

Abstract
Brain-derived neurotrophic factor (BDNF) is involved in many functions such as neuronal growth, survival, synaptic plasticity and memorization. Altered expression levels are associated with many pathological situations such as depression, epilepsy, Alzheimer’s, Huntington’s and Parkinson’s diseases. Glucocorticoid receptor (GR) is also crucial for neuron functions, via binding of glucocorticoid hormones (GCs). GR actions largely overlap those of BDNF. It has been proposed that GR could be a regulator of BDNF expression, however the molecular mechanisms involved have not been clearly defined yet. Herein, we analyzed the effect of a GC agonist dexamethasone (DEX) on BDNF expression in mouse neuronal primary cultures and in the newly characterized, mouse hippocampal BZ cell line established by targeted oncogenesis. Mouse Bdnf gene exhibits a complex genomic structure with 8 untranslated exons (I to VIII) splicing onto one common and unique coding exon IX. We found that DEX significantly downregulated total BDNF mRNA expression by around 30%. Expression of the highly expressed exon IV and VI containing transcripts was also reduced by DEX. The GR antagonist RU486 abolished this effect, which is consistent with specific GR-mediated action. Transient transfection assays allowed us to define a short 275 bp region within exon IV promoter responsible for GR-mediated Bdnf repression. Chromatin immunoprecipitation experiments demonstrated GR recruitment onto this fragment, through unidentified transcription factor tethering. Altogether, GR downregulates Bdnf expression through direct binding to Bdnf regulatory sequences. These findings bring new insights into the crosstalk between GR and BDNF signaling pathways both playing a major role in physiology and pathology of the central nervous system.
Electronic supplementary material
The online version of this article (doi:10.1186/s13041-017-0295-x) contains supplementary material, which is available to authorized users.
Keywords: Glucocorticoid receptor, Brain-derived neurotrophic factor, Glucocorticoids, Promoters
Introduction
The neurotrophin brain-derived neurotrophic factor (BDNF) is a key player in neuronal function. BDNF is highly expressed throughout the brain [1, 2], yet its strongest expression level is found within the hippocampus, a limbic structure of major importance for cognitive functions, such as memorization, learning, behavior, stress, emotions, and mood [3, 4]. In the central nervous system (CNS), BDNF regulates neuronal survival [5], differentiation and growth [6]. Growing evidence indicates that BDNF is also involved in neuronal homeostasis and brain plasticity-related processes such as memory, learning [7, 8] and drug addiction [9], as well as in long term potentiation [10]. Alterations in BDNF expression levels within specific neuron subpopulations have been associated with various pathologies, including depression, epilepsy, Alzheimer’s, Huntington’s and Parkinson’s diseases [11–16]. BDNF mainly functions by binding to its high-affinity receptor, tropomyosin-related kinase B (TrkB) activating several pathways such as MAP kinase, PI3 kinase and Phospholipase C [17]. Rodent Bdnf gene exhibits a complex genomic structure comprising of at least 9 exons (I to IX), which are alternatively spliced to generate exon-specific BDNF transcript variants with one common and unique coding exon IX at the 3’ terminal end [18]. Generation of a large set of transcript isoforms is probably of biological significance as in rat hippocampal neuronal cultures, it has been demonstrated that BDNF mRNA variants are differentially distributed in specific dendritic compartments in order to regulate the local availability of BDNF protein [19]. Moreover, BDNF expression was reported to be reduced with aging and associated with a repressed chromatin state on some of its gene regulatory regions [20]. Along this line, epigenetic histone modifications and DNA methylation marks have recently been identified as complex and crucial mechanisms enabling modified expression of various BDNF mRNA isoforms [21]. Altogether, several layers of events driving quantitatively and qualitatively BDNF expression highlight its crucial contribution to CNS function in physiology and pathology [22–24].
Glucocorticoid hormones (GCs) also exert pleiotropic actions on neurons by binding to and activating the glucocorticoid receptor (GR, NR3C1), as well as to the mineralocorticoid receptor (MR, NR3C2) [25, 26]. The latter exhibits a high ligand affinity, and as a consequence it is almost permanently occupied by GCs, while GR is mostly activated under high circulating GC concentrations such as during stress conditions or at the circadian peak of GCs. Both receptors are highly expressed in the hippocampus, acting in balance to regulate various physiological and neurological processes such as stress responses, apoptosis survival and long term potentiation [27]. Interestingly, BDNF activation of TrkB receptors regulates positively GR activity on its target gene expression by phosphorylating two key serine residues on the receptor [28]. Mutating these BDNF-sensitive sites results in the inhibition of the neuroplasticity response to chronic stress [29], unraveling a crosstalk between GC and neurotrophin signaling pathways. On the other hand, regulation of BDNF expression by stress [30] has important consequences on the pathophysiology of mood disorders [31] and in the mechanism of action of antidepressant agents [32]. As exposure to acute or chronic stress triggers a surge of circulating GC concentrations [33, 34], a role of these hormones in modulating BDNF expression has often been suggested [35–41], but most of these reports are based on indirect evidence, and are sometimes contradictory depending on the model and the treatment timeline [42–44]. As a whole, the molecular mechanisms by which GCs regulate BDNF expression are not clearly defined. In the present study, we demonstrated that, upon exposure to the glucocorticoid agonist dexamethasone (DEX), GR directly downregulates Bdnf expression, at least in part, by its binding to a specific DNA region upstream of exon IV. Interestingly, this promoter fragment was already characterized as stimulated by synaptic activity in humans and rats [45, 46]. Along with primary cultures of fetal hippocampal neurons (PCN), we used the newly characterized BZ cell line which was previously generated by targeted oncogenesis strategy [47] from a mouse hippocampus and which expresses a high level of both BDNF and GR. Altogether, this work unravels new insights about the repression by GR of Bdnf expression, findings that may be of potential physiological importance.
Methods
Primary cultures of fetal mouse hippocampal neurons
Pregnant SWISS mice at 18 or 19 days post-fertilization were euthanized by decapitation. Dissection was performed according to a video published in the Journal of Visual Experiments [48]. Hippocampal neurons were isolated and cultured from the embryos using the Pierce Primary Neuron Isolation Kit (Thermo scientific, Courtaboeuf, France) following the manufacturer’s instructions. This kit contained a neuronal media culture supplement (reference: 88286). Cells were typically seeded on culture plates coated with 10 μg/mL poly-D-lysine (Sigma-Aldrich, Lyon, France), at the density of 2.5 × 105 cells/cm2 and grown for 9 days at 37 °C in a 5% CO2 incubator.
Cell culture and reagents
BZ and Neuro-2a (N2A, ATCC number: CCL-131) growth medium was composed of DMEM (PAA, Vélizy-Villacoublay, France) containing 10% fetal bovine serum (FBS) (AbCys SA, Paris, France), 1x nonessential amino acids (PAA), 2 mM glutamine (PAA), 100 U/ml penicillin (PAA), 100 μg/ml streptomycin (PAA), 20 mM HEPES (PAA). For BZ differentiation experiments, serum concentration was lowered to 1% for 2 days. N2A differentiation medium was similar to BZ medium but was supplemented with 5 μg/ml insulin (Sigma-Aldrich), 5 μg/ml transferrin (Sigma-Aldrich), 29 nM sodium selenate (Sigma-Aldrich) and 1 μM retinoic acid (Sigma-Aldrich). For hormonal treatment, cells were grown in Dextran Charcoal Coated-treated (DCC) FBS. Dexamethasone (DEX) (Sigma-Aldrich) and RU 486 (RU) (Sigma-Aldrich) were used at the indicated concentrations diluted in ethanol. For RNA stability analysis, 10 μg/ml of 5, 6-Dichlorobenzimidazole-1-β-D-ribofuranoside (DRB) (Sigma-Aldrich) diluted in DMSO was applied.
Immunocytochemistry
BZ cells cultured in x-well tissue culture chambers (Sarstedt, Numbrecht, Germany) were fixed with PBS containing 4% paraformaldehyde (Sigma-Aldrich) solution for 15 min at room temperature. Cells were permeabilized with 0.5% Triton X-100 (Sigma-Aldrich) for 5 min and blocked by 1% non-fat milk in PBS with 0.1% Tween-20 (PBST) for 1 h at 37 °C. Cells were incubated with anti β-tubulin III antibody TU20 (sc-51670, Santa Cruz, La Jolla, CA, [49]) or with anti-GR antibody M20 (sc-23476, Santa Cruz, [50]) overnight at 4 °C and then with secondary antibody Cy3 (Interchim, Thermo scientific) for 45 min in PBST with 1% milk. After washing, BZ cells were incubated with DAPI (4′,6′-diamidino-2-phenylindole), 1: 1000 in PBST for 2 min and observed with a fluorescence Olympus microscope AX70 (Olympus, Hambourg, Germany).
RNA extraction and quantitative real-time PCR
Gene expression was quantified by reverse transcription (RT) followed by quantitative PCR (qPCR). Cells were harvested and total RNAs were extracted with Trizol reagent (Invitrogen, Cergy-Pontoise, France) according to the manufacturer’s instructions and their concentrations were determined using a Nanodrop 2000 spectrophotometer (Thermo scientific). RNAs were reverse-transcribed and processed for real-time PCR on an ABI Step One Plus (Applied Biosystems, Courtaboeuf, France). Briefly, 1 μg of total RNA was treated by DNAse I (Invitrogen), then reverse-transcribed with 50 U MultiScribe reverse transcriptase (Applied Biosystems). After 10-fold dilution, 1/40 of the RT reaction was used for qPCR using the Fast SYBR Green PCR master mix (Applied Biosystems). Final primer concentrations were 300 nM for each primer. Primer sequences for each gene are listed in Additional file 1: Table S1. Reaction parameters were 95 °C for 20 s followed by 40 cycles at 95 °C for 3 s and 60 °C for 30 s. For plasmids used for standard curves, amplicons were purified from agarose gels and subcloned into pGEMT-easy plasmid (Promega, Charbonnières, France), then sequenced to confirm the identity of each fragment. Standard curves were generated using serial dilutions of standard plasmids, spanning six orders of magnitude and yielding correlation coefficients more than 0.98 and efficiencies of at least 0.95, in all experiments. Standard and sample values were determined in duplicate from each sample. Relative expression within a given sample was calculated as the ratio: attomol of specific transcripts/attomol of 36B4 transcripts.
Protein extraction and Western blotting
Protein expression was assessed by Western blotting. Cells were harvested from the culture plates and total cellular proteins were extracted by lysis buffer containing 1% Triton X-100, 1% proteasome inhibitor cocktail (Sigma-Aldrich). Lysates were cleared by centrifugation at 13,000 g for 20 min and protein concentrations were measured using the BC Assay Protein Quantitation Kit (Uptima, Oakland, CA). Total protein lysates (50 μg) were fractionated on 15% SDS-PAGE then transferred to nitrocellulose membranes (GE Healthcare Life Sciences, Vélizy-Villacoublay, France). Membranes were blocked for 1 h in PBST with 2.5% BSA, and then incubated with primary antibodies overnight at 4 °C, rabbit polyclonal anti-BDNF (sc-546, Santa Cruz [51]) at 1:200 and mouse monoclonal anti-α-tubulin (Clone DM1A, Sigma-Aldrich) at 1:10,000. Incubation with secondary antibodies conjugated to infrared fluorophores (goat anti-rabbit IgG Dylight 800 and anti-mouse IgG Dylight 680 at 1:10,000 from Thermo Scientific) was performed for 1 h. An Odyssey infrared imaging system (LI-COR, Bad Homburg, Germany) was used to scan membranes at a wavelength of 680 nm (anti-mouse) or 800 nm (anti-rabbit). Data were analyzed with Image Studio 1.1 software (Li-COR).
Luciferase assays
pCDNA3-GR plasmid was generated by subcloning human GR (NR3C1) into pCDNA3 vector (Invitrogen). Bdnf promoter reporter plasmids were constructed by inserting PCR fragments from Bdnf promoter regions into PGL4-basic luciferase reporter vector (Promega), see Additional file 1: Table S1 for primer sequences. These fragments, named LP6, SP6, LP4, and SP4, are localized according to the distance to exon VI transcription starting site that was set at +1 (see Fig. 4). Specific genomic sequences were amplified by PCR from mouse genomic DNA and inserted into the PGL4 cloning vector in the Xho I and Hind III restriction sites using pGEMT easy vector kit (Promega), then transformed in JM109 E.Coli. Plasmids were sequenced to confirm the identity and orientation. The renilla activities driven by the expression vector PRL-TK (Promega) or total protein concentrations were used for normalizing expression.
Fig. 4.
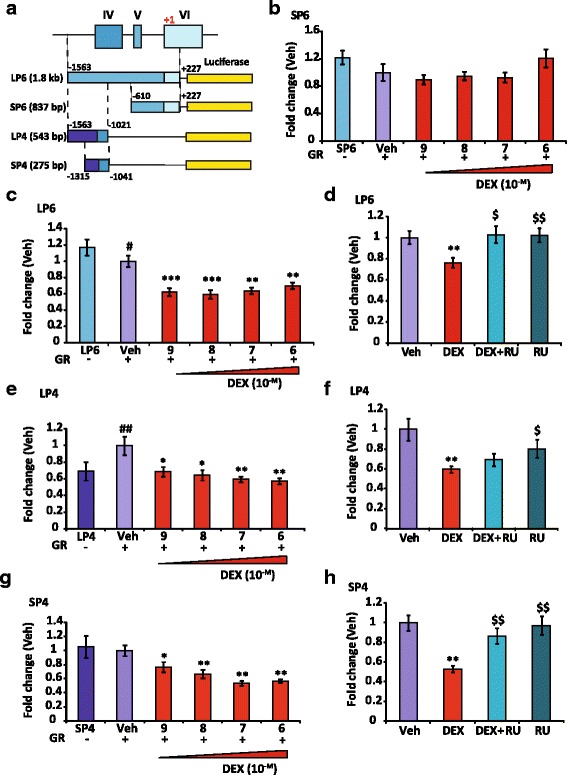
GR-dependent inhibitory effects on BDNF promoter 4 activity relies on a short sequence upstream of exon IV. a, Map of transfected constructs, LP6, SP6, LP4 and SP4 regions. These sequences were subcloned into PGL4 luciferase plasmid, setting the first base of exon VI at +1: a long (LP6, 1.8 kb, -1563, +227, containing exon IV, V and part of VI) and three short regions (SP6, 837 bp, -610, +227, partly encompassing exon VI; LP4, 543 bp, -1563, -1021; SP4, 275 bp, -1315, -1041; LP4 and SP4 are upstream of exon IV and include its transcription start site). b, DEX dose responses (Veh, Ethanol for 24 h) of SP6 transfected in N2A cells, co-transfected with GR expression vector and Renilla plasmid for normalization. Results are expressed as fold change of the luciferase/Renilla value of SP6 transfected alone. No significance was observed whatsoever (n = 16), showing SP6-driven activities were not modified by DEX. c, Same as in (b) with LP6 construct, Veh condition is set at 1; n = 16, Mean ± SEM; # p < 0.05, LP6 vs Veh; **P < 0.01, ***P < 0.005, DEX vs Veh; Mann Whitney U-tests. Luciferase activities driven by LP6 were already decreased by DEX at a low concentration. d, Transfection of LP6 in N2A cells, 24 h treatment with Veh, DEX (10-7M), RU (10-6M) and both; n = 16, Mean ± SEM; $ p < 0.05, $$ p < 0.01, vs DEX; Mann Whitney U-tests. The DEX repression is inhibited by RU. e, DEX dose responses of LP4 transfected in N2A cells. Results are expressed as fold change of the luciferase/protein value of LP4 transfected alone. Veh condition is set at 1; n = 8; ## P < 0.01, LP4 vs Veh;* P < 0.05, ** P < 0.01, DEX vs Veh; Mann Whitney U-tests. DEX reduced the LP4 activities in a dose-response manner. f, Transfection of LP4 construct plasmid in N2A cells, 24 h treatment with Veh, DEX (10-7M), RU (10-6M) or both; n = 16, Mean ± SEM; *P < 0.05 vs Veh; $ p < 0.05, vs DEX; Mann Whitney U-tests. The DEX effect was not reversed by RU. g, Same as in (e) with SP4 construct. DEX vs Veh; * P < 0.05 and ** P < 0.01. DEX repressed SP4 activity. h, SP4 fragment is repressed by GR. Same as in (f) with SP4 construct; ** P < 0.01 vs Veh; $$ P < 0.01 vs DEX
On the day prior to transfection, N2A cells were plated in 96-well plates at a density of 2 × 104 cells per well in the N2A differentiation medium. Medium was changed for OptiMEM medium (PAA) on the next day, and cells were transfected using Lipofectamine 2000 reagent (Invitrogen). Plasmid concentrations per well were 50 ng PGL4-Bdnf promoter vectors, 25 ng pCDNA3-GR or empty vector, 13.3 ng PRL-TK renilla expression vector. Six h post-transfection, OptiMEM medium was replaced by N2A DCC medium. Cells were treated with Ethanol (Veh), DEX and/or RU486 24 h after transfection and then were harvested after another 24 h with Passive Lysis Buffer from the dual luciferase reporter assay system kit (Promega). Lysates were transferred to 96-well clearbottom microplates (Fisher), and luminescence intensities were measured with a TRISTAR LB941 automatic luminometer (Berthold, Thoiry, France) with dual injectors. Normalized values were used for statistical analyses. Each experiment was performed with 8 replicates and repeated at least three times.
Chromatin immunoprecipitation (ChIP)
ChIP experiments were performed using the Ideal-ChIP for transcription factor kit (Diagenode, Liège, Belgium) following the manufacturer’s protocol. Specifically, 4 × 106 BZ cells per well of 6-well plates grown in 1% DCC medium were treated by vehicle (ethanol), Dex 10-7 M or Dex 10-7 M together with RU 10-6 M for 1 h. Cells were then fixed by a mix of formaldehyde 16% and fixation buffer from the kit (4: 1 ratio), 1/100 volume final for 8 min. ChiP experiments were performed according to Le Billan et al [52]. Chromatin samples (300 μl) were sheared by 12 cycles 30 s ON and 30 s OFF with the Diagenode Bioruptor Pico system and 2.5 μl samples were kept for input measurements. Two hundred and fifty μl of sheared chromatin samples were incubated overnight at 4 °C with 1 μg IgG from the kit as control or 5 μg anti-GR antibody H300 (Santa Cruz sc-8992-X [53]). Chromatin shearing was controlled on a 1.5% agarose gel and typically a smear was visualized ranging from 100 to 500 bp. qPCR amplifications of eluted DNA were performed with primers encompassing a short upstream sequence of exon IV, or on Per1 and Ucp1 promoters (Additional file 1: Table S1 for primer sequences). Raw data are expressed as percentage of inputs, according to the Percent of Input Method, ChIP analysis; Thermo Fischer Scientific.
Immunoprecipitation
Immunoprecipitations were performed using 30 μl protein A-coated magnetic beads (Diagenode). Beads were washed 3 times with C1 buffer of the High Cell ChIP kit (Diagenode) and incubated for 4 h with anti-GR antibody H300 (Santa Cruz) at 4 °C with protease inhibitors and 0.2% BSA. Then, 350 μg of proteins from BZ cell lysates were incubated overnight with antibody-coated beads. The following day, beads were washed 3 times with buffer C1 and once with buffer W1 (High Cell ChIP kit), and immunoprecipitated proteins were eluted from the beads with 20 μl of Laemmli buffer at 95 °C for 5 min and loaded on a 10% SDS-PAGE gel for Western blotting. Membranes were hybridized with anti-GR antibody M20 (sc-23476, Santa Cruz, [54].
Targeted mutagenesis
Targeted mutations of Bdnf promoter construct plasmids were performed using the Quickchange II XL kit (Agilent Technologies, Les Ulis, France) following the manufacturer instructions. Briefly, 10 ng of SP4 luciferase plasmid was amplified by PCR with sense and antisense mutated primers for 18 cycles. PCR reactions were transformed in XL1 blue ultracompetent E. Coli strain (Agilent, les Ulis, France) on LB Agar Ampicillin petri dishes. Bacterial colonies were picked up. Plasmid DNA were extracted and sequenced by Eurofins (Ivry sur Seine, France) to check for the introduction of the mutation and sequence integrity. Primers for mutagenesis are available in Additional file 1: Table S1. Plasmids were transfected in N2A cells as stated for the luciferase assay.
Statistical analyses
Results are expressed as mean ± SEM of at least six samples for each condition unless stated otherwise. Statistical analyses were performed using nonparametric Mann-Whitney U-tests, unless stated otherwise, using Prism 5 (GraphPad Software, Inc., San Diego, CA).
Results
GR represses Bdnf expression in primary mouse hippocampal cultures
To determine the effect of GCs on Bdnf expression in neurons, we used day 9 primary cultures of mouse hippocampal neurons (PCN), a model of high physiological relevance that expresses both BDNF and GR (Additional file 2: Figure S1a and c). Bright field microscopy at day 3 in vitro (D3 IV, Fig. 1a upper panel) already showed a nearly homogenous neuronal culture that developed in a dense neuronal network at day 9 (D9 IV, Fig. 1a, lower panel). Neuronal marker MAP2 transcripts expression measured by qPCR was in the same range in PCN than in mouse brain while glial marker GFAP expression, assessed as an estimation of astrocyte contamination displayed a more than two hundred times lower expression in PCN culture than in mouse brain (Additional file 2: Figure S1b and d), indicating a high neuronal enrichment in PCN culture. Additionally, GR mRNA and protein were clearly expressed in D9 IV cultures, as shown in qPCR (Additional file 2: Figure S1c) and in Western blot (Additional file 2: Figure S1e). One h treatment with dexamethasone (DEX), a synthetic GR agonist (10-6 M), was sufficient to significantly repress by 30% total BDNF mRNA expression (Fig. 1b), measured by qPCR on the common exon IX abundance. This inhibition was fully reversed by co-treatment with the GR antagonist RU486 (RU). These results were consistent with a previous study performed on rat PCN [42]. These experiments were performed using a neuronal media supplement (Pierce neuronal isolation kit, reference 88286) that was required for the survival of PCN and that did contain GC. We were unable to maintain the cells in culture in the absence of the supplement even for a few h. Of note, the culture medium with the supplement contains a substantial concentration of corticosterone (~2.10-8 M), as measured by LC/MS-MS (data not shown), that is sufficient to partially bind and activate GR.
Fig. 1.
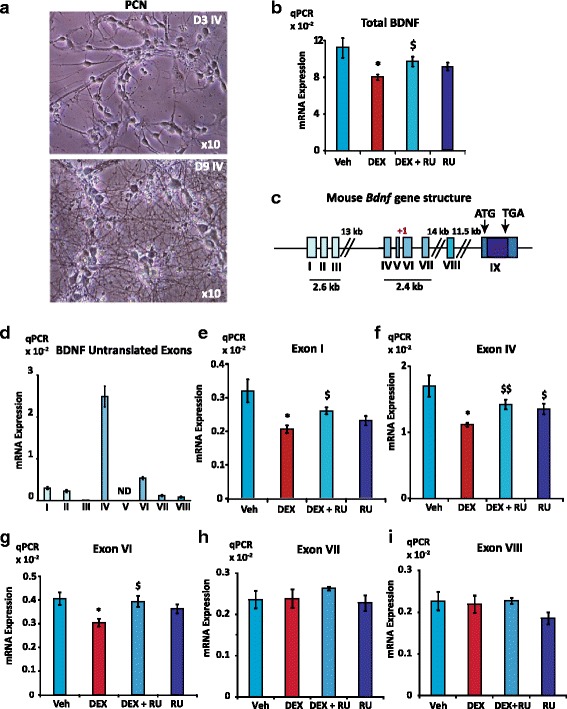
GR inhibits Bdnf expression in 9-day PCN. a, Bright field microscopy showed that PCN developed a morphology with multiple cellular processes on Day 3 in vitro (D3 IV, upper panel), while a denser neuronal network was visible on Day 9 (D9 IV); magnification x10. b, Total BDNF mRNA expression was significantly repressed by DEX. Relative expression of total BDNF mRNA assessed by qPCR on exon IX under vehicle, (Veh, ethanol) dexamethasone (DEX; 10-7 M) or DEX and GR antagonist RU486 (RU; 10-6 M) treatments (1 h). Results are expressed as attomol of BDNF mRNA/attomol of 36B4 mRNA (n = 6, Mean ± SEM); * Veh vs DEX, P < 0.05; $ DEX vs DEX + RU, P < 0.05, Mann Whitney U-tests. c, Schematic representation of mouse Bdnf gene according to Aid et al [18]. Eight untranslated exons (I to VIII) splice onto one common coding exon IX, with two regulatory regions that drive the expression of mainly 2 clusters of untranslated exons (I, II, III and IV, V, VI, respectively). d, Relative expression of Bdnf untranslated exons in 9-day PCN (n = 6, Mean ± SEM) as measured by RT-qPCR. Exons IV and VI containing transcripts are the most expressed isoforms, followed by that of exon I containing mRNA. ND: non determined. e, f, g, h, i, Expression of exon I, IV, and VI containing transcripts was downregulated by DEX while expression of exon VII and VIII containing transcripts was not modified by DEX treatment. qPCR analyses of exon I, IV VI, VII and VIII mRNA levels in the same samples than in (a), Mean ± SEM; * (P < 0.05) vs Veh; $P < 0.05; $$ P < 0.01 vs DEX; Mann Whitney U-tests
In order to better understand the mechanisms by which GCs decrease neuronal Bdnf expression, it is worth noting that mouse Bdnf gene exhibits a complex genomic structure with 8 untranslated exons (I to VIII) that splice onto the common coding exon IX and two clusters of promoters upstream of I, II, III and IV, V, VI, respectively (Fig. 1c) [18, 55]. We determined the relative expression of the untranslated exon-containing transcripts by RT-qPCR and found that exon IV containing mRNA isoform was by far the most expressed one followed by exon VI and I containing isoforms (Fig. 1d). Expression of all of these 3 exon-containing messengers was repressed by DEX while RU suppressed this effect (Fig. 1e, f and g). Conversely, DEX did not exert any effect on exons VII and VIII containing transcripts (Fig. 1h and I), highlighting an exon-specific regulation of GR on Bdnf gene. Altogether, these data showed that GR was able to repress BDNF mRNA expression in PCN by downregulating specific isoforms even in the presence of noticeable amounts of GCs in the medium.
The BZ cell line is a glucocorticoid responsive model expressing BDNF
In PCN, the presence of corticosterone in the culture supplement was likely able to interfere with investigation on GR action on BDNF expression. Nonetheless, the BZ cell line previously generated from a mouse hippocampus by targeted oncogenesis [56] displayed several interesting characteristics to study this issue. BZ cells appeared in light microscopy as slightly elongated and connected by short processes when grown in 1% fetal bovine serum (FBS, Fig. 2a, upper left panel). Immunolabeling revealed the presence of the β-tubulin III neuronal marker in BZ cells (Fig. 2a, upper right panel and lower panels), indicating they present with some neuron-like features. Moreover, GR was readily detected by immunocytochemistry as a nucleocytoplasmic labeling in BZ cells (Fig. 2b). As shown by the DAPI counterstaining, GR expression is expressed in all BZ cells. In addition, expression of Gr, Bdnf as well as the neuronal marker Map2 and the NmdaRζ1 (Grin1) isoform was observed in BZ cells as measured by RT-PCR (Fig. 2c), and compared to embryonic stem cell derived neurons (ES) and mouse brain. Quantification by qPCR of BDNF and GR mRNAs showed that their relative levels were in the same range in BZ cells and PCN (Additional file 2: Figure S1a and c), although lower when compared to those measured in the brain. While BZ cell expression of Map2 neuronal marker was comparatively lower than the one measured in PCN and brain, it was similar to that found in ES derived-neuron cultures (Additional file 2: Figure S1b). It is worth noting that the main BDNF receptor TrKB was not expressed in BZ cells (data not shown), thus any observed effect on BDNF expression is not due to a negative feedback loop resulting from TrkB receptor activation. BDNF protein was clearly detected by Western blotting on lysates from BZ cells and PCN as well as mouse brain (Fig. 2d). GR expression in BZ cells was further demonstrated by immunoprecipitation assays as revealed by the ~ 100 kDa molecular mass band observed in both input and GR immunoprecipitated complexes lanes (Fig. 2e). BZ cells were transfected with a plasmid containing two glucocorticoid response elements fused with the luciferase gene (GRE2.Luc). Although transfection efficiency was low, DEX treatment was able to transactivate GRE2.Luc starting from a dose of 10-8 M indicating that a functional GR is expressed in BZ cells (Fig. 2f). We optimized the BZ cell culture conditions by lowering fetal bovine serum concentration from 10 to 1% for 48 h and showed that this experimental condition increased the expression of the MAP2 neuronal marker mRNAs (Fig. 2g). As BDNF and GR transcripts expression are identical between all culture conditions, this effect is probably specific. Thus, BZ cells were routinely expanded using 10% FBS, plated at 12,500 cells/cm2 and grown in low serum concentration (1%) 48 h before hormonal treatments. Altogether, the BZ cell line of hippocampal origin exhibits neuronal features and expresses substantial amounts of both GR and BDNF at the messenger and protein levels, enabling to investigate the impact of GR activation on Bdnf expression.
Fig. 2.
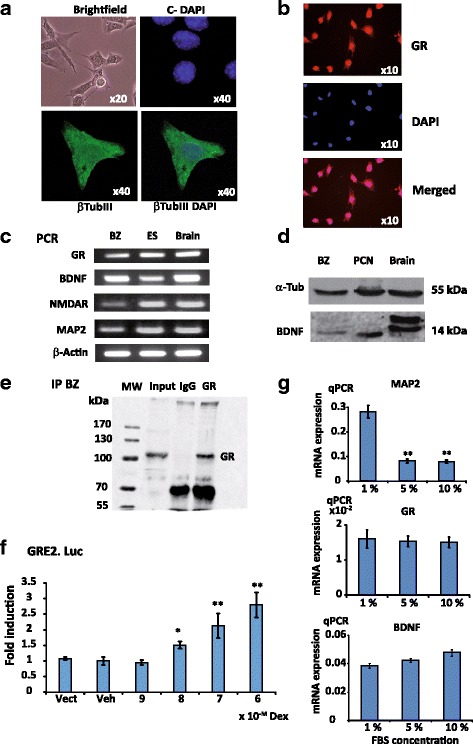
Characterization of the BZ cell line. BZ cells are cultured in DMEM supplemented with 1% FBS unless stated otherwise. a, BZ cells present with neuronal features. BZ cells display an elongated morphology harboring multiple short cellular processes, brightfield microscopy (magnification x20, upper left panel). Immunolabeling images (x40) with negative control secondary antibody and DAPI counterstaining (upper right panel), β-tubulin III (lower left panel) and merged DAPI and β-tubulin III (lower right panel). b, GR is expressed in BZ cells. Immunocytochemistry of GR staining on BZ cells grown in 10% FBS, showing a homogenous expression of GR in this cell line. GR, upper panel, DAPI, middle panel, Merged, lower panel (c), Analytical RT-PCR of GR, BDNF, NMDA receptor ζ (NMDAR) and MAP2 mRNA expression (30 cycles of amplification with DreamTaq (Thermo Scientific)) on BZ cells compared with neurons derived from mouse embryonic stem cells (ES) and mouse brain. d, Western Blot on BDNF protein in BZ cells, primary cultures of mouse hippocampal neurons (PCN), and mouse brain, showing that BDNF protein was clearly detected in all of the 3 samples. e, Immunoprecipitation in BZ cell lysates with anti-GR antibody, further demonstrating GR expression, MW: molecular weight markers in kDa, IgG: rabbit immunoglobulins, GR band is around 105 kDa. f, DEX was able to transactivate GRE2.Luc, indicating the presence of a functional GR in BZ cells. Transfection of BZ with a PGL3-Gre2.Luc plasmid under DEX dose responses (10-9 to 10-6 M, n = 8, Mean ± SEM); Vect, GRE2.Luc; Veh set at 1; * P < 0.05, ** P < 0.01, Mann Whitney U-tests. g, Increased expression of neuronal marker MAP2 transcripts at low FBS (1%) concentration. MAP2 (higher panel), GR (middle panel) and BDNF (lower panel) mRNA expression in BZ cells under various FBS concentrations from 1 to 10%; n = 6, Mean ± SEM; ** P < 0.01 vs 1% condition, Mann Whitney U- tests
GR represses Bdnf expression in BZ cells
The effect of DEX on total BDNF mRNA expression was analyzed on time course experiments, using a 10-fold lower concentration (10-7 M) than in PCN. DEX significantly decreased total BDNF transcript abundance by 30% after 3 and 4 h treatment (Fig. 3a). At this dose, no effect was observed on PCN probably because of the presence of corticosterone in the culture medium (data not shown). Parallel measurements of the GR target gene Sgk1 transcript levels revealed, as expected, a strong induction (3 to 5 fold) already detected after 1 h stimulation, thus validating GC treatment effectiveness, and the BZ cell ability to respond to GCs (Additional file 3: Figure S2a). In the presence of the transcription inhibitor DRB, there was no difference in the rate of BDNF mRNA turnover between Veh and DEX, suggesting that the DEX-induced reduction of BDNF transcript levels was related to transcriptional repression rather than RNA decay mechanisms (Fig. 3b). These findings also demonstrated that the half-life time of BDNF transcripts in BZ cells was evaluated at around 2 h, which is consistent with a rapid detection of BDNF mRNA level diminution upon GC exposure. Co-treatment with RU for 3 h abolished the effect of DEX on Bdnf expression, demonstrating that DEX repression is indeed mediated by GR (Fig. 3c). As a control, under the same experimental conditions, Sgk1 activation by DEX was fully repressed by addition of RU (Additional file 3: Figure S2b). The relative expression of Bdnf untranslated exons in BZ cells was somehow different from the one observed in PCN since exon VI-containing mRNA represented the highest expressed isoform in BZ cells, followed by exon VII and VIII then exon IV mRNA while the expression of exon I-containing transcript was negligible (Fig. 3d). Examination of exon IV and VI isoform expression after 3 h DEX treatment showed a similar pattern of repression than with total BDNF transcripts, which was fully prevented by coincubation with RU (Fig. 3e and f). Interestingly, DEX-induced repression was stronger with isoform IV and VI (around 50%) than for total BDNF transcripts. Likewise in PCN, there was no effect of DEX on exon VII and VIII (Fig. 3g and h), indicating once again the specificity of the DEX-repressive action on exon IV and VI expression. These findings suggested a GR mediated effect through a repressive mechanism on the promoter regions upstream of exon IV and/or VI (see Fig. 1c).
Fig. 3.
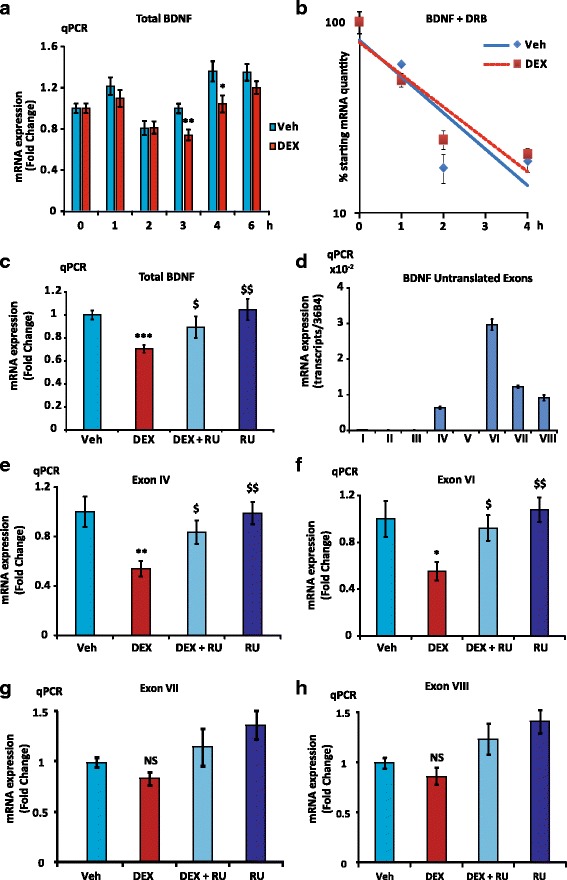
GR downregulates BDNF expression in BZ cells. a, The total BDNF transcript abundance was decreased by 30% after 3 and 4 h DEX treatment. Time course of total BDNF transcript expression under 10-7 M DEX or vehicle treatment, time 0 is set at 1, n = 12, Mean ± SEM; * P < 0.05, ** P < 0.01vs Veh, Mann Whitney U-tests. b, Repression of BDNF mRNA expression is transcriptional. Time course assessment of total BDNF mRNA expression under DRB in the presence or absence of DEX. Note the logarithmic scale of the y-axis. Total BDNF transcript half-life time was estimated at approximately 2 h. The corresponding slopes were not significantly different with ANCOVA test (P = 0.14) (c), GR effect is inhibited by RU. RT-qPCR on common exon IX, 3 h treatment with DEX 10-7 M and/or RU 10-6 M (n = 12, Mean ± SEM); Veh vs DEX ***P < 0.001, RU and DEX + RU vs DEX, $ P < 0.05, $$ P < 0.01. d, Relative expression of BDNF untranslated exons containing mRNA in 4-day BZ cultures (n = 6, Mean ± SEM) as measured by RT-qPCR. The highest expressed isoform in BZ cells is exon VI-containing transcript, followed by exon VII and VIII, then exon IV, while exon I mRNA expression was negligible. e, f, g, h, GR specifically repressed exon IV and VI expression but was ineffective on exon VII and VIII expression. qPCR on exon IV, VI, VII and VIII (n = 12, Mean ± SEM) containing transcript levels in the same samples than in (c); Veh set at 1, * (P < 0.05), and ** (P < 0.001) vs Veh; $ (P < 0.05) and $$ (P < 0.01) vs Dex, Mann Whitney U-tests
GR represses Bdnf expression by acting upstream of exon IV
Exons IV and VI are in a close vicinity on mouse Bdnf gene, located in a ~ 1.5 kbp sequence (Fig. 4a). To clarify the mechanisms by which activated GR represses neuronal Bdnf expression, we generated several Bdnf promoter luciferase constructs including four fragments of this region, LP6 (1.8 kb, -1563; +227) and SP6 (837 bp, -610; +227) flanking exon VI, LP4 (543 bp, -1593; -1021) and SP4 (275 bp -1315; -1041) flanking exon IV. The first base-pair of exon VI was arbitrarily set at +1 for the sequence numbering (see Fig. 4a). The activities of these fragments inserted upstream of the reporter luciferase gene were investigated for their responses to GCs using luciferase assays in the neuroblastoma tumor cell line N2A. This model has been preferred to BZ cells given that these latter cells exhibit a very low transfection efficiency. DEX dose response curves (10-9 to 10-6 M) were generated with the four constructs while cotransfecting a GR expression vector. Whereas the luciferase activities driven by the SP6 construct were not modified by DEX whatever the GC dose (Fig. 4b), LP6-driven activities were significantly reduced even at the lowest DEX concentration (10-9 M) (Fig. 4c). Co-treatment with RU abolished DEX inhibitory effect, stressing that this repression was mediated by GR (Fig. 4d). To determine the location of the regulatory regions on which DEX-activated GR affected Bdnf expression, we narrowed down our promoter analyses by focusing on two shorter DNA fragments, LP4 and SP4, upstream of exon IV (see Fig. 4a) since DEX did not regulate SP6 activity. Luciferase activities driven by both the two shorter fragments were significantly reduced by GR with DEX compared with vehicle, likewise what was observed with the LP6 fragment, in a dose-response manner (Fig. 4e and g). While DEX was able to repress LP4 construct, (Fig. 4f), significant antagonist effect of RU was observed with the SP4 construct (Fig. 4h) indicating that activated GR may act directly or indirectly on this short 275 bp region upstream of exon IV in an inhibitory fashion.
AP1 and CRE response elements do not mediate GR transrepression
To investigate the molecular mechanisms of the glucocorticoid-dependent repression, we analyzed in silico the GR-sensitive SP4 sequence (275 bp; -188, +87 from exon IV transcription start site) using Jaspar database (URL: http://jaspar.genereg.net/cgi-bin/jaspar_db.pl) but were not able to identify any potential GR response elements (GRE). In contrast, we found two Jun/Fos response elements, comprising two potential AP1 binding sites (positions -127 and -101), as well as two cAMP response elements (CRE), for CREB1 (position -38) and CREB3l2 (position -14), see Fig. 5a. Given that GR is able to repress SP4 sequence transactivation ability (see Fig. 4h), it was tempting to speculate that GR effect was the result of a transrepression mediated by its interaction with AP1 and/or CRE binding transcription factors as it has been previously proposed [57]. Of interest, in humans and rats, these CRE response elements, which are evolutionary conserved were characterized as positively regulated by synaptic activity [46], while constructs harboring the AP1 sites were not sensitive to a previously described BDNF-positive feedback loop [58]. To test this hypothesis, deletion mutants of AP1 (mAP1-1 and mAp1-2) and CRE (mCRE1 and mCRE2) binding sites were generated from SP4-luciferase plasmid, as well as double mutants with deletion of the two potential AP1 binding sites (dmAP1) or the two CRE binding sites (dmCRE) sites (Fig. 5a, the deleted bases are indicated in white lettering). All AP1 and CRE mutants, as well as the double mutants showed a strong reduction of their basal transcriptional activities in N2A cells compared to the WT plasmid indicating they are functional sequences in SP4 construct (Fig. 5b and d). The transcriptional response to DEX was measured setting the vehicle condition value at 1 for each construct (Fig. 5c and e). For all these mutants, as well as for AP1 and CRE double mutants (dmAP1 and dmCRE), DEX treatment still led to the repression of their transcriptional activities in a similar fashion than with the SP4 wild type fragment. In the absence of classical GRE and other response elements for transcription factors known to interact with GR in the SP4 sequence, these data excluded the possibility of an interaction between GR and AP1 and/or CRE response elements to transrepress Bdnf expression in these neuron-like cells.
Fig. 5.
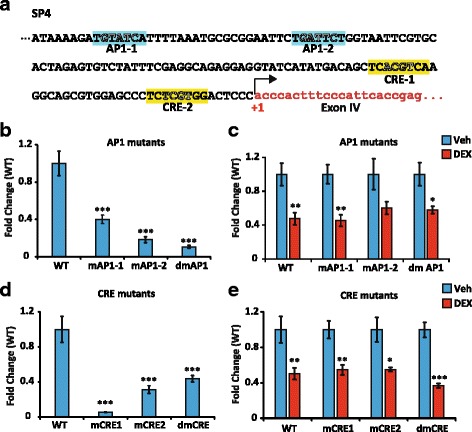
AP1 and CRE response elements do not mediate GR transrepression. a, Sequence analysis of a short fragment upstream of exon IV, and included in SP4 sequence that contains two potential AP1 binding sites, TGTATCA and TGATTCT, as well as two potential cAMP Response-Elements (CRE): CRE-1 (for CREB1, TCACGTCA) and CRE-2 (for CREB3l2, TCTCGTGG) according to the Jaspar database; +1 is the transcription start site of exon IV. b, Luciferase activity of WT and AP1 mutants. mAP1-1: construct with AP1-1 deletion, mAP1-2, construct with deletion of AP1-2 sequence, dmAP1, deletion of both sites. Deleted based are shown in white lettering in Fig. 5a. Basal transcriptional activity driven by the WT sequence was arbitrarily set at 1, Means ± SEM; *** P < 0.005; Mann Whitney U-tests, n = 8. c, Luciferase activities of promoter mutants with AP1 deletions were still repressed by DEX. * P < 0.05 DEX (red) vs vehicle (blue). Mean ± SEM; ** P < 0.01, DEX vs Veh of the same construct. Mann Whitney U-tests, n = 8; all Veh conditions were set at 1. d, Luciferase activity of WT and CRE mutants. mCRE-1: construct with CRE1-1 deletion, mCRE-2, construct with deletion of CRE-2 sequence, dmCRE, deletion of both sites. Deleted based are shown in white lettering in Fig. 5a. Basal transcriptional activity driven by the WT sequence was arbitrarily set at 1, *** P < 0.005; Mann Whitney U-tests, n = 8. e, Luciferase activities of promoter mutants with CRE deletions were still repressed by DEX. mCRE-1: construct with CRE-1 deletion, mCRE-2, construct with deletion of CRE1-2 sequence, dmCRE, deletion of both sites. * P < 0.05 DEX (red) vs vehicle (blue). Means ± SEM; ** P < 0.01, DEX vs Veh for the same construct; *** P < 0.005, DEX vs Veh for the same construct, Mann Whitney U-tests, n = 8; all Veh conditions were set at 1
GR binds upstream of exon IV upon DEX treatment
Even if no specific DNA response element was identified for DEX responses, transfection data strongly suggest a direct involvement of GR to repress SP4 transcriptional activity. To determine whether GR does bind to Bdnf SP4 promoter fragment, chromatin immunoprecipitation (ChIP) experiments were undertaken in BZ cells using specific primers (see Additional file 1: Table S1), the targeted sequence and primer pair being schematized in Fig. 6a. DEX treatment led to a ~ 3-fold enrichment of GR recruitment compared to the control condition (Veh) or IgG, while cotreatment with RU significantly reduced GR binding (Fig. 6b). As a positive control, we also examined the capacity of GR to interact with the regulatory sequence of a well-known GR target gene period circadian clock 1 (Per1) [52]. Under such experimental conditions, a 10-fold enrichment of GR upon DEX exposure was measured that was antagonized by RU (Fig. 6c). Besides, as a negative control, no GR recruitment was detected on a genomic sequence comprising a fragment of Ucp1 promoter (Kuhn et al, unpublished observations), a gene whose expression is mostly restricted to brown adipocytes [59] (Fig. 6d), further demonstrating that GR recruitment on Bdnf and Per1 promoters was specific. Taken together, ChIP experiments provided evidence that DEX inhibits Bdnf transcriptional activity, at least partly by specific GR recruitment onto a short fragment (SP4) directly upstream of exon IV.
Fig. 6.
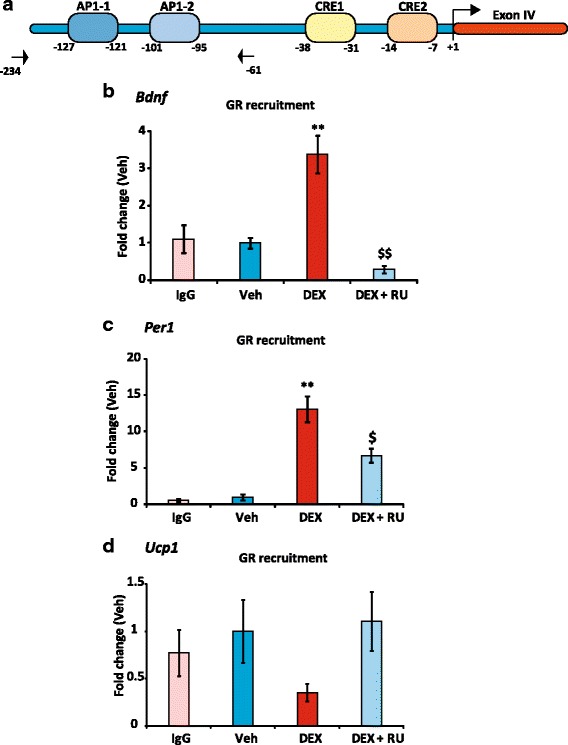
GR recruitment on Bdnf DNA sequence. a, Schematic representation of a short fragment upstream of exon IV that contains the two potential AP1 binding sites, AP1-1 and AP1-2 as well as two potential cAMP Response-Elements CRE-1 and CRE-2 according to the Jaspar database. Exon IV transcription start site is set at +1. Primers for ChIP are indicated by arrows. b, ChIP-qPCR results showing GR recruitment upstream of exon IV region in BZ cells after treatment with DEX (10-7 M) or DEX together with RU (10-6 M) for 1 h. IgG: ChIP-qPCR with IgG negative control antibodies on the same sequence. Results of 3 different experiments are pooled setting the vehicle mean value at 1 (n = 6, Mean ± SEM), and showed as fold change of the percentage of input value of Veh.** P < 0.01, DEX vs Veh; $$ P < 0.01, DEX + RU vs DEX; Mann Whitney U-tests. c, ChIP-qPCR results (same samples than in B) of GR recruitment on Per1 gene regulatory sequence used as positive control in BZ cells under DEX or DEX + RU treatment for 1 h. ** P < 0.01, DEX vs Veh. $ P < 0.05, DEX + RU vs DEX. d, same as in (c) with GR recruitment on Ucp1 gene used as negative control. Primers for genomic PCR amplification are listed in Additional file 1: Table S1
Discussion
BDNF, the main neurotrophin is a key factor regulating neuronal function in health and diseases, such as survival, growth, synaptic structure and activity. Thus, understanding the molecular mechanisms involved in the regulation of neuronal Bdnf expression is a major issue. However, the effect of glucocorticoids on Bdnf neuronal expression is still a matter of debate [60]. Given the complexity of the central nervous system anatomy and its functional organization, where neurons represent only a small fraction of the total number of cells, we focused our attention in the present study to in vitro neuronal models in order to investigate GC action on Bdnf expression. Besides, GC bind to two receptors in the brain; the high affinity MR, which is occupied even at low concentrations of hormones, and GR with a lower affinity for ligands thus more sensitive to hormonal regulation and variation [27]. In the present work, we decipher the role of GR as a mediator of GC actions and for this purpose we use the glucocorticoid agonist dexamethasone (DEX) that exhibits a low MR activating capacity [61, 62].
We have demonstrated that DEX exposure led to a marked downregulation of BDNF transcript expression in neuronal cells that was mediated by GR. More precisely, total BDNF mRNA levels were reduced after GC treatment in primary hippocampal neuron cultures, but also in the neuron-like cell line BZ. In both models, BDNF transcripts containing exons IV and VI isoforms were highly expressed and specifically repressed by GR activation. Transfection experiments were performed using a long 1.8 kb DNA fragment (LP6) encompassing the closely packed exon IV, V and VI and their 5’ regulatory sequences. LP6-driven activity was significantly repressed by DEX yet prevented by addition of GR antagonist RU. Functional characterization of the transcriptional activity of LP6 construct fragments by luciferase reporter assays led to identify a short 275 pb sequence (SP4) directly upstream of exon IV, and encompassing its transcription start site, which might account for the inhibitory effects of GCs. Analysis by Jaspar online software (URL: http://jaspar.genereg.net/cgi-bin/jaspar_db.pl), revealed that two Jun/Fos response elements, constituting AP1 binding sites, were located on this region as well as two cAMP response elements (CRE), a CREB1 and a closely related CREB3l2 response element, all being located just upstream of the exon IV transcription start site (see Fig. 6a). Chromatin immunoprecipitation assays demonstrated that GR binds to this sequence, GR recruitment being prevented by GR antagonist RU. However, promoter constructs with deletion of AP1 and CREB binding sites were still repressed by DEX excluding their involvement in GR regulation of Bdnf expression. Altogether, we propose that one of the mechanisms responsible for the repression of Bdnf expression by DEX is the binding of GR just upstream of exon IV, through ternary complexes with transcription factors that are still to be determined. Such a repression of gene expression by GR tethered to various transcription factor complexes has been reported in several studies [57, 63]. Nonetheless, no other response elements for proteins susceptible to interact with GR, such as NFκB, were identified on the SP4 sequence using Jaspar software. Another proposed mechanism of direct GR repression was uncovered recently in the form of negative GRE (nGRE) with a specific sequence (CTCCXGGAG) that clearly differed from positive GRE [64]. Anyhow, none of putative nGRE was identified in the SP4 sequence. Eventually, one might speculate that GR binds to a specific GRE outside the long LP6 fragment that lacks any GRE and acts on Bdnf promoter by a folding DNA loop, a mechanism that has been recently described for GR [65]. However, such a mechanism is very unlikely given that GR interacts with the SP4 sequence as demonstrated by transfection and ChIP assays. Nonetheless, we believe these results are of important since they excluded the involvement of specific response elements for transcription factors known to be transrepressed by GR (Jun/fos and CREB) in the SP4 sequence. This raises the hypothesis of a new mechanism involving some partners mediating GR transrepression that remains to be identified.
It is worth noting that this region upstream of exon IV, which displays a high degree of homology between mammalian species emphasizing its biological importance, has been extensively characterized in previous studies by several groups as positively regulated by calcium [45, 66, 67], synaptic activity [46] and BDNF itself [58] in humans and rats. Moreover the two CRE elements for CREB (CRE1) and CREB3l2 (CRE2) we identified in the mouse sequence are conserved and involved in this regulation, CRE2 being formerly reported as a BHLHB2 response element [46, 68]. In sum, we unraveled that GR represses the activity of exon IV promoter that was previously shown to be stimulated by synaptic activity [46]. This is consistent with the adverse effects of the overactivation of glucocorticoid pathways on brain physiology [27].
The BZ cell line was derived few years ago from a mouse hippocampus. Herein, a more detailed characterization of this neuronal cell line was undertaken and revealed that BZ cells constitute a suitable cellular tool to investigate the regulation of BDNF expression. In comparison, the commercially available N2A cell line expresses very low levels of BDNF transcripts, impairing analysis of isoform expression even if total BDNF transcripts could be measured by the sensitive qPCR technique and we found that GC were able to repress Bdnf expression in this model (data not shown). Beyond that, BZ cells appear to be a suitable cell-based system for studying GR signaling pathways in a neuron-like cell context in vitro.
In the present study, quantification of BDNF protein abundance by Western blot in PCN and BZ cells, did not show any significant change with short term (6 h) or long term (24 h) DEX treatments (data not shown). Even if we could not exclude that GCs might reduce BDNF protein expression on a different timeline, recent studies underscored that BDNF mRNA isoforms localization might be a major factor regulating its functions [69]. For instance, transcripts containing exon IV and VI in rat primary hippocampal neuron cultures were found to display a more distal localization relative to the soma than those including exon I, while they were able to stimulate dendritic branching [19]. Based on this fine-tuning spatial code, it could be proposed that the localization of BDNF mRNA isoform translation would be related to distinct BDNF sites of action. Thus, it is very likely that total expression of BDNF or secretion level of the protein might constitute inappropriate indexes to define BDNF biological function.
Altogether, we provide evidence for a functional crosstalk between activated GR and Bdnf expression in mouse neurons that is, at least in part, mediated by GR binding to a restricted sequence upstream of exon IV. In sum, high circulating glucocorticoid levels prevailing under certain circumstances such as stress may alter neuronal Bdnf expression [44] and specifically its transcripts distribution leading to a remodeling of dendrite and synapse architecture and function. This could be of physiological importance in processes such as memorization and behavior as well as causal in various pathologies associated with modification of GR and BDNF pathways.
Additional files
Primer table. (DOCX 15 kb)
Gene expression in PCN and BZ cells. (EPS 1379 kb)
GR stimulates Sgk1 expression in BZ cells. (EPS 1437 kb)
Acknowledgements
We are indebted to Patrice Penfornis for the initial derivation and subcloning of the BZ cell line (present address: Cancer Institute, University of Mississippi Medical Center, Jackson, MS, USA). We thank Fanny Jung for part of the BZ cell line characterization during her practical training for her advance technology degree (BTS) in biotechnologies. We also thank Simon Travers (Service de Génétique Moléculaire, Pharmacogénomique et Hormonologie, Bicêtre Hospital) for corticosterone measurements by LC/MS-MS.
Funding
This work was supported by grants from the Institut National de la Santé et de la Recherche Médicale, the Université Paris-Sud and Agence Nationale de la Recherche (Grant 11-BSV1-028-01). HC was funded by a China Scholarship Council (CSC) fellowship.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Authors’ contributions
ML, HC, and DL designed the study. HC and DL performed the experiments. ML, HC, and DL wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Swiss mice were housed and handled according to the National Institutes of Health Guidelines. The animal work is part of an approved project by the ethical comity CEEA 26 (#2012_021). DL has an agreement to conceive and perform animal procedure (N°R-75UPMC-F1-08) by the French state agency ‘Ministère de l’Agriculture, de l’Agroalimentaire et de la Forêt’.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- BDNF
Brain-derived neurotrophic factor
- DEX
Dexamethasone
- GCs
Glucocorticoids
- GR
Glucocorticoid receptor
- GRE
GR response element
- PCN
Primary cultures of hippocampal Neurons
- N2A
Neuro-2A cells
- RU
RU486 (Mifepristone)
Contributor Information
Hui Chen, Email: hui.chen@u-psud.fr.
Marc Lombès, Email: marc.lombes@u-psud.fr.
Damien Le Menuet, Email: damien.le-menuet@u-psud.fr.
References
- 1.Ernfors P, Wetmore C, Olson L, Persson H. Identification of cells in rat brain and peripheral tissues expressing mRNA for members of the nerve growth factor family. Neuron. 1990;5(4):511–526. doi: 10.1016/0896-6273(90)90090-3. [DOI] [PubMed] [Google Scholar]
- 2.Hohn A, Leibrock J, Bailey K, Barde YA. Identification and characterization of a novel member of the nerve growth factor/brain-derived neurotrophic factor family. Nature. 1990;344(6264):339–341. doi: 10.1038/344339a0. [DOI] [PubMed] [Google Scholar]
- 3.Kawamoto Y, Nakamura S, Nakano S, Oka N, Akiguchi I, Kimura J. Immunohistochemical localization of brain-derived neurotrophic factor in adult rat brain. Neuroscience. 1996;74(4):1209–1226. doi: 10.1016/0306-4522(96)00245-X. [DOI] [PubMed] [Google Scholar]
- 4.Yan Q, Rosenfeld RD, Matheson CR, Hawkins N, Lopez OT, Bennett L, Welcher AA. Expression of brain-derived neurotrophic factor protein in the adult rat central nervous system. Neuroscience. 1997;78(2):431–448. doi: 10.1016/S0306-4522(96)00613-6. [DOI] [PubMed] [Google Scholar]
- 5.Lipsky RH, Marini AM. Brain-derived neurotrophic factor in neuronal survival and behavior-related plasticity. Ann N Y Acad Sci. 2007;1122:130–143. doi: 10.1196/annals.1403.009. [DOI] [PubMed] [Google Scholar]
- 6.Bernd P. The role of neurotrophins during early development. Gene Expr. 2008;14(4):241–250. doi: 10.3727/105221608786883799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamada K, Mizuno M, Nabeshima T. Role for brain-derived neurotrophic factor in learning and memory. Life Sci. 2002;70(7):735–744. doi: 10.1016/S0024-3205(01)01461-8. [DOI] [PubMed] [Google Scholar]
- 8.Tyler WJ, Alonso M, Bramham CR, Pozzo-Miller LD. From acquisition to consolidation: on the role of brain-derived neurotrophic factor signaling in hippocampal-dependent learning. Learn Mem. 2002;9(5):224–237. doi: 10.1101/lm.51202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolanos CA, Nestler EJ. Neurotrophic mechanisms in drug addiction. Neuromolecular Med. 2004;5(1):69–83. doi: 10.1385/NMM:5:1:069. [DOI] [PubMed] [Google Scholar]
- 10.McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 11.Bibel M, Barde YA. Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000;14(23):2919–2937. doi: 10.1101/gad.841400. [DOI] [PubMed] [Google Scholar]
- 12.Binder DK, Scharfman HE. Brain-derived neurotrophic factor. Growth Factors. 2004;22(3):123–131. doi: 10.1080/08977190410001723308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castren E. Neurotrophins as mediators of drug effects on mood, addiction, and neuroprotection. Mol Neurobiol. 2004;29(3):289–302. doi: 10.1385/MN:29:3:289. [DOI] [PubMed] [Google Scholar]
- 14.Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington’s disease. Nat Rev Neurosci. 2005;6(12):919–930. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- 15.Murer MG, Yan Q, Raisman-Vozari R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog Neurobiol. 2001;63(1):71–124. doi: 10.1016/S0301-0082(00)00014-9. [DOI] [PubMed] [Google Scholar]
- 16.Russo-Neustadt AA, Chen MJ. Brain-derived neurotrophic factor and antidepressant activity. Curr Pharm Des. 2005;11(12):1495–1510. doi: 10.2174/1381612053764788. [DOI] [PubMed] [Google Scholar]
- 17.Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4(4):299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- 18.Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007;85(3):525–535. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baj G, Leone E, Chao MV, Tongiorgi E. Spatial segregation of BDNF transcripts enables BDNF to differentially shape distinct dendritic compartments. Proc Natl Acad Sci U S A. 2011;108(40):16813–16818. doi: 10.1073/pnas.1014168108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palomer E, Martin-Segura A, Baliyan S, Ahmed T, Balschun D, Venero C, Martin MG, Dotti CG. Aging triggers a repressive chromatin state at bdnf promoters in hippocampal neurons. Cell Rep. 2016;16(11):2889–2900. doi: 10.1016/j.celrep.2016.08.028. [DOI] [PubMed] [Google Scholar]
- 21.Karpova NN. Role of BDNF epigenetics in activity-dependent neuronal plasticity. Neuropharmacology. 2014;76 Pt C:709–718. doi: 10.1016/j.neuropharm.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 22.Nair A, Vadodaria KC, Banerjee SB, Benekareddy M, Dias BG, Duman RS, Vaidya VA. Stressor-specific regulation of distinct brain-derived neurotrophic factor transcripts and cyclic AMP response element-binding protein expression in the postnatal and adult rat hippocampus. Neuropsychopharmacology. 2007;32(7):1504–1519. doi: 10.1038/sj.npp.1301276. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Gu F, Chen J, Dong W. Chronic antidepressant administration alleviates frontal and hippocampal BDNF deficits in CUMS rat. Brain Res. 2010;1366:141–148. doi: 10.1016/j.brainres.2010.09.095. [DOI] [PubMed] [Google Scholar]
- 24.Nair B, Wong-Riley MT. Transcriptional Regulation of Brain-derived Neurotrophic Factor Coding Exon IX: Role of Nuclear Respiratory Factor 2. J Biol Chem. 2016;291(43):22583–593. [DOI] [PMC free article] [PubMed]
- 25.McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87(3):873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- 26.Joels M, Krugers HJ, Lucassen PJ, Karst H. Corticosteroid effects on cellular physiology of limbic cells. Brain Res. 2009;1293:91–100. doi: 10.1016/j.brainres.2009.03.036. [DOI] [PubMed] [Google Scholar]
- 27.Le Menuet D, Lombes M. The neuronal mineralocorticoid receptor: from cell survival to neurogenesis. Steroids. 2014;91:11–19. doi: 10.1016/j.steroids.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 28.Lambert WM, Xu CF, Neubert TA, Chao MV, Garabedian MJ, Jeanneteau FD. Brain-derived neurotrophic factor signaling rewrites the glucocorticoid transcriptome via glucocorticoid receptor phosphorylation. Mol Cell Biol. 2013;33(18):3700–3714. doi: 10.1128/MCB.00150-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arango-Lievano M, Lambert WM, Bath KG, Garabedian MJ, Chao MV, Jeanneteau F. Neurotrophic-priming of glucocorticoid receptor signaling is essential for neuronal plasticity to stress and antidepressant treatment. Proc Natl Acad Sci U S A. 2015;112(51):15737–15742. doi: 10.1073/pnas.1509045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9(4):519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- 31.Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59(12):1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Luikart BW, Birnbaum S, Chen J, Kwon CH, Kernie SG, Bassel-Duby R, Parada LF. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron. 2008;59(3):399–412. doi: 10.1016/j.neuron.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herman JP, Cullinan WE. Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 1997;20(2):78–84. doi: 10.1016/S0166-2236(96)10069-2. [DOI] [PubMed] [Google Scholar]
- 34.Jankord R, Herman JP. Limbic regulation of hypothalamo-pituitary-adrenocortical function during acute and chronic stress. Ann N Y Acad Sci. 2008;1148:64–73. doi: 10.1196/annals.1410.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murakami S, Imbe H, Morikawa Y, Kubo C, Senba E. Chronic stress, as well as acute stress, reduces BDNF mRNA expression in the rat hippocampus but less robustly. Neurosci Res. 2005;53(2):129–139. doi: 10.1016/j.neures.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 36.Dwivedi Y, Rizavi HS, Pandey GN. Antidepressants reverse corticosterone-mediated decrease in brain-derived neurotrophic factor expression: differential regulation of specific exons by antidepressants and corticosterone. Neuroscience. 2006;139(3):1017–1029. doi: 10.1016/j.neuroscience.2005.12.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gourley SL, Kedves AT, Olausson P, Taylor JR. A history of corticosterone exposure regulates fear extinction and cortical NR2B, GluR2/3, and BDNF. Neuropsychopharmacology. 2009;34(3):707–716. doi: 10.1038/npp.2008.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith MA, Makino S, Kvetnansky R, Post RM. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci. 1995;15(3 Pt 1):1768–1777. doi: 10.1523/JNEUROSCI.15-03-01768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asadi A, Hedman E, Widen C, Zilliacus J, Gustafsson JA, Wikstrom AC. FMS-like tyrosine kinase 3 interacts with the glucocorticoid receptor complex and affects glucocorticoid dependent signaling. Biochem Biophys Res Commun. 2008;368(3):569–574. doi: 10.1016/j.bbrc.2008.01.146. [DOI] [PubMed] [Google Scholar]
- 40.Cheung LW, Leung KW, Wong CK, Wong RN, Wong AS. Ginsenoside-Rg1 induces angiogenesis via non-genomic crosstalk of glucocorticoid receptor and fibroblast growth factor receptor-1. Cardiovasc Res. 2011;89(2):419–425. doi: 10.1093/cvr/cvq300. [DOI] [PubMed] [Google Scholar]
- 41.Kumamaru E, Numakawa T, Adachi N, Kunugi H. Glucocorticoid suppresses BDNF-stimulated MAPK/ERK pathway via inhibiting interaction of Shp2 with TrkB. FEBS Lett. 2011;585(20):3224–3228. doi: 10.1016/j.febslet.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 42.Kino T, Jaffe H, Amin ND, Chakrabarti M, Zheng YL, Chrousos GP, Pant HC. Cyclin-dependent kinase 5 modulates the transcriptional activity of the mineralocorticoid receptor and regulates expression of brain-derived neurotrophic factor. Mol Endocrinol. 2010;24(5):941–952. doi: 10.1210/me.2009-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ridder S, Chourbaji S, Hellweg R, Urani A, Zacher C, Schmid W, Zink M, Hortnagl H, Flor H, Henn FA, et al. Mice with genetically altered glucocorticoid receptor expression show altered sensitivity for stress-induced depressive reactions. J Neurosci. 2005;25(26):6243–6250. doi: 10.1523/JNEUROSCI.0736-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daskalakis NP, De Kloet ER, Yehuda R, Malaspina D, Kranz TM. Early Life Stress Effects on Glucocorticoid-BDNF Interplay in the Hippocampus. Front Mol Neurosci. 2015;8:68. doi: 10.3389/fnmol.2015.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20(4):727–740. doi: 10.1016/S0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- 46.Pruunsild P, Sepp M, Orav E, Koppel I, Timmusk T. Identification of cis-elements and transcription factors regulating neuronal activity-dependent transcription of human BDNF gene. J Neurosci. 2011;31(9):3295–3308. doi: 10.1523/JNEUROSCI.4540-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Le Menuet D, Zennaro MC, Viengchareun S, Lombes M. Transgenic mouse models to study human mineralocorticoid receptor function in vivo. Kidney Int. 2000;57(4):1299–1306. doi: 10.1046/j.1523-1755.2000.00966.x. [DOI] [PubMed] [Google Scholar]
- 48.Seibenhener ML, Wooten MW: Isolation and culture of hippocampal neurons from prenatal mice. J Vis Exp. 2012;(65). doi:10.3791/3634. [DOI] [PMC free article] [PubMed]
- 49.Martinez Y, Bena F, Gimelli S, Tirefort D, Dubois-Dauphin M, Krause KH, Preynat-Seauve O. Cellular diversity within embryonic stem cells: pluripotent clonal sublines show distinct differentiation potential. J Cell Mol Med. 2012;16(3):456–467. doi: 10.1111/j.1582-4934.2011.01334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sarabdjitsingh RA, Meijer OC, de Kloet ER. Specificity of glucocorticoid receptor primary antibodies for analysis of receptor localization patterns in cultured cells and rat hippocampus. Brain Res. 2010;1331:1–11. doi: 10.1016/j.brainres.2010.03.052. [DOI] [PubMed] [Google Scholar]
- 51.Zuccotti A, Kuhn S, Johnson SL, Franz C, Singer W, Hecker D, Geisler HS, Kopschall I, Rohbock K, Gutsche K, et al. Lack of brain-derived neurotrophic factor hampers inner hair cell synapse physiology, but protects against noise-induced hearing loss. J Neurosci. 2012;32(25):8545–8553. doi: 10.1523/JNEUROSCI.1247-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Le Billan F, Khan JA, Lamribet K, Viengchareun S, Bouligand J, Fagart J, Lombes M: Cistrome of the aldosterone-activated mineralocorticoid receptor in human renal cells. FASEB J. 2015. [DOI] [PubMed]
- 53.Mifsud KR, Reul JM. Acute stress enhances heterodimerization and binding of corticosteroid receptors at glucocorticoid target genes in the hippocampus. Proc Natl Acad Sci U S A. 2016;113(40):11336–11341. doi: 10.1073/pnas.1605246113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haffner MC, Jurgeit A, Berlato C, Geley S, Parajuli N, Yoshimura A, Doppler W. Interaction and functional interference of glucocorticoid receptor and SOCS1. J Biol Chem. 2008;283(32):22089–22096. doi: 10.1074/jbc.M801041200. [DOI] [PubMed] [Google Scholar]
- 55.Salerno KM, Jing X, Diges CM, Cornuet PK, Glorioso JC, Albers KM. Sox11 modulates brain-derived neurotrophic factor expression in an exon promoter-specific manner. J Neurosci Res. 2012;90(5):1011–1019. doi: 10.1002/jnr.23010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Le Menuet D, Viengchareun S, Penfornis P, Walker F, Zennaro MC, Lombes M. Targeted oncogenesis reveals a distinct tissue-specific utilization of alternative promoters of the human mineralocorticoid receptor gene in transgenic mice. J Biol Chem. 2000;275(11):7878–7886. doi: 10.1074/jbc.275.11.7878. [DOI] [PubMed] [Google Scholar]
- 57.Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol. 2013;132(5):1033–1044. doi: 10.1016/j.jaci.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tuvikene J, Pruunsild P, Orav E, Esvald EE, Timmusk T. AP-1 Transcription Factors Mediate BDNF-Positive Feedback Loop in Cortical Neurons. J Neurosci. 2016;36(4):1290–1305. doi: 10.1523/JNEUROSCI.3360-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Penfornis P, Viengchareun S, Le Menuet D, Cluzeaud F, Zennaro MC, Lombes M. The mineralocorticoid receptor mediates aldosterone-induced differentiation of T37i cells into brown adipocytes. Am J Physiol Endocrinol Metab. 2000;279(2):E386–394. doi: 10.1152/ajpendo.2000.279.2.E386. [DOI] [PubMed] [Google Scholar]
- 60.Numakawa T, Adachi N, Richards M, Chiba S, Kunugi H. Brain-derived neurotrophic factor and glucocorticoids: reciprocal influence on the central nervous system. Neuroscience. 2013;239:157–172. doi: 10.1016/j.neuroscience.2012.09.073. [DOI] [PubMed] [Google Scholar]
- 61.Hellal-Levy C, Couette B, Fagart J, Souque A, Gomez-Sanchez C, Rafestin-Oblin M. Specific hydroxylations determine selective corticosteroid recognition by human glucocorticoid and mineralocorticoid receptors. FEBS Lett. 1999;464(1-2):9–13. doi: 10.1016/S0014-5793(99)01667-1. [DOI] [PubMed] [Google Scholar]
- 62.Lombes M, Kenouch S, Souque A, Farman N, Rafestin-Oblin ME. The mineralocorticoid receptor discriminates aldosterone from glucocorticoids independently of the 11 beta-hydroxysteroid dehydrogenase. Endocrinology. 1994;135(3):834–840. doi: 10.1210/endo.135.3.8070376. [DOI] [PubMed] [Google Scholar]
- 63.Dougherty EJ, Elinoff JM, Ferreyra GA, Hou A, Cai R, Sun J, Blaine KP, Wang S, Danner RL: Mineralocorticoid Receptor (MR) Trans-Activation of Inflammatory AP-1 Signaling: Dependence on DNA Sequence, MR Conformation, and AP-1 Family Member Expression. J Biol Chem 2016. [DOI] [PMC free article] [PubMed]
- 64.Hua G, Paulen L, Chambon P. GR SUMOylation and formation of an SUMO-SMRT/NCoR1-HDAC3 repressing complex is mandatory for GC-induced IR nGRE-mediated transrepression. Proc Natl Acad Sci U S A. 2016;113(5):E626–634. doi: 10.1073/pnas.1522821113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vockley CM, D’Ippolito AM, McDowell IC, Majoros WH, Safi A, Song L, Crawford GE, Reddy TE. Direct GR Binding Sites Potentiate Clusters of TF Binding across the Human Genome. Cell. 2016;166(5):1269–1281. doi: 10.1016/j.cell.2016.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20(4):709–726. doi: 10.1016/S0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 67.Tao X, West AE, Chen WG, Corfas G, Greenberg ME. A calcium-responsive transcription factor, CaRF, that regulates neuronal activity-dependent expression of BDNF. Neuron. 2002;33(3):383–395. doi: 10.1016/S0896-6273(01)00561-X. [DOI] [PubMed] [Google Scholar]
- 68.Jiang X, Tian F, Du Y, Copeland NG, Jenkins NA, Tessarollo L, Wu X, Pan H, Hu XZ, Xu K, et al. BHLHB2 controls Bdnf promoter 4 activity and neuronal excitability. J Neurosci. 2008;28(5):1118–1130. doi: 10.1523/JNEUROSCI.2262-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mallei A, Baj G, Ieraci A, Corna S, Musazzi L, Lee FS, Tongiorgi E, Popoli M: Expression and Dendritic Trafficking of BDNF-6 Splice Variant are Impaired in Knock-In Mice Carrying Human BDNF Val66Met Polymorphism. Int J Neuropsychopharmacol. 2015;18(12). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer table. (DOCX 15 kb)
Gene expression in PCN and BZ cells. (EPS 1379 kb)
GR stimulates Sgk1 expression in BZ cells. (EPS 1437 kb)
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and its additional files.