroblast-like synoviocytes was TNF-α-dependent [151]. These data suggest that inflammation-mediated ER stress alters proteostasis and that altered proteostasis may enhance inflammation, thereby amplifying the local inflammatory response in RA.
Yang et al. reported that RA naïve T-cells are autophagy-deficient. The authors provide evidence that RA T-cells are in an energy-deprived state and thereby rendered apoptosis-sensitive. Increased apoptosis of naive CD4+ T-cells in RA patients may lead to increased homeostatic proliferation and early senescence of the T-cell pool [154]. Thus, metabolic defects may be responsible for loss of proteostasis in RA naïve T cells and may underlie their accelerated senescence.
3.5. Altered Nutrient-Sensing
Pathways involved in nutrient-sensing are thought to have a critical role in aging, as suggested by the overall positive effects of caloric restriction on lifespan. Nutrient-sensing pathways whose dysregulation is consequential for aging include insulin and insulin-like growth factor-1 (IGF-1) signaling (IIS) and the mTOR pathway [1]. Intriguingly, IGF-1 deficiency may shorten or extend lifespan, a paradox which remains unresolved at present. Pituitary-derived growth hormone (GH) induces IGF-1 production in the liver, and IGF-1 suppresses GH in a negative feedback loop [155]. IGF-1 represents one of two ligands of the IGF family, which also consists of six IGF binding proteins (BP). Availability of IGF-1 for the IGF-1 receptor (IGF-1R) is regulated mostly by IGFBP-3 due to its highest abundance in human serum [156]. GH and IGF-1 levels decrease during normal aging [155, 157].
Most studies demonstrated decreased levels of IGF-1 [158-161] and increased levels of IGFBP-3 [156, 158, 162] in serum/plasma of RA patients, which suggests low bioavailability of IGF-1. Acute starvation in RA patients (7 day fasting period) led to a further decline of IGF-1 and reduced measures of inflammation such as ESR, CRP, tender joint count as well as T-cell counts. Interestingly, mitogen activation of CD4+ T-cells after fasting showed increased IL-4 production in vitro [163]. These data link nutrient-sensing pathways to altered T-cell activation in RA. However, Matsumoto et al. showed that reduced serum levels of IGF-1 in RA did not correlate with clinical parameters of the disease but were negatively correlated with the age of the patients [158]. Moreover, several studies in RA patients failed to detect a change in IGF-1 [164-166] and IGFBP-3 levels [164], and in addition, also observed decreased IGFB-3 levels [160, 166]. In SF, both IGF-1 and IGFBP-3 levels were shown to be increased [167-169]. Thus, the picture is not unequivocal in RA.
Similar contrasting data were obtained for levels of GH [155]. Available data show either decreased [166] or increased [159, 164] GH levels in the periphery of RA patients. A study in newly diagnosed RA patients demonstrated impaired GH production and, similar to IGF-1, a role of pro-inflammatory cytokines (i.e. IL-1) in the further suppression of GH [170].
Components of the nutrient-sensing machinery, found to be important in aging, such as Akt (protein kinase B), Foxo and mTOR, have been implicated in the local inflammatory process in RA and were studied mostly in joint-derived FLS [171-176]. In RA circulating T-cells, expression of nutrient-sensing proteins (AMPK and mTOR) was not different compared with controls [154].
In summary, alterations of nutrient-sensing pathways in RA seem to be linked to the increased inflammatory status.
3.6. Mitochondrial Dysfunction
Mitochondrial dysfunction is a feature of aging and may be caused by the accumulation of somatic mutations within the mitochondrial genome (mtDNA), and/or by respiratory chain dysfunction, oxidation of mitochondrial proteins and lipids. Relevant to RA pathogenesis are the induction of ROS and mtDNA mutations in synovium. The increase of mtDNA mutations were found only when compared to osteoarthritis (OA) control tissue [177], but not when compared to psoriatic arthritis (PsA) tissue [178]. Elevated mtDNA mutation frequency in RA was independent of age. MtDNA mutations positively correlated with macroscopic synovitis, vascularity and with synovial fluid TNF-α and IFN-γ levels. As with other alterations observed within the inflammatory synovial environment, cause-effect relationships are challenging to study. Currently available data suggest involvement of TNF-α, and TNF-α-associated ROS generation in induction of mtDNA mutations [177, 178]. Mitochondrial dysfunction as a result of compromised metabolism induced by the ATP synthase inhibitor oligomycin, has been shown to promote ROS- and NF-κB-dependent induction of inflammatory markers by normal human synoviocytes [179].
Moreover, cell-free mtDNA has been detected in RA synovial fluid and plasma at levels significantly higher than in healthy subjects [180, 181], likely as the result of the increased tissue damage in RA. A study by Collins et al. demonstrated that cell-free oxidized mtDNA induces development of arthritis upon its intra-articular injection in mice [181].
Chronic inflammation was found to affect the metabolic competence of T-cells in RA. Their capacity to mobilize aerobic glycolysis for ATP generation was significantly reduced [154].
3.7. Cellular Senescence
Various cell intrinsic and extrinsic stressors can induce cellular senescence. P16ink4a and p53 play major roles in the cellular senescence signaling program [182]. Both proteins are central in the regulation of cell cycling and apoptosis. It has been proposed that the p53 and Rb pathways are responsible for the stress-induced growth arrest, while permanent expression of p16ink4a and p21WAF/CIP1 maintain the senescence state [183]. P53 was overexpressed in synovial tissues from both early and late-stage RA when compared to OA and reactive arthritis, while the cells in normal synovial tissue showed significantly less p53 expression [184]. Besides wild-type p53, also mutated p53 has been detected in RA synovium [185]. Yamanishi et al. analysed p53 mutations in RA synovium and demonstrated preferential survival of FLS with the mutated p53 allele. Interestingly, regions with high p53 expression levels also contained high levels of IL-6 transcripts [185]. This can be explained by the loss of IL-6 suppression dependent on the wild-type p53. Induction of p53 mutations in synovium has been suggested to represent an event secondary to chronic oxidative stress and the continuous need for DNA repair [184-186].
Upregulation of p16 in synovial fibroblasts leads to permanent growth arrest, a feature associated with the senescent phenotype. The in vitro upregulation of p16 in macrophages inhibited IL-6 expression [187]. Thus, local induction of p16 expression may have beneficial effects and has been proposed as a possible treatment strategy for RA [188, 189].
Interestingly, downregulation of p53 and other proteins from the DDR pathway in naive CD4+ T-cells has been implicated in T-cell senescence. Conversely, p16 and p53 were highly expressed in CD56+CD28- T-cells [13].
Another marker of cellular senescence is SA-β-gal. SA-β-gal was found to be endogenous lysosomal β-gal [190]. Although SA-β-gal was detected in OA cartilage, we are not aware of any studies reporting on SA-β-gal expression in RA synovium.
3.8. Stem Cell Exhaustion
Several studies suggest that alterations at the level of hematopoietic stem cells (HSC) underlie premature aging of the peripheral T-cell pool in RA.
Cells derived from the bone marrow of RA patients showed less effective colony formation potency when compared to HC-derived bone marrow cells [191-193]. Further studies confirmed the reduced proliferative capacity of CD34+ HSC from RA patients [194-198]. HSC in RA also demonstrated an increased apoptosis rate. Concomitantly, the numbers of CD34+ stem cells were found to be decreased in the bone marrow [194-196] and PB in RA [197].
Colmegna et al. [198] demonstrated a dampened ERK signaling pathway in HSC of RA patients. ERK signaling is involved in the growth, differentiation and prevention of apoptosis [199]. More specifically, alteration in RA HSC involved decreased interaction between K-Ras and B-Raf. Ras-induced translocation of Raf to the cellular membrane is necessary for activation of the downstream events of the Raf/MEK/ERK pathway [199]. Defects in K-Ras and B-Raf co-localization (and consequently reduced level of activated pERK) have been linked to senescence-associated impairment of the HSC proliferative response in RA.
Impaired proliferative capacity of HSC in RA has also been associated with decreased telomere length. As HSC are the precursors of T-cells, accelerated telomere shortening in RA HSC may underlie the previously reported premature telomere attrition in RA T-cells [5, 200]. Interestingly, while naive CD4+ T-cells from RA patients were characterized by both telomere erosion and defective telomerase activity, RA HSC demonstrated an increased telomerase activity, suggesting activation of a compensatory pathway [200]. Yet, this did not suffice to prevent telomere erosion in HSC of RA patients carrying the RA-associated HLA-DRB1*04 alleles and led to the hypothesis of genetically-driven premature immunosenescence in RA [5]. Senescence-associated changes within HSC of RA patients were independent from disease duration or disease activity [197] and did not differ between treated and non-treated patients [194, 197]. However, Papadaki et al. showed that increased bone marrow levels of TNF-α in RA patients is associated with increased apoptosis and a numerical decrease of CD34+ HSC [194].
3.9. Altered Intercellular Communication Leading to Inflammaging
A low-grade inflammation develops with age, while specific immunity wanes. This persistent inflammation has been termed sterile inflammation due to the lack of a defined pathogenic trigger, or inflammaging. Inflammaging has been associated with functional changes of senescent or terminally differentiated cells giving rise to the senescence-associated secretory phenotype (SASP) [3].
Both elderly individuals and patients with RA show increased systemic levels of pro-inflammatory cytokines including, among others, IL-6, IL-8 and TNF-α [201]. RA-derived FLS, which undergo replicative senescence induced by serial passage, produce significantly more IL-6, IL-8, vascular endothelial growth factor (VEGF) and prostaglandin E2 (PGE2) in response to IL-1β, when compared to early-passage FLS [202]. Analysis of the molecular mechanisms underlying SASP revealed a critical role of persistent DNA damage response involving increased levels of proteins from the ATM pathway (ATM, NBS1, CHK2) [203, 204]. In contrast, activated p53 may suppress the senescence-associated cytokine (IL-6, IL-8) secretion, while loss of p53 may amplify the SASP [203-205]. Persistent DNA damage has also been suggested as an underlying factor of T-cell replicative senescence in RA. Thus, despite the fact that SASP-inducing ATM and NSB1 were found to be decreased in RA T-cells [77], other proteins involved in the persistent DDR could contribute to SASP in the context of RA. One candidate may involve p53 which was found decreased in RA-derived naive CD4+ T-cells [77].
Hallmarks of aging reflect biological aging but may also be the consequence of inflammation. * The culprit phenotype in RA is represented by pro-inflammatory CD28- T-cells. Pro-inflammatory cytokine production by these cells resembles the SASP.
In conclusion, multiple hallmarks of aging have been identified in early and/or late stage RA patients and are summarized in Table 1.
Table 1. Hallmarks of aging in early and late RA and their modulation upon anti-TNF-α treatment.
| Aging hallmark | Early RA | Reference | Late RA | Reference |
Normalization upon
anti-TNF-α treatment |
Reference | |
|---|---|---|---|---|---|---|---|
| 1. | Genomic instability | yes | [77] | yes | [77, 78, 81] | yes | [206, 207] |
| 2. | Loss of telomeres | yes | [91, 93, 200] | yes | [90, 91, 200] | nd | |
| 3. | Gene regulation | nd | yes | nd | |||
| 3.1. | Histone modifications | nd | yes | [105, 112] | contradictory results | [112, 208] | |
| 3.2. | DNA methylation | nd | yes | [16, 19] | nd | ||
| 3.3. | ncRNAs | nd | yes | [123] | nd | ||
| 4. | Proteostasis | nd | yes | [128, 130, 154] | nd | ||
| 5. | Altered nutrient sensing | yes | [170] | yes | [158-161, 164] | nd | |
| 6. | Mitochondrial dysfunction | nd | yes | [178,180] | yes | [178] | |
| 7. | Cellular senescence | yes | [184] | yes | [184] | nd | |
| 8. | Stem cell exhaustion | yes | [194, 197] | yes | [194, 196, 197] | yes | [194, 195, 209] |
| 9. | Inflammaging* | nd | yes | [14, 18, 19] | contradictory results | [22, 24] |
1-4 hallmarks of aging, 5-7 antagonistic hallmarks, 8-9 culprit phenotype. nd = not determined.
4. Effects of RA treatment on the aged profile in RA
Biological hallmarks of aging seem to converge in the pre-aged phenotype as seen in RA (Table and Figure). Yet, this premature aging may be inflammation-driven. To gain further insight in cause and consequence, we reviewed the effects of anti-inflammatory treatment on the pre-aged phenotype in RA patients. Several researchers addressed the issue of reversibility of premature T-cell aging upon treatment in RA patients. First, in vitro studies demonstrated that chronic TNF-α stimulation-induced CD28 loss by T-cells was partly restored by TNF-α inhibition [210, 211]. Effects of TNF-α blockade on the number of CD28- T-cells in patients, however, are inconclusive. While some studies reported on a reduction of the number of circulating CD4+CD28- T-cells [22, 24], others did not observe any change [37]. Also, anti-TNF-α treatment did not affect the CMV-specific IFN-γ response of CD4+CD28- T-cells [37]. The absolute number of CD4+CD28- T-cells was not different between RA patients receiving abatacept (CTLA-4Ig) or DMARDs [212]. Others demonstrated reduction of CD8+CD28- and late-stage CD8 effector memory, but not CD4+CD28- T-cells after 48 weeks of abatacept treatment. The decreases of CD28- cells among both CD4+ and CD8+ T lymphocyte populations after abatacept therapy were correlated with the clinical response as measured by DAS28/CRP [213]. Interestingly, the absolute numbers of CD28- T-cells (before treatment) were found to predict the response to abatacept [214].
More importantly, beneficial effects of treatment on premature HSC aging, a putative underlying cause of accelerated T-cell senescence in RA, were reported. The bone marrow niche of RA patients showed decreased apoptosis rates of the CD34+ fraction, increased cell growth potential (assessed by the colony formation assay) and an increased frequency of CD34+ cells after anti-TNF-α treatment [194, 195, 209]. Besides the positive effect on the number of HSC, blocking of TNF-α also resulted in an increase in the levels of recent thymic emigrants, defined as CD31+CD4+ T-cells [209]. Parameters of bone marrow function were not different between RA patients who were or were not on concomitant MTX [196], suggesting that TNF-α- but not MTX-mediated suppression of inflammation, is important for rejuvenation of the HSC pool in RA.
Telomere erosion and reduced telomerase activity in CD4+ T-cells were not affected by treatment of RA patients with MTX or prednisone [91, 200]. Similarly, accelerated telomere erosion in HSC was seen in both treated and not (yet) treated RA patients in cross-sectional analyses only [197]. To the best of our knowledge, the influence of anti-TNF-α agents on telomere length and telomerase activity in RA has not yet been addressed. Neutralization of TNF-α in vitro was associated with up regulation of telomerase activity in CD8+ T-cells [215].
Effects of DMARD treatment on cellular senescence, markers of DNA damage and epigenetic alterations relevant to RA were studied only in cell line models. In vitro-induced senescence in human fibroblast and cancer cell lines demonstrated a glucocorticoid (GC)-mediated reduction of SASP, such as reduced IL-6 and IL-8 expression. GC did not affect persistent DNA damage, the underlying cause of SASP, nor growth arrest or expression of SA-β-gal [216].
Longitudinal clinical studies demonstrated reduction of the markers of oxidative stress, including 8-hydroxydeoxyguanosine upon treatment with MTX or anti-TNF-α agents. These findings suggest that DMARD treatment reduces ROS-induced DNA damage [206, 207, 217]. Also mtDNA mutations frequency, regarded as a marker of mitochondrial dysfunction, was significantly lower in patients with low disease activity (DAS28 < 3.2) after TNF-α blocking therapy [178].
Studies investigating effects of anti-TNF-α treatment in RA patients on the activity of HAT and HDAC, regulating the acetylation status of histones, yielded contradictory results. HDAC activity, significantly higher in RA at baseline, did not change upon etanercept treatment [112]. Others showed an increase in HAT and a decrease in HDAC activity in a group of RA patients receiving various TNF-α inhibitors and increase of both HAT and HDAC upon RTX treatment [208]. Contradictory results were attributed to the use of different biologicals in these studies.
Thus, the combined evidence suggests that inflammation involving TNF-α, drives premature HSC aging, a putative cause of accelerated T-cell senescence in RA. To further substantiate the notion of reversibility of the senescent phenotype by TNF-α blocking agents or DMARDs, longitudinal clinical studies in well-defined patient cohorts before and after treatment are needed to clarify this issue.
5. Premature aging in RA: cause or consequence?
It has been suggested that premature aging of the immune system may be causal to RA. Telomere shortening of CD4+ T-cells was accelerated in RA patients carrying the RA-associated HLA-DRB1*04 alleles, which led to the hypothesis of genetically-driven premature immunosenescence in RA [5]. However, in light of more recent discoveries, we suggest that the causes of premature senescence in RA may be inflammation-driven. New knowledge of how environmental factors interact with susceptibility genes and the immune system in the development of seropositive RA (ACPA+ and/or RF+) has emerged. ACPA were found to develop more profoundly in individuals carrying the RA-associated HLA-DRB1 shared epitope-containing alleles and can be detected years before symptoms become manifest [218]. Presence of ACPA and RF in pre-RA patients was associated with elevated levels of pro-inflammatory cytokines [219, 220]. ACPA and/or RF may trigger the inflammatory response via an FcγR-mediated mechanism [221, 222]. Thus, the autoantibody-mediated inflammatory state, that precedes RA development by many years, may be responsible for premature immunosenescence in RA.
6. Conclusions and future perspectives
In order to evaluate the notion that premature immunosenescence in RA is inflammation-driven, we propose to study immunosenescence features in prospective, longitudinal cohort studies. Inclusion of individuals who are at risk for future RA development (not yet receiving DMARD treatment) would allow to assess the association between inflammatory markers and the immunonesenescent phenotype in the preclinical phase of RA, at disease onset and beyond. Cohorts to be used to assess the preclinical alterations putatively implicated in RA pathogenesis are seropositive arthralgia patients (SAP) and patients with early, undifferentiated arthritis (UA). Both UA and SAP have 30% chance of progressing towards classifiable RA [223, 224]. Furthermore, we propose to study immunesenescence features in late onset seropositive and seronegative RA.
As normal aging is associated with an overall decline of immune function, it will be important to compare healthy age-matched controls, excluding elderly subjects with co-morbidities. Assessment of immunosenescence parameters together with disease activity and pro-inflammatory markers at the preclinical stage and at the switch to clinical synovitis, are expected to provide insight into the mechanisms of their mutual regulation.
ACKNOWLEDGEMENTS
Declared none.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
PATIENT’S CONSENT
Declared None.
References
- 1.Lopez-Otin C., Blasco M.A., Partridge L., Serrano M., Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boots A.M., Maier A.B., Stinissen P., Masson P., Lories R.J., De Keyser F. The influence of ageing on the development and management of rheumatoid arthritis. Nat. Rev. Rheumatol. 2013;9(10):604–613. doi: 10.1038/nrrheum.2013.92. [DOI] [PubMed] [Google Scholar]
- 3.Weyand C.M., Yang Z., Goronzy J.J. T-cell aging in rheumatoid arthritis. Curr. Opin. Rheumatol. 2014;26(1):93–100. doi: 10.1097/BOR.0000000000000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crowson C.S., Matteson E.L., Myasoedova E., et al. The lifetime risk of adult-onset rheumatoid arthritis and other inflammatory autoimmune rheumatic diseases. Arthritis Rheum. 2011;63(3):633–639. doi: 10.1002/art.30155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schonland S.O., Lopez C., Widmann T., et al. Premature telomeric loss in rheumatoid arthritis is genetically determined and involves both myeloid and lymphoid cell lineages. Proc. Natl. Acad. Sci. USA. 2003;100(23):13471–13476. doi: 10.1073/pnas.2233561100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Namekawa T., Snyder M.R., Yen J.H., et al. Killer cell activating receptors function as costimulatory molecules on CD4+CD28null T cells clonally expanded in rheumatoid arthritis. J. Immunol. 2000;165(2):1138–1145. doi: 10.4049/jimmunol.165.2.1138. [DOI] [PubMed] [Google Scholar]
- 7.Warrington K.J., Takemura S., Goronzy J.J., Weyand C.M. CD4+, CD28- T cells in rheumatoid arthritis patients combine features of the innate and adaptive immune systems. Arthritis Rheum. 2001;44(1):13–20. doi: 10.1002/1529-0131(200101)44:1<13::AID-ANR3>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 8.Snyder M.R., Muegge L.O., Offord C., et al. Formation of the killer Ig-like receptor repertoire on CD4+CD28null T cells. J. Immunol. 2002;168(8):3839–3846. doi: 10.4049/jimmunol.168.8.3839. [DOI] [PubMed] [Google Scholar]
- 9.Broux B., Markovic-Plese S., Stinissen P., Hellings N. Pathogenic features of CD4+CD28- T cells in immune disorders. Trends Mol. Med. •••;18(8):446–453. doi: 10.1016/j.molmed.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Groh V., Bruhl A., El-Gabalawy H., Nelson J.L., Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA. 2003;100(16):9452–9457. doi: 10.1073/pnas.1632807100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y., Mariuzza R.A. Structural basis for recognition of cellular and viral ligands by NK cell receptors. Front. Immunol. 2014;5:123. doi: 10.3389/fimmu.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Michel J.J., Turesson C., Lemster B., et al. CD56-expressing T cells that have features of senescence are expanded in rheumatoid arthritis. Arthritis Rheum. 2007;56(1):43–57. doi: 10.1002/art.22310. [DOI] [PubMed] [Google Scholar]
- 13.Lemster B.H., Michel J.J., Montag D.T., et al. Induction of CD56 and TCR-independent activation of T cells with aging. J. Immunol. 2008;180(3):1979–1990. doi: 10.4049/jimmunol.180.3.1979. [DOI] [PubMed] [Google Scholar]
- 14.Fasth A.E., Cao D., Van Vollenhoven R., Trollmo C., Malmstrom V. CD28nullCD4+ T cells--characterization of an effector memory T-cell population in patients with rheumatoid arthritis. Scand. J. Immunol. •••;60(1-2):199–208. doi: 10.1111/j.0300-9475.2004.01464.x. [DOI] [PubMed] [Google Scholar]
- 15.Lee W.W., Yang Z.Z., Li G., Weyand C.M., Goronzy J.J. Unchecked CD70 expression on T cells lowers threshold for T cell activation in rheumatoid arthritis. J. Immunol. 2007;179(4):2609–2615. doi: 10.4049/jimmunol.179.4.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y., Chen Y., Richardson B. Decreased DNA methyltransferase levels contribute to abnormal gene expression in “senescent” CD4(+)CD28(-) T cells. Clin. Immunol. 2009;132(2):257–265. doi: 10.1016/j.clim.2009.03.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y., Gorelik G.J., Strickland F.M., Richardson B.C. Decreased ERK and JNK signaling contribute to gene overexpression in “senescent” CD4+CD28- T cells through epigenetic mechanisms. J. Leukoc. Biol. 2010;87(1):137–145. doi: 10.1189/jlb.0809562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thewissen M., Somers V., Hellings N., Fraussen J., Damoiseaux J., Stinissen P. CD4+CD28null T cells in autoimmune disease: pathogenic features and decreased susceptibility to immunoregulation. J. Immunol. 2007;179(10):6514–6523. doi: 10.4049/jimmunol.179.10.6514. [DOI] [PubMed] [Google Scholar]
- 19.Pieper J., Johansson S., Snir O., et al. Peripheral and site-specific CD4(+) CD28(null) T cells from rheumatoid arthritis patients show distinct characteristics. Scand. J. Immunol. 2014;79(2):149–155. doi: 10.1111/sji.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fasth A.E., Snir O., Johansson A.A., et al. Skewed distribution of proinflammatory CD4+CD28null T cells in rheumatoid arthritis. Arthritis Res. Ther. 2007;9(5):R87. doi: 10.1186/ar2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martens P.B., Goronzy J.J., Schaid D., Weyand C.M. Expansion of unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheum. 1997;40(6):1106–1114. doi: 10.1002/art.1780400615. [DOI] [PubMed] [Google Scholar]
- 22.Pawlik A., Ostanek L., Brzosko I., et al. The expansion of CD4+CD28- T cells in patients with rheumatoid arthritis. Arthritis Res. Ther. 2003;5(4):R210–R213. doi: 10.1186/ar766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goronzy J.J., Matteson E.L., Fulbright J.W., et al. Prognostic markers of radiographic progression in early rheumatoid arthritis. Arthritis Rheum. 2004;50(1):43–54. doi: 10.1002/art.11445. [DOI] [PubMed] [Google Scholar]
- 24.Gerli R., Schillaci G., Giordano A., et al. CD4+CD28- T lymphocytes contribute to early atherosclerotic damage in rheumatoid arthritis patients. Circulation. 2004;109(22):2744–2748. doi: 10.1161/01.CIR.0000131450.66017.B3. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt D., Goronzy J.J., Weyand C.M. CD4+ CD7- CD28- T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J. Clin. Invest. 1996;97(9):2027–2037. doi: 10.1172/JCI118638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Namekawa T., Wagner U.G., Goronzy J.J., Weyand C.M. Functional subsets of CD4 T cells in rheumatoid synovitis. Arthritis Rheum. 1998;41(12):2108–2116. doi: 10.1002/1529-0131(199812)41:12<2108::AID-ART5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 27.Warrington K.J., Vallejo A.N., Weyand C.M., Goronzy J.J. CD28 loss in senescent CD4+ T cells: reversal by interleukin-12 stimulation. Blood. 2003;101(9):3543–3549. doi: 10.1182/blood-2002-08-2574. [DOI] [PubMed] [Google Scholar]
- 28.Kim W., Min S., Cho M., et al. The role of IL-12 in inflammatory activity of patients with rheumatoid arthritis (RA). Clin. Exp. Immunol. 2000;119(1):175–181. doi: 10.1046/j.1365-2249.2000.01095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawai H., Park Y.W., Roberson J., Imai T., Goronzy J.J., Weyand C.M. T cell costimulation by fractalkine-expressing synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2005;52(5):1392–1401. doi: 10.1002/art.21140. [DOI] [PubMed] [Google Scholar]
- 30.Sawai H., Park Y.W., He X., Goronzy J.J., Weyand C.M. Fractalkine mediates T cell-dependent proliferation of synovial fibroblasts in rheumatoid arthritis. Arthritis Rheum. 2007;56(10):3215–3225. doi: 10.1002/art.22919. [DOI] [PubMed] [Google Scholar]
- 31.Thewissen M., Somers V., Venken K., et al. Analyses of immunosenescent markers in patients with autoimmune disease. Clin. Immunol. 2007;123(2):209–218. doi: 10.1016/j.clim.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 32.Van Bergen J., Kooy-Winkelaar E.M., Van Dongen H., et al. Functional killer Ig-like receptors on human memory CD4+ T cells specific for cytomegalovirus. J. Immunol. 2009;182(7):4175–4182. doi: 10.4049/jimmunol.0800455. [DOI] [PubMed] [Google Scholar]
- 33.Fletcher J.M., Vukmanovic-Stejic M., Dunne P.J., et al. Cytomegalovirus-specific CD4+ T cells in healthy carriers are continuously driven to replicative exhaustion. J. Immunol. 2005;175(12):8218–8225. doi: 10.4049/jimmunol.175.12.8218. [DOI] [PubMed] [Google Scholar]
- 34.Pawelec G., McElhaney J.E., Aiello A.E., Derhovanessian E. The impact of CMV infection on survival in older humans. Curr. Opin. Immunol. 2012;24(4):507–511. doi: 10.1016/j.coi.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 35.Lewis D.E., Merched-Sauvage M., Goronzy J.J., Weyand C.M., Vallejo A.N. Tumor necrosis factor-alpha and CD80 modulate CD28 expression through a similar mechanism of T-cell receptor-independent inhibition of transcription. J. Biol. Chem. 2004;279(28):29130–29138. doi: 10.1074/jbc.M402194200. [DOI] [PubMed] [Google Scholar]
- 36.Pierer M., Rothe K., Quandt D., et al. Association of anticytomegalovirus seropositivity with more severe joint destruction and more frequent joint surgery in rheumatoid arthritis. Arthritis Rheum. 2012;64(6):1740–1749. doi: 10.1002/art.34346. [DOI] [PubMed] [Google Scholar]
- 37.Davignon J.L., Boyer J.F., Jamard B., Nigon D., Constantin A., Cantagrel A. Maintenance of cytomegalovirus-specific CD4pos T-cell response in rheumatoid arthritis patients receiving anti-tumor necrosis factor treatments. Arthritis Res. Ther. 2010;12(4):R142. doi: 10.1186/ar3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murayama T., Jisaki F., Ayata M., et al. Cytomegalovirus genomes demonstrated by polymerase chain reaction in synovial fluid from rheumatoid arthritis patients. Clin. Exp. Rheumatol. 1992;10(2):161–164. [PubMed] [Google Scholar]
- 39.Tamm A., Ziegler T., Lautenschlager I., et al. Detection of cytomegalovirus DNA in cells from synovial fluid and peripheral blood of patients with early rheumatoid arthritis. J. Rheumatol. 1993;20(9):1489–1493. [PubMed] [Google Scholar]
- 40.Scotet E., Peyrat M.A., Saulquin X., et al. Frequent enrichment for CD8 T cells reactive against common herpes viruses in chronic inflammatory lesions: towards a reassessment of the physiopathological significance of T cell clonal expansions found in autoimmune inflammatory processes. Eur. J. Immunol. 1999;29(3):973–985. doi: 10.1002/(SICI)1521-4141(199903)29:03<973::AID-IMMU973>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 41.Edinger J.W., Bonneville M., Scotet E., Houssaint E., Schumacher H.R., Posnett D.N. EBV gene expression not altered in rheumatoid synovia despite the presence of EBV antigen-specific T cell clones. J. Immunol. 1999;162(6):3694–3701. [PubMed] [Google Scholar]
- 42.Stahl H.D., Hubner B., Seidl B., et al. Detection of multiple viral DNA species in synovial tissue and fluid of patients with early arthritis. Ann. Rheum. Dis. 2000;59(5):342–346. doi: 10.1136/ard.59.5.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mehraein Y., Lennerz C., Ehlhardt S., Remberger K., Ojak A., Zang K.D. Latent Epstein-Barr virus (EBV) infection and cytomegalovirus (CMV) infection in synovial tissue of autoimmune chronic arthritis determined by RNA- and DNA-in situ hybridization. Mod. Pathol. 2004;17(7):781–789. doi: 10.1038/modpathol.3800119. [DOI] [PubMed] [Google Scholar]
- 44.Moura R.A., Weinmann P., Pereira P.A., et al. Alterations on peripheral blood B-cell subpopulations in very early arthritis patients. Rheumatology (Oxford) 2010;49(6):1082–1092. doi: 10.1093/rheumatology/keq029. [DOI] [PubMed] [Google Scholar]
- 45.Moura R.A., Cascao R., Perpetuo I., et al. Cytokine pattern in very early rheumatoid arthritis favours B-cell activation and survival. Rheumatology (Oxford) 2011;50(2):278–282. doi: 10.1093/rheumatology/keq338. [DOI] [PubMed] [Google Scholar]
- 46.Bosello S., Youinou P., Daridon C., et al. Concentrations of BAFF correlate with autoantibody levels, clinical disease activity, and response to treatment in early rheumatoid arthritis. J. Rheumatol. 2008;35(7):1256–1264. [PubMed] [Google Scholar]
- 47.Gottenberg J.E., Miceli-Richard C., Ducot B., Goupille P., Combe B., Mariette X. Markers of B-lymphocyte activation are elevated in patients with early rheumatoid arthritis and correlated with disease activity in the ESPOIR cohort. Arthritis Res. Ther. 2009;11(4):R114. doi: 10.1186/ar2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morbach H., Eichhorn E.M., Liese J.G., Girschick H.J. Reference values for B cell subpopulations from infancy to adulthood. Clin. Exp. Immunol. 2010;162(2):271–279. doi: 10.1111/j.1365-2249.2010.04206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van der Geest K.S., Abdulahad W.H., Chalan P., et al. Disturbed B cell homeostasis in newly diagnosed giant cell arteritis and polymyalgia rheumatica. Arthritis Rheumatol. 2014;66(7):1927–1938. doi: 10.1002/art.38625. [DOI] [PubMed] [Google Scholar]
- 50.Rubtsov A.V., Rubtsova K., Fischer A., et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood. 2011;118(5):1305–1315. doi: 10.1182/blood-2011-01-331462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Duggal N.A., Upton J., Phillips A.C., Sapey E., Lord J.M. An age-related numerical and functional deficit in CD19(+) CD24(hi) CD38(hi) B cells is associated with an increase in systemic autoimmunity. Aging Cell. 2013;12(5):873–881. doi: 10.1111/acel.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gayoso I., Sanchez-Correa B., Campos C., et al. Immunosenescence of human natural killer cells. J. Innate Immun. 2011;3(4):337–343. doi: 10.1159/000328005. [DOI] [PubMed] [Google Scholar]
- 53.Mariani E., Meneghetti A., Formentini I., et al. Telomere length and telomerase activity: effect of ageing on human NK cells. Mech. Ageing Dev. 2003;124(4):403–408. doi: 10.1016/s0047-6374(03)00015-0. [DOI] [PubMed] [Google Scholar]
- 54.Mariani E., Meneghetti A., Formentini I., et al. Different rates of telomere shortening and telomerase activity reduction in CD8 T and CD16 NK lymphocytes with ageing. Exp. Gerontol. 2003;38(6):653–659. doi: 10.1016/s0531-5565(03)00058-5. [DOI] [PubMed] [Google Scholar]
- 55.Ouyang Q., Baerlocher G., Vulto I., Lansdorp P.M. Telomere length in human natural killer cell subsets. Ann. N. Y. Acad. Sci. 2007;1106:240–252. doi: 10.1196/annals.1392.001. [DOI] [PubMed] [Google Scholar]
- 56.Borrego F., Alonso M.C., Galiani M.D., et al. NK phenotypic markers and IL2 response in NK cells from elderly people. Exp. Gerontol. 1999;34(2):253–265. doi: 10.1016/s0531-5565(98)00076-x. [DOI] [PubMed] [Google Scholar]
- 57.Krishnaraj R. Senescence and cytokines modulate the NK cell expression. Mech. Ageing Dev. 1997;96(1-3):89–101. doi: 10.1016/s0047-6374(97)00045-6. [DOI] [PubMed] [Google Scholar]
- 58.Chidrawar S.M., Khan N., Chan Y.L., Nayak L., Moss P.A. Ageing is associated with a decline in peripheral blood CD56bright NK cells. Immun. Ageing. 2006;3:10. doi: 10.1186/1742-4933-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Solana R, Mariani E. NK. Vaccine. 2000;18(16):1613–1620. doi: 10.1016/s0264-410x(99)00495-8. [DOI] [PubMed] [Google Scholar]
- 60.Shibatomi K., Ida H., Yamasaki S., et al. A novel role for interleukin-18 in human natural killer cell death: high serum levels and low natural killer cell numbers in patients with systemic autoimmune diseases. Arthritis Rheum. 2001;44(4):884–892. doi: 10.1002/1529-0131(200104)44:4<884::AID-ANR145>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 61.Aggarwal A., Sharma A., Bhatnagar A. Role of cytolytic impairment of natural killer and natural killer T-cell populations in rheumatoid arthritis. Clin. Rheumatol. 2014;33(8):1067–1078. doi: 10.1007/s10067-014-2641-z. [DOI] [PubMed] [Google Scholar]
- 62.Conigliaro P., Triggianese P., Perricone C., et al. Restoration of peripheral blood natural killer and B cell levels in patients affected by rheumatoid and psoriatic arthritis during etanercept treatment. Clin. Exp. Immunol. 2014;177(1):234–243. doi: 10.1111/cei.12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perez-Mancera P.A., Young A.R., Narita M. Inside and out: the activities of senescence in cancer. Nat. Rev. Cancer. 2014;14(8):547–558. doi: 10.1038/nrc3773. [DOI] [PubMed] [Google Scholar]
- 64.Sagiv A., Krizhanovsky V. Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology. 2013;14(6):617–628. doi: 10.1007/s10522-013-9473-0. [DOI] [PubMed] [Google Scholar]
- 65.Ziegler-Heitbrock H.W., Fingerle G., Strobel M., et al. The novel subset of CD14+/CD16+ blood monocytes exhibits features of tissue macrophages. Eur. J. Immunol. 1993;23(9):2053–2058. doi: 10.1002/eji.1830230902. [DOI] [PubMed] [Google Scholar]
- 66.Ziegler-Heitbrock L. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J. Leukoc. Biol. 2007;81(3):584–592. doi: 10.1189/jlb.0806510. [DOI] [PubMed] [Google Scholar]
- 67.Merino A., Buendia P., Martin-Malo A., Aljama P., Ramirez R., Carracedo J. Senescent CD14+CD16+ monocytes exhibit proinflammatory and proatherosclerotic activity. J. Immunol. 2011;186(3):1809–1815. doi: 10.4049/jimmunol.1001866. [DOI] [PubMed] [Google Scholar]
- 68.Debacq-Chainiaux F., Erusalimsky J.D., Campisi J., Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009;4(12):1798–1806. doi: 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]
- 69.Kawanaka N., Yamamura M., Aita T., et al. CD14+,CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheum. 2002;46(10):2578–2586. doi: 10.1002/art.10545. [DOI] [PubMed] [Google Scholar]
- 70.Seidler S, Zimmermann HW, Bartneck M, Trautwein C, Tacke F. Age-dependent alterations of monocyte subsets and monocyterelated chemokine pathways in healthy adults. . BMC Immunol. 2010;11 doi: 10.1186/1471-2172-11-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krasselt M., Baerwald C., Wagner U., Rossol M. CD56+ monocytes have a dysregulated cytokine response to lipopolysaccharide and accumulate in rheumatoid arthritis and immunosenescence. Arthritis Res. Ther. 2013;15(5):R139. doi: 10.1186/ar4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Naylor R.M., Baker D.J., van Deursen J.M. Senescent cells: a novel therapeutic target for aging and age-related diseases. Clin. Pharmacol. Ther. 2013;93(1):105–116. doi: 10.1038/clpt.2012.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat. Rev. Cancer. 2008;8(7):512–522. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- 74.Kuilman T., Michaloglou C., Mooi W.J., Peeper D.S. The essence of senescence. Genes Dev. 2010;24(22):2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bashir S., Harris G., Denman M.A., Blake D.R., Winyard P.G. Oxidative DNA damage and cellular sensitivity to oxidative stress in human autoimmune diseases. Ann. Rheum. Dis. 1993;52(9):659–666. doi: 10.1136/ard.52.9.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bhusate L.L., Herbert K.E., Scott D.L., Perrett D. Increased DNA strand breaks in mononuclear cells from patients with rheumatoid arthritis. Ann. Rheum. Dis. 1992;51(1):8–12. doi: 10.1136/ard.51.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shao L., Fujii H., Colmegna I., Oishi H., Goronzy J.J., Weyand C.M. Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. J. Exp. Med. 2009;206(6):1435–1449. doi: 10.1084/jem.20082251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shao L., Goronzy J.J., Weyand C.M. DNA-dependent protein kinase catalytic subunit mediates T-cell loss in rheumatoid arthritis. EMBO Mol. Med. 2010;2(10):415–427. doi: 10.1002/emmm.201000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McConnell J.R., Crockard A.D., Cairns A.P., Bell A.L. Neutrophils from systemic lupus erythematosus patients demonstrate increased nuclear DNA damage. Clin. Exp. Rheumatol. 2002;20(5):653–660. [PubMed] [Google Scholar]
- 80.Jikimoto T., Nishikubo Y., Koshiba M., et al. Thioredoxin as a biomarker for oxidative stress in patients with rheumatoid arthritis. Mol. Immunol. 2002;38(10):765–772. doi: 10.1016/s0161-5890(01)00113-4. [DOI] [PubMed] [Google Scholar]
- 81.Ogawa M, Matsuda T, Ogata A, et al. DNA damage in rheumatoid arthritis: an age-dependent increase in the lipid peroxidationderived DNA adduct, heptanone-etheno-2'-deoxycytidine. . Autoimmune Dis . 2013; 2013 :183–487. doi: 10.1155/2013/183487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Harris G., Asbery L., Lawley P.D., Denman A.M., Hylton W. Defective repair of 0(6)-methylguanine in autoimmune diseases. Lancet. 1982;2(8305):952–956. doi: 10.1016/s0140-6736(82)90159-3. [DOI] [PubMed] [Google Scholar]
- 83.Lawley P.D., Topper R., Denman A.M., Hylton W., Hill I.D., Harris G. Increased sensitivity of lymphocytes from patients with systemic autoimmune diseases to DNA alkylation by the methylating carcinogen N-methyl-N-nitrosourea. Ann. Rheum. Dis. 1988;47(6):445–451. doi: 10.1136/ard.47.6.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Colaco C.B., Harris G., Lawley P.D., Lydyard P.M., Roitt I.M. Deficient repair of O6-methylguanine in lymphocytes from rheumatoid arthritis families may be an acquired defect. Clin. Exp. Immunol. 1988;72(1):15–19. [PMC free article] [PubMed] [Google Scholar]
- 85.Djojosubroto M.W., Choi Y.S., Lee H.W., Rudolph K.L. Telomeres and telomerase in aging, regeneration and cancer. Mol. Cells. 2003;15(2):164–175. [PubMed] [Google Scholar]
- 86.Beliveau A., Bassett E., Lo A.T., et al. P53-Dependent Integration of Telomere and Growth Factor Deprivation Signals. Proc. Natl. Acad. Sci. USA. 2007;104(11):4431–4436. doi: 10.1073/pnas.0700260104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mason P.J., Perdigones N. Telomere biology and translational research. Transl. Res. 2013;162(6):333–342. doi: 10.1016/j.trsl.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Herbig U., Jobling W.A., Chen B.P., Chen D.J., Sedivy J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell. 2004;14(4):501–513. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 89.Hodes R.J., Hathcock K.S., Weng N.P. Telomeres in T and B cells. Nat. Rev. Immunol. 2002;2(9):699–706. doi: 10.1038/nri890. [DOI] [PubMed] [Google Scholar]
- 90.Pedersen-Lane J.H., Zurier R.B., Lawrence D.A. Analysis of the thiol status of peripheral blood leukocytes in rheumatoid arthritis patients. J. Leukoc. Biol. 2007;81(4):934–941. doi: 10.1189/jlb.0806533. [DOI] [PubMed] [Google Scholar]
- 91.Koetz K., Bryl E., Spickschen K., O'Fallon W.M., Goronzy J.J., Weyand C.M. T cell homeostasis in patients with rheumatoid arthritis. Proc. Natl. Acad. Sci. USA. 2000;97(16):9203–9208. doi: 10.1073/pnas.97.16.9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yudoh K., Matsuno H., Nezuka T., Kimura T. Different mechanisms of synovial hyperplasia in rheumatoid arthritis and pigmented villonodular synovitis: the role of telomerase activity in synovial proliferation. Arthritis Rheum. 1999;42(4):669–677. doi: 10.1002/1529-0131(199904)42:4<669::AID-ANR9>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 93.Thewissen M., Linsen L., Geusens P., Raus J., Stinissen P. Impaired activation-induced telomerase activity in PBMC of early but not chronic rheumatoid arthritis patients. Immunol. Lett. 2005;100(2):205–210. doi: 10.1016/j.imlet.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 94.Feil R., Fraga M.F. Epigenetics and the environment: emerging patterns and implications. Nat. Rev. Genet. 2012;13(2):97–109. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- 95.Meda F., Folci M., Baccarelli A., Selmi C. The epigenetics of autoimmunity. Cell. Mol. Immunol. 2011;8(3):226–236. doi: 10.1038/cmi.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ballestar E. Epigenetic alterations in autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2011;7(5):263–271. doi: 10.1038/nrrheum.2011.16. [DOI] [PubMed] [Google Scholar]
- 97.Zufferey F., Williams F.M., Spector T.D. Epigenetics and methylation in the rheumatic diseases. Semin. Arthritis Rheum. 2014;43(5):692–700. doi: 10.1016/j.semarthrit.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 98.Firestein G.S. The disease formerly known as rheumatoid arthritis. Arthritis Res. Ther. 2014;16(3):114. doi: 10.1186/ar4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.de la Rica L., Urquiza J.M., Gomez-Cabrero D., et al. Identification of novel markers in rheumatoid arthritis through integrated analysis of DNA methylation and microRNA expression. J. Autoimmun. 2013;41:6–16. doi: 10.1016/j.jaut.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 100.Karouzakis E., Gay R.E., Michel B.A., Gay S., Neidhart M. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2009;60(12):3613–3622. doi: 10.1002/art.25018. [DOI] [PubMed] [Google Scholar]
- 101.Neidhart M., Rethage J., Kuchen S., et al. Retrotransposable L1 elements expressed in rheumatoid arthritis synovial tissue: association with genomic DNA hypomethylation and influence on gene expression. Arthritis Rheum. 2000;43(12):2634–2647. doi: 10.1002/1529-0131(200012)43:12<2634::AID-ANR3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 102.Takami N., Osawa K., Miura Y., et al. Hypermethylated promoter region of DR3, the death receptor 3 gene, in rheumatoid arthritis synovial cells. Arthritis Rheum. 2006;54(3):779–787. doi: 10.1002/art.21637. [DOI] [PubMed] [Google Scholar]
- 103.Grabiec A.M., Reedquist K.A. Histone deacetylases in RA: epigenetics and epiphenomena. Arthritis Res. Ther. 2010;12(5):142. doi: 10.1186/ar3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Huber L.C., Brock M., Hemmatazad H., et al. Histone deacetylase/acetylase activity in total synovial tissue derived from rheumatoid arthritis and osteoarthritis patients. Arthritis Rheum. 2007;56(4):1087–1093. doi: 10.1002/art.22512. [DOI] [PubMed] [Google Scholar]
- 105.Wendling D., Vidon C., Abbas W., Guillot X., Toussirot E., Herbein G. Sirt1 activity in peripheral blood mononuclear cells from patients with rheumatoid arthritis. Joint Bone Spine. 2014;81(5):462–463. doi: 10.1016/j.jbspin.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 106.Maciejewska-Rodrigues H., Karouzakis E., Strietholt S., et al. Epigenetics and rheumatoid arthritis: the role of SENP1 in the regulation of MMP-1 expression. J. Autoimmun. 2010;35(1):15–22. doi: 10.1016/j.jaut.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 107.Meinecke I., Cinski A., Baier A., et al. Modification of nuclear PML protein by SUMO-1 regulates Fas-induced apoptosis in rheumatoid arthritis synovial fibroblasts. Proc. Natl. Acad. Sci. USA. 2007;104(12):5073–5078. doi: 10.1073/pnas.0608773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stanczyk J., Pedrioli D.M., Brentano F., et al. Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 2008;58(4):1001–1009. doi: 10.1002/art.23386. [DOI] [PubMed] [Google Scholar]
- 109.Grabiec A.M., Reedquist K.A. The ascent of acetylation in the epigenetics of rheumatoid arthritis. Nat. Rev. Rheumatol. 2013;9(5):311–318. doi: 10.1038/nrrheum.2013.17. [DOI] [PubMed] [Google Scholar]
- 110.Kawabata T., Nishida K., Takasugi K., et al. Increased activity and expression of histone deacetylase 1 in relation to tumor necrosis factor-alpha in synovial tissue of rheumatoid arthritis. Arthritis Res. Ther. 2010;12(4):R133. doi: 10.1186/ar3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Niederer F., Ospelt C., Brentano F., et al. SIRT1 overexpression in the rheumatoid arthritis synovium contributes to proinflammatory cytokine production and apoptosis resistance. Ann. Rheum. Dis. 2011;70(10):1866–1873. doi: 10.1136/ard.2010.148957. [DOI] [PubMed] [Google Scholar]
- 112.Gillespie J., Savic S., Wong C., et al. Histone deacetylases are dysregulated in rheumatoid arthritis and a novel histone deacetylase 3-selective inhibitor reduces interleukin-6 production by peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Rheum. 2012;64(2):418–422. doi: 10.1002/art.33382. [DOI] [PubMed] [Google Scholar]
- 113.Richardson B. DNA methylation and autoimmune disease. Clin. Immunol. 2003;109(1):72–79. doi: 10.1016/s1521-6616(03)00206-7. [DOI] [PubMed] [Google Scholar]
- 114.Richardson B. Impact of aging on DNA methylation. Ageing Res. Rev. 2003;2(3):245–261. doi: 10.1016/s1568-1637(03)00010-2. [DOI] [PubMed] [Google Scholar]
- 115.Deaton A.M., Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25(10):1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Attwood J.T., Yung R.L., Richardson B.C. DNA methylation and the regulation of gene transcription. Cell. Mol. Life Sci. 2002;59(2):241–257. doi: 10.1007/s00018-002-8420-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lopatina N., Haskell J.F., Andrews L.G., Poole J.C., Saldanha S., Tollefsbol T. Differential maintenance and de novo methylating activity by three DNA methyltransferases in aging and immortalized fibroblasts. J. Cell. Biochem. 2002;84(2):324–334. doi: 10.1002/jcb.10015. [DOI] [PubMed] [Google Scholar]
- 118.So A.Y., Jung J.W., Lee S., Kim H.S., Kang K.S. DNA methyltransferase controls stem cell aging by regulating BMI1 and EZH2 through microRNAs. PLoS One. 2011;6(5):e19503. doi: 10.1371/journal.pone.0019503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nile C.J., Read R.C., Akil M., Duff G.W., Wilson A.G. Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum. 2008;58(9):2686–2693. doi: 10.1002/art.23758. [DOI] [PubMed] [Google Scholar]
- 120.Trenkmann M., Brock M., Gay R.E., et al. Expression and function of EZH2 in synovial fibroblasts: epigenetic repression of the Wnt inhibitor SFRP1 in rheumatoid arthritis. Ann. Rheum. Dis. 2011;70(8):1482–1488. doi: 10.1136/ard.2010.143040. [DOI] [PubMed] [Google Scholar]
- 121.Miao C.G., Yang Y.Y., He X., et al. New advances of microRNAs in the pathogenesis of rheumatoid arthritis, with a focus on the crosstalk between DNA methylation and the microRNA machinery. Cell. Signal. 2013;25(5):1118–1125. doi: 10.1016/j.cellsig.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 122.Harries L.W. MicroRNAs as Mediators of the Ageing Process. Genes (Basel) 2014;5(3):656–670. doi: 10.3390/genes5030656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Feng Z.T., Li J., Ren J., Lv Z. Expression of miR-146a and miR-16 in peripheral blood mononuclear cells of patients with rheumatoid arthritis and their correlation to the disease activity. Nan Fang Yi Ke Da Xue Xue Bao. 2011;31(2):320–323. [PubMed] [Google Scholar]
- 124.Nakamachi Y., Kawano S., Takenokuchi M., et al. MicroRNA-124a is a key regulator of proliferation and monocyte chemoattractant protein 1 secretion in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Rheum. 2009;60(5):1294–1304. doi: 10.1002/art.24475. [DOI] [PubMed] [Google Scholar]
- 125.Harries L.W., Hernandez D., Henley W., et al. Human aging is characterized by focused changes in gene expression and deregulation of alternative splicing. Aging Cell. 2011;10(5):868–878. doi: 10.1111/j.1474-9726.2011.00726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.van Kouwenhove M., Kedde M., Agami R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat. Rev. Cancer. 2011;11(9):644–656. doi: 10.1038/nrc3107. [DOI] [PubMed] [Google Scholar]
- 127.Sugihara M., Tsutsumi A., Suzuki E., et al. Effects of infliximab therapy on gene expression levels of tumor necrosis factor alpha, tristetraprolin, T cell intracellular antigen 1, and Hu antigen R in patients with rheumatoid arthritis. Arthritis Rheum. 2007;56(7):2160–2169. doi: 10.1002/art.22724. [DOI] [PubMed] [Google Scholar]
- 128.Edwards C.J., Feldman J.L., Beech J., et al. Molecular profile of peripheral blood mononuclear cells from patients with rheumatoid arthritis. Mol. Med. 2007;13(1-2):40–58. doi: 10.2119/2006-000056.Edwards. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Blass S., Union A., Raymackers J., et al. The stress protein BiP is overexpressed and is a major B and T cell target in rheumatoid arthritis. Arthritis Rheum. 2001;44(4):761–771. doi: 10.1002/1529-0131(200104)44:4<761::AID-ANR132>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 130.Corrigall V.M., Bodman-Smith M.D., Fife M.S., et al. The human endoplasmic reticulum molecular chaperone BiP is an autoantigen for rheumatoid arthritis and prevents the induction of experimental arthritis. J. Immunol. 2001;166(3):1492–1498. doi: 10.4049/jimmunol.166.3.1492. [DOI] [PubMed] [Google Scholar]
- 131.Martin C.A., Carsons S.E., Kowalewski R., Bernstein D., Valentino M., Santiago-Schwarz F. Aberrant extracellular and dendritic cell (DC) surface expression of heat shock protein (hsp)70 in the rheumatoid joint: possible mechanisms of hsp/DC-mediated cross-priming. J. Immunol. 2003;171(11):5736–5742. doi: 10.4049/jimmunol.171.11.5736. [DOI] [PubMed] [Google Scholar]
- 132.Kurzik-Dumke U., Schick C., Rzepka R., Melchers I. Overexpression of human homologs of the bacterial DnaJ chaperone in the synovial tissue of patients with rheumatoid arthritis. Arthritis Rheum. 1999;42(2):210–220. doi: 10.1002/1529-0131(199902)42:2<210::AID-ANR2>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 133.Yokota S.I., Hirata D., Minota S., et al. Autoantibodies against chaperonin CCT in human sera with rheumatic autoimmune diseases: comparison with antibodies against other Hsp60 family proteins. Cell Stress Chaperones. 2000;5(4):337–346. doi: 10.1379/1466-1268(2000)005<0337:aaccih>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tsoulfa G., Rook G.A., Bahr G.M., et al. Elevated IgG antibody levels to the mycobacterial 65-kDa heat shock protein are characteristic of patients with rheumatoid arthritis. Scand. J. Immunol. 1989;30(5):519–527. doi: 10.1111/j.1365-3083.1989.tb02459.x. [DOI] [PubMed] [Google Scholar]
- 135.Bodman-Smith M.D., Corrigall V.M., Berglin E., et al. Antibody response to the human stress protein BiP in rheumatoid arthritis. Rheumatology (Oxford) 2004;43(10):1283–1287. doi: 10.1093/rheumatology/keh312. [DOI] [PubMed] [Google Scholar]
- 136.Goeb V., Thomas-L'Otellier M., Daveau R., et al. Candidate autoantigens identified by mass spectrometry in early rheumatoid arthritis are chaperones and citrullinated glycolytic enzymes. Arthritis Res. Ther. 2009;11(2):R38. doi: 10.1186/ar2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Auger I., Roudier J. Interaction between HSP73 and HLA-DRB1*0401: implications for the development of rheumatoid arthritis. Immunol. Res. 2005;31(3):261–266. doi: 10.1385/IR:31:3:261. [DOI] [PubMed] [Google Scholar]
- 138.Bodman-Smith M.D., Corrigall V.M., Kemeny D.M., Panayi G.S. BiP, a putative autoantigen in rheumatoid arthritis, stimulates IL-10-producing CD8-positive T cells from normal individuals. Rheumatology (Oxford) 2003;42(5):637–644. doi: 10.1093/rheumatology/keg204. [DOI] [PubMed] [Google Scholar]
- 139.Barbera A., Lorenzo N., Garrido G., et al. APL-1, an altered peptide ligand derived from human heat-shock protein 60, selectively induces apoptosis in activated CD4+ CD25+ T cells from peripheral blood of rheumatoid arthritis patients. Int. Immunopharmacol. 2013;17(4):1075–1083. doi: 10.1016/j.intimp.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 140.Cuervo A.M. Autophagy and aging: keeping that old broom working. Trends Genet. 2008;24(12):604–612. doi: 10.1016/j.tig.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Vicencio J.M., Galluzzi L., Tajeddine N., et al. Senescence, apoptosis or autophagy? When a damaged cell must decide its path--a mini-review. Gerontology. 2008;54(2):92–99. doi: 10.1159/000129697. [DOI] [PubMed] [Google Scholar]
- 142.Xu K., Xu P., Yao J.F., Zhang Y.G., Hou W.K., Lu S.M. Reduced apoptosis correlates with enhanced autophagy in synovial tissues of rheumatoid arthritis. Inflamm. Res. 2013;62(2):229–237. doi: 10.1007/s00011-012-0572-1. [DOI] [PubMed] [Google Scholar]
- 143.Shin Y.J., Han S.H., Kim D.S., et al. Autophagy induction and CHOP under-expression promotes survival of fibroblasts from rheumatoid arthritis patients under endoplasmic reticulum stress. Arthritis Res. Ther. 2010;12(1):R19. doi: 10.1186/ar2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kato M., Ospelt C., Gay R.E., Gay S., Klein K. Dual role of autophagy in stress-induced cell death in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol. 2014;66(1):40–48. doi: 10.1002/art.38190. [DOI] [PubMed] [Google Scholar]
- 145.Connor A.M., Mahomed N., Gandhi R., Keystone E.C., Berger S.A. TNFalpha modulates protein degradation pathways in rheumatoid arthritis synovial fibroblasts. Arthritis Res. Ther. 2012;14(2):R62. doi: 10.1186/ar3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lin N.Y., Stefanica A., Distler J.H. Autophagy: a key pathway of TNF-induced inflammatory bone loss. Autophagy. 2013;9(8):1253–1255. doi: 10.4161/auto.25467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Izumi T., Fujii R., Izumi T., et al. Activation of synoviolin promoter in rheumatoid synovial cells by a novel transcription complex of interleukin enhancer binding factor 3 and GA binding protein alpha. Arthritis Rheum. 2009;60(1):63–72. doi: 10.1002/art.24178. [DOI] [PubMed] [Google Scholar]
- 148.Kawaida R., Yamada R., Kobayashi K., et al. CUL1, a component of E3 ubiquitin ligase, alters lymphocyte signal transduction with possible effect on rheumatoid arthritis. Genes Immun. 2005;6(3):194–202. doi: 10.1038/sj.gene.6364177. [DOI] [PubMed] [Google Scholar]
- 149.Negi S., Kumar A., Thelma B.K., Juyal R.C. Association of Cullin1 haplotype variants with rheumatoid arthritis and response to methotrexate. Pharmacogenet. Genomics. 2011;21(9):590–593. doi: 10.1097/FPC.0b013e3283492af7. [DOI] [PubMed] [Google Scholar]
- 150.Amano T., Yamasaki S., Yagishita N., et al. Synoviolin/Hrd1, an E3 ubiquitin ligase, as a novel pathogenic factor for arthropathy. Genes Dev. 2003;17(19):2436–2449. doi: 10.1101/gad.1096603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Toh M.L., Marotte H., Blond J.L., et al. Overexpression of synoviolin in peripheral blood and synoviocytes from rheumatoid arthritis patients and continued elevation in nonresponders to infliximab treatment. Arthritis Rheum. 2006;54(7):2109–2118. doi: 10.1002/art.21926. [DOI] [PubMed] [Google Scholar]
- 152.Yamasaki S., Yagishita N., Tsuchimochi K., Nishioka K., Nakajima T. Rheumatoid arthritis as a hyper-endoplasmic-reticulum-associated degradation disease. Arthritis Res. Ther. 2005;7(5):181–186. doi: 10.1186/ar1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Takada K., Nasu H., Hibi N., et al. Serum concentrations of free ubiquitin and multiubiquitin chains. Clin. Chem. 1997;43(7):1188–1195. [PubMed] [Google Scholar]
- 154.Yang Z., Fujii H., Mohan S.V., Goronzy J.J., Weyand C.M. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J. Exp. Med. 2013;210(10):2119–2134. doi: 10.1084/jem.20130252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Denko C.W., Malemud C.J. Role of the growth hormone/insulin-like growth factor-1 paracrine axis in rheumatic diseases. Semin. Arthritis Rheum. 2005;35(1):24–34. doi: 10.1016/j.semarthrit.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 156.Neidel J. Changes in systemic levels of insulin-like growth factors and their binding proteins in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2001;19(1):81–84. [PubMed] [Google Scholar]
- 157.Sherlock M., Toogood A.A. Aging and the growth hormone/insulin like growth factor-I axis. Pituitary. 2007;10(2):189–203. doi: 10.1007/s11102-007-0039-5. [DOI] [PubMed] [Google Scholar]
- 158.Matsumoto T., Tsurumoto T. Inappropriate serum levels of IGF-I and IGFBP-3 in patients with rheumatoid arthritis. Rheumatology (Oxford) 2002;41(3):352–353. doi: 10.1093/rheumatology/41.3.352. [DOI] [PubMed] [Google Scholar]
- 159.Ishigami S., Nakajima A., Tanno M., Matsuzaki T., Suzuki H., Yoshino S. Effects of mirthful laughter on growth hormone, IGF-1 and substance P in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2005;23(5):651–657. [PubMed] [Google Scholar]
- 160.Blackman M.R., Muniyappa R., Wilson M., et al. Diurnal secretion of growth hormone, cortisol, and dehydroepiandrosterone in pre- and perimenopausal women with active rheumatoid arthritis: a pilot case-control study. Arthritis Res. Ther. 2007;9(4):R73. doi: 10.1186/ar2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dhaunsi G.S., Uppal S.S., Haider M.Z. Insulin-like growth factor-1 gene polymorphism in rheumatoid arthritis patients. Scand. J. Rheumatol. 2012;41(6):421–425. doi: 10.3109/03009742.2012.691177. [DOI] [PubMed] [Google Scholar]
- 162.Lee H.S., Woo S.J., Koh H.W., et al. Regulation of apoptosis and inflammatory responses by IGFBP-3 in fibroblast-like synoviocytes and experimental animal models of rheumatoid arthritis. Arthritis Rheum. 2013 doi: 10.1002/art.38303. [DOI] [PubMed] [Google Scholar]
- 163.Fraser D.A., Thoen J., Reseland J.E., Forre O., Kjeldsen-Kragh J. Decreased CD4+ lymphocyte activation and increased interleukin-4 production in peripheral blood of rheumatoid arthritis patients after acute starvation. Clin. Rheumatol. 1999;18(5):394–401. doi: 10.1007/s100670050125. [DOI] [PubMed] [Google Scholar]
- 164.Toussirot E., Nguyen N.U., Dumoulin G., Aubin F., Cedoz J.P., Wendling D. Relationship between growth hormone-IGF-I-IGFBP-3 axis and serum leptin levels with bone mass and body composition in patients with rheumatoid arthritis. Rheumatology (Oxford) 2005;44(1):120–125. doi: 10.1093/rheumatology/keh421. [DOI] [PubMed] [Google Scholar]
- 165.Suzuki S., Morimoto S., Fujishiro M., et al. Inhibition of the insulin-like growth factor system is a potential therapy for rheumatoid arthritis. Autoimmunity. 2015;48(4):251–258. doi: 10.3109/08916934.2014.976631. [DOI] [PubMed] [Google Scholar]
- 166.Lee S.D., Chen L.M., Kuo W.W., et al. Serum insulin-like growth factor-axis and matrix metalloproteinases in patients with rheumatic arthritis or rheumatic heart disease. Clin. Chim. Acta. 2006;367(1-2):62–68. doi: 10.1016/j.cca.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 167.Tavera C., Abribat T., Reboul P., et al. IGF and IGF-binding protein system in the synovial fluid of osteoarthritic and rheumatoid arthritic patients. Osteoarthritis Cartilage. 1996;4(4):263–274. doi: 10.1016/s1063-4584(05)80104-9. [DOI] [PubMed] [Google Scholar]
- 168.Neidel J., Blum W.F., Schaeffer H.J., et al. Elevated levels of insulin-like growth factor (IGF) binding protein-3 in rheumatoid arthritis synovial fluid inhibit stimulation by IGF-I of articular chondrocyte proteoglycan synthesis. Rheumatol. Int. 1997;17(1):29–37. doi: 10.1007/pl00006847. [DOI] [PubMed] [Google Scholar]
- 169.Matsumoto T., Yamashita S., Rosenfeld R.G. Increased levels of IGF-I and IGFBP-3 in synovial fluids of patients with rheumatoid arthritis. Endocr. J. 1998;45(Suppl.):S141–S144. doi: 10.1507/endocrj.45.suppl_s141. [DOI] [PubMed] [Google Scholar]
- 170.Templ E., Koeller M., Riedl M., Wagner O., Graninger W., Luger A. Anterior pituitary function in patients with newly diagnosed rheumatoid arthritis. Br. J. Rheumatol. 1996;35(4):350–356. doi: 10.1093/rheumatology/35.4.350. [DOI] [PubMed] [Google Scholar]
- 171.Volin M.V., Huynh N., Klosowska K., Chong K.K., Woods J.M. Fractalkine is a novel chemoattractant for rheumatoid arthritis fibroblast-like synoviocyte signaling through MAP kinases and Akt. Arthritis Rheum. 2007;56(8):2512–2522. doi: 10.1002/art.22806. [DOI] [PubMed] [Google Scholar]
- 172.Garcia S., Liz M., Gomez-Reino J.J., Conde C. Akt activity protects rheumatoid synovial fibroblasts from Fas-induced apoptosis by inhibition of Bid cleavage. Arthritis Res. Ther. 2010;12(1):R33. doi: 10.1186/ar2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Grabiec A.M., Angiolilli C., Hartkamp L.M., van Baarsen L.G., Tak P.P., Reedquist K.A. JNK-dependent downregulation of FoxO1 is required to promote the survival of fibroblast-like synoviocytes in rheumatoid arthritis. Ann. Rheum. Dis. 2014 doi: 10.1136/annrheumdis-2013-203610. [Epub ahead of Print]. [DOI] [PubMed] [Google Scholar]
- 174.Ludikhuize J., de Launay D., Groot D., et al. Inhibition of forkhead box class O family member transcription factors in rheumatoid synovial tissue. Arthritis Rheum. 2007;56(7):2180–2191. doi: 10.1002/art.22653. [DOI] [PubMed] [Google Scholar]
- 175.Malemud C.J. Intracellular signaling pathways in rheumatoid arthritis. J. Clin. Cell. Immunol. 2013;4:160. doi: 10.4172/2155-9899.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Saxena A., Raychaudhuri S.K., Raychaudhuri S.P. Interleukin-17-induced proliferation of fibroblast-like synovial cells is mTOR dependent. Arthritis Rheum. 2011;63(5):1465–1466. doi: 10.1002/art.30278. [DOI] [PubMed] [Google Scholar]
- 177.Da Sylva T.R., Connor A., Mburu Y., Keystone E., Wu G.E. Somatic mutations in the mitochondria of rheumatoid arthritis synoviocytes. Arthritis Res. Ther. 2005;7(4):R844–R851. doi: 10.1186/ar1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Harty L.C., Biniecka M., O'Sullivan J., et al. Mitochondrial mutagenesis correlates with the local inflammatory environment in arthritis. Ann. Rheum. Dis. 2012;71(4):582–588. doi: 10.1136/annrheumdis-2011-200245. [DOI] [PubMed] [Google Scholar]
- 179.Valcarcel-Ares M.N., Riveiro-Naveira R.R., Vaamonde-Garcia C., et al. Mitochondrial dysfunction promotes and aggravates the inflammatory response in normal human synoviocytes. Rheumatology (Oxford) 2014;53(7):1332–1343. doi: 10.1093/rheumatology/keu016. [DOI] [PubMed] [Google Scholar]
- 180.Hajizadeh S., DeGroot J., TeKoppele J.M., Tarkowski A., Collins L.V. Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res. Ther. 2003;5(5):R234–R240. doi: 10.1186/ar787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Collins L.V., Hajizadeh S., Holme E., Jonsson I.M., Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc. Biol. 2004;75(6):995–1000. doi: 10.1189/jlb.0703328. [DOI] [PubMed] [Google Scholar]
- 182.van Deursen J.M. The role of senescent cells in ageing. Nature. 2014;509(7501):439–446. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Alexander E., Hildebrand D.G., Kriebs A., et al. IkappaBzeta is a regulator of the senescence-associated secretory phenotype in DNA damage- and oncogene-induced senescence. J. Cell Sci. 2013;126(Pt 16):3738–3745. doi: 10.1242/jcs.128835. [DOI] [PubMed] [Google Scholar]
- 184.Tak P.P., Smeets T.J., Boyle D.L., et al. P53 Overexpression in synovial tissue from patients with early and longstanding rheumatoid arthritis compared with patients with reactive arthritis and osteoarthritis. Arthritis Rheum. 1999;42(5):948–953. doi: 10.1002/1529-0131(199905)42:5<948::AID-ANR13>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 185.Yamanishi Y., Boyle D.L., Rosengren S., Green D.R., Zvaifler N.J., Firestein G.S. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA. 2002;99(15):10025–10030. doi: 10.1073/pnas.152333199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lee C.S., Portek I., Edmonds J., Kirkham B. Synovial membrane p53 protein immunoreactivity in rheumatoid arthritis patients. Ann. Rheum. Dis. 2000;59(2):143–145. doi: 10.1136/ard.59.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Murakami Y., Mizoguchi F., Saito T., Miyasaka N., Kohsaka H. p16(INK4a) exerts an anti-inflammatory effect through accelerated IRAK1 degradation in macrophages. J. Immunol. 2012;189(10):5066–5072. doi: 10.4049/jimmunol.1103156. [DOI] [PubMed] [Google Scholar]
- 188.Taniguchi K., Kohsaka H., Inoue N., et al. Induction of the p16INK4a senescence gene as a new therapeutic strategy for the treatment of rheumatoid arthritis. Nat. Med. 1999;5(7):760–767. doi: 10.1038/10480. [DOI] [PubMed] [Google Scholar]
- 189.Carson D.A., Haneji N. Fighting arthritis with a senescence gene. Nat. Med. 1999;5(7):731–732. doi: 10.1038/10447. [DOI] [PubMed] [Google Scholar]
- 190.Lee B.Y., Han J.A., Im J.S., et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006;5(2):187–195. doi: 10.1111/j.1474-9726.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 191.Harvey A.R., Clarke B.J., Chui D.H., Kean W.F., Buchanan W.W. Anemia associated with rheumatoid disease. Inverse correlation between erythropoiesis and both IgM and rheumatoid factor levels. Arthritis Rheum. 1983;26(1):28–34. doi: 10.1002/art.1780260105. [DOI] [PubMed] [Google Scholar]
- 192.Reid C.D., Prouse P.J., Baptista L.C., Gumpel J.M., Chanarin I. The mechanism of the anaemia in rheumatoid arthritis: effects of bone marrow adherent cells and of serum on in-vitro erythropoiesis. Br. J. Haematol. 1984;58(4):607–615. doi: 10.1111/j.1365-2141.1984.tb06107.x. [DOI] [PubMed] [Google Scholar]
- 193.Sugimoto M., Wakabayashi Y., Hirose S., Takaku F. Immunological aspects of the anemia of rheumatoid arthritis. Am. J. Hematol. 1987;25(1):1–11. doi: 10.1002/ajh.2830250102. [DOI] [PubMed] [Google Scholar]
- 194.Papadaki H.A., Kritikos H.D., Gemetzi C., et al. Bone marrow progenitor cell reserve and function and stromal cell function are defective in rheumatoid arthritis: evidence for a tumor necrosis factor alpha-mediated effect. Blood. 2002;99(5):1610–1619. doi: 10.1182/blood.v99.5.1610. [DOI] [PubMed] [Google Scholar]
- 195.Papadaki H.A., Kritikos H.D., Valatas V., Boumpas D.T., Eliopoulos G.D. Anemia of chronic disease in rheumatoid arthritis is associated with increased apoptosis of bone marrow erythroid cells: improvement following anti-tumor necrosis factor-alpha antibody therapy. Blood. 2002;100(2):474–482. doi: 10.1182/blood-2002-01-0136. [DOI] [PubMed] [Google Scholar]
- 196.Porta C., Caporali R., Epis O., et al. Impaired bone marrow hematopoietic progenitor cell function in rheumatoid arthritis patients candidated to autologous hematopoietic stem cell transplantation. Bone Marrow Transplant. 2004;33(7):721–728. doi: 10.1038/sj.bmt.1704407. [DOI] [PubMed] [Google Scholar]
- 197.Colmegna I., Diaz-Borjon A., Fujii H., Schaefer L., Goronzy J.J., Weyand C.M. Defective proliferative capacity and accelerated telomeric loss of hematopoietic progenitor cells in rheumatoid arthritis. Arthritis Rheum. 2008;58(4):990–1000. doi: 10.1002/art.23287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Colmegna I., Pryshchep S., Oishi H., Goronzy J.J., Weyand C.M. Dampened ERK signaling in hematopoietic progenitor cells in rheumatoid arthritis. Clin. Immunol. 2012;143(1):73–82. doi: 10.1016/j.clim.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.McCubrey J.A., Steelman L.S., Chappell W.H., et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta. 2007;1773(8):1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Fujii H., Shao L., Colmegna I., Goronzy J.J., Weyand C.M. Telomerase insufficiency in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA. 2009;106(11):4360–4365. doi: 10.1073/pnas.0811332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Masi A.T., Rehman A.A., Elmore K.B., Aldag J.C. Serum acute phase protein and inflammatory cytokine network correlations: comparison of a pre-rheumatoid arthritis and non-rheumatoid arthritis community cohort. J. Innate Immun. 2013;5(2):100–113. doi: 10.1159/000345700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Park J., Choi H.M., Yang H.I., Yoo M.C., Kim K.S. Increased expression of IL-1 receptors in response to IL-1beta may produce more IL-6, IL-8, VEGF, and PGE(2) in senescent synovial cells induced in vitro than in presenescent cells. Rheumatol. Int. 2012;32(7):2005–2010. doi: 10.1007/s00296-011-1891-1. [DOI] [PubMed] [Google Scholar]
- 203.Rodier F., Coppe J.P., Patil C.K., et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009;11(8):973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Correia-Melo C, Hewitt G, Passos JF. Telomeres, oxidative stress and inflammatory factors: partners in cellular senescence? 2014. [DOI] [PMC free article] [PubMed]
- 205.Fumagalli M., d'Adda di Fagagna F. SASPense and DDRama in cancer and ageing. Nat. Cell Biol. 2009;11(8):921–923. doi: 10.1038/ncb0809-921. [DOI] [PubMed] [Google Scholar]
- 206.Kageyama Y., Takahashi M., Nagafusa T., Torikai E., Nagano A. Etanercept reduces the oxidative stress marker levels in patients with rheumatoid arthritis. Rheumatol. Int. 2008;28(3):245–251. doi: 10.1007/s00296-007-0419-1. [DOI] [PubMed] [Google Scholar]
- 207.Kageyama Y., Takahashi M., Ichikawa T., Torikai E., Nagano A. Reduction of oxidative stress marker levels by anti-TNF-alpha antibody, infliximab, in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2008;26(1):73–80. [PubMed] [Google Scholar]
- 208.Toussirot E., Wendling D., Herbein G., CIC-1431 Biological treatments given in patients with rheumatoid arthritis or ankylosing spondylitis modify HAT/HDAC (histone acetyltransferase/histone deacetylase) balance. Joint Bone Spine. 2014;81(6):544–545. doi: 10.1016/j.jbspin.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 209.Wagner U., Schatz A., Baerwald C., Rossol M. Brief report: deficient thymic output in rheumatoid arthritis despite abundance of prethymic progenitors. Arthritis Rheum. 2013;65(10):2567–2572. doi: 10.1002/art.38058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Bryl E., Vallejo A.N., Matteson E.L., Witkowski J.M., Weyand C.M., Goronzy J.J. Modulation of CD28 expression with anti-tumor necrosis factor alpha therapy in rheumatoid arthritis. Arthritis Rheum. 2005;52(10):2996–3003. doi: 10.1002/art.21353. [DOI] [PubMed] [Google Scholar]
- 211.Bryl E., Vallejo A.N., Weyand C.M., Goronzy J.J. Down-regulation of CD28 expression by TNF-alpha. J. Immunol. 2001;167(6):3231–3238. doi: 10.4049/jimmunol.167.6.3231. [DOI] [PubMed] [Google Scholar]
- 212.Gomez-Garcia L., Ramirez-Assad C., Vargas A., et al. Reduced numbers of circulating CD28-negative CD4+ cells in patients with rheumatoid arthritis chronically treated with abatacept. Int. J. Rheum. Dis. 2013;16(4):469–471. doi: 10.1111/1756-185X.12056. [DOI] [PubMed] [Google Scholar]
- 213.Scarsi M., Ziglioli T., Airo P. Decreased circulating CD28-negative T cells in patients with rheumatoid arthritis treated with abatacept are correlated with clinical response. J. Rheumatol. 2010;37(5):911–916. doi: 10.3899/jrheum.091176. [DOI] [PubMed] [Google Scholar]
- 214.Scarsi M., Ziglioli T., Airo' P. Baseline numbers of circulating CD28-negative T cells may predict clinical response to abatacept in patients with rheumatoid arthritis. J. Rheumatol. 2011;38(10):2105–2111. doi: 10.3899/jrheum.110386. [DOI] [PubMed] [Google Scholar]
- 215.Parish S.T., Wu J.E., Effros R.B. Modulation of T lymphocyte replicative senescence via TNF-{alpha} inhibition: role of caspase-3. J. Immunol. 2009;182(7):4237–4243. doi: 10.4049/jimmunol.0803449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Laberge R.M., Zhou L., Sarantos M.R., et al. Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell. 2012;11(4):569–578. doi: 10.1111/j.1474-9726.2012.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kageyama Y., Takahashi M., Nagafusa T., Torikai E., Nagano A. Methotrexate reduces the levels of pentosidine and 8-hydroxy-deoxy guanosine in patients with rheumatoid arthritis. Mod. Rheumatol. 2007;17(5):398–402. doi: 10.1007/s10165-007-0607-6. [DOI] [PubMed] [Google Scholar]
- 218.Klareskog L., Catrina A.I., Paget S. Rheumatoid arthritis. Lancet. 2009;373(9664):659–672. doi: 10.1016/S0140-6736(09)60008-8. [DOI] [PubMed] [Google Scholar]
- 219.Kokkonen H., Soderstrom I., Rocklov J., Hallmans G., Lejon K., Rantapaa Dahlqvist S. Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum. 2010;62(2):383–391. doi: 10.1002/art.27186. [DOI] [PubMed] [Google Scholar]
- 220.Sokolove J., Johnson D.S., Lahey L.J., et al. Rheumatoid factor as a potentiator of anti-citrullinated protein antibody-mediated inflammation in rheumatoid arthritis. Arthritis Rheumatol. 2014;66(4):813–821. doi: 10.1002/art.38307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Clavel C., Nogueira L., Laurent L., et al. Induction of macrophage secretion of tumor necrosis factor alpha through Fcgamma receptor IIa engagement by rheumatoid arthritis-specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Rheum. 2008;58(3):678–688. doi: 10.1002/art.23284. [DOI] [PubMed] [Google Scholar]
- 222.Laurent L., Clavel C., Lemaire O., et al. Fcgamma receptor profile of monocytes and macrophages from rheumatoid arthritis patients and their response to immune complexes formed with autoantibodies to citrullinated proteins. Ann. Rheum. Dis. 2011;70(6):1052–1059. doi: 10.1136/ard.2010.142091. [DOI] [PubMed] [Google Scholar]
- 223.van de Stadt L.A., Witte B.I., Bos W.H., van Schaardenburg D. A prediction rule for the development of arthritis in seropositive arthralgia patients. Ann. Rheum. Dis. 2012 doi: 10.1136/annrheumdis-2012-202127. [DOI] [PubMed] [Google Scholar]
- 224.van der Helm-van Mil A.H. Risk estimation in rheumatoid arthritis: from bench to bedside. Nat. Rev. Rheumatol. 2014;10(3):171–180. doi: 10.1038/nrrheum.2013.215. [DOI] [PubMed] [Google Scholar]