

Abstract
The identification of protein biomarkers for acute myeloid leukemia (AML) that could find applications in AML diagnosis and prognosis, treatment and the selection for bone marrow transplant requires substantial comparative analyses of the proteomes from AML patients. In the past years, several studies have suggested some biomarkers for AML diagnosis or AML classification using methods for sample preparation with low proteome coverage and low resolution mass spectrometers. However, most of the studies did not follow up, confirm or validate their candidates with more patient samples. Current proteomics methods, new high resolution and fast mass spectrometers allow the identification and quantification of several thousands of proteins obtained from few tens of μg of AML cell lysate. Enrichment methods for posttranslational modifications (PTM), such as phosphorylation, can isolate several thousands of site-specific phosphorylated peptides from AML patient samples, which subsequently can be quantified with high confidence in new mass spectrometers. While recent reports aiming to propose proteomic or phosphoproteomic biomarkers on the studied AML patient samples have taken advantage of the technological progress, the access to large cohorts of AML patients to sample from and the availability of appropriate control samples still remain challenging.
Keywords: Acute myeloid leukemia, biomarker, mass spectrometry, proteomics, phosphoproteomics, diagnosis, prognosis
1. Introduction
1.1. Acute Myeloid Leukemia
AML is an aggressive hematopoietic disease diagnosed primarily in the elderly population, with a median age of approximately 65 years [1]. Although 40-50% of the older patients achieve complete remission, the 5-year survival rate for patients above 65 years is 5% [1] due to high rates of relapse [2, 3]. The disease is highly heterogeneous, and is sub-classified according to cellular morphology, hematopoietic lineage as well as common translocations and mutations [4]. Cytogenetic analyses, revealing large chromosomal aberrations and gene translocations, are still regarded to give the most relevant information on prognosis [5, 6]. However, these aberrations, with the exception of acute promyeloid leukemia associated with the t(15;17)(q22;q12) translocation giving the PML-RARA fusion protein [7], seldom guide the choice of treatment offered. Therefore, more sophisticated and sensitive methods have been developed to discover and validate protein biomarkers that might both be clinically useful in prognostication as well as choosing therapy strategy.
Proteomics is the study of proteins at large scale. Proteomic strategies involve mass spectrometry (MS), protein chips and reverse-phase protein microarrays. However, MS-based proteomics has become very popular in the last decade and proteomics currently refer to the MS-based analysis of proteins from biological samples. The speed and sensitivity of current mass spectrometers allow the identification and quantification of several thousand of proteins, including those of low abundance and their PTM accurately. Proteomic studies of biological events can describe key proteins involved in relevant signaling pathways.
Recently, a review focused on MS aspects and leukemia research has been published [8]. In the following, MS-based proteomics applied on AML for disease-related biomarker discovery will be presented, with emphasis on sample considerations and new proteomic protocols. Our review of proteomic studies will describe in detail the most relevant reports on AML MS-based proteomics and phosphosproteomics published recently.
1.2. The Clinical Potential of AML Biomarkers
Proteomic research for finding biomarkers in AML has been a subject since the 1980s, boosted by Hanash et al. who found different protein abundance patterns of AML blasts and acute lymphoid leukemia (ALL), using 2 dimensional (2D) electrophoresis [9]. Compared to other leukemias such as ALL, the prognosis for survival of AML patients has improved in the last 30 years [1]. The five-year relative survival has increased from 6.2 (1975-1977) to 25.4% (2004-2010). Still, AML is among the cancers with highest mortality, thus it exists a pressing need for the discovery of new biomarkers that could meliorate the clinical outcome for these patients.
For hematological clinicians a biomarker or biomarker panel for AML would be desirable for many aspects: diagnosis, risk stratification and prognosis, treatment selection, prediction of chemoresistance, selection for allogeneic stem cell transplantation and therapy monitoring. A consensus defining biomarkers for these categories does not exist, nor does a consensus defining the optimal or most practical sample type for clinical assessment of a biomarker. As conventional chemotherapy is highly toxic and not appropriate for all AML patients, biomarkers could ultimately enable personalized treatment, in which a diagnostic test could identify patients who would benefit from conventional chemotherapy and those who would benefit from alternative therapies, such as allogeneic stem cell transplantation.
A high-throughput technique such as proteomics rise as a promising tool to achieve this aim, as it enables large-scale protein and PTM analysis of clinical samples. In a heterogeneous and complex malignancy like AML, proteins transcribed from mutated genes can be valuable clinical biomarkers measurable by MS-proteomics [10]. Also, PTM on proteins or altered mRNA splicing leading to irregular protein isoforms can provide new measurable features to the protein such as altered activity or cause localization in different compartments of the cell.
2. Sample considerations
2.1. AML Cell Lines as Models
Since the development of the first hematopoietic cell line, the Raji cell line cultured from a Nigerian patient with Burkitt’s lymphoma in 1963 [11], the use of cell line systems has become increasingly popular. Investigating the commercial database of the Leibniz-Institut DSMZ-Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, 51 different AML cell lines are available (http://www.dsmz.de/catalogues/catalogue-human-and-animal-cell-lines.html, accessed 26.02. 2015). Although some of these represent derivatives of the original cell lines, it is obvious that the availability of models of AML is large. Still, a subgroup of these are preferred (Table 1) [12-35] and are often chosen to represent AML patients in both drug development and protein signaling analyses.
Table 1.
Currently used AML cell lines.
| Cell Linea | FAB$ | AML Relevant Mutations and Translocations a | Cancer Relevant Mutations§ | ||
|---|---|---|---|---|---|
| HL-60 | [12, 13] | M2 | Amplification of MYC TP53 null |
[14] [15] |
CDKN2A p.R80* DNMT3B p.R537Q MLL3 p.? (c.2769+7C>T) MLLT4 p.M1226I NRAS p.Q61L |
| KASUMI-1 | [16] | M2 | t(8;21)(q22;q22) giving RUNX1-RUNX1T1 (AML1-ETO) KIT N822K mutation |
[17] [18] |
ASXL1 p.G646fs*12 CREBBPp.?(c.1676+3_1676+12delaagaccctgt) TP53 p.R248Q |
| KG-1 | [19] | M0 | ins(12;8)(p11;p11p22) giving FGFR1OP2-FGFR1 fusion | [20] | BCR p.? (c.3322+9T>C) EP300 p.? (c.730-10T>C) JUN p.Q202P MLL3 p.R907Q, p.? (c.2872-6C>A) NOTCH2 p.K1514R TP53 p.? (c.672+1G>A) |
| MOLM-13 | [21] | M5 | ins(11;9((q23;p22p23)) giving MLL-AF9 fusion FLT3 ITD CBL deltaExon8 mutant |
[21] [22] [23] |
BRCA2 p.Q819R CBL p.? (c.1226_1227+12delAGgtacggatctaa) FLT3p.E598_Y599insFDRFREYE MLL p.A1650T MLL3 p.M741T, p.C394Y MTOR p.T571K PML p.R689W |
| MONOMAC-6 | [24] | M5 | t(9;11)(p22;q23) giving MLL-AF9 fusion | [25, 26] | ASXL1 p.L1393fs*30 FLT3 p.V592A RUNX1 p.A134V TP53 p.R273H |
| MV4-11 | [27] | M5 | t(4;11) giving MLL-AF4 FLT3 ITD |
[27] [22] |
FLT3 p.D600_L601ins10 MECOM p.E35K |
| NB-4 | [28] | M3 | t(15;17)(q22;q11-12.1) giving PML-RARa fusion | [28, 29] | KRAS p.A18D RUNX1T1 p.R157C TP53 p.R248Q |
| OCI-AML | [30] | M4 | Npm1 mutation (type A) DNMT3A R882C |
[30] [31] |
NPM1 p.W288fs*12 NRAS p.Q61L |
| THP-1 | [32] | M5 | t(9;11)(p21;q23) giving MLL-MLLT3 (MLL-AF9) fusion | [33] | MLLT4 p.A765T NRAS p.G12D TP53 p.R174fs*3 |
$FAB – French-American- British classification system for AML divides patients in subgroups M0-M7 based on morphology of the leukemic cells [34].
§The mutation data was obtained from the Sanger Institute Catalogue of Somatic Mutations in Cancer (COSMIC) web site (http://www.sanger.ac.uk/cosmic) [35].
aThe references in this table are numbered following the order of the main text.
Although most laboratories use immortalized cell lines in basal research, there has been much debate about the actual relevance of these models. As the cells are immortalized, there has been a concern that the cells no longer resemble their origin, and cannot therefore accurately represent the disease. There is therefore a general opinion that the use of cell lines is inferior to analyses of patient material. General guidelines have therefore recently been constructed to ensure the correct development and maintenance of cell lines to prevent common errors as contamination, misidentification and instability [36]. Regarding AML, this concern might be considered more relevant compared to certain solid tumors, as the patients with AML themselves are very heterogeneous. The question is therefore whether AML cell lines can be good models for the study of the disease. There are only a few studies set out to explore if hematopoietic cell lines indeed are relevant and reliable as model systems. However, the development of DNA microarray-based technologies has provided a method for investigating this more thoroughly by both DNA copy number alterations and gene expression changes. Rücker et al. investigated 17 different myeloid leukemia cell lines by DNA microarray and compared the expression profiles with previously published AML patient data from 116 patients as well as cell line data [37] to interrogate the fidelity of cell lines concerning alterations during cultivation as well as relevance compared to patient cells [38]. Cytogenetic signatures were found to be conserved by investigating the 717 genes best characterizing cytogenetic subgroups as determined by significance analysis of microarrays. The analysis of the cell lines and AML patients based on these 717 genes showed co-clustering of groups carrying identical cytogenetic aberrations, including t(8;21), inv(16) and t(15;17). Unsupervised hierarchical cluster analysis also confirmed that cell lines analyzed at different times in different laboratories showed stable gene expression patterns, indicating stability of cell lines after culturing [38]. However, Gillet et al. [39] investigated multidrug resistance (MDR) mechanisms in 59 of 60 cancer cell lines defined as the NCI-60 panel, and compared their results to a series of primary cancer cells from matched diseases (ovarian serous carcinoma, glioblastoma, colorectal cancer, AML and T-ALL, metastatic melanoma and breast cancer). The authors used Taqman-based RT-qPCR to compare expression profiles of 380 genes linked to MDR. Hierarchical clustering indicated that the cultured ovarian cancer cell lines showed significantly up-regulation of 225 of the 380 genes compared to primary ovarian serous carcinoma and effusion samples from ascites fluid, suggesting to be a result of selection pressure and culture conditions allowing the cell lines to grow in their in vitro environment. Gillet et al. suggested that the cancer cell lines were selected during establishment for expression of genes associated with MDR. Their analysis of T-ALL and AML cell lines revealed the same tendencies, and showed that AML cell lines and AML patient cells did not cluster together. Hierarchical clustering of protein expression in five AML cell lines (compared and characterized in [40]) and twenty-seven AML patient samples from unpublished shotgun proteomics data from our lab showed that 560 of the 1410 proteins quantified in all thirty-two samples were significantly differently expressed in the cell lines compared to primary cells. Proteins involved in processes such as translational initiation and elongation were higher expressed in the cell lines, while proteins involved in or part of the mitochondrion were lower expressed in the cell lines, compared to the primary patient cells. Principal component analysis (PCA) plots of the total protein batch and the significantly regulated proteins showed a clear separation of the cell lines (Fig. 1A), while the PCA plot of the not significantly regulated protein subset (Fig. 1B), showed heterogeneity among all samples. This strongly implies that some proteins have altered expression in the cell lines, but that approximately 60% of the protein levels remains stable. The advantages of using cell lines in AML research are their unlimited supply, worldwide availability and near
Fig. (1).
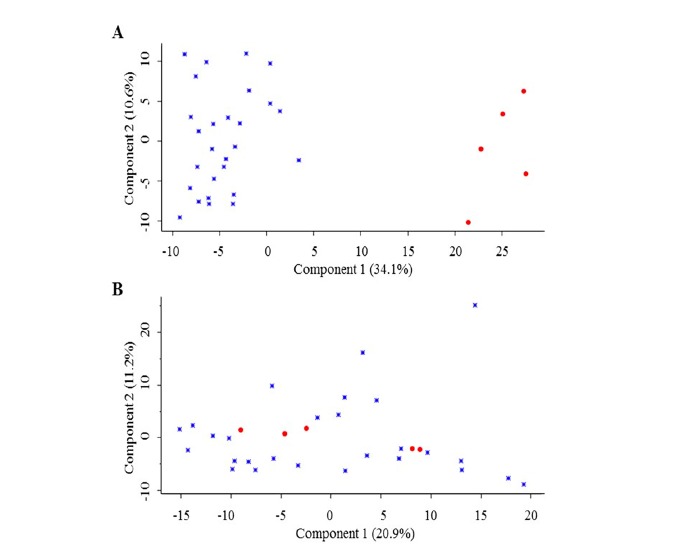
PCA plots based on the proteomes of 27 primary AML samples (blue diamonds) and 5 AML cell lines (red circles). 1410 proteins were quantified in all samples and included for PCA analysis. (A) PCA plot of significantly regulated proteins (n=560; p<0.05). (B) PCA plot of not significantly regulated proteins (n=850).
infinite viable storability in liquid nitrogen. However it is essential to be aware of the limitations of the model. Thus it is important to keep in mind that the cell lines can be an approximate representation of AML, and that they cannot be used to fully describe the biological complexity of AML patient samples. Moreover, the cytogenetic status, stage of disease and mutational status are heterogeneous from patient to patient and difficult to mimic using cell line models.
2.2. Specimen Source
AML is a disease originated in the bone marrow and affects the production of circulating blood cells of a patient. Leukemic cells exist as leukemic stem-like cells (LSC) - the malignant version of the normal hematopoietic stem cells (HSC), and as undifferentiated blast cells of the myeloid linage in the bone marrow (BM) and peripheral blood (PB). The LSC population is CD34+CD38- and can potentiate colony-forming progenitors [41]. It has been hypothesized to be the reason for leukemia regrowth, and can thus be important for chemoresistance and relapse [42, 43]. The LSC population may differ between individuals, as indicated by quantitative proteomic profiling of two primary mouse leukemias, in terms of LSC frequency (1 LSC/1.4 cells versus 1 LSC/>100 cells) [20]. The study revealed more than 400 differentially expressed proteins, of which some were associated with stem cell fate. The LSC population has been described as significantly different compared to other AML and CD34+ subpopulations [43], and has been the preferred material in some proteomic and transcriptomic studies [44, 45]. However, the LSC population is less abundant and therefore harder to obtain, thus other studies are based on AML blast cells isolated from PB [46-48] or BM [49-53], sometimes referred to as the bulk population. Which of these is most favorable is also debatable; the BM is in close proximity to the hematopoietic stem cells, but sample collection is more invasive and separation of BM-derived leukemic cells from other BM-derived cells is more complicated than blast separation from most PB samples. Good correlation between matched BM and PB samples has been found when comparing 51 proteins with reverse phase protein array (RPPA) technology [54]. The study indicated seven proteins as significantly altered, pointing to two phosphoproteins: proto-oncogen tyrosine-protein kinase Src (SRC) with a higher level in blood, and ribosomal protein S6 (RS6) with higher levels in marrow. The authors suggested that blood cells might be in a less proliferative and transcriptional state compared to leukemic cells in the BM [54]. Single cell network profiling of intracellular signaling nodes of leukemic paired BM and PB blasts before and after treatment has demonstrated that the specimen source is not significantly affecting the proteome signaling [55]. As BM aspiration is a considerably more invasive procedure than collection of PB, blast samples from PB are more practical for biomarker assessment in a clinical setting – also in terms of therapy monitoring, where collection of BM might not be clinically excused [55].
Cell sorting of cell populations expressing distinct surface markers has also been used, including studies on single AML cells [56]. AML involves different clonal cells, and it is important for the biological understanding to also look at the clones individually. However, for a biomarker to have clinical value it should be measureable in an easily accessible sample type, thus potential markers from such studies should also be tested in PB samples.
2.3. Pre-analytical Sample Preparation
Leukemic cells as sample are a valuable and important source for clinical biomarker studies. Sample collection, storage and preparation are all steps where bias can be introduced, thus a standardized protocol for these procedures is of outmost importance. In studies including blast cells, patients with high blast counts in PB are preselected for the AML bio-bank to minimize the amount of contaminant non-leukemic cells, ensuring over 95% blasts after separation [57]. The advantage of this pre-selection is the certainty of having the purest leukemic cell preparations for analysis, but on the other hand, this might skew the patient demographics used in clinical studies, as factors such as nucleophosmin-1 (NPM) and Fms-like tyrosine kinase 3 internal tandem duplication (FLT3-ITD) mutations are linked to higher blast percentage and leukocyte count [58]. For proteomic analysis, no standardized consensus has been published, but sample preparation procedures have been summarized, reviewed and proposed [59]. Ficoll-Hypaque separation (density gradient separation) is commonly used to isolate the mononuclear cell fraction, which contains the leukemic blast population (leukemic PBMC). The mononuclear fraction can either be depleted of CD3+/CD19+ B and T cells by magnetic antibody-conjugated sorting or cryopreserved directly in liquid nitrogen, with 80-90% viability when re-thawed [59, 60]. The latter approach ensures less sample preparation steps that can influence the proteome, however, the leukemic blast population may be contaminated with some B and T cells.
Storage conditions together with freeze-cycles can affect the sample stability [61, 62]. Tibes et al. did not find any significant changes in their analysis of eight phosphoepitopes after one and three freeze-thaw cycles (cryopreserved) compared to fresh samples [60]. However, delayed sample storage has been found to alter the mRNA expression of two transcripts in mononuclear cells of BM [63] and similar changes could be expected at protein level.
To date, no large-scale proteomics studies on leukemic cells have been performed to examine these important aspects more thoroughly. A standardized bio-banking protocol to ensure identical sample handling of all samples is crucial for larger biomarker studies. The ultimate goal should be to use fresh cells to easily assess a diagnostic or prognostic marker, and it would therefore be important to compare the results from protocols with cryopreserved cells as used for biomarker studies today to the results from a protocol that uses fresh AML cells. The logistics of obtaining sufficient fresh sample amount for statistical significance has probably been a limiting factor, but new opportunities in proteomic sample preparation enables sodium dodecyl sulphate (SDS) cell lysing of fresh material, which can then be frozen as a “fresh” cell lysate.
2.4. Finding a Suitable Cell Specimen Control
A major collective challenge for AML proteomic studies is the selection of control cells to include in the experimental design. Since leukemic blasts are immature cells where differentiation is blocked, it is hard to identify a healthy comparable counterpart. The tendencies in leukemia research are to use cell types (such as mononuclear cells) isolated from PB of healthy volunteers, hematopoietic CD34+ cells from BM and/or leukemia cells from ALL or chronic myeloid leukemia (CML) as controls. Why a certain control has been included in a study should however be discussed to a larger degree. The choice of the experimental control would depend on the biological and clinical aspect of the study being conducted, as the control group might differ in a therapeutic screening compared to a diagnostic biomarker study. We here summarize the most commonly used controls.
PBMC may be the most frequently used control. This collective term comprises cells with one round nucleus, such as lymphocytes, monocytes and macrophages, and excessively leukemic blast cells in case of AML. This cell subset can be separated from the whole peripheral venous blood, which is easily collected from healthy volunteers. The whole PB contains the immunized, differentiated lymphocytes (B cells, T cells, NK cells and regulatory T-cells), dendritic cells, granulocytes, hematopoietic stem and progenitor cells, mononuclear cells, myeloid cells, monocytes (incompletely differentiated precursor to macrophage and myeloid dendritic cells) and plasma cells [64]. The monocytes and macrophages have the same common myeloid progenitor as the AML blast cells, while the common lymphoid progenitors are precursors of lymphocytes. Proteomic-based comparisons of AML mononuclear cells derived from PB and BM could not identify differences between the two leukemic cell samples, but differences in these hematopoietic cell compartments were found in comparison to healthy volunteer samples after stem cell mobilization [65]. These results were also observed with the flow cytometry technique [66].
Normal leukocytes/white blood cells (granulocytes, lymphocytes and monocytes) have also been used as control, and can be isolated from the PB by gradient separation [67]. Forty-four proteins differently expressed in acute leukemias (AML and ALL) and normal white blood cells have been identified by matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF)-MS [68], of which several proteins were suggested to be involved in leukemogenesis and one in leukemia classification. Although, the availability of leukocytes as control group is advantageous, the cells are quite different from the leukemic blast when it comes to both function and maturation. However, proteins upregulated in AML compared to both ALL and leukocytes may reflect malignant transformation of AML [68]. ALL is acute leukemia of the lymphoid linage, and it is dominated by immature malignant hematopoietic cells, like AML. ALL as a control therefore suits as a lineage-independent control, but with similar blockage of differentiation. Defining the proteins that are differentially expressed in AML and ALL may aid for diagnostic purposes. Proteins with similar expression in AML and ALL, but different to more mature cells, can be related to differentiation stop.
All blood cells are originated from a common pluripotent HSC, a rare cell type (1:100.000 to 1:10 million [42]) located in specialized niches in the BM [64]. Human primary CD34+ cells isolated from BM mononuclear cells include both hematopoietic stem and progenitor cells and should be characterized by flow cytometry after isolation. The cells can be isolated immunomagnetically from the BM using positive selection. For therapeutic purposes, knowledge regarding which proteins are expressed on leukemic cells compared to HSC can be utilized to develop drugs targeting only AML expressed proteins. This cell population as control therefore has an important role in proteomic biomarker studies. Kornblau et al. set to compare five different AML subsets relative to normal CD34+ cells (HSC was unobtainable) and found many cancer-related proteins, such as cyclin-dependent kinase inhibitor 1B (CDN1B), signal transducer and activator of transcription 3 (STAT3) and cellular tumor antigen p53 (P53), to have more than 60% higher expression in the LSC (CD34+ CD38-) subset [43]. Foss et al. included CD34+ cells isolated from PB as healthy controls for blockage of differentiation stage, in addition to mononuclear cells as control for healthy monocyte contamination [52]. For mobilization of the CD34+ cells into the blood, the patients were stimulated with granulocyte colony-stimulating factor (CSF3), which induces production of myeloid precursors. Thus these cells resembled the AML blast cells compared to ALL cells, mononuclear cells and expectedly the stem cell CD34+ population. Seventeen of 639 proteins were reported to have significantly different expression in CD34+ versus AML.
The difficulties of defining an appropriate control group for AML blast cells is probably the reason that some research groups avoids including a control, and in some studies paired samples – for instance before and after therapy – may serve as internal control [53]. In addition to carefully selecting the appropriate cell population as control sample, considerations should be put into which healthy volunteers to include. The matching of patients and controls in terms of sex, ethnicity and age is usually not discussed. Healthy volunteers donating BM samples are often medical students recruited and do not reflect the AML patient cohort whose median age is 60-70 years. PBMC control samples are often collected from blood donors at the local blood bank, and thus might reflect a more diverse range of age groups.
3. MS-based methodologies for the study the AML proteome
MS-based approaches to identify and quantify proteins and peptides in complex biological samples are characterized by a multistep workflow that includes sample preparation, liquid chromatography tandem mass spectrometry (LC-MS/MS), data analysis and result interpretation. During sample preparation the cells are lysed, the disulphide bonds from the released proteins are reduced, free cysteines are alkylated and the proteins are digested into peptides. A desalting step to remove chemicals and undigested proteins is performed before LC-MS/MS. The peptide sample can then be analyzed with no further processing (single-shot approach) or fractionated into several subsamples before loading onto the LC-MS/MS system to improve signal-to-noise ratios and increase proteome coverage. Although current mass spectrometers have a wide dynamic range, more low abundant proteins in complex mixtures would be identified and quantified after fractionation. Figure 2 illustrates the main steps for sample preparation that are described bellow. The most popular methods to prepare biological samples prior to MS-analysis are one dimensional polyacrylamide gel electrophoresis (1D-PAGE) [69], in-solution digestion [70] and filter-assisted sample preparation (FASP) [71, 72]. With the 1D-PAGE method, the proteins from typically detergent-lysed samples are separated and the lane containing the protein sample is usually excised into several sections for processing. In-solution digestion approaches use denaturing reagents (urea, guanidinium hydrochloride, trifluoroethanol or sodium deoxycholate) at high concentration to solubilize the biological sample. Digestion can be performed with more than one protease. In fact, a serial Lys-C/trypsin proteolysis was found most efficient to yield fully cleaved peptides while reducing the percentage of miscleavage [73]. In the FASP method, the sample is processed in a standard filtration device. As described on the in-solution digestion method, FASP can be performed using a consecutive digestion with LysC and then trypsin to increase the population of peptides notably [74, 75]. An enhanced version of the original FASP method (eFASP) has been described to increase proteome coverage and sample recovery [76] by activation treatment of the filter before use, and use of deoxycholic acid as digestion buffer. However, a recent comparative reevaluation of the classic FASP and eFASP methods by LC-MS/MS has revealed no significant difference at protein level [77]. 2D-PAGE and 2D-differential in-gel electrophoresis (DIGE), which uses up to three different fluorescent dyes to compare different protein samples within a single 2D-gel platform, have been used in AML biomarker research as common methods to process AML patient or AML cell line samples.
Fig. (2).
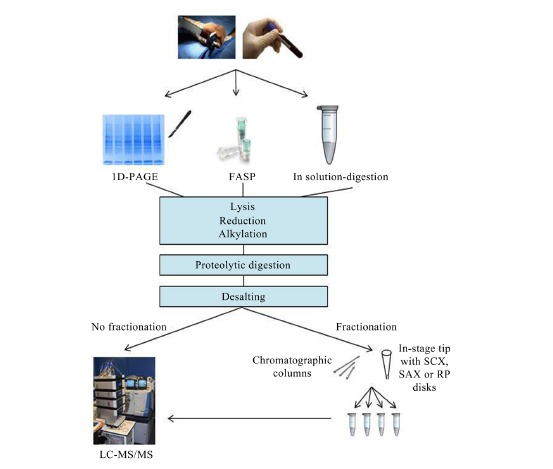
Proteomic workflow comprising three of the most popular techniques for sample preparation. For complex samples, the fractionation option is shown.
In the past few months, optimized in-solution methods using trifluoroethanol as protein denaturant [78] or guanidinium hydrochloride containing tris(2-carboxyethyl)phosphine and chloroacetamide to perform simultaneous reduction and alkylation [79] along with a new in-solution/FASP hybrid approach named in-StageTip method [80] have been described to report deep proteome coverage and excellent quantification accuracy using fast and highly sensitive benchtop quadrupole ultra-high-field orbitrap MS. In our lab, we use the FASP method [71, 75], with Lys-C and trypsin as digestion enzymes, to process AML patient samples. We are able to identify and quantify 3100 proteins from 20 μg of sample using the FASP method analyzed on a Linear Trap Quadrupole (LTQ) Orbitrap Elite MS (unpublished data).
Peptide samples of high complexity are often fractionated using chromatographic columns or self-packed microcolumns in pipet tips [81] taking advantage of peptide properties such as charge, polarity, hydrophobicity, isoelectric point and size. Of the many types of fractionation techniques [82-84], strong cation exchange (SCX), strong anion exchange
(SAX) and reverse phase (RP) chromatography are most commonly used in proteomic workflows. The recent report by Kulak et al. [80] has shown that the mixed mode reverse phase/cation exchange (MM) identified and quantified more proteins than SCX or SAX. In our lab, we also tested the MM versus SCX and SAX in stage tips to fractionate AML proteomes digested in solution and we observed a 6% and 12% increase in identified and quantified proteins with the MM when compared to the SCX and SAX, respectively, using an LTQ Orbitrap Elite MS.
To quantify the proteome, a labeling strategy could be included in the proteomic workflow. The differently labeled samples should be processed with the same proteomic workflow and combined to be comparable. The main approaches for quantitative proteomics can be grouped in label free, metabolic labeling, isobaric labeling and targeted methods [85]. Label free techniques can be used on any soluble biological sample and unlimited number of samples can be compared. The label free approach avoids use of expensive chemicals for labeling, but requires more analysis time on the MS. However, it can handle a high dynamic range. As quantification relies on MS peptide ion intensities or MS/MS spectral counting, sample preparation, LC and MS conditions must show high reproducibility, which could be challenging when fractionation is included. Stable isotope labeling by amino acids (SILAC) is a metabolic labeling technique that introduces light and heavy versions of amino acids (the most commonly used are arginine and lysine) to whole cells through the growth medium containing the desired amino acid form. Although the quantification of different SILAC samples can be very precise, this technique is not applicable to non-metabolically active samples. For such purposes, the super-SILAC approach [86] - which comprises of a mixture of SILAC labeled disease-representative cell lines as internal standard - is a suitable alternative, as it allows protein quantification of patient derived tissue and cell lysate. It should be emphasized that protein quantification using SILAC relies on the presence of proteins in both the patient sample and the internal standard. Thus, there is a risk of losing quantification of proteins only present in the patient sample. However, in our hands, the percentage of proteins in AML patient samples that could not be quantified with a super-SILAC mix of 5 AML cell lines [40] never exceeded 2% (unpublished data). With isobaric labeling techniques targeting the N-terminus and side chain amines of peptides such as isobaric tags for relative and absolute quantification (iTRAQ) and tandem mass tag (TMT) it is possible to compare up to eight and ten samples, respectively. Because of the possibility to multiplex, these techniques are often preferred for in depth quantitative biomarker discovery studies where SILAC is not applicable [87]. If specific peptides in biological samples need to be quantified with high confidence, multiple reaction monitoring (MRM; also referred to as selected reaction monitoring (SRM)) [88], can be performed using triple quadrupole or hybrid triple quadrupole/linear ion trap MS. Moreover, MRM can do absolute quantification if the target peptide is accompanied by known concentrations of its isotopically labelled synthetic form. The introduction of new quadrupole-orbitrap instruments such as the Q Exactive allow analyses to be operated in parallel reaction monitoring (PRM) mode [89]. Compared to MRM on the triple quadrupole, this high resolution and accurate mass instrument allows for even higher accuracy and selectivity, which minimize interfering from co-eluting peptides.
4. PTM, phosphoproteomics and MS-based methodologies for the study of the AML phosphoproteome
Functional protein properties can be changed by PTM, in which a chemical group is covalently attached to an amino acid side chain. Although more than 300 types of protein PTM can occur in living organisms [90], phosphorylation, glycosylation, ubiquitination, acetylation, methylation, nitrosylation and oxidation represent the most frequent modifications. Protein PTM regulate most of the biological processes and are involved in cancerogenesis, neurodegenerative disease and diabetes [91, 92]. The low abundance and low stoichiometry of many PTM makes characterization of the modification sites a challenging task. PTM analysis typically requires large amounts of the biological sample; a specific PTM enrichment step, usually before or after peptide fractionation in the general proteomic workflow; and fast and sensitive mass spectrometers equipped with different peptide fragmentation techniques such as collision-induced dissociation (CID), higher-energy collision dissociation (HCD) and electron-transfer dissociation (ETD) [93].
Phosphorylation is one of the most ubiquitous PTM of proteins and plays a crucial role in cell signaling and regulation. Five hundred eighteen protein kinase genes are found in the human genome [94]. Nearly 30% of all proteins can reversibly be phosphorylated on serine, threonine and tyrosine residues during the cell cycle [95]. This means that thousands of phosphorylation sites can be identified from cell lysates and describe the regulatory mechanisms of the biological process under investigation. The development of phosphopeptide enrichment techniques can be found in recent PTM reviews by Beltran and Cutillas [96] and Huang et al. [97]. Phosphoproteomics has made a great progress in the last ten years. Besides optimized enrichment methods and advanced mass spectrometers, several computational methods are freely available and described [98]. Thus, phosphorylation is the most studied PTM in biological samples with MS methods. Current approaches are capable of mapping more than 50.000 phosphopeptides in a single human cancer cell line [99].
Currently, popular enrichment protocols of phosphopeptides from digested samples are chromatography-based and involve the use of titanium dioxide beads -metal oxide affinity chromatography (MOAC)- or iron (III) ions chelated to a nitrilotriacetic matrix-immobilized metal ion affinity chromatography (IMAC)- or the sequential use of both materials in the SIMAC (sequential elution from IMAC) strategy to separate mono-phosphorylated and multiply-phosphorylated peptide pools [100] (Fig. 3). A new generation of IMAC materials have recently been developed using a monodisperse microsphere-based immobilized titanium (IV) ion matrix [101]. Even metal immobilized magnetic nanoparticles have been synthesized for a highly selective and sensitive enrichment of phosphopeptides [102]. Furthermore, the identification of phosphotyrosine-peptides, which are usually less abundant than phosphoserine- or phosphothreonine-peptides, requires a specific immunoaffinity purification step using antiphospho-tyrosine antibodies (such as 4G10, P-Tyr-100 or P-Tyr-1000, separately or in combination) that typically precede the general phosphopeptide enrichment [103].
Fig. (3).
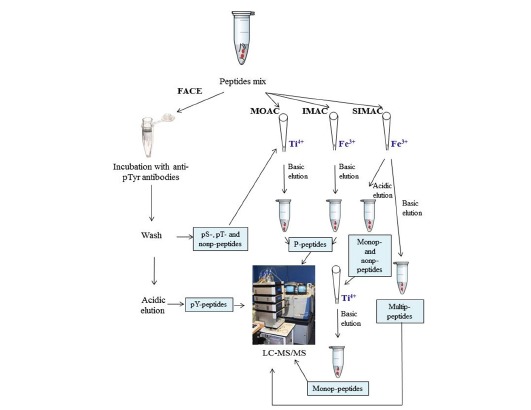
Phosphoproteomic workflow. FACE, MOAC, IMAC and SIMAC p-peptide enrichment steps are shown. Peptides with an attached red circle represent phospho-peptides (p-peptides).
It is well known that each approach leads to the enrichment of a different phosphopeptide set. The performance of an enrichment technique also depends on the nature and complexity of the sample. Samples containing many thousands of phosphopeptides might require peptide fractionation before or after phosphopeptide enrichment [104]. Therefore, an initial test of the individual or combined enrichment techniques on the biological sample to be analyzed might be beneficial to identify the phosphoproteomic workflow giving the desired balance between sample throughput, the amount of consumed sample material and coverage of phosphorylated peptides. In our lab, we use the FASP method, with Lys-C and trypsin as digestion enzymes, followed by a separate TiO2-beads enrichment of the two peptide pools, to identify and quantify 2.900 phosphorylation sites from only 250 μg of AML sample using an LTQ Orbitrap Elite MS (unpublished data).
5. Proteomic contributions to unravel the AML proteome
As more advanced proteomic techniques have been developed in the recent years, proteomic research has in general moved from 2D-PAGE combined with MALDI-TOF or surface-enhanced laser desorption-ionization (SELDI)-TOF to LC-MS/MS-based proteomics of large-scale protein analysis and accurate targeted approaches. A decade ago, the contribution of proteomics to the study of AML proteins was very poor. AML cells could be accessed from BM or PB and purified in large by gradient separation [59]. However, partial-coverage proteomic protocols and low sensitive mass spectrometers could not cope with the high complexity and large dynamic range of AML samples, resulting in few reported proteins as potential AML biomarkers. The earliest report detected 10 differentially expressed proteins from AC133+ hematopoietic stem cell-like fractions isolated from different leukemic disorders, including AML [44]. Few years later, MS-based quantitative proteomic tools such as SILAC, iTRAQ and isotope-coded affinity tag (ICAT) in addition to DIGE, became popular proteomic approaches for cancer biomarker discovery using upgraded high sensitive MALDI-MS/MS, MALDI-TOF, SELDI-TOF, electrospray ionization
(ESI)-MS/MS and nanoESI-MS/MS mass spectrometers. Despite the fast progress of MS-based proteomic methods, the challenges in finding large patient cohorts and working with limited amounts of AML samples have hindered the use of shotgun proteomics (based on LC-MS/MS spectrometry) for the global characterization of the AML proteome comprised of several thousand of proteins. In the following section, most of the proteomic findings in AML research regarding improved diagnostics, risk stratification and prognostics in the last decade will be reported while all the potential biomarkers identified from these studies will be listed in Table 2.
Table 2.
List of published potential AML biomarkers described using a MS-based proteomic approach. Only verification/ validation of biomarkers performed in patient cohorts has been considered.
| Name* | Uniprot ID | Marker specificity | Sample type | Number of samples (AML/ control) |
MS-based
methodology |
Verification/
Validation |
Ref. a |
|---|---|---|---|---|---|---|---|
| NUMA1 | Q14980 | AML diagnosis | BM | 9/4L | 2D MALDI-TOF | RT-PCR (n= 20) | [44] |
| ITA6 | P23229 | AML development | BM and PB | 2/0 | SCX LC-MS/MS | FACS (n= 9-12) and long term stromal coculture assay (n=2) | [45] |
| UBA1, FIBA, PLF4 | P22314, P02671, P02776 | AML diagnosis; refractory AML; relapsed AML | PB | 139/72HV | MB-WCX MALDI-TOF; LC-MS/MS |
Western blot (n=3) | [46] |
| FETUA, CLUS, RET4, APOC3, immunoglobulin heavy-chain variant, PRS4, HPT | P02765, P10909, P02753, P02656, Q9NPP6, P62191, P00738 | AML diagnosis | PB | 12/12HV | 2D MALDI-TOF; ESI-TOF | - | [47] |
| ANXA3, PLSL, 6PGD, CATA, PRDX6, ANXA1, ACTG, GSTO1, ESTD | P12429, P13796, P52209, P04040, P30041, P04083, P63261, P78417, P10768 | AML subtypes; therapy response | BM and PB | 38/17HV | 2D MALDI-TOF; ESI-MS/MS | Western blot (n= 32) |
[48] |
| HNRH1, CALR, ROA2 | P31943, P27797, P22626 | AML prognosis | BM | 42/1CD | 2D and DIGE MALDI-TOF | Western blot (n=10) | [49] |
| S10A8, S10A9, CATG, NDKA, PERM, UP1 (derivative of ROA1) | P05109, P06702 P08311, P15531, P05164, P09651 | AML subtypes; AML diagnosis; AML prognosis | BM | 51/10L/8HV | 2D MALDI-TOF; ESI-MS/MS | - | [50, 68] |
| UBC9 | P63279 | AML diagnosis | K562 (CEBPAp30-ER), WT K562 as CML cell lines; BM from AML patients containing CEBPAp30 | 11/0 | 2D MALDI-TOF | - | [51] |
| NICA, AL1A1, THIK, GLU2B, MPCP | Q92542, P00352, P09110, P14314, Q00325 | AML diagnosis | BM | 4/5L/8HV | LC-MS/MS (Undescribed fractionation) | - | [52] |
| EEPD1, BC11A, RANB3, RPGR, LMNA | Q7L9B9, Q9H165, Q9H6Z4, Q92834, P02545 | AML quizartinib-therapy response | BM | 6 AML quizartinib-responders and 6 AML quizartinib-non-responders | SCX IMAC LC-MS/MS | 6 extra AML quizartinib-responders and 3 extra AML quizartinib–non responders | [53] |
| ENOA, GDIR2, ANXA1, ANX10, CATA, PRDX2, TPM3 | P06733, P52566, P04083, Q9UJ72, P04040, P32119, P06753 | AML subtypes; AML prognosis | BM | 13/10HV | 2D MALDI-TOF | - | [105] |
| MOES, EZRI, AIFM1 | P26038, P15311, O95831 | AML diagnosis (pediatric) | BM and PB | 5/3HV | 2D MALDI-TOF MS | Western blot (n=4) | [106] |
| LEO1, TP4A3 | Q8WVC0, O75365 | AML development | TF1 (an erythroleukemia cell line)-derived cell lines | - | SILAC-based LC-MS/MS (Unfractionated) | Western blot (n=24) | [107] |
| CD166 | Q13740 | AML diagnosis; therapeutic development of AML antibody-based treatment | HL60, THP1, NB4 and PLB985 AML cell lines; K562 CML cell line | - | Biotinylation/affinity chromatography MALDI-TOF | FACS (n= 4) | [108] |
| Name* | Uniprot ID | Marker specificity | Sample type | Number of samples (AML/ control) |
MS-based methodology |
Verification/ Validation |
Ref.a |
| S10A8 | P05109 | AML prognosis | BM and PB | 54/0 | SAX/SCX SELDI-TOF; MALDI-TOF/TOF; LC-MS/MS |
Western blot (n=12) | [109] |
| BTK | Q06187 | AML therapy response | KG-1 and MV4-11 AML cell lines, BM from AML patients | 28/0 | Immunoprecipitation and SILAC-based LC-MS/MS | - | [110] |
| PTN6 | P29350 | AML development | BaF3 (a pro-B cell line) cells (FLT3-ITD, WT FLT3, FLT3-D835Y, WT FLT3/FL) | - | Immunoprecipitation and iTRAQ-based LC-MS/MS | - | [111] |
| PIN1 | Q13526 | AML development | K562 (a CML cell line)-CEBPA-p30-ER cells | - | 2D MALDI-TOF | mRNA assay (n= 6) | [112] |
| ESDT, ACTG | P10768, P63261 | AML M1/M2 prognosis; therapeutic targets | BM and PB | 33/17HV | 2D MALDI-TOF; ESI-MS/MS | - | [113] |
| PPIA, CATA, NPM, PCNA, TCPA, ROA2, ENOA, PRDX1 | P62937, P04040, P06748, P12004, P17987, P22626, P06733, Q06830 | AML-treatment response with DNA methyltransferase inhibitors | AML1/ETO AML cell line | - | 2D MALDI-TOF | Western blot (n= 2) | [114] |
| PRKDC, PK3CA, CSK21, CDK1, PAK1, MK01, PDPK1, CDC7, ABL1, LCK, SRC, CDK1 | P78527, P42336, P68400, P06493, Q13153, P28482, O15530, O00311, P00519, P06239, P12931, P06493 | AML diagnosis; AML therapy response with PI3K and mTOR inhibitors | P31/Fuj and Kasumi-1 AML cell lines; PB from AML patients | 39/5HV | MOAC LC-MS/MS | - | [115] |
| BCR-ABL fusion protein$, TEL-ARG fusion protein¤, JAK2 |
Q8NEY0, O60674 | AML therapeutic targets | HEL, HT-93 and KMB-3 AML cell lines | - | Immunoprecipitation LC -MS/MS | - | [116] |
| KSYK | P43405 | AML therapeutic targets | HL-60 AML cell line | - | Immunoprecipitation LC-MS/MS (in combination with shRNA screening) | Western blot and in vitro inhibition studies (n= 14) | [117] |
| PTN11, PTN6, RUNX1, STA5A |
Q06124, P29350, Q01196, P42229 |
AML therapeutic targets | MV4-11, Molm 14, Marimo, Me-F2, KY821, OCI/AML3, Nomo-1 and ML-1 AML cell lines; SEM, RS4-11, and REH ALL cell lines; BM from AML patients | 6/0 | Immunoprecipitation LC-MS/MS | - | [118] |
| SRC, BTK | P12931, Q06187 | AML erlotinib- and gefinitib-treatment targets | KG1 AML cell line | - | SCX IMAC LC-MS/MS | - | [119] |
*Biomarker names are shown as protein abbreviations.
$The Uniprot ID for the largest fusion protein is reported.
¤No Uniprot ID was found for this fusion protein.
HV stands for healthy volunteer as control.
L stands for another type of leukemia different from AML as control.
CD stands for CD34+ cells as control.
aThe reference numbers of the described studies of this table refer to the references cited in the main text.
5.1. Diagnostic Markers and Global Protein Expression Profiling
In the clinic today, cytogenetic and molecular markers are used to diagnose AML patients according to the WHO system [4]. However, the present guidelines are not adequate to differentiate between all subtypes and do not always predict the clinical outcome. The fact that nearly 50% of the AML patients show no cytogenetically abnormality discourages the use of cytogenetic markers [6]. Therefore, protein expression profiling of AML subtypes could become a frequent tool to be used in diagnostics and prognostics of AML patients, where a specific protein or protein signature, within a certain set of proteins, either carrying PTM or not, is expressed in a distinct pattern related to a specific clinical feature. The protein or PTM signature can potentially act as a biomarker itself or reveal upstream or downstream biomarkers being part of the pathway(s) related to the protein pattern.
For AML diagnostic research, Kwak et al. used 2D-MS analysis to compare the serum of 12 AML patients and 12 healthy controls. They found eight proteins, including alpha-2-HS-glycoprotein (FETUA) and immunoglobulin heavy-chain variant, differentially expressed in the AML group [47]. The same technology was applied on blast cells isolated from BM of 13 AML patients classified according to the WHO system. They identified seven proteins, including Rho GFP-dissociation inhibitor 2 (GDIR2), catalase (CATA), annexin 1 (ANXA1) and annexin A10 (ANX10), as altered in the AML blast population compared to normal mononuclear cells [105]. Despite the potential use of the proteins as markers for prognosis or disease outcome, no further validation or application has been reported.
Braoudaki et al. [106] aimed to distinguish AML from myelodyslastic syndrome and suggested moesin (MOES) and ezrin (EZRI) as potential diagnostic biomarkers for AML. The expression of MOES was validated with Western blot, but only four patient samples were included. In another attempt to define molecular profiles of FAB-based AML classification and ALL versus AML, Cui et al. analyzed 61 patient samples using 2D-MS. The authors identified 27 proteins
differently expressed between AML and ALL, and 23 proteins differently expressed between the granulocytic and monocytic linages of AML [50, 68]. Among them, protein S100-A8 (S10A8) and S100-A9 (S10A9) were found to differentiate AML from ALL, and nucleoside diphosphate kinase A (NDKA) described to be absent in the M3a subtype. Proteomic profiling aiming to classify the different AML subtypes has also been done with RPPA by Kornblau et al. [54] in a study including 265 patients. They were able to separate the myeloid subtypes (M0-M2), the monocytic subtypes (M4-M5), erythroleukemia and megakaryocytic leukemia from a subset of 24 differentially expressed proteins and phosphoproteins. In a recent study using SELDI-TOF-MS technology, a protein profile classification model was constructed for characterization of acute leukemia subgroups, resulting in five distinct proteomic signatures which could serve as a new diagnose approach [120]. The identity of the proteins included in the proteomic signatures has, however, not been revealed.
In an early 2D and DIGE MALDI-TOF study, Balkhi et al. reported specific PTM to be associated with cytogenetic risks in a patient cohort of 42 AML patients with various cytogenetic aberrations [49]. β-O-linked N-acetyl glucosamine was found in heterogeneous nuclear ribonucleoprotein H (HNRH1) of patients with 11q23 translocation, acetylation of calreticulin (CALR) was associated with t(8;21) translocation and methylation of heterogeneous nuclear ribonucleoprotein A2/B1 (ROA2) was found with both t(8;21) translocation and inv(16). Abnormalities in chromosome 11q23 is usually associated with poor prognosis [121], while translocation of both t(8;21) and inv(16) are associated with good prognosis [122]. Different cytogenetic groups were all found to alter the proteome, and proteins contributing to cytogenetic differences were reported and used to create biological protein networks. However, regardless of identical risk stratifying markers or cytogenetic aberrations, patients are known to respond differently to the given therapy. This might explain why four phosphoprotein signatures were found to correlate to prognosis and treatment response, but not to the cytogenetics in a study by Irish et al., where they applied multiparameter flow cytometry on single cells [56]. To date, there is no published global proteomic data on AML PTM such as glycosylation, ubiquitination, acetylation, methylation and redox modifications. However, the phosphorylation state in several AML cell lines as well as in AML patients has been studied (Table 2).
Using advanced LC-MS/MS spectrometry, Foss et al. described deeper coverage of the AML proteome, demonstrating that the use of label free alignment algorithms enabled differentiation between known classes of acute leukemias (AML and ALL), in addition to healthy mobilized CD34+ controls [52]. Of more than 600 quantified proteins, the authors found 91, 71 and 17 proteins that distinguished ALL from CD34+, ALL from AML, and AML from CD34+, respectively. One of the most promising biomarker candidate was nicastrin (NICA), a component of the gamma secretase complex, which separated AML from both ALL and CD34+ samples. Retinal dehydrogenase 1 (AL1A1), NICA and 3-ketoacyl CoA thiolase (THIK) were proposed as biomarkers distinguishing AML from CD34+. In a follow-up study on the same dataset by Elo et al. [123], an advanced statistical approach called “ROTS” (reproducibility optimized test statistics) was applied on the dataset to account for high experimental variability and missing quantifications. They concluded that the label free-aligned algorithm identified more potential biomarkers involved in known leukemic processes, compared to transcriptomics.
Another type of proteomic screening or profiling where MS-based proteomics can reveal new biological insight and markers is over-expression or knock-down of oncogenes in cell lines followed by protein quantification of the induced biological activities, as demonstrated in a study of protein tyrosine phosphatase type IVA 3 (TP4A3) by Chong et al. [107]. By screening the quantified proteins in the downstream signaling pathway of TP4A3, 398 proteins were significantly altered and RNA polymerase-associated protein Leo1 was identified as a novel mediator of the oncogenic functions of TP4A3 in leukemia. This approach can ultimately be used for drug development, targeting specific pathways or proteins otherwise hard to identify.
Antibody drug-conjugates can be used as therapy for cell surface proteins. In a recent study by Strassberg et al. the cell surface proteome of four AML cell lines, one CML cell line and normal blood cells were characterized by label free MS-based proteomics [108]. Of 823 proteins, 320 were annotated as membrane proteins and were clustered into eight groups based on their relative quantification, differentiating among the AML cell lines, normal granulocytes and the CML cell line. The report focused on CD166 antigen as it was upregulated in all AML cell lines and AML samples from some patients. A duocarmycin derivate was used for the development of an antibody-drug conjugate, which killed HL60 AML cells in a vitro assay with an IC50 of 8 nM.
5.2. Predictive and Prognostic Markers
Since many AML patients exhibit no risk-stratifying factors, meaning clinical features that predict risk and outcome, and guides clinicians in medical decision-making, many research groups aim at finding new prognostic and risk stratifying biomarkers to complement the morphological, cytogenetic and molecular risk factors. Several groups have used proteomics to refine the cytogenetic classes. Nicolas et al. found a proteomic classification that refines the cytogenetic subgroups, as the protein profiles could subdivide the intermediate and unfavorable cytogenetic classes into subgroups with significantly different survival rates [109]. For instance, patients with intermediate cytogenetic risk could be separated into two proteomic profiles, where patients of one profile had similar survival rates as patients with favorable cytogenetic risk, while the other proteomic profile had similar survival rates as the unfavorable cytogenetic group. The most discriminating protein between survival and death was S10A8, which was verified as a biomarker for poor-prognosis in a different patient cohort, and found to predict death (during the follow-up period of 57 months) with 85% sensitivity and 72% specificity. Increased expression of this protein may be used as a high-risk prognostic AML biomarker, but it might not represent a therapeutic target, as it was found to be decreased in AML compared to normal neutrophils in another study [68]. Profiling of serum peptides has been performed on samples from 72 AML patients, 72 healthy controls, 37 AML patients with complete remission (CR) and 30 refractory and relapse AML patients [46]. The samples were analyzed with MALDI-TOF and LC-ESI-MS/MS spectrometry to identify candidate biomarkers, followed by immunoblotting for validation studies. Three proteins, ubiquitin-like modifier activating enzyme 1 (UBA1), isoform 1 of fibrinogen alpha chain precursor (FIBA) and platelet factor 4 (PLF4) correlated with AML clinical outcome and could possibly be used for predicting AML relapse, monitoring minimal residual disease and predicting prognosis in clinical practice.
Using an LC-MS/MS phosphoproteomics strategy combined with interactome analysis and transcriptome sequencing, Oellerich et al. showed that FLT3-ITD-positive AML differed from FLT3-ITD-negative AML in regard to tyrosine-protein kinase BTK-dependent signaling [110]. In FLT3-ITD-positive subtypes, BTK mediated FLT3-ITD-dependent activation of myc proto-oncogen protein (MYC) and signal transducer and activator of transcription 5 (STAT5), while BTK couples Toll-like receptor 9 (TLR9) activation to nuclear factor NF-kappa B (NFKB1) and STAT5 in the FLT3-ITD-negative subtype, pointing towards BTK inhibitors as subtype-specific treatment strategies, either as a single agent or in combination with other drugs. The BTK inhibitor ibrutinib has been approved by the Food and Drug Administration (FDA) for treatment of chronic lymphocytic leukemia patients, inducing cellular apoptosis and reduced growth [124]. The FLT3 kinase has been a subject of extensive research, and despite the fact that phosphorylation of tyrosine residues counts for less than 2% of the total phosphosites, Zhang et al. were able to quantify 371 unique phosphotyrosine peptides on 276 proteins with an LC-MS/MS spectrometer [111]. Comparison of FLT3 wild type, ITD and D835Y mutant showed that the two mutated proteins caused different signaling events, measured by different phosphorylation status of tyrosine-protein phosphatase non-receptor type 6 (PTN6), tyrosine-protein kinase JAK2 and STAT5. Interestingly, the authors found that the constitutive FLT3 activation in the D835Y phenotype lead to phosphorylation of PTN6 and down-regulation of JAK2-STAT5 signaling.
Mutation of the transcription factor CCAAT/enhancer-binding protein alpha (CEBPA-p30) is observed in approximately 10% of AML patients and is involved in leukemia development [125, 126]. In a proteomic screen, it was found that AML patients with the CEBPA-p30 mutation had increased expression of SUMO-conjugating enzyme UBC9, causing sumoylation of CEBPA-p42, which inhibits granulocytic differentiation [51]. The AML subgroup having the CEBPA-p30 mutation is also related to increased expression of peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (PIN1) with CEBPA-p30 binding site in the promoter region [112]. Silencing of PIN1 led to granulocytic differentiation, and the authors suggested PIN1 inhibition as a potential strategy for AML treatment of the patients harboring the CEBPA-p30 mutation.
5.3. Therapy-related Markers
Altered signal transduction pathways and mutated proteins with altered activation are potential therapeutic targets, and known oncogenic proteins, such as FLT3, have been extensively targeted and inhibited in clinical trials [127-130]. Although such treatment has been successful for some patients, it has not lead to a revolution in AML treatment. Chemotherapy induces massive cell death in the patient, and studies on therapy-induced proteomic changes have been performed [131]. Chemoresistance is a major problem in the treatment of many AML patients, and biomarkers predicting therapy response are highly needed. High concentrations of gamma 1 actin (ACTG) was highlighted as a potential biomarker predicting resistance by Kazmierczak et al. [113], based on a study where they correlated clinical, cytogenetic and molecular data to the results of induction chemotherapy. However, no follow-up studies on this marker have been published so far. Recently, 2D and MALDI-TOF spectrometry was used again to study drug-targeted protein expression in AML1/ETO cell line [114]. Treatment with DNA methyltransferase inhibitor azacitidine induced downregulation of peptidyl-prolyl cis-trans isomerase A (PPIA), CATA, NPM and proliferating cell nuclear antigen (PCNA) whereas decitabine induced downregulation of alpha-enolase (ENOA) and peroxiredoxin-1 (PRDX1). Downregulation of T-complex protein 1 subunit alpha (TCPA) and ROA2 was observed with the two drugs. To date, there are few studies on AML-therapy-related markers. Proteomic analysis of current and future induction and post remission drugs on AML patients could provide valuable insights into the molecular changes under treatment to evaluate drug response.
5.4. Major MS-based Proteomics Contributions to Identify AML Biomarkers
A detailed proteomic study by Luczak et al. [48] on the identification of AML biomarkers from 38 AML patients and 17 healthy volunteers addressed the protein profiling of two AML subtypes (M1 and M2); the correlation between some protein biomarkers and therapeutic outcome or relapse time; and the potential use of BM and PB samples for the purpose of discovering useful biomarkers. Using samples from BM and PB at time of diagnosis (T0), at time of CR (T1) and at time of disease recurrence (T2), the authors showed that the proteomes of AML-M1/M2 patients were not significantly different between the two samples at the different time points. Thus, this comparison encourages the use of PB samples, which requires a less complicated collecting procedure than aspiration, for AML research. Proteomic analysis of combined AML-M1/M2-T0 BM and PB samples with PB and BM samples from healthy volunteers, separately, identified 21 differentially expressed proteins in each data set. Seventeen out of the 21 proteins from each comparison overlapped. However, the expression changes were different in each set. This was not surprising as the comparative proteomic analysis of the PB and BM samples from healthy volunteers revealed numerous quantitative and qualitative differences. The group of nine proteins that differed between AML-M1/M2-T0 and both PB and BM from healthy volunteers included histone-binding protein RBBP4, α-actinin 1 (ACTN1), a 14-3-3 protein, transketolase (TKT), pyruvate kinase (KPYR), protein deglycase DJ-1 (PARK7), F-actin capping protein alpha-1 (CAZA1), annexin A4 (ANXA4) and MOES. They represented potential biomarkers for differentiating AML-M1/M2 patients from healthy controls. Interestingly, the authors did not find significant differences in the proteome comparisons between AML-M1/M2-T0 and AML-M1/M2-T1, and between AML-M1/M2-T0 and AML-M1/M2-T2. Significant expression differences of ANXA1, glutathione S-transferase omega-1 (GSTO1) and esterase D/formyglutathione hydrolase (ESTD) were observed between the proteomes of AML-M1/M2-T0 and the proteome from patients who responded to treatment and achieved remission (AML-M1/M2-T0-CR) and from patients resistant to treatment (AML-M1/M2-T0-RES). In addition, ACTG was found to be differentially expressed between short-term and long-term remission patients and showed a similar high expression pattern in the short-term remission and AML-M1/M2-T0-RES groups. For the first time, four biomarker candidates were introduced to differentiate patients who were treatment-responsive from those who showed short-term response or resistance to the treatment. A final analysis of the AML-M1/M2-T0 group involving both BM and PB samples performing hierarchical clustering was enabled to discriminate proteins from AML-M1 and AML-M2 subtypes. The classification was consistent with the clinical diagnosis in 81% of the cases. Five proteins, annexin A3 (ANXA3), 6-phosphogluconate dehydrogenase (6PGD), CATA, one of the L-plastin isoforms (PLSL) and peroxiredoxin-6 (PRDX6), exhibited differential expression and could be of potential use to diagnose AML patients into the M1 and M2 subtypes.
Two important contributions to the understanding of phosphorylation in AML are described next. The study by Casado et al. [115] described a computational strategy, named kinase-substrate enrichment analysis (KSEA), to infer the activation of given kinase pathways from MS-based phosphoproteomic data of AML cells. Firstly, the phosphopeptides were arranged into substrate groups which contained phosphorylation sites known as substrates of specific kinases or sharing specific phosphorylation motifs when the fidelity of kinase-substrate databases was compromised. Then, the statistical significance of the enrichment of these groups relative to the phosphoproteomics data was calculated. P31/Fuj and Kasumi-1 AML cell lines with and without treatment with inhibitors of phosphatidylinositol 4,5-biphosphate 3-kinase catalytic subunit alpha isoform (PK3CA) and serine/threonine-protein kinase mTOR (AZ123, Ku-794 and PI-103) were used to evaluate the computational modeling. The KSEA results showed an activation of DNA-dependent protein kinase catalytic subunit (PRKDC) as a consequence of inhibiting PK3CA and mTOR and were in agreement with those obtained by Western blotting analysis. KSEA analysis of the phosphoproteomic data from AML patients showed an enrichment of phosphorylated substrate groups downstream of PK3CA in 55% of the cases. Phosphorylated substrates of casein kinase 2α (CSK21), cyclin-dependent kinases (CDK) and serine/threonine-protein kinases PAK were significantly enriched in nearly 40% of the AML cases. Phosphorylated substrates of mitogen-activated protein kinase 1 (MK01 or MAPK1), 3-phosphoinositide-dependent protein kinase-1 (PDPK1) and cell division cycle 7 (CDC7) were enriched in AML patient samples that showed resistance to inhibition of PI3K-mTOR signaling by AZ123, whereas substrates of tyrosine-protein kinase ABL1, tyrosine-protein kinase Lck, SRC and CDK1 were enriched in AZ123-sensitive AML patient samples. The use of KSEA to predict the responses of AML cells to drugs that target kinase pathways may be beneficial to estimate therapy success and reassess the clinical use of kinase inhibitors as AML targets. KSEA is not available online yet. More validation studies of the script with different AML data sets would provide key information on the accuracy of mathematical modelling in the prediction of therapy success based on kinase inhibitors. However, another modeling study by the same group on AML (along with lymphoma and multiple myeloma) cell lines found a good correlation between the phosphoproteomics data and the phenotypic responses of these cancer cells to kinase inhibitors [132], supporting the potential use of modeling for the identification of novel markers of resistance or sensitivity to drugs that inhibit key enzymes such as kinases.
The first global phosphoproteome analysis of human BM samples was published by Schaab et al. last year [53]. AC220 (quizartinib), a tyrosine kinase inhibitor, is a FLT3 inhibitor and it has been used in the treatment of patients with relapsed/refractory AML. The CR rate in FLT3-ITD-positive patients was 54% and the partial remission (PR) rate was 17% [133]. The phosphoproteome of six quizartinib-responders (patients with CR and PR) and six quizartinib-non-responders was characterized using SCX/IMAC and SILAC-based quantitative MS. More than 13.000 phosphosites were identified and nearly 8.000 were confidently assigned to serine, threonine and tyrosine amino acids with a localization probability higher than 0.75. Further analysis of this data set found three significantly differently expressed phosphosites at a FDR of 10% from endonuclease/exonuclease/ phosphatase family domain-containing protein 1 (EEPD1), B-cell lymphoma/leukemia 11A (BC11A) and Ran-binding protein 3 (RANB3). BC11A is a myeloid and B-cell proto-oncogene and it is associated with a poor outcome of AML patients [134]. RNAB3, regulated by Ras/MAPK1/RSK and the PK3CA/AKT signaling pathways, is involved in the nuclear export of mothers against decapentaplegic homolog 2 and 3 (SMAD2/3) and inhibits transforming growth factor beta-1 (TGFB1) signaling [135]. Additional bioinformatics analysis using the mean-rank test and the ensemble feature selection method [136], resulted in a final phospho-signature consisting of five phosphorylation sites significantly regulated between responder and non-responder groups. These included the three sites described earlier and the x-linked retinitis pigmentosa GTPase regulator (RPGR) and pre-lamin A/C (LMNA), which form the nuclear lamina and it is involved in gene transcription. Validation of the phospho-peptide signature in a new group of six quizartinib-responders and three quizartinib-non-responders predicted the observed drug response at 78% accuracy. Moreover, the authors showed that the phosphorylation of the LMNA site was correlated with the expression of the protein, offering the option of simple measuring LMNA protein expression to predict quizartinib response.
These reports highlight the importance of PTM discovery and computational approaches to identify phospho-signature to predict drug response of AML patients. Future discovery of patient phospho-signatures of available AML drugs might be of great help with the choice of successful AML treatments to improve remission rates.
6. Challenges
Although proteomics hold all opportunities for finding new biomarkers, clinical proteomic studies are often reflected by lack of proper experimental design, few biological samples and proper control samples, resulting in poor statistical power and very few FDA-approved protein biomarkers [137, 138]. In 2013, Skates et al. published a statistical design for identifying the proper number of biological samples (case and control) required in discovery (n=50) and verification (n=250) studies to reach a high probability of a potential biomarker to reach the clinical validation stage [137]. They also highlighted that the gap between suggested biomarkers in discovery studies compared to clinically validated biomarkers is large, referring to the many research groups who have published biomarker candidates from discovery studies without following verification with another technology or with an independent patient cohort and without decisive testing on clinical trials. This is the case for AML (Table 2) and for other disease-related studies [139].
The choice of control samples represents a major difficulty in the experimental design of AML proteomic projects. This task becomes even more challenging when long-term survivors or patients with CR or relapse are included in the analysis.
Previously, time-consuming proteomics protocols have limited the number of samples included in studies. However, today one can prepare ten-folds of patient samples for proteomic analysis in one or two days, depending on the protocol and potential fractionation chosen. Nowadays, the time-dependent factor is rather the LC-MS/MS time used to obtain maximum peptide separation and sequencing.
7. Future perspectives
Discovery and verification studies of AML proteomes could provide a substantial number of new biomarkers in a near future using current proteomic methods and mass spectrometers. The increasing number of AML patient samples available in current biobanks will support such a progress. Moreover, efforts to share AML samples from the different biobanks might develop a universal approach to validate AML biomarkers using accurate MRM and PRM tools within different populations. The use of peptide and modified-peptide signatures based on MS data to diagnose the different AML subtypes and predict prognosis or treatment response might become a standard procedure in clinical AML strategies [140]. Moreover, the development of new methodologies to study multiple PTM simultaneously would make it possible to look at the cross-talk of several PTM that might provide us with a more realistic and complex picture of the biological events that dictate AML development and progress.
The current effort of MS-based research on finding new AML biomarkers might hopefully lead the development of future fast procedures to process fresh AML patient samples and informatics tools to provide accurate data on the diagnosis, prognosis and treatment success of the patients.
ACKNOWLEDGEMENTS
All the authors have contributed similarly to the design and reporting of this review. EA, RBF and MHV were involved in the initial review’s drafting. The authors would like to thank the Norwegian Cancer Society and Øyvinn Mølbach-Petersens Fond for the funding of this work.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
References
- 1.Cancer Statistics Review S.E. SEER Cancer Statistics Review, 1975-2011. http://seer.cancer.gov/csr/ 1975_2011/ (accessed April 16, 2015).
- 2.Yanada M., Garcia-Manero G., Borthakur G., Ravandi F., Kantarjian H., Estey E. Relapse and death during first remission in acute myeloid leukemia. Haematologica. 2008;93(4):633–634. doi: 10.3324/haematol.12366. [DOI] [PubMed] [Google Scholar]
- 3.Estey E.H. Acute myeloid leukemia: 2014 update on risk-stratification and management. Am. J. Hematol. 2014;89(11):1063–1081. doi: 10.1002/ajh.23834. [DOI] [PubMed] [Google Scholar]
- 4.Vardiman J.W., Thiele J., Arber D.A., Brunning R.D., Borowitz M.J., Porwit A., Harris N.L., Le Beau M.M., Hellstrom-Lindberg E., Tefferi A., Bloomfield C.D. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood. 2009;114(5):937–951. doi: 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- 5.Grimwade D., Hills R.K., Moorman A.V., Walker H., Chatters S., Goldstone A.H., Wheatley K., Harrison C.J., Burnett A.K. National Cancer Research Institute Adult Leukaemia Working, G., Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116(3):354–365. doi: 10.1182/blood-2009-11-254441. [DOI] [PubMed] [Google Scholar]
- 6.Grimwade D., Walker H., Oliver F., Wheatley K., Harrison C., Harrison G., Rees J., Hann I., Stevens R., Burnett A., Goldstone A. The importance of diagnostic cytogenetics on outcome in AML: Analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood. 1998;92(7):2322–2333. [PubMed] [Google Scholar]
- 7.Tallman M.S., Altman J.K. How I treat acute promyelocytic leukemia. Blood. 2009;114(25):5126–5135. doi: 10.1182/blood-2009-07-216457. [DOI] [PubMed] [Google Scholar]
- 8.Roboz J., Roboz G.J. Mass spectrometry in leukemia research and treatment. Expert Rev. Hematol. 2015;8(2):225–235. doi: 10.1586/17474086.2015.1018889. [DOI] [PubMed] [Google Scholar]
- 9.Hanash S., Baier L. Two-dimensional gel electrophoresis of cellular proteins reveals myeloid origin of blasts in two children with otherwise undifferentiated leukemia. Cancer. 1986;57(8):1539–1543. doi: 10.1002/1097-0142(19860415)57:8<1539::aid-cncr2820570817>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 10.Wang Q., Chaerkady R., Wu J., Hwang H., Papadopoulos N., Kopelovich L., Maitra A., Matthaei H., Eshleman J., Hruban R., Kinzler K., Pandey A., Vogelstein B. Mutant proteins as cancer-specific biomarkers. Proc. Natl. Acad. Sci. USA. 2011;108(6):2444–2449. doi: 10.1073/pnas.1019203108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pulvertaft J.V. Cytology of Burkitt's Tumour (African Lymphoma). Lancet. 1964;1(7327):238–240. doi: 10.1016/s0140-6736(64)92345-1. [DOI] [PubMed] [Google Scholar]
- 12.Collins S.J., Gallo R.C., Gallagher R.E. Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature. 1977;270(5635):347–349. doi: 10.1038/270347a0. [DOI] [PubMed] [Google Scholar]
- 13.Dalton W.T., Jr, Ahearn M.J., McCredie K.B., Freireich E.J., Stass S.A., Trujillo J.M. HL-60 cell line was derived from a patient with FAB-M2 and not FAB-M3. Blood. 1988;71(1):242–247. [PubMed] [Google Scholar]
- 14.Dalla-Favera R., Westin E., Gelmann E.P., Martinotti S., Bregni M., Wong-Staal F., Gallo R.C. The human onc gene c-myc: structure, expression, and amplification in the human promyelocytic leukemia cell line HL-60. Haematol. Blood Transfus. 1983;28:247–254. doi: 10.1007/978-3-642-68761-7_47. [DOI] [PubMed] [Google Scholar]
- 15.Ju J.F., Banerjee D., Lenz H.J., Danenberg K.D., Schmittgen T.C., Spears C.P., Schonthal A.H., Manno D.J., Hochhauser D., Bertino J.R., Danenberg P.V. Restoration of wild-type p53 activity in p53-null HL-60 cells confers multidrug sensitivity. Clin. Cancer Res. 1998;4(5):1315–1322. [PubMed] [Google Scholar]
- 16.Asou H., Tashiro S., Hamamoto K., Otsuji A., Kita K., Kamada N. Establishment of a human acute myeloid leukemia cell line (Kasumi-1) with 8; 21 chromosome translocation. Blood. 1991;77(9):2031–2036. [PubMed] [Google Scholar]
- 17.Kwong Y.L., Chan V., Wong K.F., Chan T.K. Use of the polymerase chain reaction in the detection of AML1/ETO fusion transcript in t(8;21). Cancer. 1995;75(3):821–825. doi: 10.1002/1097-0142(19950201)75:3<821::aid-cncr2820750312>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 18.Beghini A., Magnani I., Ripamonti C.B., Larizza L. Amplification of a novel c-Kit activating mutation Asn(822)-Lys in the Kasumi-1 cell line: a t(8;21)-Kit mutant model for acute myeloid leukemia. Hematol. J. 2002;3(3):157–163. doi: 10.1038/sj.thj.6200168. [DOI] [PubMed] [Google Scholar]
- 19.Koeffler H.P., Golde D.W. Acute myelogenous leukemia: a human cell line responsive to colony-stimulating activity. Science. 1978;200(4346):1153–1154. doi: 10.1126/science.306682. [DOI] [PubMed] [Google Scholar]
- 20.Gu T.L., Goss V.L., Reeves C., Popova L., Nardone J., Macneill J., Walters D.K., Wang Y., Rush J., Comb M.J., Druker B.J., Polakiewicz R.D. Phosphotyrosine profiling identifies the KG-1 cell line as a model for the study of FGFR1 fusions in acute myeloid leukemia. Blood. 2006;108(13):4202–4204. doi: 10.1182/blood-2006-06-026666. [DOI] [PubMed] [Google Scholar]
- 21.Matsuo Y., MacLeod R.A., Uphoff C.C., Drexler H.G., Nishizaki C., Katayama Y., Kimura G., Fujii N., Omoto E., Harada M., Orita K. Two acute monocytic leukemia (AML-M5a) cell lines (MOLM-13 and MOLM-14) with interclonal phenotypic heterogeneity showing MLL-AF9 fusion resulting from an occult chromosome insertion, ins(11;9)(q23;p22p23). Leukemia. 1997;11(9):1469–1477. doi: 10.1038/sj.leu.2400768. [DOI] [PubMed] [Google Scholar]
- 22.Quentmeier H., Reinhardt J., Zaborski M., Drexler H.G. FLT3 mutations in acute myeloid leukemia cell lines. Leukemia. 2003;17(1):120–124. doi: 10.1038/sj.leu.2402740. [DOI] [PubMed] [Google Scholar]
- 23.Caligiuri M.A., Briesewitz R., Yu J., Wang L., Wei M., Arnoczky K.J., Marburger T.B., Wen J., Perrotti D., Bloomfield C.D., Whitman S.P. Novel c-CBL and CBL-b ubiquitin ligase mutations in human acute myeloid leukemia. Blood. 2007;110(3):1022–1024. doi: 10.1182/blood-2006-12-061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ziegler-Heitbrock H.W., Thiel E., Futterer A., Herzog V., Wirtz A., Riethmuller G. Establishment of a human cell line (Mono Mac 6) with characteristics of mature monocytes. Int. J. Cancer. 1988;41(3):456–461. doi: 10.1002/ijc.2910410324. [DOI] [PubMed] [Google Scholar]
- 25.MacLeod R.A., Voges M., Drexler H.G. Mono Mac 6: A mature monoblastic leukemia cell line with t(9;11)(p21;q23). Blood. 1993;82(10):3221–3222. [PubMed] [Google Scholar]
- 26.Super H.J., Martinez-Climent J., Rowley J.D. Molecular analysis of the Mono Mac 6 cell line: detection of an MLL-AF9 fusion transcript. Blood. 1995;85(3):855–856. [PubMed] [Google Scholar]
- 27.Lange B., Valtieri M., Santoli D., Caracciolo D., Mavilio F., Gemperlein I., Griffin C., Emanuel B., Finan J., Nowell P. Growth factor requirements of childhood acute leukemia: Establishment of GM-CSF-dependent cell lines. Blood. 1987;70(1):192–199. [PubMed] [Google Scholar]
- 28.Lanotte M., Martin-Thouvenin V., Najman S., Balerini P., Valensi F., Berger R. NB4, a maturation inducible cell line with t(15;17) marker isolated from a human acute promyelocytic leukemia (M3). Blood. 1991;77(5):1080–1086. [PubMed] [Google Scholar]
- 29.Mozziconacci M.J., Rosenauer A., Restouin A., Fanelli M., Shao W., Fernandez F., Toiron Y., Viscardi J., Gambacorti-Passerini C., Miller W.H., Jr, Lafage-Pochitaloff M. Molecular cytogenetics of the acute promyelocytic leukemia-derived cell line NB4 and of four all-trans retinoic acid-resistant subclones. Genes Chromosomes Cancer. 2002;35(3):261–270. doi: 10.1002/gcc.10117. [DOI] [PubMed] [Google Scholar]
- 30.Quentmeier H., Martelli M.P., Dirks W.G., Bolli N., Liso A., Macleod R.A., Nicoletti I., Mannucci R., Pucciarini A., Bigerna B., Martelli M.F., Mecucci C., Drexler H.G., Falini B. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia. 2005;19(10):1760–1777. doi: 10.1038/sj.leu.2403899. [DOI] [PubMed] [Google Scholar]
- 31.Hollink I.H., Feng Q., Danen-van Oorschot A.A., Arentsen-Peters S.T., Verboon L.J., Zhang P., de Haas V., Reinhardt D., Creutzig U., Trka J., Pieters R., van den Heuvel-Eibrink M.M., Wang J., Zwaan C.M. Low frequency of DNMT3A mutations in pediatric AML, and the identification of the OCI-AML3 cell line as an in vitro model. Leukemia. 2012;26(2):371–373. doi: 10.1038/leu.2011.210. [DOI] [PubMed] [Google Scholar]
- 32.Tsuchiya S., Yamabe M., Yamaguchi Y., Kobayashi Y., Konno T., Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer. 1980;26(2):171–176. doi: 10.1002/ijc.2910260208. [DOI] [PubMed] [Google Scholar]
- 33.Odero M.D., Zeleznik-Le N.J., Chinwalla V., Rowley J.D. Cytogenetic and molecular analysis of the acute monocytic leukemia cell line THP-1 with an MLL-AF9 translocation. Genes Chromosomes Cancer. 2000;29(4):333–338. [PubMed] [Google Scholar]
- 34.Bennett J.M., Catovsky D., Daniel M.T., Flandrin G., Galton D.A., Gralnick H.R., Sultan C. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br. J. Haematol. 1976;33(4):451–458. doi: 10.1111/j.1365-2141.1976.tb03563.x. [DOI] [PubMed] [Google Scholar]
- 35.Bamford S., Dawson E., Forbes S., Clements J., Pettett R., Dogan A., Flanagan A., Teague J., Futreal P.A., Stratton M.R., Wooster R. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer. 2004;91(2):355–358. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geraghty R.J., Capes-Davis A., Davis J.M., Downward J., Freshney R.I., Knezevic I., Lovell-Badge R., Masters J.R., Meredith J., Stacey G.N., Thraves P., Vias M. Cancer Research, U.K. Guidelines for the use of cell lines in biomedical research. Br. J. Cancer. 2014;111(6):1021–1046. doi: 10.1038/bjc.2014.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bullinger L., Dohner K., Bair E., Frohling S., Schlenk R.F., Tibshirani R., Dohner H., Pollack J.R. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N. Engl. J. Med. 2004;350(16):1605–1616. doi: 10.1056/NEJMoa031046. [DOI] [PubMed] [Google Scholar]
- 38.Rucker F.G., Sander S., Dohner K., Dohner H., Pollack J.R., Bullinger L. Molecular profiling reveals myeloid leukemia cell lines to be faithful model systems characterized by distinct genomic aberrations. Leukemia. 2006;20(6):994–1001. doi: 10.1038/sj.leu.2404235. [DOI] [PubMed] [Google Scholar]
- 39.Gillet J.P., Calcagno A.M., Varma S., Marino M., Green L.J., Vora M.I., Patel C., Orina J.N., Eliseeva T.A., Singal V., Padmanabhan R., Davidson B., Ganapathi R., Sood A.K., Rueda B.R., Ambudkar S.V., Gottesman M.M. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc. Natl. Acad. Sci. USA. 2011;108(46):18708–18713. doi: 10.1073/pnas.1111840108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aasebo E., Vaudel M., Mjaavatten O., Gausdal G., Van der Burgh A., Gjertsen B.T., Doskeland S.O., Bruserud O., Berven F.S., Selheim F. Performance of super-SILAC based quantitative proteomics for comparison of different acute myeloid leukemia (AML) cell lines. Proteomics. 2014;14(17-18):1971–1976. doi: 10.1002/pmic.201300448. [DOI] [PubMed] [Google Scholar]
- 41.Lapidot T., Sirard C., Vormoor J., Murdoch B., Hoang T., Caceres-Cortes J., Minden M., Paterson B., Caligiuri M.A., Dick J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 42.Bonnet D., Dick J. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997;3(7):730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 43.Kornblau S., Qutub A., Yao H., York H., Qiu Y., Graber D., Ravandi F., Cortes J., Andreeff M., Zhang N., Coombes K. Proteomic profiling identifies distinct protein patterns in acute myelogenous leukemia CD34+CD38- stem-like cells. PLoS One. 2013;8(10) doi: 10.1371/journal.pone.0078453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ota J., Yamashita Y., Okawa K., Kisanuki H., Fujiwara S-I., Ishikawa M., Lim Choi Y., Ueno S., Ohki R., Koinuma K., Wada T., Compton D., Kadoya T., Mano H. Proteomic analysis of hematopoietic stem cell-like fractions in leukemic disorders. Oncogene. 2003;22(36):5720–5728. doi: 10.1038/sj.onc.1206855. [DOI] [PubMed] [Google Scholar]
- 45.Bonardi F., Fusetti F., Deelen P., van Gosliga D., Vellenga E., Schuringa J. A proteomics and transcriptomics approach to identify leukemic stem cell (LSC) markers. Mol. Cell. Proteomics. 2013;12(3):626–637. doi: 10.1074/mcp.M112.021931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bai J., He A., Zhang W., Huang C., Yang J., Yang Y., Wang J., Zhang Y. Potential biomarkers for adult acute myeloid leukemia minimal residual disease assessment searched by serum peptidome profiling. Proteome Sci. 2013;11:39. doi: 10.1186/1477-5956-11-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kwak J.Y., Ma T.Z., Yoo M.J., Choi B.H., Kim H.G., Kim S.R., Yim C.Y., Kwak Y.G. The comparative analysis of serum proteomes for the discovery of biomarkers for acute myeloid leukemia. Exp. Hematol. 2004;32(9):836–842. doi: 10.1016/j.exphem.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 48.Luczak M., Kazmierczak M., Handschuh L., Lewandowski K., Komarnicki M., Figlerowicz M. Comparative proteome analysis of acute myeloid leukemia with and without maturation. J. Proteomics. 2012;75(18):5734–5748. doi: 10.1016/j.jprot.2012.07.030. [DOI] [PubMed] [Google Scholar]
- 49.Balkhi M.Y., Trivedi A.K., Geletu M., Christopeit M., Bohlander S.K., Behre H.M., Behre G. Proteomics of acute myeloid leukaemia: Cytogenetic risk groups differ specifically in their proteome, interactome and post-translational protein modifications. Oncogene. 2006;25(53):7041–7058. doi: 10.1038/sj.onc.1209689. [DOI] [PubMed] [Google Scholar]
- 50.Cui J-W., Wang J., He K., Jin B-F., Wang H-X., Li W., Kang L-H., Hu M-R., Li H-Y., Yu M., Shen B-F., Wang G-J., Zhang X-M. Proteomic analysis of human acute leukemia cells: insight into their classification. Clin. Cancer Res. 2004;10(20):6887–6896. doi: 10.1158/1078-0432.CCR-04-0307. [DOI] [PubMed] [Google Scholar]
- 51.Geletu M., Balkhi M., Peer Zada A., Christopeit M., Pulikkan J., Trivedi A., Tenen D., Behre G. Target proteins of C/EBPalphap30 in AML: C/EBPalphap30 enhances sumoylation of C/EBPalphap42 via up-regulation of Ubc9. Blood. 2007;110(9):3301–3309. doi: 10.1182/blood-2007-01-071035. [DOI] [PubMed] [Google Scholar]
- 52.Foss E.J., Radulovic D., Stirewalt D.L., Radich J., Sala-Torra O., Pogosova-Agadjanyan E.L., Hengel S.M., Loeb K.R., Deeg H.J., Meshinchi S., Goodlett D.R., Bedalov A. Proteomic classification of acute leukemias by alignment-based quantitation of LC-MS/MS data sets. J. Proteome Res. 2012;11(10):5005–5010. doi: 10.1021/pr300567r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schaab C., Oppermann F., Klammer M., Pfeifer H., Tebbe A., Oellerich T., Krauter J., Levis M., Perl A., Daub H., Steffen B., Godl K., Serve H. Global phosphoproteome analysis of human bone marrow reveals predictive phosphorylation markers for the treatment of acute myeloid leukemia with quizartinib. Leukemia. 2014;28(3):716–719. doi: 10.1038/leu.2013.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kornblau S., Tibes R., Qiu Y., Chen W., Kantarjian H., Andreeff M., Coombes K., Mills G. Functional proteomic profiling of AML predicts response and survival. Blood. 2009;113(1):154–164. doi: 10.1182/blood-2007-10-119438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cesano A., Rosen D.B., O'Meara P., Putta S., Gayko U., Spellmeyer D.C., Cripe L.D., Sun Z., Uno H., Litzow M.R., Tallman M.S., Paietta E. Functional pathway analysis in acute myeloid leukemia using single cell network profiling assay: Effect of specimen source (bone marrow or peripheral blood) on assay readouts. Cytometry B Clin. Cytom. 2012;82(3):158–172. doi: 10.1002/cyto.b.21007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Irish J.M., Hovland R., Krutzik P.O., Perez O.D., Bruserud Ø., Gjertsen B.T., Nolan G.P. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell. 2004;118(2):217–228. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 57.Bruserud O., Tore Gjertsen B., Brustugun O.T., Bassoe C.F., Nesthus I., Espen Akselsen P., Buhring H.J., Pawelec G. Effects of interleukin 10 on blast cells derived from patients with acute myelogenous leukemia. Leukemia. 1995;9(11):1910–1920. [PubMed] [Google Scholar]
- 58.Su L., Li W., Cui J.W., Tan Y.H., Yang Y., Liu X.L., Yu P., Hu R.P., Wang L.L., Gao S.J. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2013;21(3):571–575. doi: 10.7534/j.issn.1009-2137.2013.03.007. [Correlation of NPM1, FLT3-ITD mutations with leukocyte count and myeloblasts percentage in AML patients with normal karyotype]. [DOI] [PubMed] [Google Scholar]
- 59.Gjertsen B.T., Oyan A.M., Marzolf B., Hovland R., Gausdal G., Doskeland S.O., Dimitrov K., Golden A., Kalland K.H., Hood L., Bruserud O. Analysis of acute myelogenous leukemia: Preparation of samples for genomic and proteomic analyses. J. Hematother. Stem Cell Res. 2002;11(3):469–481. doi: 10.1089/15258160260090933. [DOI] [PubMed] [Google Scholar]
- 60.Tibes R., Qiu Y., Lu Y., Hennessy B., Andreeff M., Mills G., Kornblau S. Reverse phase protein array: Validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol. Cancer Ther. 2006;5(10):2512–2521. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- 61.Fry L., Giner S., Gomez S., Green M., Anderson S., Horder J., McArdle S., Rees R., Madrigal J. Avoiding room temperature storage and delayed cryopreservation provide better postthaw potency in hematopoietic progenitor cell grafts. Transfusion. 2013;53(8):1834–1842. doi: 10.1111/trf.12006. [DOI] [PubMed] [Google Scholar]
- 62.Germann A., Oh Y-J., Schmidt T., Schön U., Zimmermann H., von Briesen H. Temperature fluctuations during deep temperature cryopreservation reduce PBMC recovery, viability and T-cell function. Cryobiology. 2013;67(2):193–200. doi: 10.1016/j.cryobiol.2013.06.012. [DOI] [PubMed] [Google Scholar]
- 63.Bach E., Krahl R., Lange T., Schuler F., Al-Ali H., Buchner T., Haferlach T., Dolken G., Niederwieser D., Cross M. Delayed processing of bone marrow samples reveals a prognostic pattern of NME mRNA expression in cytogenetically normal acute myeloid leukemia. Leuk. Lymphoma. 2012;53(8):1561–1568. doi: 10.3109/10428194.2012.676176. [DOI] [PubMed] [Google Scholar]
- 64.Abbas A.K., Lichtman A.H., Pillai S. Cellular and molecular immunology Seventh . Philadelphia, : Elsevier Saunders:; 2012. [Google Scholar]
- 65.Luczak M., Kaźmierczak M., Handschuh L., Lewandowski K., Komarnicki M., Figlerowicz M. Comparative proteome analysis of acute myeloid leukemia with and without maturation. J. Proteomics. 2012;75(18):5734–5748. doi: 10.1016/j.jprot.2012.07.030. [DOI] [PubMed] [Google Scholar]
- 66.Rezaei A., Adib M., Mokarian F., Tebianian M., Nassiri R. Leukemia markers expression of peripheral blood vs bone marrow blasts using flow cytometry. Med. Sci. Monit. 2003;9(8):62. [PubMed] [Google Scholar]
- 67.Gemmell M.A., Anderson N.L. Lymphocyte, monocyte, and granulocyte proteins compared by use of two-dimensional electrophoresis. Clin. Chem. 1982;28(4 Pt 2):1062–1066. [PubMed] [Google Scholar]
- 68.Cui J.W., Wang J., He K., Jin B.F., Wang H.X., Li W., Kang L.H., Hu M.R., Li H.Y., Yu M., Shen B.F., Wang G.J., Zhang X.M. Two-dimensional electrophoresis protein profiling as an analytical tool for human acute leukemia classification. Electrophoresis. 2005;26(1):268–279. doi: 10.1002/elps.200406124. [DOI] [PubMed] [Google Scholar]
- 69.Gundry R.L., White M.Y., Murray C.I., Kane L.A., Fu Q., Stanley B.A., Van Eyk J.E. Curr. Protoc. Mol. Biol., 2009, Chapter 10, 2009. Preparation of proteins and peptides for mass spectrometry analysis in a bottom-up proteomics workflow. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leon I.R., Schwammle V., Jensen O.N., Sprenger R.R. Quantitative assessment of in-solution digestion efficiency identifies optimal protocols for unbiased protein analysis. Mol. Cell. Proteomics. 2013;12(10):2992–3005. doi: 10.1074/mcp.M112.025585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Manza L.L., Stamer S.L., Ham A.J., Codreanu S.G., Liebler D.C. Sample preparation and digestion for proteomic analyses using spin filters. Proteomics. 2005;5(7):1742–1745. doi: 10.1002/pmic.200401063. [DOI] [PubMed] [Google Scholar]
- 72.Wisniewski J.R., Zougman A., Nagaraj N., Mann M. Universal sample preparation method for proteome analysis. Nat. Methods. 2009;6(5):359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 73.Glatter T., Ludwig C., Ahrne E., Aebersold R., Heck A.J., Schmidt A. Large-scale quantitative assessment of different in-solution protein digestion protocols reveals superior cleavage efficiency of tandem Lys-C/trypsin proteolysis over trypsin digestion. J. Proteome Res. 2012;11(11):5145–5156. doi: 10.1021/pr300273g. [DOI] [PubMed] [Google Scholar]
- 74.Wisniewski J.R., Mann M. Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal. Chem. 2012;84(6):2631–2637. doi: 10.1021/ac300006b. [DOI] [PubMed] [Google Scholar]
- 75.Wisniewski J.R., Rakus D. Multi-enzyme digestion FASP and the 'Total Protein Approach'-based absolute quantification of the Escherichia coli proteome. J. Proteomics. 2014;109:322–331. doi: 10.1016/j.jprot.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 76.Erde J., Loo R.R., Loo J.A. Enhanced FASP (eFASP) to increase proteome coverage and sample recovery for quantitative proteomic experiments. J. Proteome Res. 2014;13(4):1885–1895. doi: 10.1021/pr4010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nel A.J., Garnett S., Blackburn J.M., Soares N.C. Comparative reevaluation of FASP and enhanced FASP methods by LC-MS/MS. J. Proteome Res. 2015 doi: 10.1021/pr501266c. [DOI] [PubMed] [Google Scholar]
- 78.Scheltema R.A., Hauschild J.P., Lange O., Hornburg D., Denisov E., Damoc E., Kuehn A., Makarov A., Mann M. The Q Exactive HF, a Benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Mol. Cell. Proteomics. 2014;13(12):3698–3708. doi: 10.1074/mcp.M114.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kelstrup C.D., Jersie-Christensen R.R., Batth T.S., Arrey T.N., Kuehn A., Kellmann M., Olsen J.V. Rapid and deep proteomes by faster sequencing on a benchtop quadrupole ultra-high-field Orbitrap mass spectrometer. J. Proteome Res. 2014;13(12):6187–6195. doi: 10.1021/pr500985w. [DOI] [PubMed] [Google Scholar]
- 80.Kulak N.A., Pichler G., Paron I., Nagaraj N., Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods. 2014;11(3):319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- 81.Rappsilber J., Ishihama Y., Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003;75(3):663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- 82.Issaq H.J., Conrads T.P., Janini G.M., Veenstra T.D. Methods for fractionation, separation and profiling of proteins and peptides. Electrophoresis. 2002;23(17):3048–3061. doi: 10.1002/1522-2683(200209)23:17<3048::AID-ELPS3048>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 83.Gan C.S., Reardon K.F., Wright P.C. Comparison of protein and peptide prefractionation methods for the shotgun proteomic analysis of Synechocystis sp. PCC 6803. Proteomics. 2005;5(9):2468–2478. doi: 10.1002/pmic.200401266. [DOI] [PubMed] [Google Scholar]
- 84.Manadas B., Mendes V.M., English J., Dunn M.J. Peptide fractionation in proteomics approaches. Expert Rev. Proteomics. 2010;7(5):655–663. doi: 10.1586/epr.10.46. [DOI] [PubMed] [Google Scholar]
- 85.Wasinger V.C., Zeng M., Yau Y. Current status and advances in quantitative proteomic mass spectrometry. . Int. J. Proteomics, 2013. [DOI] [PMC free article] [PubMed]
- 86.Geiger T., Cox J., Ostasiewicz P., Wisniewski J., Mann M. Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat. Methods. 2010;7(5):383–385. doi: 10.1038/nmeth.1446. [DOI] [PubMed] [Google Scholar]
- 87.Simpson K.L., Whetton A.D., Dive C. Quantitative mass spectrometry-based techniques for clinical use: biomarker identification and quantification. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009;877(13):1240–1249. doi: 10.1016/j.jchromb.2008.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Geisler C., Gaisa N.T., Pfister D., Fuessel S., Kristiansen G., Braunschweig T., Gostek S., Beine B., Diehl H.C., Jackson A.M., Borchers C.H., Heidenreich A., Meyer H.E., Knuchel R., Henkel C. Identification and validation of potential new biomarkers for prostate cancer diagnosis and prognosis Using 2D-DIGE and MS. . Biomed. Res. Int. 2015. [DOI] [PMC free article] [PubMed]
- 89.Peterson A.C., Russell J.D., Bailey D.J., Westphall M.S., Coon J.J. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteomics. 2012;11(11):1475–1488. doi: 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Witze E.S., Old W.M., Resing K.A., Ahn N.G. Mapping protein post-translational modifications with mass spectrometry. Nat. Methods. 2007;4(10):798–806. doi: 10.1038/nmeth1100. [DOI] [PubMed] [Google Scholar]
- 91.Krueger K.E., Srivastava S. Posttranslational protein modifications: Current implications for cancer detection, prevention, and therapeutics. Mol. Cell. Proteomics. 2006;5(10):1799–1810. doi: 10.1074/mcp.R600009-MCP200. [DOI] [PubMed] [Google Scholar]
- 92.Fukushima N., Furuta D., Hidaka Y., Moriyama R., Tsujiuchi T. Post-translational modifications of tubulin in the nervous system. J. Neurochem. 2009;109(3):683–693. doi: 10.1111/j.1471-4159.2009.06013.x. [DOI] [PubMed] [Google Scholar]
- 93.Silva A.M., Vitorino R., Domingues M.R., Spickett C.M., Domingues P. Post-translational modifications and mass spectrometry detection. Free Radic. Biol. Med. 2013;65:925–941. doi: 10.1016/j.freeradbiomed.2013.08.184. [DOI] [PubMed] [Google Scholar]
- 94.Manning G., Whyte D.B., Martinez R., Hunter T., Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 95.Ficarro S.B., McCleland M.L., Stukenberg P.T., Burke D.J., Ross M.M., Shabanowitz J., Hunt D.F., White F.M. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 2002;20(3):301–305. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- 96.Beltran L., Cutillas P.R. Advances in phosphopeptide enrichment techniques for phosphoproteomics. Amino Acids. 2012;43(3):1009–1024. doi: 10.1007/s00726-012-1288-9. [DOI] [PubMed] [Google Scholar]
- 97.Huang J., Wang F., Ye M., Zou H. Enrichment and separation techniques for large-scale proteomics analysis of the protein post-translational modifications. J. Chromatogr. A. 2014;1372C:1–17. doi: 10.1016/j.chroma.2014.10.107. [DOI] [PubMed] [Google Scholar]
- 98.Chen Y.A., Eschrich S.A. Computational methods and opportunities for phosphorylation network medicine. Transl. Cancer Res. 2014;3(3):266–278. [PMC free article] [PubMed] [Google Scholar]
- 99.Sharma K., D'Souza R.C., Tyanova S., Schaab C., Wisniewski J.R., Cox J., Mann M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Reports. 2014;8(5):1583–1594. doi: 10.1016/j.celrep.2014.07.036. [DOI] [PubMed] [Google Scholar]
- 100.Thingholm T.E., Jensen O.N., Robinson P.J., Larsen M.R. SIMAC (sequential elution from IMAC), a phosphoproteomics strategy for the rapid separation of monophosphorylated from multiply phosphorylated peptides. Mol. Cell. Proteomics. 2008;7(4):661–671. doi: 10.1074/mcp.M700362-MCP200. [DOI] [PubMed] [Google Scholar]
- 101.Zhou H., Ye M., Dong J., Corradini E., Cristobal A., Heck A.J., Zou H., Mohammed S. Robust phosphoproteome enrichment using monodisperse microsphere-based immobilized titanium (IV) ion affinity chromatography. Nat. Protoc. 2013;8(3):461–480. doi: 10.1038/nprot.2013.010. [DOI] [PubMed] [Google Scholar]
- 102.Zhang L., Zhao Q., Liang Z., Yang K., Sun L., Zhang L., Zhang Y. Synthesis of adenosine functionalized metal immobilized magnetic nanoparticles for highly selective and sensitive enrichment of phosphopeptides. Chem. Commun. (Camb.) 2012;48(50):6274–6276. doi: 10.1039/c2cc31641b. [DOI] [PubMed] [Google Scholar]
- 103.Wisniewski J.R., Nagaraj N., Zougman A., Gnad F., Mann M. Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J. Proteome Res. 2010;9(6):3280–3289. doi: 10.1021/pr1002214. [DOI] [PubMed] [Google Scholar]
- 104.Engholm-Keller K., Birck P., Storling J., Pociot F., Mandrup-Poulsen T., Larsen M.R. TiSH--a robust and sensitive global phosphoproteomics strategy employing a combination of TiO2, SIMAC, and HILIC. J. Proteomics. 2012;75(18):5749–5761. doi: 10.1016/j.jprot.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 105.López-Pedrera C., Villalba J., Siendones E., Barbarroja N., Gómez-Díaz C., Rodríguez-Ariza A., Buendía P., Torres A., Velasco F. Proteomic analysis of acute myeloid leukemia: Identification of potential early biomarkers and therapeutic targets. Proteomics. 2006;6(Suppl. 1):9. doi: 10.1002/pmic.200500384. [DOI] [PubMed] [Google Scholar]
- 106.Braoudaki M., Tzortzatou-Stathopoulou F., Anagnostopoulos A., Papathanassiou C., Vougas K., Karamolegou K., Tsangaris G. Proteomic analysis of childhood de novo acute myeloid leukemia and myelodysplastic syndrome/AML: correlation to molecular and cytogenetic analyses. Amino Acids. 2011;40(3):943–951. doi: 10.1007/s00726-010-0718-9. [DOI] [PubMed] [Google Scholar]
- 107.Chong P., Zhou J., Cheong L-L., Liu S-C., Qian J., Guo T., Sze S., Zeng Q., Chng W. LEO1 is regulated by PRL-3 and mediates its oncogenic properties in acute myelogenous leukemia. Cancer Res. 2014;74(11):3043–3053. doi: 10.1158/0008-5472.CAN-13-2321. [DOI] [PubMed] [Google Scholar]
- 108.Strassberger V., Gutbrodt K., Krall N., Roesli C., Takizawa H., Manz M., Fugmann T., Neri D. A comprehensive surface proteome analysis of myeloid leukemia cell lines for therapeutic antibody development. J. Proteomics. 2014;99:138–151. doi: 10.1016/j.jprot.2014.01.022. [DOI] [PubMed] [Google Scholar]
- 109.Nicolas E., Ramus C., Berthier S., Arlotto M., Bouamrani A., Lefebvre C., Morel F., Garin J., Ifrah N., Berger F., Cahn J.Y., Mossuz P. Expression of S100A8 in leukemic cells predicts poor survival in de novo AML patients. Leukemia. 2011;25(1):57–65. doi: 10.1038/leu.2010.251. [DOI] [PubMed] [Google Scholar]
- 110.Oellerich T., Mohr S., Corso J., Beck J., Döbele C., Braun H., Cremer A., Münch S., Wicht J., Oellerich M., Bug G., Bohnenberger H., Perske C., Schütz E., Urlaub H., Serve H. FLT3-ITD and TLR9 employ Bruton's tyrosine kinase to activate distinct transcriptional programs mediating AML cell survival and proliferation. Blood. 2015 doi: 10.1182/blood-2014-06-585216. [DOI] [PubMed] [Google Scholar]
- 111.Zhang Y., Askenazi M., Jiang J., Luckey C., Griffin J., Marto J. A robust error model for iTRAQ quantification reveals divergent signaling between oncogenic FLT3 mutants in acute myeloid leukemia. Mol. Cell. Proteomics. 2010;9(5):780–790. doi: 10.1074/mcp.M900452-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pulikkan J., Dengler V., Peer Zada A., Kawasaki A., Geletu M., Pasalic Z., Bohlander S., Ryo A., Tenen D., Behre G. Elevated PIN1 expression by C/EBPalpha-p30 blocks C/EBPalpha-induced granulocytic differentiation through c-Jun in AML. Leukemia. 2010;24(5):914–923. doi: 10.1038/leu.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kaźmierczak M., Luczak M., Lewandowski K., Handschuh L., Czyż A., Jarmuż M., Gniot M., Michalak M., Figlerowicz M., Komarnicki M. Medical Oncol. England: 2013. Esterase D and gamma 1 actin level might predict results of induction therapy in patients with acute myeloid leukemia without and with maturation. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Buchi F., Spinelli E., Masala E., Gozzini A., Sanna A., Bosi A., Ferrari G., Santini V. Proteomic analysis identifies differentially expressed proteins in AML1/ETO acute myeloid leukemia cells treated with DNMT inhibitors azacitidine and decitabine. Leuk. Res. 2012;36(5):607–618. doi: 10.1016/j.leukres.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 115.Casado P., Rodriguez-Prados J.C., Cosulich S.C., Guichard S., Vanhaesebroeck B., Joel S., Cutillas P.R. Kinase-substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci. Signal. 2013;6(268):rs6. doi: 10.1126/scisignal.2003573. [DOI] [PubMed] [Google Scholar]
- 116.Walters D., Goss V., Stoffregen E., Gu T-L., Lee K., Nardone J., McGreevey L., Heinrich M., Deininger M., Polakiewicz R., Druker B. Phosphoproteomic analysis of AML cell lines identifies leukemic oncogenes. Leuk. Res. 2006;30(9):1097–1104. doi: 10.1016/j.leukres.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 117.Hahn C., Berchuck J., Ross K., Kakoza R., Clauser K., Schinzel A., Ross L., Galinsky I., Davis T., Silver S., Root D., Stone R., DeAngelo D., Carroll M., Hahn W., Carr S., Golub T., Kung A., Stegmaier K. Proteomic and genetic approaches identify Syk as an AML target. Cancer Cell. 2009;16(4):281–294. doi: 10.1016/j.ccr.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gu T-l., Nardone J., Wang Y., Loriaux M., Villén J., Beausoleil S., Tucker M., Kornhauser J., Ren J., MacNeill J., Gygi S., Druker B., Heinrich M., Rush J., Polakiewicz R. Survey of activated FLT3 signaling in leukemia. PLoS One. 2011;6(4) doi: 10.1371/journal.pone.0019169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Weber C., Schreiber T.B., Daub H. Dual phosphoproteomics and chemical proteomics analysis of erlotinib and gefitinib interference in acute myeloid leukemia cells. J. Proteomics. 2012;75(4):1343–1356. doi: 10.1016/j.jprot.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 120.Xu Y., Zhuo J., Duan Y., Shi B., Chen X., Zhang X., Xiao L., Lou J., Huang R., Zhang Q., Du X., Li M., Wang D., Shi D. Construction of protein profile classification model and screening of proteomic signature of acute leukemia. Int. J. Clin. Experiment. Pathol. 2014;7(9):5569–5581. [PMC free article] [PubMed] [Google Scholar]
- 121.Chen Y., Kantarjian H., Pierce S., Faderl S., O'Brien S., Qiao W., Abruzzo L., de Lima M., Kebriaei P., Jabbour E., Daver N., Kadia T., Estrov Z., Garcia-Manero G., Cortes J., Ravandi F. Prognostic significance of 11q23 aberrations in adult acute myeloid leukemia and the role of allogeneic stem cell transplantation. Leukemia. 2013;27(4):836–842. doi: 10.1038/leu.2012.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Grimwade D., Walker H., Harrison G., Oliver F., Chatters S., Harrison C., Wheatley K., Burnett A., Goldstone A. Medical Research Council Adult Leukemia Working, P., The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood. 2001;98(5):1312–1320. doi: 10.1182/blood.v98.5.1312. [DOI] [PubMed] [Google Scholar]
- 123.Elo L., Karjalainen R., Ohman T., Hintsanen P., Nyman T., Heckman C., Aittokallio T. Statistical detection of quantitative protein biomarkers provides insights into signaling networks deregulated in acute myeloid leukemia. Proteomics. 2014;14(21-22):2443–2453. doi: 10.1002/pmic.201300460. [DOI] [PubMed] [Google Scholar]
- 124.Byrd J.C., Furman R.R., Coutre S.E., Flinn I.W., Burger J.A., Blum K.A., Grant B., Sharman J.P., Coleman M., Wierda W.G., Jones J.A., Zhao W., Heerema N.A., Johnson A.J., Sukbuntherng J., Chang B.Y., Clow F., Hedrick E., Buggy J.J., James D.F., O'Brien S. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2013;369(1):32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Preudhomme C., Sagot C., Boissel N., Cayuela J.M., Tigaud I., de Botton S., Thomas X., Raffoux E., Lamandin C., Castaigne S., Fenaux P., Dombret H., Group A. Favorable prognostic significance of CEBPA mutations in patients with de novo acute myeloid leukemia: a study from the Acute Leukemia French Association (ALFA). Blood. 2002;100(8):2717–2723. doi: 10.1182/blood-2002-03-0990. [DOI] [PubMed] [Google Scholar]
- 126.Gombart A.F., Hofmann W.K., Kawano S., Takeuchi S., Krug U., Kwok S.H., Larsen R.J., Asou H., Miller C.W., Hoelzer D., Koeffler H.P. Mutations in the gene encoding the transcription factor CCAAT/enhancer binding protein alpha in myelodysplastic syndromes and acute myeloid leukemias. Blood. 2002;99(4):1332–1340. doi: 10.1182/blood.v99.4.1332. [DOI] [PubMed] [Google Scholar]
- 127.Fischer T., Stone R.M., Deangelo D.J., Galinsky I., Estey E., Lanza C., Fox E., Ehninger G., Feldman E.J., Schiller G.J., Klimek V.M., Nimer S.D., Gilliland D.G., Dutreix C., Huntsman-Labed A., Virkus J., Giles F.J. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J. Clin. Oncol. 2010;28(28):4339–4345. doi: 10.1200/JCO.2010.28.9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Stone R.M., Fischer T., Paquette R., Schiller G., Schiffer C.A., Ehninger G., Cortes J., Kantarjian H.M., DeAngelo D.J., Huntsman-Labed A., Dutreix C., del Corral A., Giles F. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia. 2012;26(9):2061–2068. doi: 10.1038/leu.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Smith B.D., Levis M., Beran M., Giles F., Kantarjian H., Berg K., Murphy K.M., Dauses T., Allebach J., Small D. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103(10):3669–3676. doi: 10.1182/blood-2003-11-3775. [DOI] [PubMed] [Google Scholar]
- 130.Konig H., Levis M. Targeting FLT3 to treat leukemia. Expert Opin. Ther. Targets. 2015;19(1):37–54. doi: 10.1517/14728222.2014.960843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Forshed J., Pernemalm M., Tan C.S., Lindberg M., Kanter L., Pawitan Y., Lewensohn R., Stenke L., Lehtio J. Proteomic data analysis workflow for discovery of candidate biomarker peaks predictive of clinical outcome for patients with acute myeloid leukemia. J. Proteome Res. 2008;7(6):2332–2341. doi: 10.1021/pr070482e. [DOI] [PubMed] [Google Scholar]
- 132.Casado P., Alcolea M.P., Iorio F., Rodriguez-Prados J.C., Vanhaesebroeck B., Saez-Rodriguez J., Joel S., Cutillas P.R. Phosphoproteomics data classify hematological cancer cell lines according to tumor type and sensitivity to kinase inhibitors. Genome Biol. 2013;14(4):R37. doi: 10.1186/gb-2013-14-4-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cortes J.E., Dombret H., Kayser S., Steffen B., Rousselot P., Martinelli G., Estey E.H., Burnett A.K., Gammon G., Trone D., Leo E., Levis M.J. Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in Patients 60 Years of Age with FLT3 ITD positive or negative relapsed/ refractory acute myeloid leukemia. Blood, (ASH Annual Meeting Abstracts) 2012, 120, Abstract 48. ; 2012. [Google Scholar]
- 134.Yin B., Delwel R., Valk P.J., Wallace M.R., Loh M.L., Shannon K.M., Largaespada D.A. A retroviral mutagenesis screen reveals strong cooperation between Bcl11a overexpression and loss of the Nf1 tumor suppressor gene. Blood. 2009;113(5):1075–1085. doi: 10.1182/blood-2008-03-144436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dai F., Lin X., Chang C., Feng X.H. Nuclear export of Smad2 and Smad3 by RanBP3 facilitates termination of TGF-beta signaling. Dev. Cell. 2009;16(3):345–357. doi: 10.1016/j.devcel.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Abeel T., Helleputte T., Van de Peer Y., Dupont P., Saeys Y. Robust biomarker identification for cancer diagnosis with ensemble feature selection methods. Bioinformatics. 2010;26(3):392–398. doi: 10.1093/bioinformatics/btp630. [DOI] [PubMed] [Google Scholar]
- 137.Skates S., Gillette M., LaBaer J., Carr S., Anderson L., Liebler D., Ransohoff D., Rifai N., Kondratovich M., Težak Ž., Mansfield E., Oberg A., Wright I., Barnes G., Gail M., Mesri M., Kinsinger C., Rodriguez H., Boja E. Statistical design for biospecimen cohort size in proteomics-based biomarker discovery and verification studies. J. Proteome Res. 2013;12(12):5383–5394. doi: 10.1021/pr400132j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Polanski M., Anderson N.L. A list of candidate cancer biomarkers for targeted proteomics. Biomark. Insights. 2007;1:1–48. [PMC free article] [PubMed] [Google Scholar]
- 139.Kroksveen A.C., Opsahl J.A., Guldbrandsen A., Myhr K.M., Oveland E., Torkildsen O., Berven F.S. Cerebrospinal fluid proteomics in multiple sclerosis. Biochim. et Biophysic. Acta, 2014. [DOI] [PubMed]
- 140.Alcolea M.P., Casado P., Rodriguez-Prados J.C., Vanhaesebroeck B., Cutillas P.R. Phosphoproteomic analysis of leukemia cells under basal and drug-treated conditions identifies markers of kinase pathway activation and mechanisms of resistance. Mol. Cell. Proteomics. 2012;11(8):453–466. doi: 10.1074/mcp.M112.017483. [DOI] [PMC free article] [PubMed] [Google Scholar]