

ABSTRACT
Maltodextrin is a mixture of maltooligosaccharides, which are produced by the degradation of starch or glycogen. They are mostly composed of α-1,4- and some α-1,6-linked glucose residues. Genes presumed to code for the Enterococcus faecalis maltodextrin transporter were induced during enterococcal infection. We therefore carried out a detailed study of maltodextrin transport in this organism. Depending on their length (3 to 7 glucose residues), E. faecalis takes up maltodextrins either via MalT, a maltose-specific permease of the phosphoenolpyruvate (PEP):carbohydrate phosphotransferase system (PTS), or the ATP binding cassette (ABC) transporter MdxEFG-MsmX. Maltotriose, the smallest maltodextrin, is primarily transported by the PTS permease. A malT mutant therefore exhibits significantly reduced growth on maltose and maltotriose. The residual uptake of the trisaccharide is catalyzed by the ABC transporter, because a malT mdxF double mutant no longer grows on maltotriose. The trisaccharide arrives as maltotriose-6″-P in the cell. MapP, which dephosphorylates maltose-6′-P, also releases Pi from maltotriose-6″-P. Maltotetraose and longer maltodextrins are mainly (or exclusively) taken up via the ABC transporter, because inactivation of the membrane protein MdxF prevents growth on maltotetraose and longer maltodextrins up to at least maltoheptaose. E. faecalis also utilizes panose and isopanose, and we show for the first time, to our knowledge, that in contrast to maltotriose, its two isomers are primarily transported via the ABC transporter. We confirm that maltodextrin utilization via MdxEFG-MsmX affects the colonization capacity of E. faecalis, because inactivation of mdxF significantly reduced enterococcal colonization and/or survival in kidneys and liver of mice after intraperitoneal infection.
IMPORTANCE Infections by enterococci, which are major health care-associated pathogens, are difficult to treat due to their increasing resistance to clinically relevant antibiotics, and new strategies are urgently needed. A largely unexplored aspect is how these pathogens proliferate and which substrates they use in order to grow inside infected hosts. The use of maltodextrins as a source of carbon and energy was studied in Enterococcus faecalis and linked to its virulence. Our results demonstrate that E. faecalis can efficiently use glycogen degradation products. We show here that depending on the length of the maltodextrins, one of two different transporters is used: the maltose-PTS transporter MalT, or the MdxEFG-MsmX ABC transporter. MdxEFG-MsmX takes up longer maltodextrins as well as complex molecules, such as panose and isopanose.
KEYWORDS: enterococci, maltodextrin, phosphotransferase system, ABC transporter, host colonization
INTRODUCTION
Maltose, maltotriose, and higher maltodextrins represent important sources of carbon and energy for numerous bacteria, including several human pathogens, such as Streptococcus pyogenes (1), Streptococcus pneumoniae (2), Listeria monocytogenes (3), and Neisseria meningitidis (4). Maltose and maltodextrins are produced by the degradation of starch or glycogen and are therefore abundant in decaying plants and in the intestine and oral cavity of humans and many animals. Maltose catabolism in Enterococcus faecalis requires the proteins encoded by the five genes malP, pgcM, malM, malT, and mapP (5–7). The first three genes are organized in an operon, and they encode a maltose phosphorylase, a β-phosphoglucomutase, and an aldose-1-epimerase, respectively. The malT gene encodes a maltose-specific permease belonging to the glucose family of phosphoenolpyruvate (PEP):carbohydrate phosphotransferase system (PTS) transporters; it has the domain order EIICBAMal (6, 7). The malT gene is located upstream from malP but oriented in the opposite direction. MalT and the two general cytoplasmic PTS components EI and HPr form a phosphorylation cascade in order to catalyze the uptake and phosphorylation of maltose. EI autophosphorylates at a conserved histidine by using PEP as a phosphoryl donor (8). The phosphoryl group is then transferred via His-15 in HPr (9) and presumably His-645 in the EIIA domain (10) to Cys-492 in the EIIB domain of MalT. In the last step, the P∼Cys-EIIBMal domain donates its phosphoryl group to a maltose molecule bound to the membrane-spanning EIICMal domain. Phosphorylation at the 6′ position of the disaccharide probably lowers its affinity for the EIICMal domain, and maltose-6′-P is released into the cytoplasm (11).
E. faecalis uses MapP, the gene of which is located just downstream from malT, to dephosphorylate maltose-6′-P formed during PTS-catalyzed transport (7). Subsequently, MalP phosphorolyzes intracellular maltose to α-d-glucose and glucose-1-P. The β-phosphoglucomutase PgcM transforms glucose-1-P into glucose-6-P (7).
In addition to the PTS permease MalT, enterococci and streptococci contain an ATP binding cassette (ABC) transporter, which takes up maltooligosaccharides (2, 12, 13) and, in some organisms, maltose as well (14, 15). In E. faecalis, the residual maltose uptake activity observed for the malT mutant was proposed to be mediated by the ABC transporter (6, 7). The maximum chain length of maltodextrins taken up via ABC transporters varies between 8 and 10 (16). The maltodextrin-specific ABC transporters are composed of the substrate binding protein MdxE and the two integral membrane proteins MdxF and MdxG. Their genes are organized in an operon (Fig. 1). The MdxEFG transporter is completed by an ATP binding protein called either MalK or MsmX. MsmX serves as ATP hydrolase for other ABC transport systems (17) and, in contrast to malK, its gene is usually not associated with the mdxEFG operon. Interestingly, the E. faecalis genes presumed to encode MdxEFG and MsmX were recently reported to be strongly induced during E. faecalis infection using a mouse peritonitis model (18).
FIG 1.

The E. faecalis JH2-2 chromosomal region containing the mdxEFG and the upstream operon, which both contain genes required for maltodextrin utilization. (A) Presented are the three genes of the mdxEFG operon and the three upstream genes. They encode the two membrane components of the ABC transporter and the periplasmic binding protein and two α-glucosidases and a glucosyl transferase, respectively (P. Joyet, N. Sauvageot, A. Hartke, and J. Deutscher, unpublished data). The two operons are transcribed from two divergent promoters (small arrows). (B) A major part of our JH2-2 stock carries a mutation in the intergenic region leading to the replacement of the T in the sixth position of the −10 promoter region with a G (dashed box). The −10 promoter region of the mdxEFG operon is underlined, the transcription initiation point is marked with a small arrow, and the ribosome binding site (RBS) is written in italics. The start codon of mdxE is written in bold letters. The T-to-G mutation prevents the utilization of maltotetraose and longer maltooligosaccharides, as well as of panose and isopanose. The E. faecalis strain V583 and all other strains for which the genome has been completely sequenced, have the CATAAT −10 promoter sequence for the mdxEFG operon. However, these strains lack the T directly following the −10 promoter in JH2-2.
While most Enterococcus and Streptococcus species seem to primarily use the PTS permease MalT and the maltose-6′-P phosphatase MapP for the uptake and first catabolic step of maltose, the transport of maltotriose and maltotetraose, the two smallest maltooligosaccharides, seems to vary from one species to another. For example, S. pyogenes takes up maltose, maltotriose, and higher maltooligosaccharides mainly via an efficient ABC transport system. A mutant defective in MalE grew slowly on maltose and maltotriose (1, 19). The slow uptake of these two compounds is catalyzed by MalT. In contrast to S. pyogenes, the Streptococcus mutans PTS permease MalT (SMU.2046c) was reported to efficiently transport maltose and maltotriose and to also contribute to maltotetraose uptake. Deletion of malT prevented utilization of maltose, strongly diminished maltotriose consumption, and significantly slowed growth on maltotetraose (20). Finally, S. pneumoniae takes up maltose primarily via the PTS permease MalT (SP0758), while maltotriose is transported with similar efficiencies by MalT and the ABC transporter MalXCD. Maltooligosaccharides are almost exclusively taken up by the ABC transporter. The residual maltose uptake observed for the malT mutant is not catalyzed by the ABC transporter (2).
The variations in maltotriose and maltotetraose uptake in the three described streptococci, which use the same set of transport enzymes, prompted us to carry out a detailed study of maltooligosaccharide transport in E. faecalis. We found that maltotriose is mainly taken up via the enterococcal PTS permease MalT and that maltotriose-6″-P is also dephosphorylated by the phosphatase MapP. In contrast, maltotetraose and higher maltodextrins are mainly taken up via the ABC transporter MdxEFG, which also catalyzes the transport of panose and isopanose, two maltotriose isomers formed during glycogen degradation. Interestingly, genes encoding enzymes involved in maltodextrin metabolism were reported to be strongly expressed during mouse soft tissue infection with S. pyogenes (21). A similar observation was recently made during intraperitoneal infection of mice with E. faecalis (18), suggesting that maltodextrin metabolism might be important for E. faecalis virulence. We here confirm this hypothesis by demonstrating that deletion of the ABC transporter but not of the PTS permease MalT significantly reduced mouse organ colonization of E. faecalis.
RESULTS AND DISCUSSION
E. faecalis transports maltotriose mainly via the PTS permease MalT.
In order to determine to which extent the ABC and the PTS transport systems of E. faecalis contribute to the uptake of maltotriose, the smallest maltodextrin, we carried out growth studies with mutants devoid of either the maltose-specific PTS permease MalT or the ABC transport protein MdxF. We observed that the wild-type strain grew with similar efficiencies on maltotriose, maltose, and glucose. In contrast, the malT mutant grew significantly slower on maltose and maltotriose (Fig. 2). These results therefore suggest that E. faecalis transports and phosphorylates the smallest maltodextrin mainly via the PTS permease MalT, which has previously been shown to transport and phosphorylate maltose (6, 7). Indeed, a mutant deleted for mdxF, which encodes one of the transmembrane components of the ABC transporter, exhibited a growth rate on maltotriose similar to that of the wild-type strain (compare Fig. 2 and 3). We also constructed a malT mdxF double mutant and tested its growth on the three above-mentioned carbon sources. The double mutant grew normally on glucose but was no longer able to grow on maltotriose (Fig. 3), indicating that the ABC transporter MdxEFG-MsmX is responsible for the residual growth on the trisaccharide observed for the malT mutant (Fig. 2). In contrast, the slow growth on maltose observed for the malT mutant (Fig. 2) was not altered in the malT mdxF double mutant (Fig. 3). It therefore seems that an unknown carbohydrate transporter, possibly an ABC transporter or a PTS permease, is responsible for the residual growth on maltose observed for the malT mutant. An identical observation was made for S. pneumoniae, where, similarly to the malT single mutant, the malT mdxE double mutant was also able to grow slowly on maltose (2). However, in contrast to S. pneumoniae, which takes up maltotriose equally well by MalT and the ABC transporter, E. faecalis transports the trisaccharide primarily by the PTS and only slowly by MdxEFG-MsmX.
FIG 2.
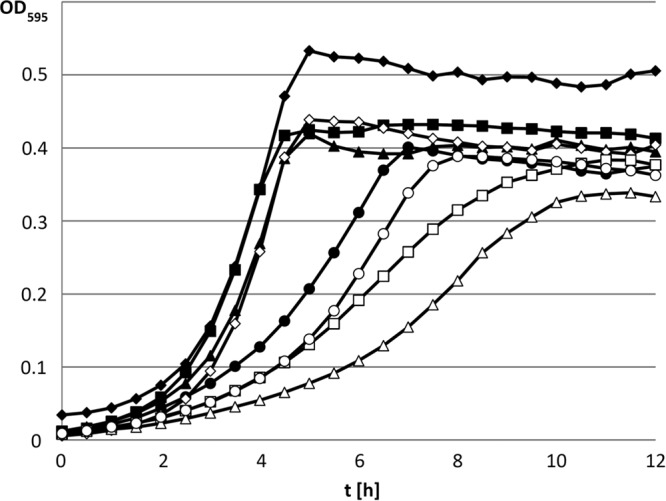
Growth studies with the maltotetraose-positive E. faecalis strain JH2-2 (filled symbols) and the malT mutant derived from it (open symbols). Strains were grown in M17cc medium supplemented with 0.3% glucose (diamonds), maltose (squares), maltotriose (triangles), or maltotetraose (circles). Growth studies were carried out as described in Materials and Methods. t, time.
FIG 3.
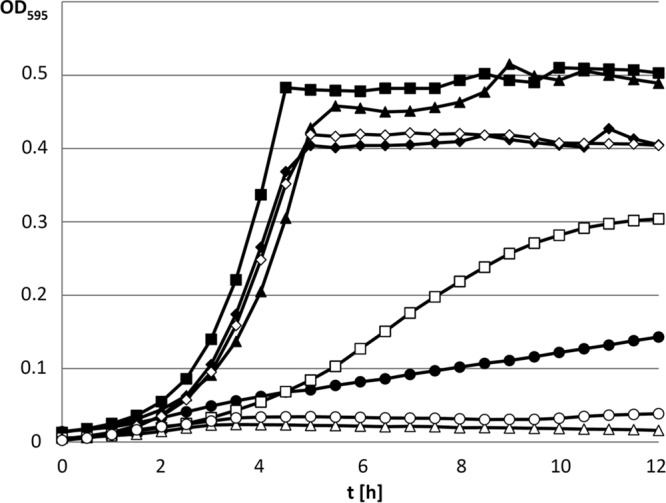
Growth studies with the E. faecalis mdxF mutant (filled symbols) and the malT mdxF double mutant (open symbols). The two strains were grown in M17cc medium supplemented with 0.3% glucose (diamonds), maltose (squares), maltotriose (triangles), or maltotetraose (circles).
E. faecalis transports maltotetraose and higher maltodextrins mainly via the ABC transporter MdxEFG-MsmX.
We also tested whether E. faecalis transports longer maltooligosaccharides ranging from maltotetraose to maltoheptaose as well as the mixture of oligosaccharides present in a commercially available maltodextrin via the PTS permease MalT or the ABC transporter MdxEFG-MsmX. Growth studies revealed that the wild-type E. faecalis strain JH2-2 is able to utilize maltotetraose (Fig. 2) and maltopentaose, maltohexaose, and maltoheptaose (Fig. 4), as well as maltodextrin (data not shown). However, growth on maltotetraose and longer maltooligosaccharides was significantly slower than growth on glucose, maltose, or maltotriose. Compared to JH2-2, the mdxF mutant grew significantly slower on maltotetraose (Fig. 3), maltodextrin (data not shown), and maltopentaose (Fig. 4) and did not grow at all on maltohexaose and maltoheptaose (Fig. 4). In contrast, the wild-type strain and the malT mutant grew with identical efficiencies on the above-mentioned carbon sources (data not shown). These results establish that E. faecalis takes up maltooligosaccharides composed of more than three glucose residues mainly or exclusively via MdxEFG-MsmX.
FIG 4.
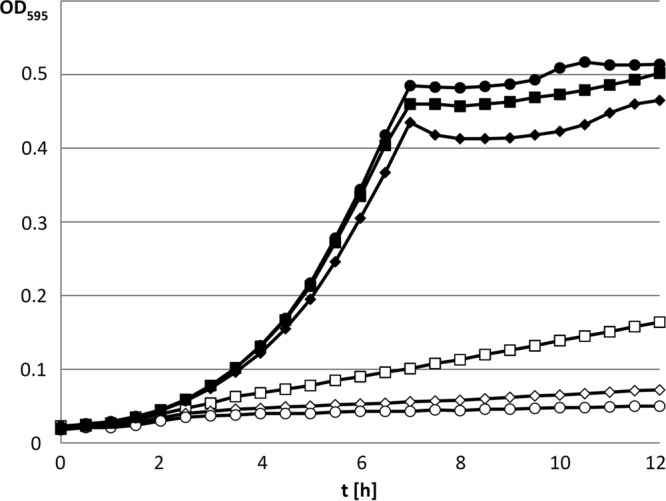
Growth studies with the maltotetraose-positive E. faecalis JH2-2 strain (filled symbols) and the mdxF mutant derived from it (open symbols). Cells were grown in M17cc medium supplemented with 0.3% (wt/vol) maltopentaose (squares), maltohexaose (diamonds), or maltoheptaose (circles).
We also complemented the mdxF mutant with the mdxF wild-type allele cloned into plasmid pAGEnt, in which the inserted genes are expressed from the agmatine-inducible aguB promoter (22). Only slight maltotetraose utilization was observed for the mdxF mutant transformed with empty pAGEnt (Fig. 5). In contrast, when the mdxF mutant was transformed with pAGEnt carrying the mdxF gene, growth on maltotetraose was restored to half the level observed for the wild-type strain.
FIG 5.
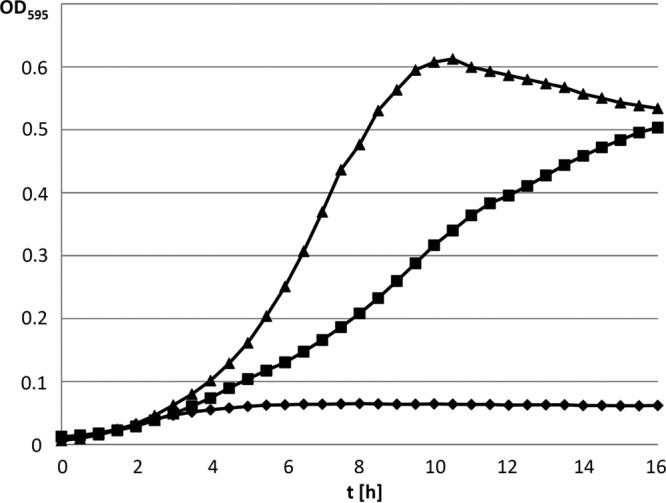
Growth studies with the complemented E. faecalis mdxF mutant. The maltotetraose-positive E. faecalis strain JH2-2 with the empty plasmid pAGEnt (▲) and the mdxF mutant transformed with either the empty plasmid pAGEnt (◆) or pAGEnt carrying the mdxF allele (pAGEnt:66c) (■) were grown in M17cc medium supplemented with 0.3% maltotetraose.
E. faecalis MapP dephosphorylates both maltotriose-6″-P and maltotetraose-6‴-P.
Because the PTS phosphorylates its oligosaccharide substrates at the 6-position of the glycosyl residue at the nonreducing end, maltotriose arrives as maltotriose-6″-P in the cell. Similarly, maltose enters the cytoplasm as maltose-6′-P, and due to the lack of a 6-P-α-glucosidase in enterococci, it is intracellularly dephosphorylated to maltose by the enzyme MapP (6, 7). To investigate whether MapP dephosphorylates the phosphorylated trisaccharide, we synthesized maltotriose-6″-P, as described in Materials and Methods, and carried out mass spectrometry and malachite green assays. Mass spectrometry revealed that MapP indeed dephosphorylates maltotriose-6″-P. The synthesized maltotriose-6″-P sample produced a major peak corresponding to maltotriose-6″-P carrying one Na+ ion (m/z 607.14), a minor peak with the mass of maltotriose-6″-P coordinated with two Na+ ions (m/z 629.12), and a very small peak corresponding to unphosphorylated maltotriose carrying one Na+ ion (m/z 527.16) (Fig. 6A). After incubation for 2 h at 37°C with MapP, the peaks corresponding to maltotriose-6″-P had disappeared, and a strong peak with the m/z value of unphosphorylated maltotriose carrying one Na+ ion became visible (Fig. 6B).
FIG 6.

MapP-catalyzed dephosphorylation of maltotriose-6″-P, as evidenced by mass spectrometry. Dephosphorylation assays of maltotriose-6″-P with E. faecalis MapP and subsequent analysis by mass spectrometry were carried out as described in Materials and Methods. (A) Untreated maltotriose-6″-P: the weak peak with a mass m/z 527.16 corresponds to unphosphorylated maltotriose containing one Na+ ion. The two strongest peaks at m/z 604.14 and 629.12 correspond to maltotriose-6″-P with one [(M+H)+Na+] and two [(M+H)+2Na+] Na+ ions, respectively. (B) MapP-treated maltotriose-6″-P: the two strong peaks in panel A completely disappeared after treatment with MapP, and a very strong single peak appeared at m/z 527.19, which corresponds to maltotriose carrying one Na+ adduct.
Using the semiquantitative malachite green assay, which detects inorganic phosphate (23), we were able to confirm that MapP efficiently dephosphorylates maltotriose-6″-P. The maximum reaction rate was about 80% of that observed with maltose-6′-P (Table 1). It is therefore likely that after its uptake and phosphorylation by the PTS, maltotriose-6″-P is dephosphorylated to intracellular maltotriose. Indeed, an enzyme catalyzing the hydrolysis of unphosphorylated maltotriose to maltose and glucose has been identified (P. Joyet, N. Sauvageot, A. Hartke, and J. Deutscher, unpublished data).
TABLE 1.
Specific activity of MapP determined with the malachite green assay in the presence of its substrates maltose-6′-P, maltotriose-6″-P, and maltotetraose-6‴-P
| Substrate | Sp act (mean ± SD) (μmol · min−1 · mg of protein−1) (%) with substrate at concn: |
|
|---|---|---|
| 2 mM | 4 mM | |
| Maltose-6′-P | 161 ± 15 (100) | 173 ± 28 (100) |
| Maltotriose-6″-P | 123 ± 21 (76) | 141 ± 22 (81) |
| Maltotetraose-6‴-P | 133 ± 17 (82) | 139 ± 18 (80) |
Because MalT of S. mutans had been suggested to catalyze the slow transport of maltotetraose, we hypothesized that the slow growth of the E. faecalis mdxF mutant on maltotetraose (Fig. 3) and maltopentaose (Fig. 4) might be due to their slow uptake by MalT. In fact, only when the malT gene was also deleted did the resulting double mutant completely lose its ability to grow on maltotetraose (Fig. 3). It was therefore likely that similar to maltose-6′-P and maltotriose-6″-P, maltotetraose-6‴-P produced during slow PTS-catalyzed uptake is also dephosphorylated by MapP in order to allow its further catabolism. We therefore also synthesized maltotetraose-6‴-P and tested its dephosphorylation by MapP. Mass spectrometry revealed that similar to what was observed with maltotriose-6″-P, MapP also completely dephosphorylated maltotetraose-6‴-P to maltotetraose carrying one Na+ ion. After incubation for 2 h at 37°C with MapP, the two peaks corresponding to maltotetraose-6‴-P and maltotetraose-6‴-P carrying one Na+ ion disappeared, and a strong peak with the mass of unphosphorylated maltotetraose coordinated with one Na+ ion became visible (data not shown). According to the malachite green assay, dephosphorylation of maltotetraose-6‴-P was slightly slower than dephosphorylation of maltose-6′-P but as fast as the dephosphorylation of maltotriose-6″-P (Table 1). These results support the concept that maltotetraose is not only efficiently taken up by the maltodextrin transporter MdxEFG-MsmX but is also taken up slowly by the PTS permease MalT. In addition, the fast dephosphorylation of maltotetraose-6‴-P by MapP indicates that the rate-limiting step of the observed slow maltotetraose utilization in the mdxF mutant must be the MalT-catalyzed transport and phosphorylation of maltotetraose. The slow utilization of maltopentaose by the mdxF mutant (Fig. 4) might also be mediated by MalT. Maltotetraose (20) and maltopentaose are the longest oligosaccharides suggested to be transported by the PTS.
A spontaneous mutation in the mdxEFG promoter also prevents the utilization of longer maltooligosaccharides.
When carrying out control experiments with E. faecalis JH2-2-derived mutants affected in genes not related to maltose and maltodextrin metabolism, we noticed that most of them did not grow on maltotetraose and higher maltooligosaccharides (data not shown). By carrying out a more extensive study, we observed that only 12.5% of the bacteria in our stock of E. faecalis JH2-2 were able to grow on maltotetraose. We isolated several maltotetraose-positive clones, grew them for about 100 generations in glucose-containing M17 medium, and found that the maltotetraose-positive phenotype was stable. We subsequently sequenced the entire mdxEFG operon and its upstream region in several maltotetraose-positive and -negative clones and found that the only difference between them was a single base modification in the intergenic region between the mdxE gene and the EFT4_1964 gene (Fig. 1). We determined the transcription start site of the mdxEFG operon by rapid amplification of cDNA ends (RACE) PCR and found that the −10 promoter region of the mdxEFG operon was changed from CATAAT in maltotetraose-utilizing strains to CATAAG in strains not able to grow on maltodextrin (Fig. 1). The T-for-G exchange probably strongly decreases the expression of the mdxEFG operon and thus prevents growth on maltotetraose and higher maltooligosaccharides. Quantitative real-time PCR (qRT-PCR) revealed indeed that growth on maltodextrin caused 400- to 1,000-fold stronger induction of the mdxE and mdxF genes in the maltotetraose-positive strain than in the maltotetraose-negative strain (Fig. 7). In the presence of glucose, the strong induction of mdxE and mdxF by maltodextrin was not significantly altered in the maltotetraose-positive strain, indicating that the mdxEFG operon is not submitted to catabolite repression (Fig. 7). To determine whether the capacity of enterococci to grow on maltooligosaccharides is the norm or an exception, we tested 21 different E. faecalis strains and found that they were all able to grow on maltotetraose (data not shown). Only the TX4000 (JH2-2) clone used for genome sequencing (http://www.ncbi.nlm.nih.gov/genome/808?genome_assembly_id=168551) has the CATAAG −10 promoter sequence and should therefore also fail to grow on maltotetraose and higher maltooligosaccharides. A comparison with the genome sequences of different E. faecalis strains revealed that all contain an mdxEFG −10 promoter sequence identical to that of the maltotetraose-positive JH2-2 strain. Finally, it is important to mention that all mutants used in this study were derived from a maltotetraose-positive JH2-2 clone.
FIG 7.
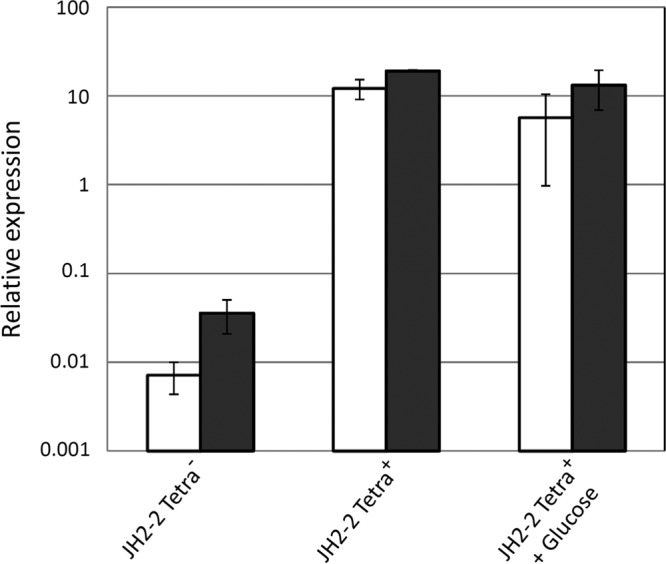
Relative transcription of the mdxE gene (black bars) and mdxF gene (white bars) in maltotetraose-negative (Tetra−) and maltotetraose-positive (Tetra+) JH2-2 clones. The strains were grown in M17cc medium containing either 0.5% maltodextrin or 0.5% maltodextrin plus 0.5% glucose. The mean values calculated from three independent experiments are presented. Standard deviations are indicated by error bars.
E. faecalis transports panose and isopanose via the ABC transporter MdxEFG-MsmX.
Maltodextrins produced from branched-chain polysaccharides, such as starch and glycogen, usually contain linkage isomers of maltotriose, such as panose and isopanose. Panose is also produced in large amounts during the degradation of pullulan by the enzyme neopullulanase (α-amylase family 13) (EC 3.2.1.135) (24). Maltotriose and the maltotriose isomers panose and isopanose differ in the linkages of the three glucose residues. In maltotriose, the three glucose residues are linked with α-1,4 glycosidic bonds. However, in panose, the first and second glucose residues (from the nonreducing end) are linked with an α-1,6 glycosidic bond. In isopanose, the α-1,6 linkage is located between the two glucose residues at the reducing end (25). The E. faecalis strain V583 has been shown to grow on panose (26), and several other bacteria were reported to utilize both panose and isopanose (27–29). We found that maltotetraose-positive E. faecalis JH2-2 can also grow on both maltotriose isomers (Fig. 8). However, in contrast to maltotriose, its two linkage isomers are not taken up by the PTS permease MalT but are transported by the MdxEFG-MsmX ABC transporter. While the malT mutant grew with efficiency similar to that of the wild-type strain on panose and isopanose, the mdxF mutant was not able to utilize the two trisaccharides (Fig. 8A), indicating that the ABC transporter has a broad substrate specificity which is not limited to α-1,4 glycosidic linear forms of polysaccharides. Therefore, we propose that the function of this ABC transporter is dedicated to recovery of the products of degradation from starch or, in a context of infection, from glycogen.
FIG 8.
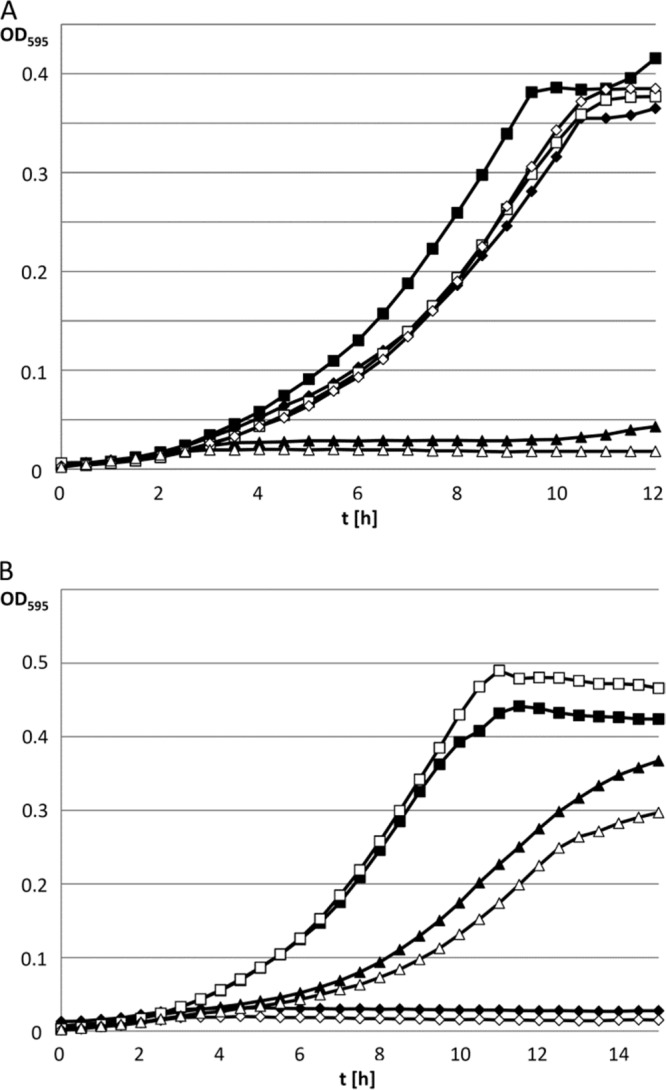
Growth studies with E. faecalis strains on panose and isopanose. (A) Growth was followed for the maltotetraose-positive JH2-2 strain (squares), the mdxF mutant (triangles), and the malT mutant (diamonds) in M17cc medium supplemented with 0.3% (wt/vol) panose (open symbols) or isopanose (filled symbols). (B) The maltotetraose-positive strain JH2-2 (squares) and the mdxF mutant derived from it (diamonds) were transformed with empty plasmid pADGEnt. The mdxF mutant was also transformed with pADGEnt:66c, which contains the mdxF gene under the control of the agmatine-inducible aguB promoter (triangles). The three strains were grown in M17cc medium supplemented with 0.3% (wt/vol) panose (open symbols) or isopanose (filled symbols).
No growth was obtained with isomaltotriose, an α-1,6-linked trisaccharide (data not shown). The main difference between maltotriose, panose or isopanose, and isomaltotriose is that isomaltotriose contains two consecutive α-1,6 linkages. Molecules with this structure might not be recognized by the E. faecalis ABC transporter. Complementation of the mdxF mutant with the mdxF wild-type allele under the control of the agmatine-inducible aguB promoter restored growth on panose and isopanose to about half the level observed for the wild-type strain (Fig. 8B).
E. faecalis strain JH2-2 was not able to grow on β-cyclodextrin (α-1,4-linked cyclic dextrin composed of seven glucose residues) and grew only very slowly on α-cyclodextrin (composed of six glucose residues). The slow transport of α-cyclodextrin seems to be catalyzed by the ABC transporter, because only the malF, but not the malT, mutant had lost the capacity to slowly grow on the cyclic maltohexaose (data not shown).
The mdxF mutant exhibits reduced colonization of mouse organs.
Increased expression of maltodextrin genes was observed during soft tissue infection with S. pyogenes (21). Similarly, a transcriptome study with cells of the E. faecalis strain V19 (a plasmid-cured derivative of strain V583) isolated from the peritoneum of infected mice had revealed that both the mdxEFG operon and the msmX gene are strongly expressed during infection. In contrast, the transcription level of the malT gene was not increased (18). These results suggested that the maltodextrin-specific ABC transporter might play a role in E. faecalis virulence. We therefore tested whether deletion of the mdxF gene would affect the survival of E. faecalis in a mouse organ colonization model. A mutant deleted for the tpx gene, which encodes a thiolperoxidase that plays a role in enterococcal virulence (30), and the malT mutant served as positive- and negative-control strains, respectively. Twenty-four hours after intraperitoneal injection, the bacterial load was determined in the peritoneum, kidneys, liver, and spleen, as described in Materials and Methods. No significant differences were observed between the wild-type strain and the malT mutant in the three organs and the peritoneum (Fig. 9A). In contrast, colonization of kidneys and liver by the tpx mutant was less efficient than that by the wild-type strain (Fig. 9B), which confirmed its previously suggested implication in enterococcal virulence using a 50% lethal dose (LD50) mouse model (30). Interestingly, compared to the wild-type strain, the mdxF mutant also exhibited significantly lower CFU numbers in kidneys (almost 5-fold lower) and in liver (16-fold lower) (Fig. 9C).
FIG 9.
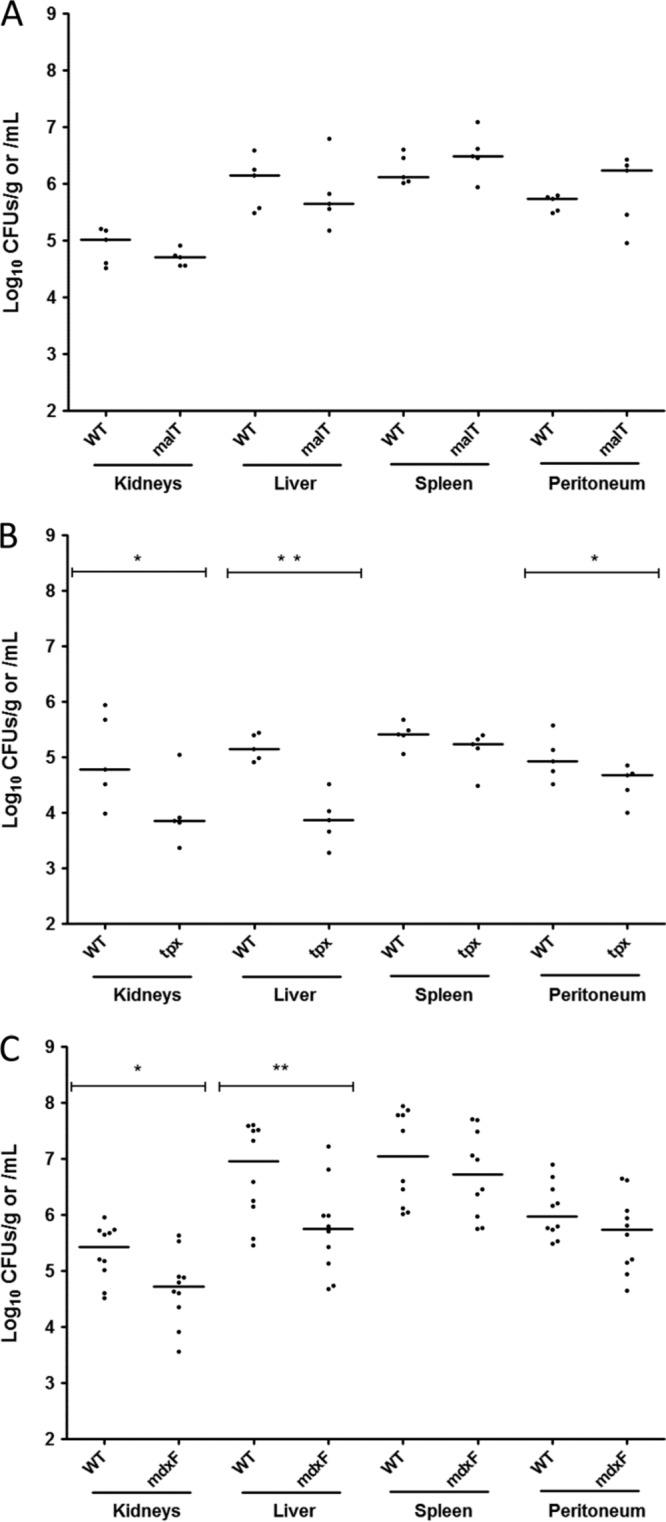
Colonization of mouse organs by various E. faecalis strains. Standardized inocula of 8 × 108 CFU of the maltotetraose-positive E. faecalis strain JH2-2 and the malT mutant (A), the tpx mutant (B), and the mdxF mutant (C) derived from it were intraperitoneally injected into Swiss white mice. After 24 h (malT and mdxF mutant) or 72 h (tpx mutant), the survival rate was determined for each strain by bacterial counts in kidneys, liver, and spleen (CFU per gram of tissue) and in the peritoneum (CFU per milliliter of washing fluid). The experiments were carried out with 5 mice per group. The experiment with the mdxF mutant was performed twice, and the results obtained with the 10 mice are shown (C). The numbers obtained for each individual mouse (dots) are presented as log10. The results are expressed in CFU per gram (for the organs) and as CFU per milliliter (for the peritoneal wash); mean values are presented as horizontal bars. To compare the results obtained for two different groups, we used the Mann-Whitney test. The calculated P values of <0.05 (*) and <0.005 (**) reflect the overall difference of the CFU values determined in kidney or liver, respectively, between the groups of mice infected with the mdxF mutant and the ones infected with the wild-type strain.
The link between carbohydrate utilization and virulence has been studied in pathogens, like group A streptococci (GAS), or more recently in Streptococcus suis using transcriptome analyses or mouse oropharynx colonization experiments (1, 31). The combined results demonstrate that maltodextrin acquisition is likely to be a key factor in the ability of GAS to successfully infect the oropharynx. Our results extend the importance of maltodextrin metabolism to deep-tissue infections. Indeed, the colonization of murine organs, especially the liver, by E. faecalis was less efficient in a mutant deficient in maltodextrin transport. The greatest impact on colonization was observed in the liver. This is of special interest, as this organ is the storage site for glycogen, which can be degraded to maltodextrins. Glycogen released from host tissues may serve as a substrate for the growth of E. faecalis. However, cultures realized on glycogen or starch as the substrate showed that E. faecalis JH2-2 does not grow on these polysaccharides, which is in agreement with the absence of genes related to glycogen degradation or encoding amylases in the genome of enterococci. Hence, the step between glycogen release and the formation of maltodextrins able to be metabolized by E. faecalis remains unclear. In conclusion, we demonstrate that E. faecalis uses the same enzymes (MalT and MapP) for efficient transport and the first catabolic steps for maltose and maltotriose. MapP can also dephosphorylate maltotetraose-6‴-P, suggesting that the enzyme recognizes primarily the phosphorylated glucose residue at the nonreducing end of maltooligosaccharides. While the transport system responsible for the residual uptake of maltose in the malT mutant remains unknown, maltotriose was found also to be slowly taken up by the MdxEFG-MsmX ABC transporter (Fig. 10). The ABC transporter functions as the major uptake system not only for maltotetraose and longer maltooligosaccharides but also for the maltotriose linkage isomers panose and isopanose. E. faecalis strain JH2-2 is not able to utilize cyclic maltodextrins. The PTS-catalyzed transport of maltose and maltotriose was found to be more efficient than the MdxEFG-MsmX-catalyzed uptake of maltotetraose and longer maltooligosaccharides. However, the utilization of maltotriose by the malT mutant via the ABC transporter was less efficient than that of maltotetraose, but it was similarly efficient as the utilization of the two trisaccharides panose and isopanose by the wild-type strain. In conclusion, E. faecalis transports maltotriose and maltotetraose, which are usually present in maltodextrin, by mechanisms and transporters which strongly resemble those reported for S. mutans (20) but which exhibit significant differences from those reported for other streptococci, such as S. pyogenes (19) and S. pneumoniae (2). To the best of our knowledge, the transport of the maltotriose linkage isomers panose and isopanose, which are usually also present in small amounts in maltodextrin, has not been studied in bacteria. In E. faecalis, they are exclusively transported by the MdxEFG-MsmX ABC transporter.
FIG 10.

Schematic presentation of the E. faecalis transport systems catalyzing the uptake of maltose, maltotriose, maltotetraose, and the maltotriose linkage isomers panose and isopanose. While maltose and maltotriose are primarily transported by the PTS permease MalT, maltotetraose and longer maltooligosaccharides, as well as panose and isopanose, are taken up by the ABC transporter MdxEFG-MsmX. When substrates or arrows are in gray, this indicates that they are transported with low efficiency. This applies for the transport of maltotriose by the ABC transporter MdxEFG-MsmX and the uptake of maltose by a yet-unknown transport protein.
Interestingly, while deletion of the PTS permease had no effect on E. faecalis virulence, inactivation of the membrane-spanning MalF significantly reduced the colonization ability of the pathogen in kidneys and liver in a mouse infection model. With the liver and, to a lesser extent, the kidney being the place for the storage and breakdown of glycogen, it is not surprising that inactivation of the maltodextrin ABC transporter affects the fitness of the pathogen in these organs.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The E. faecalis wild-type strain used in this study is a maltotetraose-positive clone isolated from our stock of strain JH2-2, which is also called TX4000. The malT and mdxF mutants used in this study were derived from the maltotetraose-positive clone (Table 2). E. faecalis strains were routinely grown at 37°C without shaking in 100-ml sealed bottles filled with 20 to 50 ml of Luria-Bertani (LB) medium (Difco, NJ) containing 0.5% (wt/vol) glucose. Growth studies were carried out with carbon-depleted M17MOPS (M17cc) medium, which was prepared as previously described (32). It was supplemented with 0.3% (wt/vol) glucose, maltose, maltotriose, maltotetraose, maltopentaose, maltohexaose, maltoheptaose, panose, or isopanose. Erythromycin, chloramphenicol, and tetracycline were added when appropriate at concentrations of 150 μg · ml−1, 10 μg · ml−1, and 5 μg · ml−1, respectively.
TABLE 2.
Bacterial strains and plasmids used in this study
| Strain, mutant, or plasmid | Relevant characteristicsa | Source or reference |
|---|---|---|
| Bacterial strains | ||
| Enterococcus faecalis | ||
| JH2-2 | Fusr Rifr, plasmid free, maltotetraose-positive wild-type strain | 40 |
| mdxF mutant | JH2-2 carrying stop codons at beginning of mdxF gene | This study |
| malT mutant | JH2-2 carrying stop codons at beginning of malT gene | This study |
| JH2-2/pAGEnt | JH2-2 harboring empty pAGEnt | This study |
| mdxF/pAGEnt strain | mdxF mutant harboring empty pAGEnt | This study |
| mdxF/pAGEnt:66c strain | mdxF mutant harboring pAGEnt with mdxF gene under control of agmatine-inducible aguB promoter | This study |
| Lactococcus lactis IL1403 | Wild-type strain | 41 |
| Escherichia coli | ||
| TOP10F′ | F′ [lacIq Tn10 (Tetr)] mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara-leu)7697 galU galK rpsL (Strr) endA1 nupG | Invitrogen |
| NM522(pQE30-mapP) | supE thi-1 Δ(lac-proAB) Δ(mcrB-hsdSM) (rK− mK−) [F′ proAB lacIqZ ΔGM15] [pQE30-mapP] | 7 |
| Plasmids | ||
| pMAD | Origin pE194ts; Ermr Ampr bgaB | 36 |
| pAGEnt | Modified pNZ8048 plasmid, Cmr, contains aguR PaguB (promoter inducible with agmatine) | 22 |
| pAGEnt:66c | pAGEnt plasmid with the mdxF gene under control of agmatine-inducible aguB promoter | This study |
Fusr, fusidic acid resistance; Rifr, rifampin resistance; Tetr, tetracycline resistance; Strr, streptomycin resistance; Ermr, erythromycin resistance; Ampr, ampicillin resistance; Cmr, chloramphenicol resistance.
Escherichia coli strains NM522 and TOP10F′ used for protein purification and cloning experiments, respectively (Table 2), were grown aerobically by gyratory shaking at 250 rpm in LB medium at 37°C and transformed by electroporation with a Gene Pulser apparatus (Bio-Rad Laboratories). Growth was followed by measuring the absorption at 600 nm in a Novaspec II spectrophotometer. When appropriate, erythromycin was added at a final concentration of 150 μg · ml−1.
The Lactococcus lactis strain IL1403 was used as a host during plasmid construction for complementation experiments and was grown in M17 medium at 30°C.
General molecular methods.
Restriction endonucleases, shrimp alkaline phosphatase, and T4 DNA ligase were obtained from Promega and used according to the manufacturer's instructions. PCR experiments were performed with the MasterCycler Gradient thermocycler (Eppendorf) using GoTaq or Phusion DNA polymerase (Promega). The primers used for the PCR experiments are listed in Table 3. If needed, PCR products were purified using the NucleoSpin extract II kit (Macherey-Nagel). E. coli was transformed by electroporation with the Gene Pulser apparatus (Bio-Rad Laboratories), as described in reference 33, and E. faecalis or L. lactis, as described in reference 34. Plasmids were extracted from E. coli by using the NucleoSpin plasmid kit (Macherey-Nagel). RNA extraction was performed as previously described (35). The 5′ end of the mRNA corresponding to the mdxEFG operon (Fig. 1) was mapped on a 5′-RACE PCR product obtained by using the 3′/5′-RACE kit (Thermo Fisher), as well as the primer race1_41965 (Table 3) for the reverse transcriptase reaction and primer race2_41965 for the PCR after poly(A) tailing. The PCR product was subsequently purified and sequenced with the primer race3_41965.
TABLE 3.
Primers used in this study
| Primer | Sequence (5′ to 3′)a | Orientationb |
|---|---|---|
| pMAD_41760_for | CCGGGCCATGGAATGAAAAAAATGTTTAGTTTTG | + |
| pMAD_41760_rev | GGTGGATCCTAGTTGATTAATAACTAC | − |
| mut41760_for | CGTTAGAATTCTAGTTTATGTTTGCTG | + |
| mut41760_rev | ACATGAATTCTTACGGCTCTGTCACAC | − |
| Verif_41760 | CGCCGGAAGCTAGTAAATCTTC | − |
| pMAD_41966_for | GTCAGTGGATCCTGCACCGTAGAAGTTAG | − |
| pMAD_41966_rev | AAAATCGGATCCAATTTTGGCGAAGATGGC | + |
| mut41966_for | AGGAACACTAGTGACCTAACAGCAAGGATTAG | + |
| mut41966_rev | GGGTCGCTAGCGTTCATAGCATCATTAATGCG | − |
| Verif_mut_41966 | TGTTAGGTCACTAGCGTTCA | − |
| pAGEnt_66_forkit | TTTTAGGAGGAACACATCATGTTCAAGAAAAAGAAAGC | + |
| pAGEnt_66_revkit | AAGCAACACGTGCTGTAATTTTACGCCTCCTTAAATGAATTC | − |
| RPV_AGEnt_RBS | CATGATGTGTTCCTCCTAAAAG | |
| FRV_AGEnt_Term | AATTACAGCACGTGTTGCTTT | |
| race1_41965 | TCTTTTTCAAAATCGGCGAC | − |
| race2_41965 | CCCATAATTTTACGTCACCAGCTG | − |
| race3_41965 | TTCGTTTTTTCTGCGCCACC | − |
| EF1344R | TCATTAATGCGTGGAAACCA | − |
| EF1344L | AAACTTCGCCAACAAGCAGT | + |
| EF1345R | ATGCCTTCCTTTCTGGTCCT | + |
| EF1345L | CCATTGCCAAAATCAATGGT | − |
Underlined sequences correspond to restriction sites, and bold letters indicate stop codons.
+, oriented in the direction of transcription; −, oriented against the direction of transcription.
For complementation of the E. faecalis mdxF mutant, the wild-type mdxF gene was cloned into vector pAGEnt (22). In this plasmid, genes inserted at the multiple-cloning site are expressed from the agmatine-inducible aguB promoter in the presence of 40 mM agmatine, because the plasmid also contains the gene for the repressor AguR of the aguBDAC operon. The mdxF gene and the plasmid were amplified with the pAGEnt_66_forkit/pAGEnt_66_revkit and RPV_AGEnt_RBS/FRV_AGEnt_Term primer pairs, respectively (Table 3). The assembly of the two PCR fragments was achieved with the NEBuilder HiFi DNA assembly cloning kit (New England BioLabs). L. lactis strain IL1403 was transformed with the resulting plasmid (named pAGEnt:66c) and subsequently plated onto GM17 medium with chloramphenicol at a final concentration of 5 μg · ml−1. The correct sequence of the inserted DNA fragment was confirmed for one clone, and its plasmid was subsequently used to complement the E. faecalis mdxF mutant. For control experiments, empty pAGEnt was electroporated into the wild-type strain and the mdxF mutant.
For quantitative real-time PCR (qRT-PCR) experiments, total RNA was extracted from maltotetraose-negative and -positive strains by using the Direct-zol RNA MiniPrep kit (Zymo Research). The strains were grown for 4 h in M17cc medium containing either 0.5% (wt/vol) maltodextrin or 0.5% (wt/vol) maltodextrin and 0.5% (wt/vol) glucose. The isolated RNA was spectrophotometrically quantified, and 2 μg of the total RNA was subsequently utilized for reverse transcription with the QuantiTect reverse transcription kit (Qiagen). qRT-PCR was subsequently carried out with SYBR green fluorescence using the Cfx96 real-time PCR machine (Bio-Rad). The primers used for the qRT-PCR experiments are EF1344R and EF1344L for the mdxF gene and EF1345R and EF1345L for the mdxE gene (Table 3). Raw data were converted into expression data by the absolute quantification method using the standard curve and then normalized using the housekeeping 5S RNA gene as a reference. The mean values and standard deviations were calculated from at least 3 independent experiments.
Construction of E. faecalis malT and mdxF mutants.
E. faecalis mutants were constructed by introducing two stop codons at the beginning of each open reading frame (ORF) in order to produce short truncated proteins. For the malT mutant, two PCR products of 1,000 bp were amplified by using chromosomal DNA as the template and the two primer pairs pMAD-41760-for/Mut-41760-rev and pMAD-41760-rev/Mut-41760-for (Table 3). The 5′ ends of Mut-41760-rev and Mut-41760-for contain EcoRI restriction sites. After amplification, both PCR products were therefore cut with EcoRI and ligated. A second PCR was performed using the reaction mixture of the ligation as the DNA template and primers pMAD-41760-for/pMAD-41760-rev. The amplified 2-kb fragment was cloned into pMAD cut with BamHI and NcoI (36), and after purification from E. coli TOP10F′, the resulting plasmid was used to transform a maltotetraose-positive E. faecalis JH2-2 strain according to a previously described protocol (7). After electroporation, cells were plated on GM17 agar medium containing 50 μg/ml erythromycin and 2.35 μM X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) and incubated at 37°C. The presence of the plasmid in blue clones resistant to the antibiotic was verified by PCR using oligonucleotide Verif_41760 and a plasmid-based primer. To allow the second crossing over, integrants were first grown for 6 h at 30°C and subsequently overnight at 42°C. After 2 or 3 cycles of temperature shifting, cells were plated on solid GM17 medium containing X-Gal but no antibiotics and incubated at 37°C. In several white clones, the presence of the introduced EcoRI site in the PCR-amplified malT gene was confirmed by restriction with this enzyme, and the presence of the stop codons was confirmed by DNA sequencing. The same strategy was applied for construction of the mdxF mutant, except that we used the pMAD-41966-for/Mut-41966-rev and pMAD-41966-rev/Mut-41966-for primer pairs (Table 3). The two Mut primers contain NheI and SpeI restriction sites, two isoschizomeric enzymes. The correct sequence in the mutant was confirmed by PCR amplification using a specific primer of the modified sequence Verif-mut-41966 and subsequent DNA sequencing.
Growth studies with wild-type and mutant strains.
The media used for the growth studies were inoculated with overnight cultures, which were started from the glycerol stocks kept at −20°C. Each overnight culture was washed once with one volume of physiological solution, and the final optical densities (ODs) at 600 nm were adjusted to 2.0. A 96-well plate was filled with 200 μl of M17MOPS medium supplemented with different sugars per well and inoculated with 2 μl of the washed overnight culture. Three drops of paraffin oil were added on top of the medium to avoid desiccation and to create anaerobic conditions. A microplate reader 680 (Bio-Rad Laboratories) was used to follow growth at 37°C. The absorption at 595 nm was measured every 30 min over a total period of 12 to 16 h. Growth studies were carried out at least three times and always provided very similar results.
Purification of His-tagged E. faecalis maltose-6′-P phosphatase.
The maltose-6′-P phosphatase of the E. faecalis strain JH2-2 is encoded by the mapP (EFT41761) gene. The enzyme was overexpressed in E. coli strain NM522 from the His tag expression vector pQE30 and purified as previously described (7). The enzyme was used for dephosphorylation assays of maltotriose-6″-P and maltotetraose-6‴-P, as described below.
Synthesis of malotriose-6″-P and maltotetraose-6‴-P.
Maltotriose and maltotetraose phosphorylated at the O-6 position of the glucose moiety at their nonreducing end were prepared enzymatically via the α-glucoside-specific PTS present in Palatinose-grown cells of Klebsiella pneumoniae (37). This PTS corresponds to the α-glucoside-specific EIICB PTS component with the protein identifier (ID) KFJ74572.1, which is encoded by the aglB gene present on chromosome 1 of the Klebsiella pneumoniae type strain ATCC 13883 (38). Phosphorylated derivatives of the α-linked oligosaccharides were isolated by ethanol and Ba2+ precipitation and further purified by ion-exchange and paper chromatography. Structures and product purity were confirmed by thin-layer chromatography, mass spectrometry, and nuclear magnetic resonance (NMR) spectroscopy.
Mass spectrometric analysis of phosphorylated maltooligosaccharides after MapP treatment.
Solutions containing malotriose-6″-P or maltotetraose-6‴-P at a concentration of 2 M were prepared with 20 mM ammonium bicarbonate. Aliquots (5 μl) of these solutions were added to either 40 μl of purified MapP (0.8 mg · ml−1, dialyzed against 20 mM ammonium bicarbonate) or 40 μl of 20 mM ammonium bicarbonate and incubated for 2 h at 37°C. The samples were subsequently lyophilized and rehydrated with 10 μl of water. Aliquots of 1 μl were mixed with 9 μl of a sugar matrix solution, and 1 μl was spotted onto the matrix-assisted laser desorption ionization (MALDI) steel plate. The sugar matrix was freshly prepared and contained 100 μg/ml 2,5-dihydroxybenzoic acid dissolved in a mixture of H2O–acetonitrile–N,N-dimethylaniline (1:1:0.02 [vol/vol]). The samples were analyzed by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS; Voyager DE super STR; AB Sciex) by irradiating them with a nitrogen laser (337 nm, 10 Hz) integrated in this instrument and by recording mass spectra in the reflectron mode using a delay extraction time of 120 ns and a m/z mass range between 200 and 1,000 Da.
Determination of the phosphatase activity of MapP with the malachite green assay.
To follow the MapP-catalyzed dephosphorylation of maltose-6′-P, maltotriose-6″-P, and maltotetraose-6‴-P over various time periods, we used the malachite green assay (23). Dephosphorylation experiments were carried out in 650-μl assay mixtures containing 50 mM Tris-HCl (pH 7.4), 5 mM MgCl2, and 2 or 4 mM phosphorylated maltooligosaccharides. The reaction was started by adding MapP, and the samples were incubated for various time periods at 37°C. The amount of MapP used in the assay was 40 μg, which allowed the detection of phosphate after a few minutes of incubation. Aliquots of 100 μl were withdrawn after 0, 1.5, 3, 6, 10, and 20 min of incubation and immediately mixed with 25 μl of the malachite green reagent (23). The samples were kept for 20 min at ambient temperature before 375 μl of water was added and the optical density at 630 nm (OD630) determined. In Table 1, the specific activity (micromoles per minute per milligram of protein) determined for the MapP-catalyzed dephosphorylation of the various phospho-compounds is also expressed relative to the activity measured with maltose-6′-P, which was set to 100%.
Colonization assay of mouse organs.
The maltooligosaccharide-positive E. faecalis JH2-2 clone and the mdxF, malT, and tpx mutants derived from it were grown in GM17 broth at 37°C. The tpx mutant was used as a negative control. The tpx gene encodes a thiolperoxidase involved in stress response, and its inactivation has previously been shown to lower the survival of E. faecalis in an LD50 mouse infection model (30). All animal experiments were performed at the platform of biological resources (CURB [http://icore.unicaen.fr/plateformes/curb/curb-272372.kjsp]) at the University of Caen in accordance with the French legislation on biomedical experimentation (ethics authorizations B14118015 and N/01-09-12/20/09-15) and the European Communities Council Directive from 22 September 2010 (39). Before being used, mice were housed at 21 ± 0.5°C, in Perspex home cages with free access to food and water.
E. faecalis wild-type and mutant strains were harvested at mid-log phase and resuspended in physiological solution to reach a density of ≈8 × 108 CFU/ml. Male and female 6- to 8-week-old Swiss white mice (CURB, France) were infected intraperitoneally with 1 ml of the bacterial suspensions. The experiments were conducted with 5 mice per group. After 24 h of infection, mice were anesthetized by inhalation of 5% isoflurane in a mixture of 30% O2 and 70% N2O before euthanasia. A peritoneal wash was performed with 5 ml of cold physiological solution, and the resulting fluid was collected in sterile tubes and placed on ice for 30 min. The abdomen was subsequently opened, and kidneys, liver, and spleen were removed and placed on ice for 30 min. The organs were crushed using pestle and mortar, transferred into sterile tubes, and resuspended in 3 volumes of cold physiological solution. The suspensions were then homogenized by vortexing twice for 30 s. The peritoneal fluid was centrifuged (10 min at 4,000 × g and 4°C) and the bacterial pellet was subsequently resuspended in 1 ml of cold physiological solution. Serial dilutions of all samples were plated onto GM17 agar in order to determine the CFU. In order to determine whether there was a significant difference between two groups of mice, we used the Mann-Whitney test in the GraphPad Prism software (GraphPad Software, San Diego, CA). A P value of <0.05 was considered to be significant. All experiments were carried out twice.
ACKNOWLEDGMENTS
We thank Maria Fernandez for providing plasmid pAGEnt and Benoit Haelewyn (CURB, University of Caen) for expert technical assistance for the mouse infection model.
This work was supported by grants from the MinCyt/ECOS-Sud program (action no. A09B03) (to C.M. and J.D.), the Agencia Nacional de Promoción y Tecnológica, Argentina (AN-PCyT; contracts 2014-1513 [to C.M.] and 2014-3482 [to V.S.B.]), the Intramural Research Program of the NIDCR, National Institutes of Health, Department of Health and Human Services, Bethesda, MD (to A.P. and J.T.), and the Initiative d'Excellence program from the French government (grant DYNAMO, ANR-11-LABX-0011) (to J.D.) and by a chair of excellence at the University of Caen for C.M. financed by the Region of Normandy and the European Regional Development Fund (ERDF). A.M. received a fellowship from the Algerian government, and V.S.B., G.D.R., and C.M. are Career Investigators of CONICET (Argentina).
We declare no conflicts of interest.
REFERENCES
- 1.Shelburne SA III, Keith DB, Davenport MT, Horstmann N, Brennan RG, Musser JM. 2008. Molecular characterization of group A Streptococcus maltodextrin catabolism and its role in pharyngitis. Mol Microbiol 69:436–452. doi: 10.1111/j.1365-2958.2008.06290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bidossi A, Mulas L, Decorosi F, Colomba L, Ricci S, Pozzi G, Deutscher J, Viti C, Oggioni MR. 2012. A functional genomics approach to establish the complement of carbohydrate transporters in Streptococcus pneumoniae. PLoS One 7:e33320. doi: 10.1371/journal.pone.0033320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gopal S, Berg D, Hagen N, Schriefer E-M, Stoll R, Goebel W, Kreft J. 2010. Maltose and maltodextrin utilization by Listeria monocytogenes depend on an inducible ABC transporter which is repressed by glucose. PLoS One 5:e10349. doi: 10.1371/journal.pone.0010349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Derkaoui M, Antunes A, Nait Abdallah J, Poncet S, Mazé A, Ma Pham QM, Mokhtari A, Deghmane A-E, Joyet P, Taha M-K, Deutscher J. 2016. Transport and catabolism of carbohydrates by Neisseria meningitidis. J Mol Microbiol Biotechnol 26:320–332. doi: 10.1159/000447093. [DOI] [PubMed] [Google Scholar]
- 5.Creti R, Koch S, Fabretti F, Baldassarri L, Huebner J. 2006. Enterococcal colonization of the gastro-intestinal tract: role of biofilm and environmental oligosaccharides. BMC Microbiol 6:60. doi: 10.1186/1471-2180-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le Breton Y, Pichereau V, Sauvageot N, Auffray Y, Rincé A. 2005. Maltose utilization in Enterococcus faecalis. J Appl Microbiol 98:806–813. doi: 10.1111/j.1365-2672.2004.02468.x. [DOI] [PubMed] [Google Scholar]
- 7.Mokhtari A, Blancato VS, Repizo GD, Henry C, Pikis A, Bourand A, de Fátima Álvarez M, Immel S, Mechakra-Maza A, Hartke A, Thompson J, Magni C, Deutscher J. 2013. Enterococcus faecalis utilizes maltose by connecting two incompatible metabolic routes via a novel maltose 6′-phosphate phosphatase (MapP). Mol Microbiol 88:234–253. doi: 10.1111/mmi.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alpert CA, Frank R, Stüber K, Deutscher J, Hengstenberg W. 1985. Phosphoenolpyruvate-dependent protein kinase enzyme I of Streptococcus faecalis: purification and properties of the enzyme and characterization of its active center. Biochemistry 24:959–964. doi: 10.1021/bi00325a023. [DOI] [PubMed] [Google Scholar]
- 9.Gassner M, Stehlik D, Schrecker O, Hengstenberg W, Maurer W, Rüterjans H. 1977. The phosphoenolpyruvate-dependent phosphotransferase system of Staphylococcus aureus. 2. 1H and 31P-nuclear-magnetic-resonance studies on the phosphocarrier protein HPr, phosphohistidines and phosphorylated HPr. Eur J Biochem 75:287–296. doi: 10.1111/j.1432-1033.1977.tb11528.x. [DOI] [PubMed] [Google Scholar]
- 10.Dörschug M, Frank R, Kalbitzer HR, Hengstenberg W, Deutscher J. 1984. Phosphoenolpyruvate-dependent phosphorylation site in enzyme IIIglc of the Escherichia coli phosphotransferase system. Eur J Biochem 144:113–119. doi: 10.1111/j.1432-1033.1984.tb08438.x. [DOI] [PubMed] [Google Scholar]
- 11.Cao Y, Jin X, Levin EJ, Huang H, Zong Y, Quick M, Weng J, Pan Y, Love J, Punta M, Rost B, Hendrickson WA, Javitch JA, Rajashankar KR, Zhou M. 2011. Crystal structure of a phosphorylation-coupled saccharide transporter. Nature 473:50–54. doi: 10.1038/nature09939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puyet A, Espinosa M. 1993. Structure of the maltodextrin-uptake locus of Streptococcus pneumoniae. Correlation to the Escherichia coli maltose regulon. J Mol Biol 230:800–811. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X, Rogers M, Bierschenk D, Bonten MJM, Willems RJL, van Schaik W. 2013. A LacI-family regulator activates maltodextrin metabolism of Enterococcus faecium. PLoS One 8:e72285. doi: 10.1371/journal.pone.0072285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen J. 2013. Molecular mechanism of the Escherichia coli maltose transporter. Curr Opin Struct Biol 23:492–498. doi: 10.1016/j.sbi.2013.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monedero V, Yebra MJ, Poncet S, Deutscher J. 2008. Maltose transport in Lactobacillus casei and its regulation by inducer exclusion. Res Microbiol 159:94–102. doi: 10.1016/j.resmic.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Ferenci T. 1980. The recognition of maltodextrins by Escherichia coli. Eur J Biochem 108:631–636. doi: 10.1111/j.1432-1033.1980.tb04758.x. [DOI] [PubMed] [Google Scholar]
- 17.Ferreira MJ, de Sá-Nogueira I. 2010. A multitask ATPase serving different ABC-type sugar importers in Bacillus subtilis. J Bacteriol 192:5312–5318. doi: 10.1128/JB.00832-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muller C, Cacaci M, Sauvageot N, Sanguinetti M, Rattei T, Eder T, Giard J-C, Kalinowski J, Hain T, Hartke A. 2015. The intraperitoneal transcriptome of the opportunistic pathogen Enterococcus faecalis in mice. PLoS One 10:e0126143. doi: 10.1371/journal.pone.0126143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shelburne SA III, Fang H, Okorafor N, Sumby P, Sitkiewicz I, Keith D, Patel P, Austin C, Graviss EA, Musser JM, Chow D-C. 2007. MalE of group A Streptococcus participates in the rapid transport of maltotriose and longer maltodextrins. J Bacteriol 189:2610–2617. doi: 10.1128/JB.01539-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Webb AJ, Homer KA, Hosie AHF. 2007. A phosphoenolpyruvate-dependent phosphotransferase system is the principal maltose transporter in Streptococcus mutans. J Bacteriol 189:3322–3327. doi: 10.1128/JB.01633-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graham MR, Virtaneva K, Porcella SF, Gardner DJ, Long RD, Welty DM, Barry WT, Johnson CA, Parkins LD, Wright FA, Musser JM. 2006. Analysis of the transcriptome of group A Streptococcus in mouse soft tissue infection. Am J Pathol 169:927–942. doi: 10.2353/ajpath.2006.060112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linares DM, Perez M, Ladero V, Del Rio B, Redruello B, Martin MC, Fernandez M, Alvarez MA. 2014. An agmatine-inducible system for the expression of recombinant proteins in Enterococcus faecalis. Microb Cell Fact 13:169. doi: 10.1186/s12934-014-0169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baykov AA, Evtushenko OA, Avaeva SM. 1988. A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay. Anal Biochem 171:266–270. doi: 10.1016/0003-2697(88)90484-8. [DOI] [PubMed] [Google Scholar]
- 24.Imanaka T, Kuriki T. 1989. Pattern of action of Bacillus stearothermophilus neopullulanase on pullulan. J Bacteriol 171:369–374. doi: 10.1128/jb.171.1.369-374.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hansen PI, Larsen FH, Motawia SM, Blennow A, Spraul M, Dvortsak P, Engelsen SB. 2008. Structure and hydration of the amylopectin trisaccharide building blocks–synthesis, NMR, and molecular dynamics. Biopolymers 89:1179–1193. doi: 10.1002/bip.21075. [DOI] [PubMed] [Google Scholar]
- 26.Gilmore MS, Rauch M, Ramsey MM, Himes PR, Varahan S, Manson JM, Lebreton F, Hancock LE. 2015. Pheromone killing of multidrug-resistant Enterococcus faecalis V583 by native commensal strains. Proc Natl Acad Sci U S A 112:7273–7278. doi: 10.1073/pnas.1500553112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cha HJ, Yoon HG, Kim YW, Lee HS, Kim JW, Kweon KS, Oh BH, Park KH. 1998. Molecular and enzymatic characterization of a maltogenic amylase that hydrolyzes and transglycosylates acarbose. Eur J Biochem 253:251–262. doi: 10.1046/j.1432-1327.1998.2530251.x. [DOI] [PubMed] [Google Scholar]
- 28.Tao L, Sutcliffe IC, Russell RR, Ferretti JJ. 1993. Transport of sugars, including sucrose, by the msm transport system of Streptococcus mutans. J Dent Res 72:1386–1390. doi: 10.1177/00220345930720100701. [DOI] [PubMed] [Google Scholar]
- 29.Whiting GC, Sutcliffe IC, Russell RR. 1993. Metabolism of polysaccharides by the Streptococcus mutans dexB gene product. J Gen Microbiol 139:2019–2026. doi: 10.1099/00221287-139-9-2019. [DOI] [PubMed] [Google Scholar]
- 30.La Carbona S, Sauvageot N, Giard J-C, Benachour A, Posteraro B, Auffray Y, Sanguinetti M, Hartke A. 2007. Comparative study of the physiological roles of three peroxidases (NADH peroxidase, alkyl hydroperoxide reductase and thiol peroxidase) in oxidative stress response, survival inside macrophages and virulence of Enterococcus faecalis. Mol Microbiol 66:1148–1163. doi: 10.1111/j.1365-2958.2007.05987.x. [DOI] [PubMed] [Google Scholar]
- 31.Ferrando ML, van Baarlen P, Orrù G, Piga R, Bongers RS, Wels M, De Greeff A, Smith HE, Wells JM. 2014. Carbohydrate availability regulates virulence gene expression in Streptococcus suis. PLoS One 9:e89334. doi: 10.1371/journal.pone.0089334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bizzini A, Zhao C, Budin-Verneuil A, Sauvageot N, Giard J-C, Auffray Y, Hartke A. 2010. Glycerol is metabolized in a complex and strain-dependent manner in Enterococcus faecalis. J Bacteriol 192:779–785. doi: 10.1128/JB.00959-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dower WJ, Miller JF, Ragsdale CW. 1988. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res 16:6127–6145. doi: 10.1093/nar/16.13.6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holo H, Nes IF. 1989. High-frequency transformation, by electroporation, of Lactococcus lactis subsp. cremoris grown with glycine in osmotically stabilized media. Appl Environ Microbiol 55:3119–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sauvageot N, Beaufils S, Mazé A, Deutscher J, Hartke A. 2006. Cloning and characterization of a gene encoding a cold-shock protein in Lactobacillus casei. FEMS Microbiol Lett 254:55–62. doi: 10.1111/j.1574-6968.2005.00006.x. [DOI] [PubMed] [Google Scholar]
- 36.Arnaud M, Chastanet A, Débarbouillé M. 2004. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, Gram-positive bacteria. Appl Environ Microbiol 70:6887–6891. doi: 10.1128/AEM.70.11.6887-6891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson J, Robrish SA, Pikis A, Brust A, Lichtenthaler FW. 2001. Phosphorylation and metabolism of sucrose and its five linkage-isomeric α-d-glucosyl-d-fructoses by Klebsiella pneumoniae. Carbohydr Res 331:149–161. doi: 10.1016/S0008-6215(01)00028-3. [DOI] [PubMed] [Google Scholar]
- 38.Daligault HE, Davenport KW, Minogue TD, Bishop-Lilly KA, Bruce DC, Chain PS, Coyne SR, Frey KG, Jaissle J, Koroleva GI, Ladner JT, Lo C-C, Meincke L, Munk AC, Palacios GF, Redden CL, Johnson SL. 2014. Draft genome assembly of Klebsiella pneumoniae type strain ATCC 13883. Genome Announc 2(5):e00939–. doi: 10.1128/genomeA.00939-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.European Union. 2010. Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes. European Union, Brussels, Belgium: http://eur-lex.europa.eu/legal-content/EN/TXT/?qid=1488555645022&uri=CELEX:32010L0063. [Google Scholar]
- 40.Yagi Y, Clewell DB. 1980. Recombination-deficient mutant of Streptococcus faecalis. J Bacteriol 143:966–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chopin A, Chopin MC, Moillo-Batt A, Langella P. 1984. Two plasmid-determined restriction and modification systems in Streptococcus lactis. Plasmid 11:260–263. doi: 10.1016/0147-619X(84)90033-7. [DOI] [PubMed] [Google Scholar]