

Abstract
We established a mouse model of developmental nonalcoholic steatohepatitis (NASH) by feeding a high polyunsaturated fat liquid diet to female glutathione-S-transferase 4-4 (Gsta4−/−)/peroxisome proliferator activated receptor α (Ppara−/−) double knockout 129/SvJ mice for 12 weeks from weaning. We used it to probe the importance of lipid peroxidation in progression of NASH beyond simple steatosis. Feeding Gsta4−/−/Ppara−/− double-knockout (dKO) mice liquid diets containing corn oil resulted in a percentage fat–dependent increase in steatosis and necroinflammatory injury (P < 0.05). Increasing fat to 70% from 35% resulted in increases in formation of 4-hydroxynonenal protein adducts accompanied by evidence of stellate cell activation, matrix remodeling, and fibrosis (P < 0.05). Comparison of dKO mice with wild-type (Wt) and single knockout mice revealed additive effects of Gsta4−/− and Ppara−/− silencing on steatosis, 4-hydroxynonenal adduct formation, oxidative stress, serum alanine amino transferase, expression of tumor necrosis factor alpha, Il6, interferon mRNA, and liver pathology (P < 0.05). Induction of Cyp2e1 protein by high-fat diet was suppressed in Gsta4−/− and dKO groups (P < 0.05). The dKO mice had similar levels of markers of stellate cell activation and matrix remodeling as Ppara−/− single KO mice. These data suggest that lipid peroxidation products play a role in progression of liver injury to steatohepatitis in NASH produced by high-fat feeding during development but appear less important in development of fibrosis.
Pediatric nonalcoholic fatty liver disease is a prevalent chronic liver pathology observed in 9.6% of children and up to 38% of those that are obese.1 Fatty liver disease has been reported in morbidly obese children as young as 2 to 3 years old.1, 2 In approximately 30% of cases, pediatric liver pathology progresses from simple steatosis to nonalcoholic steatohepatitis (pNASH), characterized by necroinflammation and development of fibrosis.3 pNASH is now the major cause of liver transplant in pediatric populations.4 However, despite the emergence of pNASH as a major pediatric disease, little is known regarding the molecular mechanisms underlying the development of NASH in children and there are no good developmental animal models that mimic the natural etiological setting in which pNASH develops clinically.
A nutritional model of adult NASH was developed in Sprague-Dawley rats by Lieber et al,5 in which liver pathology developed after feeding a 71% high-fat liquid diet. This high-fat feeding model was also applied to adult mice lacking the transcription factor peroxisome proliferator receptor α (Ppara−/−), a member of the nuclear receptor superfamily that is essential for lipid metabolism via mitochondrial and peroxisomal β- and ω-oxidation pathways.6 It was shown that this knockout resulted in enhanced steatosis, lipid peroxidation, and inflammation compared to wild-type (Wt) mice fed high-fat liquid diets for 3 weeks.7 More recently, we have made mice with a double knockout of Ppara−/− and the glutathione-S-transferase enzyme Gsta4−/− (dKO).8 Gsta4 is a detoxification enzyme that eliminates reactive electrophilic α,β unsaturated aldehydes, such as the lipid peroxidation product 4-hydroxynonenal (4-Hne).9 We demonstrated that these dKO mice had enhanced hepatic Hne protein adducts, circulating autoantibodies against lipid peroxidation product-adducted proteins, necroinflammatory injury, stellate cell activation, and matrix remodeling than either wild-type mice or mice with knockout of either gene alone when fed chronically with alcohol liquid diets.8 These data suggested the importance of lipid peroxidation products in progression of alcoholic liver disease and implicated a possible role for autoimmune responses triggered by adducted proteins in this process. Recent studies by Sutti et al10 suggest a similar autoimmune process related to generation of reactive lipid peroxidation products may underlie development of inflammation in NASH. In the current study, we used these dKO mice to establish a developmental model of NASH produced as a result of feeding high polyunsaturated fat diets beginning in early development and to examine the role of lipid peroxidation in progression of liver pathology in mice fed these diets.
Materials and Methods
Animals and Experimental Design
All of the animal studies described below were approved by the Institutional Animal Care and Use Committee at the University of Arkansas for Medical Sciences. All animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals11 at an American Association for Accreditation of Laboratory Animal Care–approved animal facility at the University of Arkansas for Medical Sciences. Sv 129/J mice and 129S4/SvJae-Pparatm1Gonz/J (stock number 003580) were purchased from Jackson Laboratories (Bar Harbor, ME). Gsta4−/− mice were generated as previously described.12 dKO mice were generated by breeding the single knockout strains to produce offspring heterozygous for both genes and subsequent breeding of the F1 generation, as previously described.8 In experiment 1, to establish a developmental NASH model, female dKO mice were weaned onto standard rodent chow (n = 5), onto liquid diets containing 35% energy from polyunsaturated fat (corn oil), 18% from protein (casein), and 47% from carbohydrates (dextrose/maltodextrin, 4:1) (35% fat) (n = 7) or liquid diets containing 70% energy from polyunsaturated fat, 18% from protein, and 12% from carbohydrate (70% fat) (n = 7) and fed ad libitum for 12 weeks. Vitamin and mineral composition was based on National Research Council recommendations for growing mice.13 In experiment 2, female Sv 129 Wt; Gsta4−/−, Ppara−/−; and dKO mice (n = 6 to 8 per group) were weaned onto standard rodent chow or 70% fat liquid diets and fed ad libitum for 12 weeks. All groups in both experiments had ad libitum access to water. Animal body weights were measured weekly. At sacrifice, serum was collected, liver was weighed, and pieces were formalin fixed and frozen for further analysis. Serum alanine aminotransferase levels were assessed as a measure of liver damage by using the Infinity alanine aminotransferase liquid stable reagent (Thermo Electron, Waltham, MA), as per manufacturer's instructions. Accumulation of hepatic lipid droplets was measured by quantitation of oil red O staining.8, 14 In addition, triglycerides were extracted from serum and liver homogenates with chloroform/methanol (2:1, v/v), and triglyceride concentrations were assayed using commercially available reagents (Cayman Chemical, Ann Arbor, MI). Overall hepatic oxidative stress was also assessed by measuring oxidized and reduced glutathione concentrations (reduced glutathione/oxidized glutathione) using a kit from Cayman Chemical.
Lipid Peroxidation
Liver 4-Hne adducts were detected immunohistochemically and quantified, as previously described by Shearn et al.15 Immunohistochemical characterization was performed using rabbit polyclonal anti–4-Hne and goat anti-rabbit polyclonal antibodies Vectastain ABC IHC kit (Vector Labs, Burlingame, CA). Images were taken on a NIKON Eclipse TE300 (Nikon Instruments Inc., Melville, NY) at ×100 magnification using a DS-Fi2 camera (Nikon Instruments Inc.). Quantification was done using NIS Elements version 4.13.04 (Nikon Instruments Inc.) and three measurements per zone (centrilobular or periportal), four exposures per slide, and four animals per condition. Exposure time was 24 milliseconds, and the area of measurement was 100 × 100 pixels. Overall changes in staining were quantified by using the ratio of staining in the periportal region compared to the centrilobular region (zone 1/zone 3). The pathological significance of hepatocellular inflammation was assessed using immunohistochemistry and antibodies directed against CD3 (goat anti-rabbit polyclonal; Dako, Carpinteria, CA), B220 (rat–anti-mouse polyclonal; BD Biosciences, San Jose, CA), and F4/80 (rat–anti-mouse polyclonal; ABD Serotec Biorad, Hercules, CA). Colocalization studies were performed in liver sections from dKO mice to determine whether 4-Hne adducts occurred in Kupffer cells or stellate cells in addition to hepatocytes. Serial sections were stained for 4-HNE, F4/80, or the appearance of α-smooth muscle actin (Sigma, St. Louis, MO) (a marker of activated stellate cells).
Liver Pathology
Liver samples were fixed in 10% neutral buffered formalin and processed, and paraffin-embedded sections were stained with hematoxylin and eosin. Hematoxylin and eosin–stained liver sections were scored for steatosis, inflammation (macrophage infiltration), and necrosis by a veterinary pathologist (L.H.) with no prior knowledge of the treatment groups. Steatosis was scored as the percentage of parenchymal cells containing fat (microsteatosis or macrosteatosis) as <5% indicates 0; 5% to 33%, 1; >33% to 66%, 2; and >66%, 3.14 Presence of contiguous patches of microsteatosis was given a weighted score of 1. The presence of inflammation based on infiltration by polymorphonuclear cells, leukocytes, and mononuclear cells was evaluated using a scale where no inflammation indicates 0; <2 foci of inflammatory cells per ×200 field, 1; 2 to 4 foci per ×200 field, 2; and >4 foci per ×200 field, 3. For scoring of necrosis, the presence of necrotic foci was assessed using a scale where 0 indcates none; 1, few; and 2, many.14 Fibrosis was detected histologically by picrosirius red staining of collagen.8
Gene Expression and Western Blotting
Expression of mRNAs coding for genes involved in oxidative stress, lipid homeostasis, inflammation and macrophage infiltration, immune cell subclasses, matrix remodeling, and fibrosis were assessed by real-time RT-PCR. Briefly, total RNA was isolated from liver using Tri reagent (MRC, Cincinnati, OH), as per manufacturer's protocol, and reversed transcribed using iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA). Gene expression was determined by use of SYBR Green and an ABI 7500 sequence detection system (Applied Biosystems, Foster City, CA). Results were quantified using ΔCt method relative to glyceraldehyde-3-phosphate dehydrogenase, and then to Wt controls. Gene-specific primers are presented in Table 1. Livers from experiment 2 were also analyzed for Cyp2e1 and Cyp4a apoprotein expression by Western immunoblot using polyclonal antibodies against rat Cyp2e1 and Cyp4a, which cross-react with their mouse homologues, as previously described.14 Protein bands were quantified using a densitometer, and band densities were corrected for total protein loaded by staining the membrane with 0.1% amido black.
Table 1.
Real-Time RT-PCR Primer Sequences
| Gene | Forward sequence | Reverse sequence |
|---|---|---|
| Fatty acid homeostasis | ||
| Fatp2 | 5′-CGGGAGAGCAAACTACCTACA-3′ | 5′-AACCTCACCTTTGGGGACT-3′ |
| Fasn | 5′-TGACCTCGTGATGAACGTGTAC-3′ | 5′-GGGTGAGGACGTTTACAAAGG-3′ |
| Cyp4a10 | 5′-CCCTGATGGACGCTCTTTAC-3′ | 5′-GGGTCAAACACCTCTGGATT-3′ |
| Cyp4a14 | 5′-GCCAGGAACTGCATTGGGAAACA-3′ | 5′-TTTGGACTTCAGCACAAATCGGGC-3′ |
| Pparg | 5′-CACAATGCCATCAGGTTTGG-3′ | 5′-GCTGGTCGATATCAGTGGAGATC-3′ |
| Cpt1a | 5′-TGGACCCAAATTGCAGTGGT-3′ | 5′-GCATCTCCATGGCGTAGTAGT-3′ |
| Pnpla3 | 5′-TAACAAGTGCATCAGAGACGGGC-3′ | 5′-TGGAATGGAAGCCAGACACCAGC-3′ |
| Plin2 | 5′-ATTGCGGTTGCCAATACCTATGCC-3′ | 5′-TCGGACGTTGGCTGGTTCAGAATA-3′ |
| ApoB | 5′-ATACCACGTTTGCAAGCAGAAGCC-3′ | 5′-TGTTGAGCCGTAAGCTGTAGCAGA-3′ |
| ROS generation | ||
| Cyp2e1 | 5′-AACAGAGACCAGCACAACTCT-3′ | 5′-TCTCATGCACTACAGCGTCCATGT-3′ |
| Nox2 | 5′-GGGATGAATCTCAGGCCAAT-3′ | 5′-GCCGTCCATACAGAGTCTTC-3′ |
| Gsta4 | 5′-GCTGCAGGCATTTAAGACAAG-3′ | 5′-GTCCTGACCACCTCAACATAG-3′ |
| Aldh2 | 5′-AGATCCTCGGCTACATCAAATC-3′ | 5′-CGGTGGGCTGGATAAAGTAG-3′ |
| Nox4 | 5′-CTGCATCTGTCCTGAACCTCAA-3′ | 5′-TCTCCTGCTAGGGACCTTCTGT-3′ |
| Inflammation and infiltration | ||
| Tnfa | 5′-GACGTGGAACTGGCAGAAGAG-3′ | 5′-GCCACAAGCAGGAATGAGAAG-3′ |
| Il6 | 5′-CTTCACAAGTCGGAGGCTTAAT-3′ | 5′-GCAAGTGCATCATCGTTGTTC-3′ |
| Cd14 | 5′-CTAAGTATTGCCCAAGCACACTCA-3′ | 5′-CCCAACTCAGGGTTGTCAGACA-3′ |
| Cd68 | 5′-TTCTCCAGCTGTTCACCTTGACCT-3′ | 5′-GTTGCAAGAGAAACATGGCCCGAA-3′ |
| Cd4 | 5′-TCCCACTCACCCTCAAGATA-3′ | 5′-ATAACCACCAGGTTCACTTC-3′ |
| B220 | 5′-CCCTTCTTCTGCCTCAAAGT-3′ | 5′-CACCTGGATGATATGTGGTCTC-3′ |
| Cd138 | 5′-ATGCGTACAACAGGGTATGG-3′ | 5′-GAGGTGGCTATTCCACAGTATC-3′ |
| Stellate cell activation, matrix remodeling, and fibrosis | ||
| Tgfb | 5′-CTGCGGCAGCTGTACATTGACTTT-3′ | 5′-TGTACTGTGTGTCCAGGCTCCAAA-3′ |
| Col1a1 | 5′-AGGCTTTGATGGACGCAATG-3′ | 5′-GCGGCTCCAGGAAGACC-3′ |
| Pdgfrb | 5′-CCTCGGCCTGTGACTAGAAG-3′ | 5′-GGACGAGGGGAACAACATTA-3′ |
| Mmp13 | 5′-TGGCTTAGAGGTGACTGGCAAACT-3′ | 5′-TATTCACCCACATCAGGCACTCCA-3′ |
| Timp1 | 5′-TCCCTTGCAAACTGGAGAGTGACA-3′ | 5′-AAGCAAAGTGACGGCTCTGGTAGT-3′ |
ROS, reactive oxygen species.
Statistical Analysis
Data are presented as means ± SEM. In experiment 1, the effect of increasing dietary fat content in dKO mice was determined by one-way analysis of variance, followed by Student-Newman-Keuls post hoc analysis. In experiment 2, the Wt, Gsta4−/−, Ppara−/−, and dKO genotype, of chow and 70% fat groups and the interaction thereof were determined using a four- by two-way analysis of variance, followed by Student-Newman-Keuls post hoc analysis. Statistical significance was set at P < 0.05. SigmaPlot software package version 11.0 (Systat Software, Inc., San Jose, CA) was used to perform all statistical tests.
Results
Development of pNASH in dKO Mice Fed 70% Polyunsaturated Fat during Early Development
In experiment 1, 12 weeks of high-fat feeding had a significant impact on weight gain compared to chow controls, 20.28 ± 0.64 g and 20.14 ± 0.50 g for 35% fat group and 70% fat group, respectively, versus 17.6 ± 0.92 g in the chow controls (P < 0.05). We observed no difference in body weight between dKO fed mice at 35% or 70% fat. Hepatic steatosis was evident in both the 35% and 70% fat groups compared to chow-fed dKO mice (P < 0.05), and biochemical analysis of triglyceride content revealed a doubling in the 35% fat group and a sixfold increase in the 70% fat group compared to chow feeding (P < 0.05) (Table 2). Liver weight, as a percentage of body weight, was also significantly increased in the 70% fat compared to other groups (P < 0.05). As seen in Figure 1A, the pattern of steatosis was primarily centrilobular macrosteatosis, in some cases extending out to the portal triad. Around the portal triad, the steatosis that was evident was primarily microsteatosis. In addition to steatosis, necroinflammatory injury developed in dKO mice fed both liquid polyunsaturated fat diets. Pathology scores were elevated in both the 35% and 70% fat groups compared to chow-fed dKO mice (P < 0.05) (Table 2). In addition, a fat concentration-dependent increase was observed in serum alanine amino transferase activities (a biochemical marker of hepatic necrosis) and in expression of the proinflammatory cytokine tumor necrosis factor mRNA (P < 0.05) (Table 2). In the 70% fat group, significant increases in expression of mRNAs encoding reactive oxygen species–generating enzymes Cyp2e1 and NADPH oxidase (Nox)2 relative to chow or 35% fat-fed mice (P < 0.05) was accompanied by higher expression of periportal 4-Hne protein adducts detected by immunohistochemistry (P < 0.05) (Figure 1, A–D). In Figure 1E, picrosirius red staining of livers revealed areas in which collagen fibers appeared to form networks surrounding individual steatotic parenchymal cells in both 35% and 70% fat fed groups of mice that were absent in the chow group (data not shown). Although extensive fibrosis was not evident in any treatment group, the 70% fat group did display evidence of increased stellate cell activation compared to the 35% fat or chow-fed groups: increased expression of platelet-derived growth factor receptor b mRNA was observed accompanied by increased expression of α collagen mRNA (P < 0.05) (Figure 1, F and G).
Table 2.
Effects of High-Fat Feeding on Nonfibrotic Liver Pathology in Ppara−/−/Gsta4−/− Double-Knockout Female Mice Receiving a 35% or 70% Fat Liquid Diet or Chow Fed from Weaning to Adulthood
| Group | Pathology score |
Serum ALT (U/L) | TNF-α mRNA expression | Liver weight (g) | Liver triglycerides (g) | ||
|---|---|---|---|---|---|---|---|
| Steatosis | Inflammation/necrosis | Total | |||||
| Chow | 0.2 ± 0.20 | 0.1 ± 0.08 | 0.3 ± 0.18 | 27.6 ± 0.60 | 1.0 ± 0.33 | 0.72 ± 0.05 | 17.2 ± 2.6 |
| 35% fat | 2.4 ± 0.24∗ | 2.1 ± 0.81∗ | 4.5 ± 1.00∗ | 46.7 ± 11.1∗ | 3.6 ± 1.04∗ | 0.79 ± 0.04 | 40.3 ± 1.5 |
| 70% fat | 2.7 ± 0.18∗ | 2.5 ± 0.32∗ | 5.2 ± 0.2∗ | 90.4 ± 13.7∗† | 5.7 ± 0.73∗† | 0.97 ± 0.03∗† | 111.0 ± 2.5∗† |
Data are expressed as means ± SEM. Total liver pathology was assessed in hematoxylin and eosin–stained sections by a veterinary pathologist, as described in Materials and Methods. Serum ALT values were determined as described in Materials and Methods. Triglyceride concentrations were biochemically measured, as previously described.16 Statistical significance for the effects associated by increasing fat intake in the diet was determined by one-way analysis of variance, followed by Student-Newman-Keuls post hoc analysis. n = 7 animals per liquid diet group; n = 5 animals per chow group.
ALT, alanine aminotransferase; TNF-α, tumor necrosis factor-α.
P < 0.05 versus chow.
P < 0.05 versus 35% high-fat diet.
Figure 1.

Lipid peroxidation product adducts; reactive oxygen species generation; and markers of stellate cell activation, fibrosis, and matrix remodeling in livers of Ppara−/−/gsta4−/− double knockout (dKO) mice fed chow, 35%, or 70% polyunsaturated fat (HF) diets. A: Representative immunohistochemistry of 4-hydroxynonenal (4-Hne) adducts in liver sections. B: Quantification of Hne protein adducts. C: Cyp2e1 mRNA expression. D: Nox2 mRNA expression. E: Representative sections of picrosirius red staining of collagen in HF livers (oil immersion objective). F: α Collagen (Col1a1) mRNA expression. G: Platelet-derived growth factor receptor b (Pdgfrb) mRNA expression. Statistical significance was determined by one-way analysis of variance followed by Student-Newman-Keuls post hoc analysis. Data are expressed as means ± SEM (B–D, F, and G). n = 5 mice (chow); n = 7 mice (35% or 75% HF). ∗P < 0.05 for chow versus HF diet; †P < 0.05 for 35% HF diet versus 70% HF diet. Original magnification, ×100 (E). Nox, NADPH oxidase.
Effect of Ppara−/− and Gsta4−/− Genotype Alone or Together on Liver Pathology after Chronic Feeding of Diets High in Polyunsaturated Fat
To distinguish the effects of ablation of Ppara−/− versus Gsta4−/− on NASH development in this model and determine the role reactive electrophilic α,β unsaturated aldehyde products of lipid peroxidation on liver pathology, we conducted a second experiment. Female Wt mice and female mice with knockout of one gene were compared with dKO mice in the developmental high-fat feeding model. In experiment 2, no significant group differences in weight gain, which ranged from 20 to 22 g, were associated with 12 weeks' feeding of the 70% fat diet from weaning. Liver weight, as a percentage of body weight, was elevated relative to chow-fed mice in both the 70% fat Ppara−/− and the dKO groups compared to the Wt and Gsta4−/− mice (P < 0.05) (Table 3). Liver triglyceride concentrations determined biochemically were elevated by 70% fat diets compared to chow-fed mice in all groups. However, liver triglyceride concentrations were greater in the 70% fat Ppara−/− group compared to Wt or Gsta4−/− groups (P < 0.05) and higher in the 70% fat dKO group than any other group (P < 0.05). Increased liver triglycerides were accompanied by significant reductions in serum triglyceride levels in both the Ppara−/− and dKO groups (P < 0.05) (Table 3). Examination of the appearance of hepatic lipid droplets by oil red O staining revealed a similar pattern to biochemical measures of triglyceride content, with a higher level of droplets in 70% fat dKO mice compared to either single knockout group (Figure 2, A and B). Moreover, the size of the lipid droplets was significantly greater in the 70% fat dKO mice compared to single KOs (Figure 2C). Pathological examination revealed the presence of inflammatory infiltrates and necrotic foci in both the 70% high-fat (HF) Ppara−/− and dKO groups but not in the Wt or Gsta4−/− groups. Overall liver pathology was higher in the 70% HF Ppara−/− group than 70% HF Wt or Gsta4−/− mice and was even more pronounced in the 70% HF dKO mice (Table 3).
Table 3.
Biochemical Analysis of Serum and Hepatic Lipid Concentrations and Liver Pathology in Chow-Fed and 70% Fed Wt, Gsta4−/−, Ppara−/−, and Ppara−/−/Gsta4−/− dKO Mice
| Group | Liver/BW (%) | Hepatic triglycerides | Serum triglycerides | Pathology score |
|||
|---|---|---|---|---|---|---|---|
| Total | Steatosis | Inflammation | Necrosis | ||||
| Wt chow | 3.70 ± 0.1 | 67.8 ± 12.4 | 100.6 ± 8.4 | 0.66 ± 0.40 | 0.66 ± 0.42 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| Wt 70% HF | 3.10 ± 0.0 | 417.2 ± 72.4∗ | 62.4 ± 18.9 | 1.57 ± 0.30 | 1.57 ± 0.29 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| Gsta4−/− chow | 3.70 ± 0.1 | 21.8 ± 8.6 | 85.7 ± 8.5 | 0.37 ± 0.18 | 0.37 ± 0.18 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| Gsta4−/− 70% HF | 3.20 ± 0.1 | 359.2 ± 44.8∗ | 116.2 ± 25.7 | 0.50 ± 0.22 | 0.50 ± 0.22 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| Ppara−/− chow | 4.10 ± 0.1 | 98.9 ± 14.1 | 191.5 ± 35.7† | 0.62 ± 0.18 | 0.62 ± 0.18 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| Ppara−/− 70% HF | 5.60 ± 0.4∗‡ | 1055.1 ± 119∗‡ | 94.3 ± 11.6∗ | 4.25 ± 0.49∗‡ | 3.00 ± 0.00 | 0.53 ± 0.18 | 0.77 ± 0.51 |
| dKO chow | 4.00 ± 0.1 | 55.7 ± 16.6 | 157.1 ± 19.5† | 0.62 ± 0.26 | 0.62 ± 0.26 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| dKO 70% HF | 5.50 ± 0.1∗‡ | 1313.0 ± 104∗§ | 100.1 ± 10.6∗ | 4.00 ± 0.37∗‡ | 2.8 ± 0.13 | 0.37 ± 0.18 | 0.75 ± 0.31 |
| P values | |||||||
| Genotype | <0.001 | <0.001 | 0.025 | <0.001 | |||
| Diet | 0.002 | <0.001 | 0.008 | <0.001 | |||
| Interaction | <0.001 | <0.001 | 0.029 | <0.001 | |||
Data are expressed as means ± SEM. Triglyceride concentrations were biochemically measured, as stated in Materials and Methods.16 Total liver pathology was assessed in hematoxylin and eosin–stained sections by a veterinary pathologist, as described in Materials and Methods. Statistical significance was determined by two-way analysis of variance, followed by Student-Newman-Keuls post hoc analysis. n = 6 animals per wild-type group; n = 8 animals per knockout group.
∗P < 0.05 for chow versus 70% HF diet (within each genotype).
†P < 0.05 (between genotypes for chow diet).
‡P < 0.05 (between genotypes for 70% HF diet).
§P < 0.01 (between genotypes for 70% HF diet).
BW, body weight; dKO, double knockout; HF, high fat; Wt, wild type.
Figure 2.

Steatosis and genes regulating fatty acid import, synthesis, and degradation lipid homeostasis in Wt, Gsta4−/−, Ppara−/−, and Ppara−/−/gsta4−/− double-knockout (dKO) mice fed chow or 70% polyunsaturated fat (HF) diets. A: Representative sections of oil red O (ORO)–stained lipid droplets in HF livers. B: Quantitation of ORO staining. C: Distribution of lipid droplet size in Ppara−/− and Ppara−/−/gsta4−/− double-knockout (dKO) mice fed HF diets. D: Fatty acid transport protein (Fatp2) mRNA expression. E: Fatty acid synthase (Fasn) mRNA expression. F: Peroxisome proliferation-activated receptor γ (Pparg) mRNA expression. G: Cyp4a10 mRNA expression. H: Carnitine palmitoyltransferase (Cpt1) mRNA expression. I: Patatin-like phospholipase domain containing 3 (Pnpla3) mRNA expression. Statistical significance was determined by two-way analysis of variance, followed by Student-Newman-Keuls post hoc analysis. Data are expressed as means ± SEM (B and D–I). n = 6 to 8 per group (B and D–I). ∗P < 0.05 for chow (C) versus 70% HF diet (within each genotype); †P < 0.05 (between genotypes for chow diet); ‡P < 0.05, ‡‡P < 0.01 (between genotypes for 70% HF diet). Original magnification, ×20 (A).
Effect of Ppara−/− and Gsta4−/− Genotype Alone or Together on Hepatic Lipid Homeostasis after Chronic Feeding of Diets High in Polyunsaturated Fat
To determine the effects of the Gsta4−/− and Ppara−/− genotype on lipid homeostasis after feeding of high polyunsaturated fat diets, we examined mRNA expression of genes involved in fatty acid uptake (fatty acid transport protein), fatty acid synthesis (fatty acid synthase and peroxisome proliferator-activated receptor γ), fatty acid degradation [cytochrome P450 Cyp4a10 (Cyp4a10) and carnitine palmitoyltransferase], triglyceride mobilization from lipid droplets (patatin-like phospholipase domain containing 3), and with very-low-density lipoprotein formation (apolipoprotein B100). As seen in Figure 2D, fatty acid transport protein expression in both chow-fed and high fat–fed mice was significantly lower in Ppara−/− and dKO mice than in Wt or Gsta4−/− mice (P < 0.05). Feeding of 70% HF diets increased expression of fatty acid transport protein mRNA in all genotypes, except Ppara−/− mice, relative to feeding chow (P < 0.05). In contrast, no effects of genotype were observed on expression of fatty acid synthase mRNA in chow-fed mice, but the significant suppression of fatty acid synthase mRNA expression observed in 70% HF fed Wt and dKO mice relative to chow-fed controls was not observed in either 70% HF-fed Gsta4−/− or Ppara−/− mice (Figure 2E). Peroxisome proliferator-activated receptor γ expression increased significantly after HF feeding irrespective of genotype (Figure 2F). Expression of the Ppara−/− target gene Cyp4a10, which catalyzes the ω-hydroxylation of fatty acids, was absent, as expected, from livers of Ppara−/− and dKO mice. In Wt and Gsta4−/− mice, feeding of 70% HF diets significantly increased Cyp4a10 mRNA expression (P < 0.05). Expression of mRNA encoding carnitine palmitoyltransferase, which is responsible for the rate-limiting transport of fatty acids into the mitochondria for degradation via β-oxidation, was significantly increased in 70% HF-fed groups of all genotypes relative to chow feeding (P < 0.05) and expression was higher in 70% HF-fed dKO mice than in mice with single knockdown of either gene (P < 0.05) (Figure 2, G and H). Expression of mRNA encoding patatin-like phospholipase domain containing 3 was reduced relative to chow-fed controls by 70% HF feeding only in Gsta4−/− and dKO mice (P < 0.05) (Figure 2I). No statistically significant effects of diet or genotype were observed on apolipoprotein B100 mRNA expression (data not shown).
Effect of Ppara−/−α and Gsta4−/− Genotype Alone or Together on Hepatic Oxidative Stress and Necroinflammation after Chronic Feeding of Diets High in Polyunsaturated Fat
In Figure 3, increases in lipid accumulation and liver pathology were accompanied by increased appearance of periportal 4-Hne protein adducts in liver slices in 70% fat-fed mice relative to chow-fed mice in both Gsta4−/− and Ppara−/− mice compared to the Wt group (P < 0.05) and by the highest level of adducts in the 70% fat dKO group (P < 0.05). No significant effects of genotype or diet were observed on expression of mRNA encoding aldehyde dehydrogenase 2, which, in addition to Gsta4, is the other major enzyme involved in detoxification of 4-Hne (data not shown). Coincident with increased protein carbonylation, we observed increases in the oxidized glutathione/reduced glutathione ratio in the 70% fat Ppara−/− and dKO groups relative to the Gsta4−/− and Wt groups (P < 0.05), indicative of increased oxidative stress (Table 3). Cyp2e1 mRNA was increased (P < 0.05) by feeding 70% HF diets in all genotypes, except Gsta4−/− mice (Table 4). In contrast, at the level of protein expression, two-way analysis of variance indicated genotype diet interaction (P < 0.05). Cyp2e1 was higher in Ppara−/− mice compared to Wt controls, but induction of Cyp2E1 after feeding 70% HF diets was not observed in either the Gsta4−/− or dKO groups (Figure 4). Cyp4a apoprotein expression was increased by 70% HF diets in both Wt and Gsta4−/− mice (P < 0.05), but no expression was observed in either Ppara−/− or dKO groups (Figure 4). Nox2 and Nox4 mRNAs were increased compared to chow-fed controls in both Ppara−/− and dKO mice (P < 0.05) (Table 4). Increased oxidative stress in 70% HF-fed Ppara−/− and dKO groups was accompanied by evidence of increased inflammation and necrosis. Interestingly, proinflammatory cytokines, tumor necrosis factor α, interferon γ, and Il6, mRNAs were significantly up-regulated in 70% HF-fed dKO group, and accompanied increased serum alanine aminotransferase values (P < 0.05) compared to the HF-fed Ppara−/−, Gsta4−/−, and Wt groups (Table 5). Similarly, increased expression of mRNA encoding B-cell marker B220 was also up-regulated in the 70% HF-fed dKO group compared to all other HF-fed groups (P < 0.05). Gene expression of additional immune cell markers, Cd4, Cd44, and Cd14, respectively, were elevated in both the 70% HF-fed Ppara−/− and dKO mice relative to chow-fed controls, 70% HF-fed Wt, and Gsta4−/− mice (P < 0.05) (Table 5). Consistent with these findings, we observed increased parenchymal accumulation of B220-positive cells and Cd3-positive T cells around necrotic foci in the Ppara−/− and dKO groups compared to the Wt and Gsta4−/− genotypes receiving the HF diet (Figures 5 and 6). However, there were no genotype or dietary effects on expression of the plasma cell marker CD138 in the HF-fed dKO mice compared to 70% HF-fed Ppara−/− mice.
Figure 3.

A: Representative 4-hydroxynonenal (4-Hne) adduct staining in livers of Wt, Gsta4−/−, Ppara−/−, and Ppara−/−/gsta4−/− double-knockout (dKO) mice fed 70% polyunsaturated fat (HF) diets. B: Ratio of zone 1/zone 3 staining. Statistical analysis was determined by two-way analysis of variance, followed by Student-Newman-Keuls post hoc analysis. Data are expressed as means ± SEM (B). n = 6 to 8 per group (B). ∗P < 0.05 for chow (C) versus 70% HF diet (within each genotype); ‡P < 0.05, ‡‡P < 0.01 (between genotypes for 70% HF diet).
Table 4.
Biochemical and Molecular Evaluation of Oxidative Stress and Liver Inflammation and Necrosis in Wt, Gsta4−/−, Ppara−/−, and Ppara−/−/Gsta4−/− dKO Mice Receiving a Chow or 70% HF Diet
| Group | GSSG/GSH | mRNA expression (fold change) |
||
|---|---|---|---|---|
| Cyp2e1 | Nox2 | Nox4 | ||
| Wt chow | 0.80 ± 0.10 | 1.00 ± 0.12 | 1.00 ± 0.06 | 1.00 ± 0.25 |
| Wt 70% HF | 0.89 ± 0.07 | 1.41 ± 0.13∗ | 1.43 ± 0.09 | 0.78 ± 0.09 |
| Gsta4−/− chow | 0.85 ± 0.17 | 1.24 ± 0.06 | 1.13 ± 0.01 | 1.49 ± 0.34 |
| Gsta4−/− 70% HF | 0.83 ± 0.06 | 1.30 ± 0.09 | 1.34 ± 0.15 | 0.80 ± 0.08 |
| Ppara−/− chow | 0.82 ± 0.10 | 0.86 ± 0.06 | 1.26 ± 0.04 | 1.24 ± 0.30 |
| Ppara−/− 70% HF | 1.79 ± 0.34∗† | 1.34 ± 0.09∗ | 3.68 ± 0.43∗† | 6.57 ± 0.49∗† |
| dKO chow | 0.84 ± 0.11 | 0.97 ± 0.03 | 1.03 ± 0.16 | 0.78 ± 0.18 |
| dKO 70% HF | 3.81 ± 0.79∗‡ | 1.65 ± 0.07∗† | 3.65 ± 0.54∗† | 5.43 ± 0.36∗† |
| P value | ||||
| Genotype | <0.001 | 0.081 | <0.001 | <0.001 |
| Diet | <0.001 | <0.001 | <0.001 | <0.001 |
| Interaction | <0.001 | 0.006 | <0.001 | <0.001 |
Data are expressed as means ± SEM. Hepatic GSSG/GSH ratio was determined as stated in Materials and Methods. Gene expression was assessed by real-time RT-PCR using 2−ΔΔCT method, as described in Materials and Methods. Statistical analysis was performed by two-way analysis of variance, followed by Student-Newman-Keuls post hoc analysis. n = 6 animals per wild-type group; n = 8 animals per knockout group.
∗P < 0.05 for chow versus 70% HF diet (within each genotype).
†P < 0.05 (between genotypes for 70% HF diet).
‡P < 0.01 (between genotypes for 70% HF diet).
dKO, double knockout; GSH, reduced glutathione; GSSG, oxidized glutathione; HF, high fat; Nox, NADPH oxidase; Wt, wild type.
Figure 4.

A: Western immunoblot analysis of Cyp4a and Cyp2e1 expression in livers of Wt, Gsta4−/−, Ppara−/−, and Gsta4−/−/Ppara−/− double-knockout (dKO) mice fed chow or 70% polyunsaturated fat (HF) diets. Each lane contains samples from a single mouse. B: Immunoquantification of Cyp4a expression corrected for differences in protein loading. C: Immunoquantification of Cyp2e1 expression corrected for differences in protein loading. Statistical analysis was determined by two-way analysis of variance, followed by Student-Newman-Keuls post hoc analysis. ∗P < 0.05 for chow (C) versus 70% HF diet (within each genotype); †P < 0.05 (between genotypes for chow diet); ‡P < 0.05, ‡‡P < 0.01 (between genotypes for 70% HF diet). AB, amido black staining of total protein.
Table 5.
Effects of a 70% HF Diet on Hepatic Immune Cell Response in Wt, Gsta4−/−, Ppara−/−, and Ppara−/−/Gsta4−/− Female Mice
| Group | Serum ALT (U/L) | mRNA expression (fold change) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Tnfa | Il6 | Cd14 | Cd44 | Ifng | B220 | Cd4 | Cd138 | ||
| Wt chow | 32.39 ± 8.66 | 1.00 ± 0.15 | 1.00 ± 0.14 | 1.00 ± 0.17 | 1.00 ± 0.13 | 1.00 ± 0.38 | 1.00 ± 0.16 | 1.00 ± 0.08 | 1.00 ± 0.09 |
| Wt 70% HF | 42.59 ± 2.99 | 1.99 ± 0.15 | 0.57 ± 0.17 | 1.48 ± 0.10 | 1.78 ± 0.10 | 1.42 ± 0.36 | 0.86 ± 0.11 | 1.18 ± 0.19 | 0.81 ± 0.03 |
| Gsta4−/− chow | 24.18 ± 2.64 | 0.95 ± 0.12 | 1.29 ± 0.18 | 1.47 ± 0.15 | 1.15 ± 0.13 | 2.09 ± 1.01 | 1.19 ± 0.18 | 1.35 ± 0.20 | 0.93 ± 0.10 |
| Gsta4−/− 70% HF | 52.21 ± 9.32∗† | 2.62 ± 1.05 | 0.80 ± 0.07 | 0.75 ± 0.05 | 1.80 ± 0.25 | 1.71 ± 0.75 | 1.11 ± 0.22 | 1.31 ± 0.11 | 0.84 ± 0.07 |
| Ppara−/− chow | 30.17 ± 4.38 | 1.61 ± 0.50 | 1.57 ± 0.21 | 1.48 ± 0.10 | 1.64 ± 0.13 | 1.66 ± 0.43 | 1.69 ± 0.32 | 1.40 ± 0.24 | 1.28 ± 0.10 |
| Ppara−/− 70% HF | 48.67 ± 2.34∗† | 5.73 ± 0.64∗† | 1.63 ± 0.29 | 4.33 ± 0.86∗† | 5.79 ± 1.05∗† | 3.51 ± 0.53∗† | 2.53 ± 0.18∗.† | 2.28 ± 0.22∗† | 0.85 ± 0.05∗† |
| dKO chow | 32.72 ± 7.83 | 1.61 ± 0.19 | 1.53 ± 0.23 | 1.21 ± 0.14 | 1.68 ± 0.27 | 1.03 ± 0.28 | 1.31 ± 0.27 | 1.13 ± 0.08 | 1.41 ± 0.13 |
| dKO 70% HF | 67.71 ± 5.86∗‡ | 8.33 ± 0.79∗‡ | 4.59 ± 1.83∗† | 3.90 ± 0.73∗† | 5.74 ± 0.77∗† | 7.05 ± 1.98∗‡ | 3.57 ± 0.54∗‡ | 2.06 ± 0.28∗† | 0.94 ± 0.05∗† |
| P value | |||||||||
| Genotype | 0.049 | <0.001 | 0.025 | <0.001 | <0.001 | 0.044 | <0.001 | 0.005 | 0.004 |
| Diet | 0.003 | <0.001 | 0.337 | <0.001 | <0.001 | 0.005 | <0.001 | 0.002 | <0.001 |
| Interaction | 0.553 | <0.001 | 0.080 | <0.001 | <0.001 | 0.011 | <0.001 | 0.044 | 0.101 |
Data are expressed as means ± SEM. Gene expression expressed as fold change relative to the Wt control group, as described in Materials and Methods. Statistical analysis was performed by two-way analysis of variance, followed by Student-Newman-Keuls post hoc analysis. n = 6 animals per wild-type groups; n = 8 animals per knockout groups.
∗P < 0.05 for chow versus 70% HF diet (within each genotype).
†P < 0.05 (between genotypes for 70% HF diet).
‡P < 0.01 (between genotypes for 70% HF diet).
ALT, alanine aminotransferase; dKO, double knockout; HF, high fat; Ifng, interferon; Tnfa, tumor necrosis factor; Wt, wild type.
Figure 5.

Immunohistochemical staining of B220-positive B cells in representative hematoxylin and eosin liver sections from Wt, Gsta4−/−, Ppara−/−, and double-knockout (dKO) mice receiving either chow or 70% HF diets for 12 weeks. Immunhohistochemistry was performed as described in Materials and Methods. CV, central vein; PT, portal triad.
Figure 6.

Immunohistochemical staining of Cd3-positive T cells associated with necrotic foci in representative hematoxylin and eosin liver sections from Wt, Gsta4−/−, Ppara−/−, and Gsta4−/−/Ppara−/− double-knockout (dKO) mice receiving either chow or 70% HF diets for 12 weeks. Immunhohistochemistry was performed as described in Materials and Methods. CV, central vein; PT, portal triad.
Effect of Ppara−/− and Gsta4−/− Genotype Alone or Together on Stellate Cell Activation, Fibrosis, and Matrix Remodeling after Chronic Feeding of Diets High in Polyunsaturated Fat
Parenchymal collagen deposition was assessed histologically using Picosirius red staining in chow-fed and HF-fed Wt, Gsta4−/−, Ppara−/−, and dKO genotypes. Overall, bridging fibrosis was not observed in any genotype after high-fat feeding (Supplemental Figure S1). However, mRNA expression of profibrotic markers, tumor growth factor β, α-smooth muscle actin, α collagen, and matrix remodeling enzymes, matrix metalloproteinase 13 and tissue inhibitor of metalloproteinases 1, were observed after HF feeding in Ppara−/− and dKO mice compared to Wt and Gsta4−/− mice (P < 0.05) (Table 6). Consistent with these findings, pericellular picrosirius red staining was observed and α-SMA–staining cells were present around necrotic foci in the parenchymal tissue of Ppara−/− and dKO mice receiving 70% HF diets (Supplemental Figure S2).
Table 6.
Gene Expression of Fibrotic Cytokines and Markers in Livers of Wt, Gsta4−/−, Ppara−/−, and dKO Mice Receiving a Chow or 70% HF Diet
| Group | Gene expression (fold change) |
|||||
|---|---|---|---|---|---|---|
| Tgfb | Cola1 | α-Sma | Pdgfrb | Mmp13 | Timp1 | |
| Wt chow | 1.00 ± 0.18 | 1.00 ± 0.19 | 1.00 ± 0.13 | 1.00 ± 0.28 | 1.00 ± 0.09 | 1.00 ± 0.06 |
| Wt 70% HF | 1.24 ± 0.20 | 0.89 ± 0.18 | 1.70 ± 0.20 | 1.04 ± 0.18 | 0.97 ± 0.14 | 1.01 ± 0.10 |
| Gsta4−/− chow | 0.98 ± 0.13 | 1.08 ± 0.11 | 2.29 ± 0.79 | 1.92 ± 0.51 | 1.21 ± 0.61 | 1.40 ± 0.28 |
| Gsta4−/− 70% HF | 1.18 ± 0.06 | 1.17 ± 0.12 | 3.17 ± 0.66 | 0.66 ± 0.17 | 0.66 ± 0.16 | 1.33 ± 0.18 |
| Ppara−/− chow | 1.07 ± 0.13 | 1.15 ± 0.28 | 3.12 ± 0.87 | 1.57 ± 0.62 | 1.21 ± 0.16 | 1.23 ± 0.19 |
| Ppara−/− 70% HF | 2.10 ± 0.21∗† | 4.85 ± 1.19∗† | 5.08 ± 0.83∗† | 2.34 ± 0.26∗† | 22.90 ± 4.74∗† | 19.23 ± 5.11∗† |
| dKO chow | 0.87 ± 0.08 | 1.37 ± 0.30 | 1.67 ± 0.5 | 1.55 ± 0.92 | 0.85 ± 0.14 | 1.71 ± 0.29 |
| dKO 70% HF | 1.81 ± 0.25∗† | 4.49 ± 1.58∗† | 9.72 ± 3.01∗† | 1.26 ± 0.85 | 31.61 ± 9.59∗† | 17.04 ± 5.86∗† |
| P value | ||||||
| Genotype | 0.005 | 0.006 | 0.058 | 0.001 | 0.002 | 0.003 |
| Diet | <0.001 | 0.002 | 0.005 | 0.246 | <0.001 | <0.001 |
| Interaction | 0.007 | 0.017 | 0.024 | <0.001 | 0.002 | 0.003 |
Data are expressed as means ± SEM. Gene expression as fold change relative to the Wt chow-fed group, as described in Materials and Methods. Statistical analysis was performed by two-way analysis of variance, followed by Student-Newman-Keuls post hoc analysis. n = 6 animals per Wt groups; n = 8 animals per knockout groups.
∗P < 0.05 for chow versus 70% HF diet (within each genotype).
†P < 0.05 (between genotypes for 70% HF diet).
Cola1, α collagen; dKO, double knockout; HF, high fat; Mmp, matrix metalloproteinase; Pdgfrb, platelet-derived growth factor receptor b; Tgfb, tumor growth factor β; Timp, tissue inhibitor of metalloproteinases; Wt, wild type; α-Sma, α-smooth muscle actin.
Discussion
In the current study, we used a double-knockout mouse model where Ppara and Gsta4 were deleted to examine the role of steatosis and lipid peroxidation products in the progression of NASH pathology after developmental feeding of high-fat diets. Gsta4 normally contributes in a major manner to detoxification and clearance of α,β short chain aldehyde products of lipid peroxidation, such as 4-Hne and malondialdehyde. We previously demonstrated that further deletion of Gsta4 in Ppara−/− mice, which are highly prone to development of steatosis, results in increased progression of liver injury to steatohepatitis and fibrosis after chronic alcohol exposure.8 Experimental models of clinical pediatric NASH have proved difficult to produce in rodents given the slow development of this pathology relative to the short period between weaning and adulthood in rats and mice and the rapid growth rates of these animals during this period.15, 17 Because Ppara−/− mice have previously been shown to also have increased steatosis and inflammation in a high-fat liquid diet feeding model of adult NASH, we took a similar approach to develop a new model of developmental NASH by feeding Ppara−/−/Gsta4−/− dKO mice liquid diets high in polyunsaturated fat (corn oil, rich in linoleic acid) from weaning to early adulthood.15, 17 We observed a fat dose–dependent increase in progression of liver pathology from steatosis to steatohepatitis, stellate cell activation, and initial stages of fibrosis in both Ppara−/− and dKO mice, consistent with the predicted role for both steatosis and lipid peroxidation products in development of NASH pathology and similar to the dose-responses to overfeeding of polyunsaturated fat diets we have previously reported in adult rat models of NASH.15 Pediatric NASH, as observed clinically in children, comes in two histological subtypes: type 1 is characterized by ballooning degeneration of hepatocytes,18 macrovesicular steatosis, polymorphonuclear leukocyte infiltration, and perisinusoidal fibrosis; type 2 is associated with steatosis, periportal inflammation, and periportal fibrosis but no ballooning degeneration. The mouse pathology observed in this developmental study more resembles type 1 pNASH rather than type 2.
To dissect the roles of increased steatosis and lipid peroxidation products in development of NASH in this model, we compared the effects of developmental feeding of 70% HF diets in wild-type and single knockout mice in comparison with dKO mice. As expected, there was a substantial increase in steatosis in Ppara−/− and dKO mice, consistent with the pivotal role Ppara−/− dependent signaling plays in fatty acid degradation.6 In addition, the observed decreased expression of fatty acid transport protein and Cyp4a10 mRNAs in chow-fed Ppara−/− and dKO mice is consistent with their being known Ppara target genes.6 Interestingly, knockout of Gsta4 resulted in additional accumulation of triglycerides and fat droplets in 70% HF-fed dKO compared to 70% HF-fed Ppara−/− mice. Moreover, the lipid droplets in the dKO mice were significantly larger. We have reported similar increases in steatosis in mice with Gsta4−/− and dKO genotypes compared to Wt and Ppara−/− mice fed chronically with ethanol.8 The current data are consistent with the suggested role of lipid peroxidation in disruption of fatty acid homeostasis leading to enhanced lipid accumulation.8, 12 We have previously reported an age-dependent increase in obesity in chow-fed Gsta4−/− mice on the 126/SvJ background associated with increases in hepatic and adipose tissue fatty acid synthesis related to increased expression and allosteric activation of acetyl-CoA carboxylase secondary to inhibition of aconitase and increased concentrations of the fatty acid precursor malonyl-CoA.12 In addition, it appears that loss of Gsta4 results in disruption of triglyceride mobilization from lipid droplets. Expression of patatin-like phospholipase domain containing 3 mRNA was suppressed by feeding of 70% HF diets in Gsta4−/− and dKO but not in Wt or Ppara−/− mice. Epidemiological studies of patatin-like phospholipase domain containing 3 have previously demonstrated increased incidence of fatty liver disease and NASH in individuals harboring dysfunctional mutations of this gene.16 Finally, there is evidence in the literature that increased oxidative stress and lipid peroxides associated with the hepatic accumulation of ω-3 and ω-6 polyunsaturated fatty acids has post-transcriptional effects resulting in degradation of apolipoprotein B100 protein and inhibition of very-low-density lipoprotein secretion.19, 20, 21 Reductions of serum triglyceride concentrations coincident with increased hepatic steatosis are consistent with such an effect on Ppara−/− and dKO mice.
Deletion of Ppara has previously been shown to result in progression of liver pathology beyond simple steatosis to inflammation and fibrosis in adult mouse models of alcoholic and nonalcoholic liver disease.6, 7, 22 The current data suggest a similar pattern of pathology when high polyunsaturated fat diets are fed to Ppara−/− mice beginning during early development. Increased evidence of oxidative stress, as indicated by increased oxidized glutathione/reduced glutathione ratio and 4-Hne protein adducts, was observed in Ppara−/− compared to Wt mice fed 70% HF diets. The sources of increased reactive oxygen species appear to be increased expression of Cyp2e1 and Nox2 in these mice, although it is unclear if these enzymes are under direct regulation by Ppara or if their induction is secondary to triglyceride accumulation in these animals. Interestingly, the lobular distribution of 4-Hne adducts was shifted toward zone 1 in these mice. Because Cyp2e1 has a centrilobular zone 3 localization, Cyp2e1-derived reactive oxygen species are unlikely to be the major source of 4-Hne. Lobular distribution of Nox enzymes and their potential role in stimulation of lipid peroxidation and progression of hepatic pathology during nonalcoholic fatty liver disease remains the focus of additional research. In this regard, a recent study has linked increased leptin, the essential Nox2 cofactor p47phox, and miR-21 expression with increased tumor growth factor β–Smad signaling and fibrosis in adult NASH patients and in adult mice developing NASH after high-fat feeding and treatment with the hepatotoxin bromochloromethane.23 Moreover, the presence of p47phox in liver parenchymal cells has also been shown to be a key mediator of liver pathology in alcoholic liver disease.24 In addition, recent studies from Bettaieb et al25 suggest that hepatocyte Nox4 may also play an important role in development of oxidative stress and progression of NASH pathology to inflammation and fibrosis. In the current study, knockdown of Ppara also resulted in increased inflammation, necrosis, matrix remodeling, and evidence of stellate cell activation and fibrosis after feeding 70% HF diets relative to Wt mice. These data are consistent with previous studies on development of NASH in adult Ppara−/− mice fed high-fat diets7 and similar to our previously published data on progression of alcoholic liver pathology in these mice.8 Interestingly, and consistent with previous studies of NASH progression in the rat,26 Cd44 mRNA, which encodes the major cell surface hyaluronan (HA) receptor, was significantly up-regulated in 70% HF diet-fed Ppara−/− mice. Cd44 plays a role in clearing fragmented HA, which accumulates during tissue injury.27 However, HA may also play a role in promoting tissue repair as the result of interactions with Toll-like receptors. In an acute lung injury model, overexpression of high molecular mass HA resulted in protection against injury in part through Toll-like receptor–mediated activation of Nfkb.28 It is possible that decreases in HA, as the result of increased Cd44 expression in 70% HF diet-fed Ppara−/− mice, contributes to the development of fibrosis observed in the current model.
Comparison of dKO mice with Ppara−/− mice revealed significant further increases in oxidative stress, 4-Hne adduct formation, inflammatory cytokine mRNAs encoding tumor necrosis factor, Infg, and Il6, increased B-cell infiltration associated with necrotic foci, and increased evidence of hepatocyte necrosis (alanine aminotransferase values) associated with further increases in overall lipid peroxidation resulting from additional knockout of the Gsta4−/− gene. These data are consistent with a role for lipid peroxidation products in development of necroinflammatory injury and development of autoimmune responses in NASH and with data demonstrating a role of 4-Hne in increased chemotaxis and recruitment of inflammatory cells in response to liver injury.29, 30 However, in contrast to previous results obtained in this model after adult alcohol treatment, there was no evidence from the current study that increased lipid peroxidation contributes to stellate cell activation, matrix remodeling, or development of fibrosis in pNASH.8 These data suggest that, although development of liver pathology is similar in alcoholic and nonalcoholic fatty liver disease, the molecular mechanisms underlying these two conditions are divergent.
Footnotes
Supported by NIH/National Institute on Alcohol Abuse and Alcoholism grant R37AA009300 (D.P.) and by US Department of Agriculture, Agricultural Service Current Information System, grant 6251-51000-010-05S.
M.R. and K.M. contributed equally to this work as first authors.
Disclosures: None declared.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2016.10.022.
Contributor Information
Martin Ronis, Email: mronis@lsuhsc.edu.
Kelly Mercer, Email: kmercer@uams.edu.
Supplemental Data
Supplemental Figure S1.
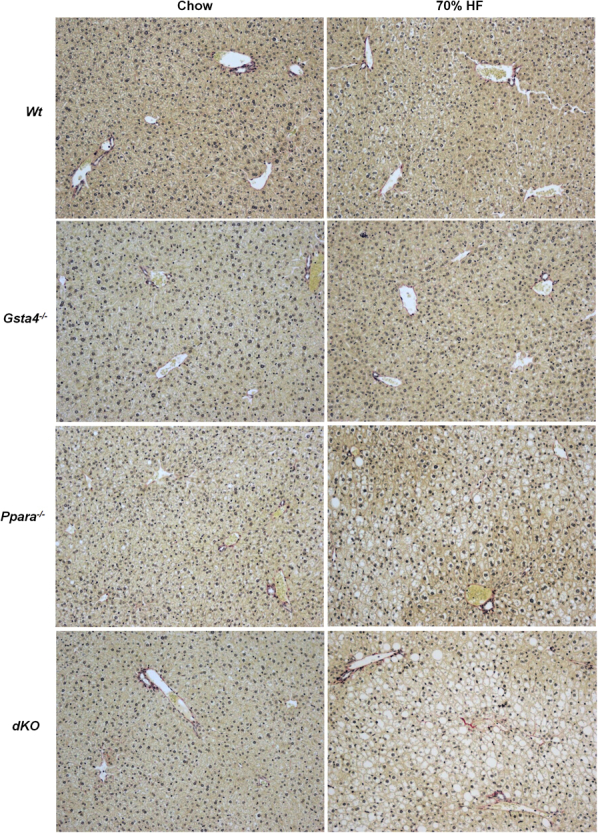
Representative liver hematoxylin and eosin sections showing fibrosis progression as depicted by collagen deposition in parenchymal tissue in Wt, Gsta4−/−, Ppara−/−, and Ppara−/−/Gsta4−/− (dKO) mice receiving a 70% HF diet or chow (light objective), picrosirius red staining as described previously.8 Original magnification, ×10.
Supplemental Figure S2.
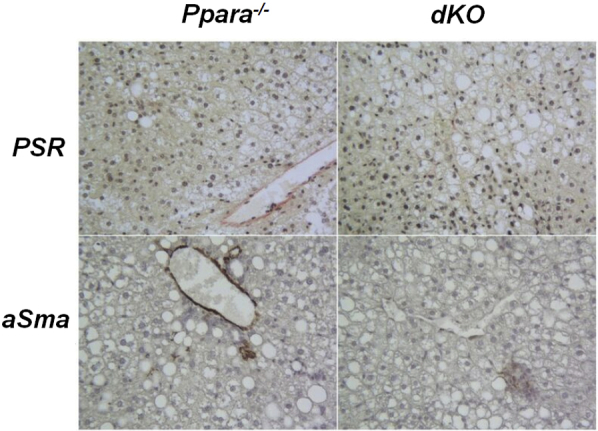
Representative liver hematoxylin and eosin sections depicting collagen deposition and activated stellate cells in parenchymal liver tissue in Ppara−/− and dKO mice receiving 70% HF diet for 12 weeks (light objective), picrosirius red staining as described previously.8 Stellate cells were identified by immunohistochemical methods, as described in Materials and Methods. Original magnification, ×200.
References
- 1.Molleston J.P., Schwimmer J.B., Yates K.P., Murray K.F., Cummings O.W., Lavine J.E., Brunt E.M., Scheimann A.O., Unalp-Arida A. Histological abnormalities in children with nonalcoholic fatty liver disease and normal or mildly elevated alanine aminotransferase levels. J Pediatr. 2014;164:707–713e3. doi: 10.1016/j.jpeds.2013.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Da Silver G.H., Coelho K.L., Coelho C.A., Escanhoela C.A. Mitochondrial alterations in nonalcoholic liver disease: pediatric case description of three submitted sequential biopsies. J Gastrointestin Liver Dis. 2009;18:215–219. [PubMed] [Google Scholar]
- 3.Singer C., Stancu P., Cosoveanu S., Botu A. Non-alcoholic fatty liver disease in children. Curr Health Sci J. 2014;40:170–176. doi: 10.12865/CHSJ.40.03.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giorgio V., Prono F., Graziano F., Nobili V. Pediatric nonalcoholic liver disease: old and new concepts on development, progression, metabolic insight and potential treatment targets. BMC Pediatr. 2013;13:40. doi: 10.1186/1471-2431-13-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lieber C.S., Leo M.A., Mak K.M., Xu Y., Cao Q., Ren C., Ponomarenko A., DeCarli L.M. Model of nonalcoholic steatohepatitis. Am J Clin Nutr. 2004;79:502–509. doi: 10.1093/ajcn/79.3.502. [DOI] [PubMed] [Google Scholar]
- 6.Pawlak M., Lefebvre P., Staels B. Molecular mechanism of Ppara−/−α action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic liver disease. J Hepatol. 2015;62:720–733. doi: 10.1016/j.jhep.2014.10.039. [DOI] [PubMed] [Google Scholar]
- 7.Abdelmegeed M.A., Yoo S.H., Henderson L.E., Gonzalez F.J., Woodcroft K.J., Song B.J. Ppara−/−α expression protects male mice from high fat-induced nonalcoholic fatty liver. J Nutr. 2011;141:603–610. doi: 10.3945/jn.110.135210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ronis M.J.J., Mercer K.E., Gannon B., Engi B., Zimniak P., Shearn C.T., Orlicky D.J., Albano E., Badger T.M., Petersen D.R. Increased 4-hydroxynonenal protein adducts in male Gsta4−/−/Ppara−/−α double knockout mice enhance injury during early stages of alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2015;308:G403–G415. doi: 10.1152/ajpgi.00154.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balogh L.M., Atkins W.M. Interactions of glutathione transferases with 4-hydroxynonenal. Drug Metab Rev. 2011;43:165–178. doi: 10.3109/03602532.2011.558092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sutti S., Jindal A., Locatelli I., Vacchiano M., Gigliotti L., Bozzola C., Albano E. Adaptive immune response triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology. 2014;59:886–897. doi: 10.1002/hep.26749. [DOI] [PubMed] [Google Scholar]
- 11.Committee for the Update of the Guide for the Care and Use of Laboratory Animals; National Research Council . ed 8. National Academies Press; Washington, DC: 2011. Guide for the Care and Use of Laboratory Animals. [Google Scholar]
- 12.Engle M.R., Singh S.P., Czernik P.J., Gaddy D., Montague D.C., Ceci J.D., Yang Y., Awasthi S., Awashi Y.C., Zimniak P. Physiological role of mGsta4−/−, a glutathione S-transferase metabolizing 4-hydroxynonenal: generation and analysis of GSTA4 null mouse. Toxicol Appl Pharmacol. 2004;194:296–308. doi: 10.1016/j.taap.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Reeves P.G., Niellsen F.H., Fahey G.C., Jr. AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J Nutr. 1993;123:1939–1951. doi: 10.1093/jn/123.11.1939. [DOI] [PubMed] [Google Scholar]
- 14.Baumgardner J.N., Shankar K., Hennings L., Badger T.M., Ronis M.J.J. A new model for nonalcoholic steatohepatitis in the rat utilizing total enteral nutrition to overfeed a high polyunsaturated fat diet. Am J Physiol Gastrointest Liver Physiol. 2008;294:G27–G38. doi: 10.1152/ajpgi.00296.2007. [DOI] [PubMed] [Google Scholar]
- 15.Shearn C.T., Smathers R.L., Bakos D.S., Reigan P., Orlicky D.J., Petersen D.R. Increased carbonylation of the lipid phosphatase PTEN contributes to Akt2 activation in a murine model of early alcohol-induced steatosis. Free Radic Biol Med. 2013;65:680–692. doi: 10.1016/j.freeradbiomed.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He S., McPhaul C., Li J.Z., Garuti R., Kinch L., Grishin N.V., Cohen J.C., Hobbs H.H. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem. 2010;285:6706–6715. doi: 10.1074/jbc.M109.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marecki J.C., Ronis M.J.J., Badger T.M. Ectopic fat disposition and insulin resistance develop after overfeeding of a high fat diet to prepubertal rats despite increased adiponectin signaling. J Nutr Biochem. 2011;22:142–152. doi: 10.1016/j.jnutbio.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 18.Manti S., Romano C., Chirico V., Filippelli M., Cuppari C., Loddo I., Salpietro C., Arrigo T. Nonalcoholic fatty liver disease/non-alcoholic steatohepatitis in childhood: endocrine-metabolic “mal-programming”. Hepat Mon. 2014;14:e17641. doi: 10.5812/hepatmon.17641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan M., Cederbaum A.I., Zhang Y.L., Ginsberg H.N., Williams K.J., Fisher E.A. Lipid peroxidation and oxidant stress regulate hepatic apoplipoprotein B degradation and VLDL production. J Clin Invest. 2004;113:1277–1287. doi: 10.1172/JCI19197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andreo U., Elkind J., Blachford C., Cederbaum A.I., Fisher E.A. Role of superoxide radical anion in the mechanism of apoB100 degradation induced by DHA in hepatic cells. FASEB J. 2011;25:3554–3560. doi: 10.1096/fj.11-182725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fisher E.A. The degradation of apolipoprotein B100: multiple opportunities to regulate VLDL triglyceride production by different proteolytic pathways. Biochem Biophys Acta. 2012;1821:778–781. doi: 10.1016/j.bbalip.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakajima T., Kamio Y., Tanaka N., Sugiyama E., Tanaka E., Kiyosawa K., Fukushima Y., Peters J.M., Gonzalez F.J., Aoyama T. Peroxisome proliferator activated receptor alpha protects against alcohol-induced liver damage. Hepatology. 2004;40:972–980. doi: 10.1002/hep.20399. [DOI] [PubMed] [Google Scholar]
- 23.Dattaroy D., Pourhoseini S., Das S., Alhasson F., Seth R.K., Nagrakatti M., Michelotti G.A., Diehl A.M., Chatterjee S. Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2015;308:G298–G312. doi: 10.1152/ajpgi.00346.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levin I., Petrasek J., Szabo G. The presence of p47phox in liver parenchymal cells is a key mediator in the pathogenesis of alcoholic liver disease. Alcohol Clin Exp Res. 2012;36:1397–1406. doi: 10.1111/j.1530-0277.2012.01739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bettaieb A., Jinag J.X., Sasaki Y., Chao T.I., Kiss Z., Chen X., Tian J., Katsuyama M., Yabe-Nishimura C., Xi Y., Szyndralewiez C., Schroder K., Shah A., Brandes R.P., Haj F.G., Torok N.J. Hepatocyte nicotinamide adenine dinucleotide phosphate reduced oxidase 4 regulates stress signaling, fibrosis and insulin sensitivity during development of steatohepatitis in mice. Gastroenterology. 2015;149:468–480. doi: 10.1053/j.gastro.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baumgardner J.N., Shankar K., Hennings L., Albano E., Badger T.M., Ronis M.J.J. N-acetylcysteine attenuates progression of liver pathology in a rat model of non-alcoholic steatohepatitis. J Nutr. 2008;138:1872–1879. doi: 10.1093/jn/138.10.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang D., Linag J., Li Y., Noble P.W. The role of Toll-like receptors in non-infectious lung injury. Cell Res. 2006;16:693–702. doi: 10.1038/sj.cr.7310085. [DOI] [PubMed] [Google Scholar]
- 28.Jiang D., Linag J., Noble P.W. Hyaluronin in tissue injury and repair. Annu Rev Cell Dev Biol. 2007;23:435–461. doi: 10.1146/annurev.cellbio.23.090506.123337. [DOI] [PubMed] [Google Scholar]
- 29.Curzio M. Interactions between neutrophils and 4-hydroxynonenals and consequences on neutrophil motility. Free Radic Res Commun. 1988;5:55–66. doi: 10.3109/10715768809066912. [DOI] [PubMed] [Google Scholar]
- 30.Rossi M.A., Curzio M., DiMauro C., Fidale F., Garramone A., Esterbauer H., Torrelli M., Diazani M.U. Experimental studies on the mechanisms of action of 4-hydroxy-2,3-transnonenal, a lipid peroxidation product displaying chemotactic activity towards rat neutrophils. Cell Biochem Funct. 1991;9:163–170. doi: 10.1002/cbf.290090304. [DOI] [PubMed] [Google Scholar]