

Abstract
Necrotizing enterocolitis (NEC) is a devastating intestinal disease that has been associated with Cronobacter sakazakii and typically affects premature infants. Although NEC has been actively investigated, little is known about the mechanisms underlying the pathophysiology of epithelial injury and intestinal barrier damage. Cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA) are important mediators and regulators of apoptosis. To test the hypothesis that C. sakazakii increases cAMP and PKA activation in experimental NEC resulting in increased epithelial apoptosis, we investigated the effects of C. sakazakii on cAMP and PKA in vitro and in vivo. Specifically, rat intestinal epithelial cells and a human intestinal epithelial cell line were infected with C. sakazakii, and cAMP levels and phosphorylation of PKA were measured. An increase in cAMP was demonstrated after infection, as well as an increase in phosphorylated PKA. Similarly, increased intestinal cAMP and PKA phosphorylation were demonstrated in a rat pup model of NEC. These increases were correlated with increased intestinal epithelial apoptosis. The additional of a PKA inhibitor (KT5720) significantly ameliorated these effects and decreased the severity of experimental NEC. Findings were compared with results from human tissue samples. Collectively, these observations indicate that cAMP and PKA phosphorylation are associated with increased apoptosis in NEC and that inhibition of PKA activation protects against apoptosis and experimental NEC.
Necrotizing enterocolitis (NEC) affects 5% of all infants in the neonatal intensive care unit, and surgical NEC survivors incur significantly greater health care costs than age-matched infants without NEC.1 NEC typically affects premature infants (90% of cases) and is fatal in up to 40% of those with the most severe disease.2, 3 As neonatal management strategies improve, many more premature infants survive, increasing the number of at-risk patients. A recent study reported an increase in NEC-related deaths in extremely premature infants and found that NEC is the leading cause of death in this population from days 15 to 60 of life.4 Survivors of NEC are often affected by long-term disabilities, including neurodevelopmental delay5 and short gut syndrome, of which NEC is the leading cause.6 Although the pathophysiology of NEC has been studied for many years, the mechanisms that drive pathogenesis remain incompletely understood.
Bacteria play an important role in the pathogenesis of NEC. Colonization of the gut with bacteria precedes the development of NEC, and it is widely recognized that NEC is associated with the presence of luminal bacteria.7 There has been a great deal of interest in the role of the microbiome in human disease, and differences in colonization patterns have been implicated in increasing susceptibility to NEC.8, 9, 10 Although no bacterium meets Koch's postulates, Cronobacter sakazakii11 is a Gram-negative bacterium that has been associated with outbreaks of NEC after ingestion of contaminated powdered formula.12 Multiple different animal models have been used to study NEC. The best established model involves gavage feedings of hyperosmolar formula along with hypoxia to induce NEC in rat pups.13, 14 Other investigators have used an intestinal ischemia model for experimental NEC.15 A variation of the hyperosmolar formula model supplements lipopolysaccharide into the formula to simulate the bacterial load that is found in neonatal infants who have NEC.16, 17, 18, 19 C. sakazakii has been demonstrated to induce NEC in an experimental rat pup model20 and has been used to augment both cell and animal models of NEC.21, 22 Furthermore, C. sakazakii resides in the same family (Enterobacteriaceae) which are the most common types of bacteria associated with clinical cultures for patients with NEC.21, 23, 24 Experimentally, C. sakazakii disrupts normal intestinal morphologic characteristics, induces epithelial inflammation, and causes increased apoptosis both in vivo and in IEC-6 cells, a nontransformed rat small intestinal crypt cell line.20 Therefore, the use of C. sakazakii to induce NEC provides a clinically relevant experimental model.
An imbalance toward increased intestinal epithelial apoptosis is an important pathogenic feature and end point in the development of NEC.25 The C. sakazakii rat pup model of NEC demonstrates increased intestinal epithelial apoptosis.20 Numerous bacterial factors are associated with increased host cellular apoptosis in disease models.26 The precise factors that allow C. sakazakii to promote epithelial apoptosis are unknown. As with almost all Gram-negative bacteria, C. sakazakii express lipopolysaccharide (LPS) on their cell wall. Virulence factors, such as LPS, act alone or in concert with other endogenous factors that may trigger epithelial and endothelial cell injury and apoptosis.27 LPS is thought to act via a Toll-like receptor 4 pathway mediating downstream NF-κB signaling.28 Evidence suggests that other pathways, specifically cyclic adenosine monophosphate (cAMP),29 are important in mediating and maintaining the inflammasome and in propagating cellular apoptosis.30 Specific pathogens, including Pseudomonas aeruginosa and Yersinia pestis, have been found to possess virulence factors that modulate intracellular cAMP and trigger human gastroenterologic disease.31, 32, 33 Therefore, we postulated that the bacteria used in our experimental model of NEC would also increase intracellular cAMP.
cAMP is a well-known secondary messenger that is present in most cells. Components of the cAMP signaling pathway, including cAMP effector protein kinase A (PKA), enhance apoptosis and have been studied as targets for cancer chemotherapeutics.34 cAMP was first recognized as playing a role in apoptosis almost 40 years ago35, 36; however, its role in many cells and tissues remains ill defined.37 Although cAMP is crucial to normal cellular homeostasis, elevated cellular cAMP is proapoptotic in several cell types and sensitizes cells to the actions of alternate proapoptotic entities, including DNA damage, acting via a non-cAMP pathway.38 We hypothesized that experimental NEC would be characterized by increased intestinal cAMP.
Although cAMP has several effects, PKA activation is the best-characterized target. PKA is a serine-threonine kinase and a primary downstream target that depends on cAMP for activation.39 PKA is accepted to depend on cAMP for phosphorylation and activation. Evidence suggests that PKA is proapoptotic through the phosphorylation of protein targets.40, 41 However, the effect of PKA activation on apoptosis is depends on cell type. It promotes cell survival in a neuronal cell line,29 while increasing apoptosis in another.42 Differences in PKA effect may also depend on the subcellular location of PKA phosphorylation. Once activated PKA phosphorylates the transcription factor cAMP response element binding protein (CREB).43 CREB binds DNA, affecting nuclear transcription, and has been implicated in pathways controlling cell survival and apoptosis.44 From these data, we hypothesized that C. sakazakii increases cAMP and PKA activation in the intestinal epithelium during experimental NEC, leading to increased apoptosis. In this study we conducted a series of experiments using both in vitro and in vivo models, as well as human tissue to determine cAMP responses in experimental NEC, and whether this is associated with PKA phosphorylation and apoptosis. We identified that cAMP and PKA activation are associated with increased apoptosis in NEC and that inhibition of PKA activation protects against apoptosis and experimental NEC.
Materials and Methods
Cells
Rat intestinal epithelial cells (IEC-6 passages 19 to 29; Sigma-Aldrich, Milwaukee, WI) were grown in Dulbecco's modified Eagle media supplemented with 10% fetal calf serum, 1 U/mL insulin, 100 U/mL penicillin G, and 100 U/mL streptomycin. Various doses (1 × 103 to 1 × 107 cfu/mL) of C. sakazakii were added to confluent monolayers of IEC-6 cells separately and incubated for 0 to 12 hours. Media were aspirated and replenished every 2 hours to limit bacterial multiplication. Experiments were repeated with 1 × 107 cfu/mL of a second cell line, FHs 74 Int cells (CCL-241; ATCC). FHs 74 Int is a nontransformed human cell line. The cells were grown in Hybri-Care Medium ATCC 46-X supplemented with 30 ng/mL epidermal growth factor, 10% fetal bovine serum. Cells were grown to 90% confluence on Chamber Slides (Nunc Lab-Tek, Naperville, IL). Cells were pretreated with PKA inhibitors (0.1–20 μmol/L) KT5720 (Cayman Chemical, Ann Arbor, MI), cAMP Dependent Protein Kinase Inhibitor (Sigma-Aldrich, St. Louis, MO), Rp-8-cAMPS (Santa Cruz Biotechnology, Dallas, TX) for 30 minutes then subjected to co-culture with C. sakazakii at various concentrations over a time course. These experiments were repeated with IEC-6 cells exposed to LPS 10 μg/mL. In addition, IEC-6 cells were treated with cAMP analogues 8-pCPT-2′-O-Me-cAMP (8C; Sigma-Aldrich), adenosine 3′,5′-cyclophosphate (Sigma-Aldrich), rat tumor necrosis factor α (TNF-α; LifeTech, Elmhurst, IL), and rat IL-6 (LifeTech). Cells were stained for apoptosis using the ApopTag Red in Situ Apoptosis Detection Kit (Chemicon, Billerica, MA).
Bacterial Strains and Endotoxins
C. sakazakii, clinical strain BAA-894 (ATCC) was grown at 37°C in Luria broth, centrifuged (3000 rpm), and washed twice before being added to cell cultures or formula. C. sakazakii growth was assessed by growing bacteria in broth with KT5720 (1 μmol/L) over a time course and by measuring optical density (Supplemental Figure S1A). C. sakazakii binding to IEC-6 cells were also performed (Supplemental Figure S1B).45 LPS from Escherichia coli clinical strain 0111:B4 (Sigma-Aldrich) was stored at 4°C. LPS was dissolved in sterile 0.9% normal saline (VWR, Radnor, PA) to achieve a stock concentration of 10 mg/mL.
Transfection
IEC-6 cells were seeded in a 12-well plate with 105 cells/well using serum-free media. Cells were transfected with Lipofectamine 2000 (LifeTech). PKA c-α siRNA (Cell Signaling, Danvers, MA), CREB siRNA (Santa Cruz Biotechnology, Dallas, TX), or control siRNA for 6 hours at 37°C and 5% CO2 were transfected. Knockdown was confirmed by Western blot analysis of phosphorylated PKA (pPKA), or quantitative RT-PCR with CREB primers supplied by Santa Cruz Biotechnology and normalized by GAPDH (forward: 5′-ATCACCATCTTCCAGGAGCG-3′; reverse: 5′-TTCTGAGTGGCAGTGAGGGC-3′). Data were analyzed by Bio-Rad CFX Manager (Bio-Rad, Hercules, CA) (Supplemental Figure S1E). After transfection the cells were changed to complete media. Thereafter, the siRNA PKA cells were treated with 1 × 107 cfu/mL C. sakazakii. Cells were harvested at times up to 6 hours after C. sakazakii addition. The siRNA CREB group was treated 24 hours after transfection.
Rat Pup Model
Female Sprague-Dawley rats with synchronized pregnancies were purchased from Charles River Laboratories (Roanoke, IL) and induced near-term at embryonic day 21 with a subcutaneous injection of oxytocin 0.1 U. Rat pups were separated from the dams and placed into experimental groups. Pups were gavaged thrice daily with formula 0.2 to 0.3 mL (15 g Similac 60/40 [Ross Pediatrics, Columbus, OH] in 75 mL of Esbilac canine milk replacer [Pet-Ag Inc., Hampshire, IL]). Pups were exposed to hypoxia (5% O2, 95% N2) for 5 minutes twice daily in a modular chamber (Billups-Rothenberg, Del Mar, CA). Experimental groups included formula-fed (FF) controls, a FF group with hypoxia (FF + H group), a FF + H group in which one of the daily feeds contained a known quantity of C. sakazakii (FF + H + C. sakazakii), and FF + H + C. sakazakii or FF + H groups pretreated with KT5720 (5 mL/kg) or vehicle (dimethyl sulfoxide) on the first day of life. Rat pups were euthanized on day 4 after birth or if they displayed symptoms of NEC (abdominal distention and discoloration), respiratory distress, or >20% weight loss. Intestine was harvested for analysis. NEC was graded microscopically by a pediatric pathologist (P.C.) blinded to groups, from grade 0 (normal) to grade 3 (severe with perforation) on the basis of pathologic manifestations, including submucosal edema, epithelial sloughing, hemorrhage, neutrophil infiltration, derangement of intestinal villus architecture, intestinal perforation, and necrosis. Animals were housed in the Northwestern University facilities that are fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. They were provided with environmental enrichment. All procedures and protocols were approved by Northwestern University Institutional Animal Care and Use Committee and were conducted in accordance with guidelines set forth by NIH’s Guide for the Care and Use of Laboratory Animals.46
Human Samples
After institutional review board approval (number 2013-15152), human intestinal tissue samples were obtained from infants undergoing bowel resection. The type of tissue obtained, reason for surgery, positive clinical cultures (blood and urine), and corrected gestational age were recorded. Intestinal specimens were categorized as active NEC (ie, the intestine was resected during surgery for perforation or sepsis) and NEC-free (eg, intestine resected during ostomy takedown or for intestinal atresia). In total 7 patients with NEC and 8 control samples were obtained. Tissue was collected in 10% buffered formalin (Cardinal Health, Dublin, OH) and processed into paraffin blocks, snap-frozen in liquid nitrogen, or preserved in optimal cutting temperature media Sakura Finetek, Torrance, CA) at −80°C.
Protein Isolation
Tissue samples from humans and rats were isolated before either being suspended in Allprotect tissue reagent (Qiagen, Valencia, CA) and stored at −80°C or snap-frozen in liquid nitrogen. The tissue was sectioned and suspended in lysis buffer (20 mmol/L Tris-HCl [pH 7.5]), 150 mmol/L NaCl, 1 mmol/L Na2 EDTA, 1 mmol/L EGTA, 1% Triton, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L glycerophosphate, 1 mmol/L Na3VO4, 1 μg/mL leupeptin; Cell Signaling Tech) and containing 1 mmol/L phenylmethylsulfonyl fluoride, protease inhibitors [1.02 mmol/L 4-(2-Aminomethyl)benzenesulfonyl fluoride hydrochloride, 0.0008 mmol/L aprotinin, 0.02 mmol/L leupeptin, 0.04 mmol/L bestatin, 0.015 mmol/L pepstatin A, 0.014 mmol/L E-64], phosphatase inhibitors (sodium vanadate, sodium molybdate, sodium tartrate, and imidazole; Sigma-Aldrich). Samples were homogenized for 3 minutes on ice using a ground glass tissue grinder. After centrifugation for 2 minutes at 14,000 × g and 4°C, supernatant fluids were collected and stored at −80°C. To isolate proteins, cultured cells were scraped into and centrifuged at 5000 rpm at 4°C for 10 minutes. Supernatant fluids were discarded, and cell pellets were resuspended in lysis buffer, drawn three times through a 27-gauge needle, and mixed on a rotating platform for 30 minutes at 4°C. Insoluble debris was then removed by centrifugation 4°C, 15 minutes, at 10,000 rpm, and supernatant fluids were stored at −80°C.
Antibodies
Antibodies were obtained from the following sources: mouse anti–β-actin (dilution 1:50,000; Sigma-Aldrich), rabbit anti-PKA C-a (dilution 1:500; Cell Signaling), rabbit anti–phospho-PKA C (dilution 1:500; Thr197; Cell Signaling), rabbit anti–phospho-CREB (dilution 1:500; Ser133; Cell Signaling), rabbit anti–caspase-3 (dilution 1:500; Cell Signaling), and rabbit anti-cleaved caspase-3 (dilution 1:500; Cell Signaling).
Protein Analysis
Samples were run on a Bio-Rad Mini-PROTEAN apparatus (Bio-Rad, Hercules, CA) at 30 mA and then transferred to a nitrocellulose membrane using standard approaches. Membranes were blocked for 1 hour at room temperature by incubation with phosphate-buffered saline (PBS)-Tween containing 5% nonfat dry milk. Primary and secondary antibodies were diluted in PBS-Tween containing 5% nonfat dry milk. Protein bands were detected using electrochemiluminescence Western blotting detection reagents (GE Health Care Life Sciences, Piscataway, NJ) on Amersham Hyperfilm electrochemiluminescence (GE Health Care Life Sciences). Bands were quantified by densitometry using Bio-Rad Image Lab software (ChemiDoc XRS+ System with Image Lab Software version 5.2.1). The ratios of selected protein/β-actin were determined for individual tissues and cell cultures in biological triplicate and reported as ± SEM.
Quantitative RT-PCR
Total RNA was extracted from rat pup intestinal tissue or cells using RNeasy Mini Kit (Qiagen). RNA was then converted to cDNA using GeneAMp RNA PCR Core Kit (Life Technologies, Carlsbad, CA). Quantitative RT-PCR was performed with equal amounts of cDNA using Bio-Rad CFX with iQ SYBR Green Supermix (Bio-Rad). Primer sequences were GAPDH: forward, 5′-ATCACCATCTTCCAGGAGCG-3′, and reverse, 5′-TTCTGAGTGGCAGTGAGGGC-3′; PKAc-α: forward, 5′-GAGCAGGAGAGCGTGAAAGA-3′, and reverse, 5′-TCCTTGTGCTTCACGAGCAT-3′.
cAMP Assay
Protein samples from IEC-6 cells, FHs 74 Int cells, rat small intestine, and human small intestine were assayed for cAMP with an enzyme immunoassay kit (Cayman Chemical). Data were normalized to β-actin protein content.
Flow Cytometry
IEC-6 and FHs 74 Int cells were grown to confluence. C. sakazakii (1 × 107 cfu/mL) was added to the experimental group and incubated for 0 to 12 hours. Media were aspirated and replenished every 2 hours to limit bacterial multiplication. After incubation, cells were gently rinsed with ice-cold PBS and then trypsinized and centrifuged at 1800 rpm at 4°C for 5 minutes for collection. To assess apoptosis, cells were stained with PE Annexin V Apoptosis Detection Kit (BD Biosciences, Franklin Lakes, NJ). Analysis was performed with a BD LSR Fortessa Analyzer.
Immunofluorescence Microscopy
IEC-6 monolayers grown on chamber slides, paraffin sections, and/or cryosections of rat and human intestine were stained for apoptosis using the ApopTag Red in Situ Apoptosis Detection Kit (Chemicon). Monolayers were washed with PBS and blocked with 10% normal goat serum in PBS with 0.1% Triton. Slides were prepared and then incubated in primary antibody pPKA (Cell Signaling), pCREB (Cell Signaling), both rabbit anti-mouse at 1:500 dilution in 1× PBS at 4°C overnight (approximately 16 hours). After incubation in primary antibody, slides were washed 4 times and incubated in secondary antibody, Alexa Fluor 488–conjugated goat anti-rabbit antibodies (Invitrogen, Carlsbad, CA), and dilution of 1:2000 for 1 hour. DAPI-conjugated fluoroshield (Sigma-Aldrich) was applied to each slide and mounted with glass coverslip. Multiphoton microscopy was perform on a Nikon A1R multiphoton microscope (Nikon, Melville, NY). All images were analyzed with ImageJ version 1.51d (NIH, Bethesda, MD). Apoptotic cells were counted using particle analysis after threshold adjustment.
Subcellular Fractionation
We used ProteoExtract Subcellular Proteome Extraction Kit (EMD Millipore Corporation, Kankakee, IL) to obtain the proteins contained in four subcellular fractions from human samples (cytosol, membrane, nucleus, and cytoskeleton). The protein concentration of these fractions were obtained using the Bio-Rad Protein Assay Kit II. Equal amounts of protein from each of the four fractions were added to wells of a SDS-PAGE gel followed by Western blot transfer and antibody detection. This technique was used rather than using β-actin, because β-actin and most other compartment markers varied as expected. Compartments were confirmed by location-specific proteins [nucleus: histone H3 (Cell Signaling), membrane: Na, K-ATPase (Cell Signaling), cytosol: GAPDH (Cell Signaling), cytoskeleton: anti-actin (Sigma-Aldrich)].
Statistical Analysis
cAMP data were analyzed using the online Cayman Chemical. Graphs were generated using Excel (Microsoft, Redman, WA) or GraphPad (La Jolla, CA), and statistical analysis (t-test and analysis of variance)9 were performed using GraphPad InStat 3 or GraphPad Prism 6. Differences were considered significant at P < 0.05. Survival data were plotted as a Kaplan-Meier survival curve, and differences were analyzed with a log-rank test.
Results
cAMP Is Increased in Experimental NEC and Precedes Intestinal Epithelial Cell Apoptosis
cAMP production and PKA activation was evaluated in nontransformed IEC-6 cells and in the embryonic, nontransformed FHs 74 Int cell line. To determine whether IEC-6 and FHs 74 Int cells release cAMP in response to C. sakazakii infection, monolayers were treated with 107 cfu of C. sakazakii for different lengths of time and compared with control cells that were untreated. We found that C. sakazakii induced an increase in intestinal epithelial cell cAMP in a bimodal fashion. An early peak was present at 5 (FHs 74 Int cells) or 15 (IEC-6 cells) minutes (P < 0.04 and P < 0.02, respectively), and a second peak was noted by 4 hours (P < 0.001 and P < 0.005) with 107 cfu/well of C. sakazakii (Figure 1A). By 12 hours, both cell lines demonstrated cAMP levels that were close to baseline (Figure 1A). Experiments were repeated in the IEC-6 cell line using LPS. LPS caused an increase in cAMP at a later time point, similar to C. sakazakii. However, the earlier (5–15 minutes) peaks were absent. In addition, cAMP levels were measured in media containing C. sakazakii bacteria without any epithelial cells. These cAMP levels were then compared with control IEC-6 and FHs 74 Int cells. There was no significant difference in cAMP levels between the IEC-6 controls and the C. sakazakii–containing media (P = 0.15). However, a significant increase was seen when comparing the C. sakazakii–containing media with the FHs 74 Int cell controls (P = 0.01) (Supplemental Figure S1C).
Figure 1.

A: cAMP is increased in a bimodal pattern in intestinal epithelial cells infected with CS. IEC-6 cells and FHs 74 Int cells were grown to confluence in triplicate and infected with CS over a time course. A: Changes in cellular cAMP in response to 107 cfu/well CS are shown. By 15 minutes of infection the IEC-6 cells show an increase in cAMP. There is a later second significant increase in cAMP that occurs after 4 hours of infection. FHs 74 Int cells demonstrated a similar bimodal peak, with a significant difference from baseline after 5 minutes of infection and a later peak reaching significance by 4 hours. In vitro experiments were repeated with the IEC-6 cell line to include a time course of LPS treatment. LPS exposure was found to increase cAMP in a bimodal pattern at 1 and 4 hours, similar to CS albeit to lower levels of cAMP. However, the earlier peak seen with CS (5–15 minutes) was not identified in cells treated with LPS alone. All values were standardized to β-actin. B: Rat pup intestine cAMP is elevated in experimental NEC. Experimental NEC was confirmed by histologic scoring by two reviewers (C.J.H., P.C.) blinded to groups. Scoring was performed after 4 experimental days of formula feeding, CS infection, and hypoxia. Levels of cAMP were significantly higher in rat pups infected with CS compared with FF + H pups and FF controls with a nearly twofold increase. C: Rat pup intestine with CS-induced NEC demonstrate marked villus sloughing and inflammatory infiltrates. Hematoxylin and eosin staining of paraffin sections of rat pup intestine demonstrates increased inflammatory infiltrate and epithelial sloughing in the NEC group compared with the control groups. Pups with a score of ≥2 are considered to have experimental NEC. The intestinal injury score for rat pups fed either clean formula or CS-infected FF + H are compared. There is a significantly higher injury score in the CS-fed pups (CS-NEC) versus controls. Data are expressed as means ± SEM. n = 10 pups per group. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001. Original magnification, ×20. cAMP, cyclic adenosine monophosphate; CS, Cronobacter sakazakii; FF, formula fed; H, hypoxic; HFs 74 Int, human small intestinal cell line; IEC-6, rat intestinal epithelial cell line; LPS, lipopolysaccharide; NEC, necrotizing enterocolitis.
The pups were divided into groups as outlined in Materials and Methods (FF, FF + H, FF + H + C. sakazakii). FF pups with and without hypoxia were used as controls. A hypoxic group was included to control for the potential effect of hypoxia on cAMP production. The FF + H group did not have a significant increase in cAMP by 4 days (Figure 1B). The FF + H + C. sakazakii group, however, demonstrated a nearly twofold increase in cAMP (P < 0.001). Rat pup intestinal injury severity was recorded based on the scoring of hematoxylin and eosin–stained intestinal segments (Figure 1C). The difference in injury severity between control pups and C. sakazakii–fed pups was statistically significant (P = 0.001). We found that pups with C. sakazakii–induced NEC had significantly greater intestinal cAMP than the control groups (P < 0.001). These data suggest that increased cAMP is associated with higher intestinal injury scores and experimental NEC.
To determine whether cAMP was present in human NEC we assayed surgical intestinal samples from patients with and without NEC. Human intestinal segments collected from patients undergoing intestinal resection for active NEC demonstrated increased inflammatory infiltrate and epithelial sloughing similar to the rat pup model of NEC (Figure 1C). Interestingly, cAMP assay on human intestinal segments revealed a lower level than controls during active perforated NEC (P < 0.008) (Supplemental Figure S1D). This may be due to a loss of viable intestine and severe epithelial sloughing characteristic of Bell's stage III NEC. The decrease of cAMP levels in Bell's stage III disease correlated with the decreased values seen at the longer time points in our in vitro experiments (ie, 12 hours). Given the importance of bacteria in the pathogenesis of NEC, corresponding human bacterial culture data were reviewed (blood and urine) for the human samples. Neither group of patients demonstrated positive cultures before operative resection of bowel. Note that all patients with NEC were on broad-spectrum antibiotics before, during, and after surgical exploration, which would be expected to affect the likelihood of obtaining positive cultures.
Increased cAMP Correlates with Increased Cellular Apoptosis
To examine whether increased cAMP levels precede downstream biochemical markers of PKA phosphorylation and apoptosis we performed an in vitro time course. IEC-6 and FHs 74 Int cells were infected with 107 cfu/mL of C. sakazakii, and markers of apoptosis were measured over a 12-hour period. We measured the change in cleavage of caspase-3, annexin V, 7-aminoactinomycin D, and DNA fragmentation. Western blot analysis revealed an increase in cleaved caspase-3 that reached statistical significance after 6 hours of C. sakazakii infection in both IEC-6 and FHs 74 Int cell lines (P < 0.001, P < 0.001) (Figure 2A). Flow cytometry revealed early apoptosis occurring in both the IEC-6 and FHs 74 Int cells (Figure 2B). In IEC-6 cells, an increase to 17.8% early apoptotic cells was seen at 1 hour of C. sakazakii treatment (P < 0.006). FHs 74 Int cells demonstrated an increase to 4.46% early apoptotic cells at this same time point (P = 0.04). Apoptosis was detected in C. sakazakii–infected IEC-6 and FHs 74 Int cells by immunofluorescence47 using the ApoTag kit measuring DNA fragmentation. We found that there was a fivefold increase in IEC-6 cell apoptosis after 6 hours of co-culture with C. sakazakii compared with controls (P < 0.001). This effect was even greater in FHs 74 Int cells in which a 20-fold increase was noted (P = 0.002) (Figure 2C). Increased markers of apoptosis occur several hours after the initial peak in cAMP which occurs in the order of minutes. In line with prior data, apoptosis was identified in the C. sakazakii rat pup model of NEC compared with controls by day 420 and confirmed apoptosis by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling staining (Figure 2D). Increased apoptosis was also seen in human intestinal segments with active NEC compared with control tissue, which is in support of the findings in our experimental models (Figure 2E). In addition, we found that increasing doses of two different cAMP analogues (adenosine 3′,5′-cyclophosphate and 8C) were associated with increased apoptosis compared with untreated controls. Inflammatory cytokines associated with intestinal inflammation and NEC were also examined. IL-6 did not induce epithelial apoptosis at the tested doses, TNF-α did produce an increase in apoptosis, albeit at a lower significance than the cAMP analogues (Figure 2F). Taken together, these data are supportive of the role of dose-specific cAMP in intestinal epithelial apoptosis.
Figure 2.

A: IEC-6 and FHs 74 Int cells show a significant increase in cleaved caspase-3 after 6 hours of CS exposure. IEC-6 cells and FHs 74 Int cells were grown to near confluence in biological triplicate and treated with 107 cfu/mL of CS over a time course (0–6 hours). Western blot analysis of caspase-3 was performed to assess apoptosis. IEC-6 cells had a significant increase in cleaved caspase-3 after 6 hours of CS treatment. Full-length caspase-3 was also decreased. Similarly, FHs 74 Int cells showed a significant reduction of full-length caspase-3 and a significant increase in cleaved caspase-3 after 6 hours of CS treatment. B: Apoptosis is increased in intestinal epithelial cells infected with CS. IEC-6 and FHs 74 Int cells were grown to near confluence in triplicate and treated with 107 cfu/mL of CS over a time course (0–12 hours). The cells were washed and stained with Annexin PE to assess apoptosis with flow cytometry. Flow cytometry revealed a significant increase in early apoptosis at the 1-hour time point in both cell lines when treated with CS. C: CS induces apoptosis in IEC-6 and FHs 74 Int cells. Both IEC-6 and FHs 74 Int cells were grown in chamber slides in triplicate and were exposed to CS for 0 to 6 hours. At 6 hours, cells treated with 107 cfu/mL of CS and were found to demonstrate increased apoptosis by immunofluorescent DNA fragmentation staining (red cells indicated by white arrows). The blue stain is nuclear DAPI. The number of apoptotic cells per 1000 cells were counted and averaged from 10 random high-power views per sample. Apoptosis met clinical significance for both the IEC-6 and FHs 74 Int cells after 6 hours of infection compared with control cells not exposed to CS. D: CS-induced rat pup NEC is associated with increased intestinal apoptosis. Rat pups were born near-term and subject to either FF three times daily or dosed daily with CS (107 cfu/mL of formula) and hypoxia. Intestinal segments were cryopreserved and stained with DAPI (blue) and ApoTag for apoptosis. The FF + H + CS group had a significant increase in apoptosis compared with controls (P = 0.0025). E: Patients with NEC have increased intestinal apoptosis. Tissue samples were obtained from patients undergoing surgical bowel resection and embedded in paraffin. Slides were stained with DAPI (blue) and ApoTag for evidence of DNA fragmentation. Total apoptotic cells per high-powered field were counted. Patients with clinical NEC demonstrated increased intestinal apoptosis compared with control subjects (P = 0.0002). F: cAMP analogues induce intestinal epithelial cell apoptosis in a dose-responsive fashion. IEC-6 cells were grown to confluence of chamber-well slides and tested with various doses of PKA activators [cAMP analogues (8C and A) and cytokines (IL-6 and TNF-α)]. Slides were stained with DAPI and ApoTag to assess apoptosis. Experiments were conducted in biological triplicate. An increase in apoptosis was found with increasing concentration of PKA activators and with TNF-α [A (100 μmol/L), 8C (50 μmol/L), and TNF-α (10 ng/mL)]. IL-6 did not increase apoptosis at doses 1 ng/mL up to 100 ng/mL. Data are expressed as means ± SEM. n = 3 experiments (A and F); n = 6 per group (D); n = 4 patients with clinical NEC (E); n = 6 control subjects (E). ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, and ∗∗∗∗P < 0.0001. Scale bars: 50 μm (D); 200 μm (E). Original magnification, ×10 (C–E). A, adenosine 3′,5′-cyclic monophosphate; cAMP, cyclic adenosine monophosphate; Cont, control; CS, Cronobacter sakazakii; FF, formula feeding; H, hypoxia; HFs 74 Int, human small intestinal cell line; HPF, high-powered field; IEC-6, rat intestinal epithelial cell line; NEC, necrotizing enterocolitis; PE, phosphatidylethanolamine; PerCP, peridinin chlorophyll; PKA, protein kinase A; Q, quadrant; TNF-α, tumor necrosis factor α; 8C, 8-(-4-Chlorophenylthio)-2′-O-methyladenosine 3′,5′-cyclic monophosphate monosodium hydrate.
Activation of PKA Is Associated with Human and Experimental NEC
To test the role of PKA activation in C. sakazakii–induced NEC, protein levels of PKA and pPKA were measured by Western blot analysis. We found that pPKA was increased in a bimodal fashion, similar to the previously described (Figure 3A) cAMP findings. An early peak was present at 1 hour (FHs 74 Int cells) or 30 minutes (IEC-6 cells) (P < 0.0006 and P < 0.0001, respectively). A later peak was noted by 6 hours for both cell types (P < 0.0003 and P < 0.0001, respectively). Experiments were conducted in the IEC-6 cell line using LPS. We found that LPS caused an increase in pPKA at a later time point, similar to the C. sakazakii bacteria. However, the earlier (5–15 minutes) peaks were absent (Figure 3A). pPKA was also increased in intestinal segments from rat pups inoculated with C. sakazakii (FF + C. sakazakii + H) compared with FF controls with or without hypoxia (FF and FF + H) (P = 0.027) (Figure 3B). Total PKA expression and total protein were similar in pups with and without experimental NEC (Figure 3B). Moreover, an increase in pPKA was noted in patient tissue samples with active NEC (P < 0.001) (Figure 3C). Immunofluorescence of rat and human small intestine samples demonstrated increased PKA activation in the intestinal tissue. pPKA was especially evident in the intestinal epithelium compared with control tissue samples. The increased expression of pPKA is especially evident in the membrane and cytoskeleton (Figure 3D). Taken together, these data suggest that PKA was activated during both experimental and human NEC.
Figure 3.

A: PKA is activated in enterocytes infected with CS or LPS. IEC-6 and FHs 74 Int cells were infected with 107 cfu/mL CS in triplicate, over a 12-hour time course. Cell lysates were collected, and Western blot analysis revealed significant increased pPKA activation. IEC-6 cells demonstrated an increase at 30 minutes and 6 hours after treatment. A similar trend was seen in the FHs 74 Int cells with a significant increase occurring at both 1 and 6 hours of co-culture (P = 0.006, P = 0.0003, respectively). β-Actin remained constant throughout the experiment. In vitro experiments were repeated in triplicate with IEC-6 cells to include a time course of LPS treatment. pPKA levels were assessed by Western blot analysis. LPS was found to increase pPKA at 30 minutes and 4 hours (P = 0.02 and P = 0.001, respectively), similar to the pattern seen in CS-treated cells. B: CS-induced experimental NEC demonstrates increased PKA activation. Rat pups were fed formula with or without CS over a 4-day time course. Rat pups without CS exposure, with and without hypoxia (FF and FF + H), did not have a significant increase in activated PKA at the conclusion of the experiment. Both expression of PKA and total protein PKA were not statistically different between control pups and pups treated with CS. However, rat small intestine samples from pups inoculated with CS (FF + H + CS) had increased PKA phosphorylation. β-Actin loading controls are shown. C: Infant intestine and experimental rat pups with NEC demonstrates increased PKA activation compared with controls. Human intestine samples were collected and were processed for Western blot analysis and histology. Western blot analysis revealed significantly greater PKA activation during NEC compared with controls. Increased intestinal epithelial pPKA was also noted in rat pups with experimental NEC compared with FF controls. β-Actin and total PKA were not statistically different between groups. D: Activated PKA was seen in the intestinal epithelium in human and rat NEC. Immunofluorescence with a pPKA primary antibody was performed on sections of intestine. Nuclei were stained with DAPI. The cell border is outlined in gray. Boxed areas are shown at higher magnification to the right. Increased activated PKA was seen in intestine with NEC compared with human and rat control samples. Activated PKA was noted to have a characteristic speculated pattern in both the rat and human samples. Subcellular fractionation was performed on control tissue and NEC tissue. Compartments were standardized using specific control proteins, and lanes were equilibrated by equal protein amounts. A representative blot demonstrates that most pPKA is located within the nucleus, and an increase in the membranous and cytoskeletal portions are found during NEC. Data are expressed as means ± SEM. n = 8 control pups (B); n = 13 pups treated with CS (B); n = 3 per group (C). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001. Scale bars: 50 μm (D, original images); 20 μm (D, magnified images). C, cytosol; CS, Cronobacter sakazakii; CTS, cytoskeleton; FF, formula-fed; H, hypoxia; HFs 74 Int, human small intestinal cell line; IEC-6, rat intestinal epithelial cell line; LPS, lipopolysaccharide; M, membrane; N, nuclear; NEC, necrotizing enterocolitis; PKA, protein kinase A; pPKA, phosphorylated PKA.
Inhibition of PKA Activation Reduces Intestinal Epithelial Apoptosis
To determine whether PKA activation mediates C. sakazakii–induced apoptosis, we examined whether it could be blocked by three known pharmacologic inhibitors of PKA phosphorylation (protein kinase inhibitor, Rp-8-cAMPS, or KT5720). IEC-6 cells were treated with inhibitors before infection with C. sakazakii or treated with PKA inhibitors alone. C. sakazakii–induced markers of apoptosis were greatly attenuated (P < 0.05), in the presence of a PKA inhibitor. Protein collected from IEC-6 cells treated with KT5720 before C. sakazakii infection demonstrated both decreased pPKA and cleaved caspase-3 compared with controls (Figure 4A). In addition, the number of apoptotic cells per high-power field detected with ApoTag Red stain was significantly decreased when IEC-6 cells were treated with PKA inhibitors compared with control and PKA-activated cells (Figure 4B). To circumvent the possibility that PKA-independent off-target effects of KT5720 were responsible for the reduction in apoptosis we used genetic inhibition and knocked-down PKA in our IEC-6 cell line before C. sakazakii infection. PKA knockdown was confirmed using Western blot analysis of PKA. There was a significant reduction in PKA in the siRNA cells compared with the wild-type cells, whereas β-actin remained stable (P < 0.001). PKA knockdown also significantly decreased caspase-3 cleavage, similar to the results found with the chemical inhibitor (P < 0.01) (Figure 4C). Furthermore, ApoTag staining of the IEC-6 cells revealed a significant reduction in apoptosis at 6 hours in those cells that underwent the PKA knockdown compared with control cells (P < 0.0014) (Figure 4D). Therefore, our findings from both the chemical inhibitor and the knockdown experiments, show that differences in epithelial apoptosis are likely a result of inhibition of PKA phosphorylation on the epithelium itself.
Figure 4.

A: PKA inhibitor (KT5720) inhibits caspase-3 activation and PKA activation in vitro. After 4 to 6 hours of infection both caspase-3 activation and pPKA were significantly increased. IEC-6 cells were treated with 1 μmol/L of KT5720 before exposure to CS demonstrated significantly less caspase-3 and PKA activation after 6 hours. B: CS-induced apoptosis is decreased by pharmacologic PKA inhibition. Immunofluorescence of DNA fragmentation and apoptosis were significantly decreased in the presence of CS even after 6 hours of infection when cells were pretreated with PKA inhibitors (KT5720, Rp-8-br-cAMPS, PKI) compared with those cells that received CS alone. The greatest decrease occurred in those cells treated with KT5720 compared with other inhibitors. IEC-6 cells were grown on chamber slides, and after various doses and exposure times they were fixed and stained with ApoTag for apoptosis and DAPI (blue). Apoptotic cells were counted per 1000 cells. C: Knockdown of PKA showed a significant reduction in caspase-3 cleavage. IEC-6 cells were transfected with lipofectamine 2000 and siRNA for PKA. Cells were then treated for 6 hours with CS. Western blot analysis of cleaved caspase-3, PKA, and β-actin were performed. PKA knockdown was confirmed with a significant reduction in PKA expression. A significant reduction in cleaved caspase 3 was also seen at the 6-hour time point. D: Knockdown of PKA protects IEC-6 cells from CS-induced apoptosis. IEC-6 cells were transfected with lipofectamine 2000 and siRNA for PKA. Cells were then treated over a time course with CS. There was a significant increase in apoptosis in the presence of CS after 6 hours. In addition, ApoTag staining revealed fewer apoptotic cells when PKA was knocked down (+siRNA) before infection with CS for 6 hours than controls (−siRNA) (P = 0.0014). ∗P < 0.05, ∗∗∗P < 0.001, and ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001. Original magnification, ×10 (B and D). CS, Cronobacter sakazakii; IEC-6, rat intestinal epithelial cell line; KD, knockdown; PKA, protein kinase A; PKI, protein kinase inhibitor; pPKA, PKA phosphorylation: WT, wild-type.
CREB Is Activated in Experimental and Human NEC
CREB is an important downstream target of cAMP and activated PKA. Given that cAMP was increased and PKA was activated in experimental NEC, we investigated whether there was an increase in phosphorylated transcription factor CREB activation. CREB activation (pCREB) has been associated with survival pathway activation in neuronal cells,48 and it plays a role in the control of cellular apoptosis. CREB was activated after infection of both IEC-6 and FHs 74 Int cells with C. sakazakii, and there were bimodal increases similar to cAMP and pPKA (Figure 5A). Phosphorylation of CREB occurred more rapidly in the FHs 74 Int cell line than in the IEC-6 cell line, and in both cases this initial increase occurred later than the first cAMP had been detected, but before our peak of pPKA. This suggests that pCREB may not be activated solely as a result of increased pPKA in our in vitro models. An early peak was present after 30 minutes in FHs 74 Int cells (P < 0.0001) and IEC-6 cells (P < 0.0001). A second peak was noted at 4 hours for the FHs 74 Int cells (P < 0.0001) and 6 hours for the IEC-6 cells (P < 0.0002). CREB activation was also identified as being significantly increased in rat pups fed C. sakazakii–inoculated formula (P < 0.0001) (Figure 5B), and pretreatment with a PKA inhibitor significantly reduced CREB activation even in the presence of C. sakazakii (P < 0.0001). This suggests that inhibition of PKA activation precedes pCREB in our animal model. Immunofluorescence identified increased CREB staining within the intestinal epithelial of rat intestine in the presence of C. sakazakii, compared with controls. Specifically, there appears to be an increased expression in the nucleus of the cells, similar to what was seen with pPKA (Figure 5C). Moreover, inhibition of PKA activation diminishes the intensity of pCREB seen in response to C. sakazakii. Humans with active NEC also demonstrated increased CREB activation compared with controls. Subcellular fractionation of human samples localize this increase to be primarily within the nuclear compartment (Figure 5D). Moreover, knockdown of CREB in a rat intestinal epithelial cell line conferred statistically significant protection against cAMP analogue (8C) and TNF-α–induced apoptosis compared with controls (Figure 5E). Taken together, these data provide support for CREB in the downstream signaling of apoptosis.
Figure 5.

A: CREB is activated in IEC-6 and FHs 74 Int cells after infection with CS. IEC-6 and FHs 74 Int cells were grown and infected with CS. Cell lysates were collected and pCREB was measured by Western blot analysis. pCREB was significantly higher than controls by 30 minutes and again at 6 hours in the IEC-6 cells. pCREB was also significantly higher than controls by 30 minutes and 4 hours in the FHs 74 Int cells of infection with CS. B: CREB was activated in experimental NEC and was diminished in the presence of a PKA inhibitor. After 4 days of FF and H, rat pups were sacrificed and intestine was harvested. Protein isolated was isolated from whole intestine samples. Western blot analysis revealed increased CREB phosphorylation in pups with CS-induced NEC compared with sterile FF controls. Pups that received a pretreatment of KT5720 had reduced CREB phosphorylation, despite the presence of CS. A representative Western blot analysis is shown. β-actin was stable. C: CREB is activated within the intestinal epithelium after CS exposure. Rat pup intestine was harvested and stored in OCT media at −80°C. Frozen sections were stained by immunofluorescence for pCREB. Imaging demonstrated increased active CREB signal in the epithelium (red stain) of rat pups fed CS-inoculated formula. This staining was less pronounced in control animals that received sterile formula (data not shown) and those pups that received a pretreatment of KT5720 (FF + Inh + CS). The blue cells are stained with DAPI. D: CREB is activated within the epithelium of human patients with active NEC. Human tissue was harvested and stored at −80°C. Frozen sections were stained by immunofluorescence for pCREB. Imaging demonstrated increased active CREB signal in the epithelium (red stain) of human patients with active NEC. The blue cells are stained with DAPI. E: CREB knockdown is protective against cAMP analogue and TNF-α–induced intestinal epithelial apoptosis. IEC-6 cells were grown on chamber slides and transfected with control siRNA or CREB siRNA. Cells were treated with doses of a cAMP analogue (8C) or TNF-α at doses to induce apoptosis. Cells were stained with DAPI and ApoTag, and apoptotic cells were quantified. CREB knockdown decreased the amount of apoptotic cells after treatment with 8C or TNF-α (P = 0.0017 and P = 0.016, respectively). Data are expressed as means ± SEM. n = 10 pups per group. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001. Scale bars = 25 μm. C, cytosol; CREB, cAMP response element binding protein; CS, Cronobacter sakazakii; CTS, cytoskeleton; FF, formula fed; H, hypoxia; HFs 74 Int, human small intestinal cell line; IEC-6, rat intestinal epithelial cell line; Inh, inhibitor; M, membrane; N, nuclear; NEC, necrotizing enterocolitis; OCT, optimal cutting temperature; pCREB, phosphorylated CREB; TNF-α, tumor necrosis factor α; 8C, 8-(-4-Chlorophenylthio)-2′-O-methyladenosine 3′,5′-cyclic monophosphate monosodium.
PKA Inhibitors Decrease the Severity of Experimental NEC
PKA inhibition has been found to inhibit apoptosis in several models of disease.49, 50 From these data suggesting that pPKA is a mediator of apoptosis in our NEC models and human NEC, we tested whether pharmacologic PKA inhibition would protect against C. sakazakii–induced NEC. Rat pups were either fed clean formula (FF), clean formula with a pretreatment of KT5720 on day of life 1 (FF + Inh), formula with once daily C. sakazakii and a pretreatment of KT5720 (FF + Inh + C. sakazakii), formula with once daily C. sakazakii (FF + C. sakazakii), or formula with a sham feed of the inhibitor's vehicle instead of the inhibitor itself (FF + Sham). Rat pups pretreated with KT5720 and then infected with C. sakazakii had a highly significant improved survival compared with rat pups that received bacteria alone (Figure 6A). These findings correlated with both gross necropsy findings (Figure 6B) and histology intestinal injury scoring (Figure 6C), where a significant decrease in experimental NEC was identified in those pups that received C. sakazakii in the presence of KT5720, compared with rat pups that did not. Immunofluorescence further confirmed a decrease in intestinal apoptosis in pups treated with KT5720, compared with rat pups that received C. sakazakii alone (Figure 6D). Taken together, these findings indicate that PKA inhibition is protective against C. sakazakii–induced NEC.
Figure 6.

A: Rat pups who received pretreatment of PKA inhibitor demonstrated improved survival from experimental NEC. After 4 days of FF and H, rat pups were sacrificed. Survival curves were generated for the individual rats pup groups. Pups that received a pretreatment of KT5720 (FF + Inhibitor + CS) demonstrated improved survival compared with control pups (FF + CS) in an experimental model of NEC (log-rank P = 0.004). B: Rat pups who received pretreatment of PKA inhibitor had decreased intestinal injury demonstrated by gross necropsy and histologic examination. After 4 days of FF and H, rat pups were sacrificed. Gross intestinal samples were photographed and compared between groups. Decreased injury was seen in pups that received a pretreatment of KT5720 (FF + Inhibitor + CS) compared with controls. C: Intestinal injury scoring was significantly decreased in experimental NEC and was seen in pups that received CS in the presence of PKA inhibitor KT5720. Rat pup intestine hematoxylin and eosin–stained slides were reviewed by a board-certified pediatric pathologist (P.C.) blinded to the experimental groups. Microscopic analysis demonstrated increased intestinal injury (sloughing, neutrophil infiltrate, and loss of normal villus architecture) in the presence of CS. A 3-point injury score was used, and there was a significant improvement in intestinal injury in pups that received KT5720 (FF + Inhibitor + CS) compared with controls (FF + CS) (P = 0.0006). Pretreatment with KT5720 diminished the degree of injury and was protective against experimental NEC. D: Rat pups that received PKA inhibitor KT5720 had a significant decrease in cellular apoptosis. After 4 days of FF and H, rat pups were sacrificed. Intestinal segments were fixed and stained with ApoTag for apoptosis and DAPI (blue). Those pups that received KT5720 (FF + Inhibitor + CS) had significantly less apoptosis than positive controls with experimental NEC (FF + CS). Data are expressed as means ± SEM. n = 19 FF pups; n = 20 FF + Inhibitor pups; n = 18 FF + Inhibitor + CS; n = 16 FF + CS pups. ∗∗∗P < 0.001. Scale bars = 25 μm. CS, Cronobacter sakazakii; FF, formula feeding; H, hypoxia; Inh, inhibitor; NEC, necrotizing enterocolitis; PKA, protein kinase A.
Discussion
NEC is a multifactorial disease process, and numerous laboratories have sought to improve on the original rat pup model described by Santulli et al.7 The original model involved enteral gavage feeding and periods of hypoxia or hypothermic stress; however, over the years several groups have found it necessary to supplement this model to enhance the incidence of experimental NEC. The addition of exogenous bacteria or LPS has been commonly described to enhance rodent NEC models.16, 51, 52 We chose to use C. sakazakii in our experimental models of NEC because this bacteria has been implicated in human outbreaks of NEC. Furthermore, C. sakazakii is representative of the most common types of pathogens found in human NEC (Enterobacteriaceae).24 C. sakazakii has been shown to induce experimental NEC in rodent models, whereas the use of other microbes such as E. coli (DH5α) were not able to replicate these effects.20 Although C. sakazakii is known to increase the release of proinflammatory cytokines and to increase epithelial apoptosis, the mechanisms underlying these effects are unknown. In this present study we have not excluded the possibility that some of our findings may be specific to C. sakazakii, although with our data in human patient samples we suspect relevance to NEC. In addition, when we treated enterocytes with LPS alone, we found similar results with both cAMP and pPKA activation.
cAMP is an important secondary mediator in many human diseases and has been shown to be induced through both activation of Toll-like receptor 453 and infection with enteric pathogens, including Vibrio cholerae, Salmonella species, E. coli, Pseudomonas aeruginosa, Campylobacter jejuni, and Shigella dysenteriae.54, 55, 56 cAMP is produced by activation of adenylate cyclase converting ADP into the active mediator and degraded by phosphodiesterase enzymes. In addition to cAMP directly effecting host homeostasis, investigators have suggested a role for cAMP in increasing pathogen epithelial binding and adherence.54 Numerous studies have implicated cAMP in inflammatory disorders,57, 58, 59 and NEC is widely regarded as a disorder of immature intestinal immunity and increased inflammation. However, the published effects of cAMP appear to differ based on the cell type and context of production.60 cAMP has been implicated in regulation of cellular apoptosis and may further sensitize cells to the proapoptotic actions of other entities that work via non-cAMP pathways.38 Some investigators have suggested a proinflammatory role for cAMP, whereas others cite dominant anti-inflammatory properties. The subcellular effect of cAMP may differ depending on the localization of effector proteins and by compartments of phosphodiesterase enzymes within the cell converting cAMP into 5′AMP.61 We have identified an increase in intestinal epithelial cAMP in two in vitro models of disease and an increase in cAMP in rat pups with experimental NEC, suggesting a role for this mediator in the pathogenesis of NEC. Note that cAMP was not increased in human tissue samples, but that this correlates with the decrease in cAMP seen in our in vitro experiments after prolonged exposure to C. sakazakii. Given the variation in our human data compared with the rat and cell data, it is possible that activation of PKA may not purely reflect cAMP levels and that other factors may be playing a role in PKA activation.
cAMP signaling activates several downstream targets, including effector proteins (ie, PKA), exchange proteins activated by cAMP (EPAC), and cyclic nucleotide-gated ion channels. Evidence supports that the effector proteins (EPAC1, EPAC2A, and EPAC2B) are localized within the cell (nuclear or cytosolic) to compartmentalize the cAMP-mediated response.62 However, PKA phosphorylation (activation) is widely considered the primary downstream effect of cAMP.57, 63 Although PKA is activated by cAMP, vasoactive peptides can activate PKA, albeit to a lesser degree.64 Treatment of intestinal epithelial cell lines (IEC-6 and T84) with cAMP analogues have been shown to activate PKA and to cause impaired migration and restitution.57 This is the first time to our knowledge that C. sakazakii infection has been shown to increase cAMP and to activate PKA. Note that, given the ubiquitous nature of cAMP and PKA, other cell types such as immune cells and the endothelium may be affected in our rat pup model. To define the role of cAMP and PKA activation in the intestinal epithelium we used two intestinal cell line models. Herein, we propose a mechanism whereby C. sakazakii leads to an increase in cAMP and activation of PKA that contributes to cellular apoptosis and experimental NEC (Figure 7).
Figure 7.

CS-induced NEC is associated with activation of PKA and epithelial apoptosis. CS bind to the intestinal epithelium. Downstream events include AC generating cyclic AMP. This facilitates activation of PKA, which may disrupt normal cellular homeostasis, allowing increased apoptosis and loss of intestinal barrier integrity. Inhibition of PKA activation is protective against experimental NEC. AC, adenylate cyclase; cAMP, cyclic adenosine monophosphate; CS, Cronobacter sakazakii; NEC, necrotizing enterocolitis; PKA, protein kinase A; p-PKA, phosphorylated PKA.
The finding that inhibition of PKA phosphorylation provides protection against apoptosis suggests that cAMP increase and PKA activation are potentially important and reversible events in the development of C. sakazakii–induced NEC. The addition of a PKA inhibitor as a pretreatment conveyed protection against both in vitro intestinal epithelial apoptosis and experimental rat pup NEC. PKA inhibition appears to protect against gut injury in our rat pup model systems. This supports its role as a potential therapeutic strategy. Because PKA specific inhibitors (H89) improved in vitro epithelial migration and restitution, inhibition of cAMP-mediated PKA activation may allow improved healing of the injured epithelium.57 PKA has also been implicated in maintenance of epithelial tight junction integrity.65 Inhibition of PKA activation in immortalized mouse epithelial mammary cells demonstrated stabilization of tight junction proteins and were protective against permeability increases due to low extracellular calcium.65 Improved barrier integrity may be important in resistance of our rat pup models to C. sakazakii–induced NEC; this represents an area of future investigation. PKA signaling cross talks with numerous other cellular pathways that have been implicated in cellular apoptosis, including mitogen-activated protein kinase (MAPK), mitogen-activated protein/extracellular signal-regulated kinase (EKR) kinase (MEK)/ERK, and receptor tyrosine kinase pathways.66, 67 Inhibition of PKA activation may be directly protective against cellular apoptosis or indirectly by affecting other downstream apoptotic pathways. Evidence supports that PKA is proapoptotic through the phosphorylation of protein targets, and studies in lymphoma cells have demonstrated that apoptosis results via an intrinsic mitochondrial-dependent mechanism.40, 68 In addition, cAMP and PKA are known to effect immune cell function, and elevations in cAMP have been associated with inhibition of T lymphocyte activity69 and IL production.70 Thus, the in vivo role of PKA inhibition may have multiple downstream effects that function in concert to protect against experimental NEC in our models. Interestingly, we found a bimodal increase in cAMP after C. sakazakii infection that decreased with prolonged exposure (>6 hours). The early rise in cAMP suggests that C. sakazakii may activate adenylate cyclase. This suggests that an increase in cAMP occurs as an early event during C. sakazakii infection, and the later peak may be as a result of a downstream cascade. However, in our human samples we found a decrease in cAMP in intestine taken from infants with advanced NEC (Bell's stage III). We hypothesize that the lower cAMP levels in the human samples may be as a result of the loss of cAMP-producing cells or that the cAMP increase had already occurred earlier in the disease process.
We have demonstrated an increased activation of CREB, which is classically consider to have prosurvival effects in a range of cell types, including neurons and breast epithelium.48, 71 There is a paucity of data on the role of CREB in the intestine. CREB has been found in phosphorylated states in gastrointestinal malignancy; however, its role in intestinal homeostasis remains unclear.72 In our experimental models cAMP elevation precedes CREB phosphorylation, as would be expected. However, the initial peak of pCREB precedes that of pPKA, suggesting an alternative mechanism of CREB activation. There is evidence that rapidly activated MAPKs may play a role in the activation of CREB, and these data could explain the timing of the early peak in CREB that we identified.73 The later peak of pCREB may be as a result of pPKA activation. It is also possible that the increase in CREB identified in our model may represent a reparative strategy, and this represents an area of future study. The data from the CREB knockdown experiments are supportive of the role of CREB in cAMP-induced apoptosis. Taken together, our data suggest that C. sakazakii is able to trigger increased cAMP in vitro and in experimental NEC and that this increase is temporally associated with both PKA activation and increased apoptosis (Figure 7). Increased PKA activation and apoptosis are also found in human intestinal samples from infants with NEC. Both downstream cell death and NEC are dramatically lessened in the presence of protein kinase inhibitors, suggesting that this pathway may play an important mechanistic role. Although CREB is activated, this phosphorylation occurs earlier than would be expected to be pPKA dependent, thus suggesting a more complex mechanism of action.
Footnotes
Supported by American Gastroenterological Association Research Scholar Award (C.J.H.) and NIH National Institute of Diabetes and Digestive and Kidney Disease grant K08DK106450 (C.J.H.). Imaging work performed at the Northwestern University Center for Advanced Microscopy was generously supported by National Cancer Institute Cancer Center Support Grant P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. Flow cytometry was performed at the Northwestern University Flow Cytometry Facility, which is supported by a Cancer Center Support grant NCI CA060553.
Disclosures: None declared.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2016.10.014.
Supplemental Data
Supplemental Figure S1.
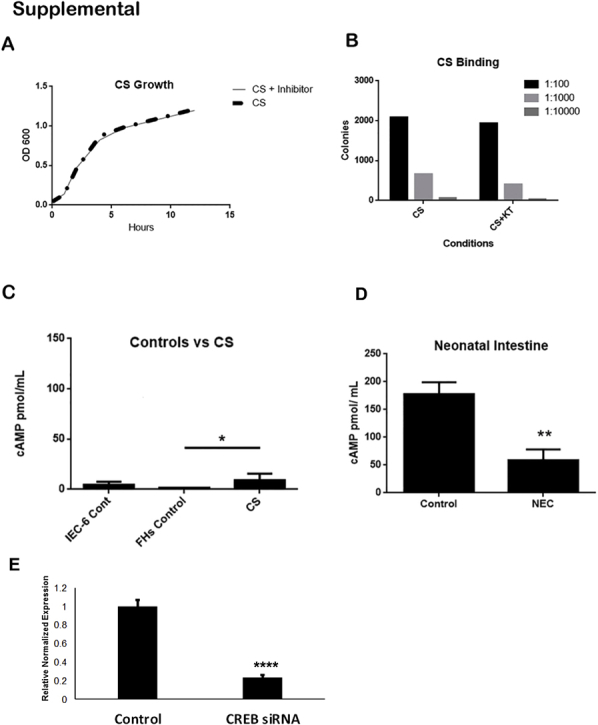
A: PKA inhibition does not affect CS growth. To determine whether PKA inhibition altered CS growth, CS bacteria were cultured in media with and without PKA inhibitor KT5720. The experiment was repeated in biological triplicate. No significant difference was identified between the groups. B: PKA inhibition does not affect CS binding. A binding assay was performed to measure whether the ability of the CS bacteria to bind to epithelial cells (IEC-6) was altered in the presence of KT 5720. The addition of PKA inhibitor KT5720 to the cell culture media was assessed at different concentrations, and no significant differences were identified (P = 0.14). This experiment was performed at three different concentrations, each in biological triplicate. C: cAMP levels in the CS bacteria do not account for the increased cAMP found in our model of NEC. cAMP levels were assessed in control IEC-6 and FHs 74 Int cell lines. These levels were then compared with cAMP levels measured in media containing CS without any epithelial cells. No significant difference was found compared with the IEC-6 cell line (P = 0.15). Although there was a significant difference compared with the FHs cell line controls (P = 0.01), the increase seen was very small compared with the increases seen in our models for NEC. Each experiment was performed in biological triplicate. D: Human intestine with active NEC demonstrate sloughing, but cAMP is not increased in whole intestine segments from patients with surgical NEC. Human intestinal tissue was collected from infants undergoing surgery for active NEC perforation. All surgical human NEC samples were obtained from patients with Bell's stage III disease, corresponding with a severity score equivalent of 3. Hematoxylin and eosin staining of paraffin sections of human intestine demonstrates increased inflammatory infiltrate and epithelial sloughing in the NEC group. cAMP was significantly lower than in whole small intestine obtained from age-matched infant control subjects (control versus NEC: P = 0.008). E: Knockdown of CREB by siRNA results in a fourfold decrease in expression. Rat intestinal epithelial cells (IEC-6) were grown to confluence and transfected with control siRNA or siRNA for CREB. RT-qPCR was performed on different samples to confirm knockdown, and a fourfold difference was identified. Data are expressed as means ± SEM. n = 7 infants undergoing surgery for active NEC perforation (D); n = 8 age-matched infant control subjects (D); n = 3 samples (E). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗∗P < 0.0001. cAMP, cyclic adenosine monophosphate; Cont, control; CREB, CREB, cAMP response element binding protein; CS, Cronobacter sakazakii; HFs 74 Int, human small intestinal cell line; IEC-6, rat intestinal epithelial cell line; NEC, necrotizing enterocolitis; PKA, protein kinase A; RT-qPCR, quantitative RT-PCR.
References
- 1.Ganapathy V., Hay J.W., Kim J.H., Lee M.L., Rechtman D.J. Long term healthcare costs of infants who survived neonatal necrotizing enterocolitis: a retrospective longitudinal study among infants enrolled in Texas Medicaid. BMC Pediatr. 2013;13:127. doi: 10.1186/1471-2431-13-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hull M.A., Fisher J.G., Gutierrez I.M., Jones B.A., Kang K.H., Kenny M., Zurakowski D., Modi B.P., Horbar J.D., Jaksic T. Mortality and management of surgical necrotizing enterocolitis in very low birth weight neonates: a prospective cohort study. J Am Coll Surg. 2014;218:1148–1155. doi: 10.1016/j.jamcollsurg.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 3.Hayakawa M., Taguchi T., Urushihara N., Yokoi A., Take H., Shiraishi J., Fujinaga H., Ohashi K., Oshiro M., Kato Y., Ohfuji S., Okuyama H. Outcomes in VLBW infants with surgical intestinal disorders at 18 months of corrected age. Pediatr Int. 2015;57:633–638. doi: 10.1111/ped.12594. [DOI] [PubMed] [Google Scholar]
- 4.Patel R.M., Kandefer S., Walsh M.C., Bell E.F., Carlo W.A., Laptook A.R., Sanchez P.J., Shankaran S., Van Meurs K.P., Ball M.B., Hale E.C., Newman N.S., Das A., Higgins R.D., Stoll B.J., Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network Causes and timing of death in extremely premature infants from 2000 through 2011. N Engl J Med. 2015;372:331–340. doi: 10.1056/NEJMoa1403489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neu J., Walker W.A. Necrotizing enterocolitis. N Engl J Med. 2011;364:255–264. doi: 10.1056/NEJMra1005408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blakely M.L., Lally K.P., McDonald S., Brown R.L., Barnhart D.C., Ricketts R.R., Thompson W.R., Scherer L.R., Klein M.D., Letton R.W., Chwals W.J., Touloukian R.J., Kurkchubasche A.G., Skinner M.A., Moss R.L., Hilfiker M.L., NEC Subcommittee of the NICHD Neonatal Research Network Postoperative outcomes of extremely low birth-weight infants with necrotizing enterocolitis or isolated intestinal perforation: a prospective cohort study by the NICHD Neonatal Research Network. Ann Surg. 2005;241:984–989. doi: 10.1097/01.sla.0000164181.67862.7f. discussion 989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santulli T.V., Schullinger J.N., Heird W.C., Gongaware R.D., Wigger J., Barlow B., Blanc W.A., Berdon W.E. Acute necrotizing enterocolitis in infancy: a review of 64 cases. Pediatrics. 1975;55:376–387. [PubMed] [Google Scholar]
- 8.Wang Y., Hoenig J.D., Malin K.J., Qamar S., Petrof E.O., Sun J., Antonopoulos D.A., Chang E.B., Claud E.C. 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. ISME J. 2009;3:944–954. doi: 10.1038/ismej.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mai V., Young C.M., Ukhanova M., Wang X., Sun Y., Casella G., Theriaque D., Li N., Sharma R., Hudak M., Neu J. Fecal microbiota in premature infants prior to necrotizing enterocolitis. PLoS One. 2011;6:e20647. doi: 10.1371/journal.pone.0020647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morowitz M.J., Poroyko V., Caplan M., Alverdy J., Liu D.C. Redefining the role of intestinal microbes in the pathogenesis of necrotizing enterocolitis. Pediatrics. 2010;125:777–785. doi: 10.1542/peds.2009-3149. [DOI] [PubMed] [Google Scholar]
- 11.Mihatsch W.A., Braegger C.P., Decsi T., Kolacek S., Lanzinger H., Mayer B., Moreno L.A., Pohlandt F., Puntis J., Shamir R., Stadtmuller U., Szajewska H., Turck D., van Goudoever J.B. Critical systematic review of the level of evidence for routine use of probiotics for reduction of mortality and prevention of necrotizing enterocolitis and sepsis in preterm infants. Clin Nutr. 2012;31:6–15. doi: 10.1016/j.clnu.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 12.van Acker J., de Smet F., Muyldermans G., Bougatef A., Naessens A., Lauwers S. Outbreak of necrotizing enterocolitis associated with Enterobacter sakazakii in powdered milk formula. J Clin Microbiol. 2001;39:293–297. doi: 10.1128/JCM.39.1.293-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barlow B., Santulli T.V., Heird W.C., Pitt J., Blanc W.A., Schullinger J.N. An experimental study of acute neonatal enterocolitis–the importance of breast milk. J Pediatr Surg. 1974;9:587–595. doi: 10.1016/0022-3468(74)90093-1. [DOI] [PubMed] [Google Scholar]
- 14.Ozdemir R., Yurttutan S., Sari F.N., Uysal B., Unverdi H.G., Canpolat F.E., Erdeve O., Dilmen U. Antioxidant effects of N-acetylcysteine in a neonatal rat model of necrotizing enterocolitis. J Pediatr Surg. 2012;47:1652–1657. doi: 10.1016/j.jpedsurg.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 15.Musemeche C.A., Baker J.L., Feddersen R.M. A model of intestinal ischemia in the neonatal rat utilizing superior mesenteric artery occlusion and intraluminal platelet-activating factor. J Surg Res. 1995;58:724–727. doi: 10.1006/jsre.1995.1114. [DOI] [PubMed] [Google Scholar]
- 16.Good M., Sodhi C.P., Egan C.E., Afrazi A., Jia H., Yamaguchi Y., Lu P., Branca M.F., Ma C., Prindle T., Jr., Mielo S., Pompa A., Hodzic Z., Ozolek J.A., Hackam D.J. Breast milk protects against the development of necrotizing enterocolitis through inhibition of Toll-like receptor 4 in the intestinal epithelium via activation of the epidermal growth factor receptor. Mucosal Immunol. 2015;8:1166–1179. doi: 10.1038/mi.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zani A., Cordischi L., Cananzi M., De Coppi P., Smith V.V., Eaton S., Pierro A. Assessment of a neonatal rat model of necrotizing enterocolitis. Eur J Pediatr Surg. 2008;18:423–426. doi: 10.1055/s-2008-1038951. [DOI] [PubMed] [Google Scholar]
- 18.Zani A., Zani-Ruttenstock E., Peyvandi F., Lee C., Li B., Pierro A. A spectrum of intestinal injury models in neonatal mice. Pediatr Surg Int. 2016;32:65–70. doi: 10.1007/s00383-015-3813-x. [DOI] [PubMed] [Google Scholar]
- 19.Zhou W., Lv H., Li M.X., Su H., Huang L.G., Li J., Yuan W.M. Protective effects of bifidobacteria on intestines in newborn rats with necrotizing enterocolitis and its regulation on TLR2 and TLR4. Genet Mol Res. 2015;14:11505–11514. doi: 10.4238/2015.September.28.2. [DOI] [PubMed] [Google Scholar]
- 20.Hunter C.J., Singamsetty V.K., Chokshi N.K., Boyle P., Camerini V., Grishin A.V., Upperman J.S., Ford H.R., Prasadarao N.V. Enterobacter sakazakii enhances epithelial cell injury by inducing apoptosis in a rat model of necrotizing enterocolitis. J Infect Dis. 2008;198:586–593. doi: 10.1086/590186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McElroy S.J., Castle S.L., Bernard J.K., Almohazey D., Hunter C.J., Bell B.A., Al Alam D., Wang L., Ford H.R., Frey M.R. The ErbB4 ligand neuregulin-4 protects against experimental necrotizing enterocolitis. Am J Pathol. 2014;184:2768–2778. doi: 10.1016/j.ajpath.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emami C.N., Mittal R., Wang L., Ford H.R., Prasadarao N.V. Recruitment of dendritic cells is responsible for intestinal epithelial damage in the pathogenesis of necrotizing enterocolitis by Cronobacter sakazakii. J Immunol. 2011;186:7067–7079. doi: 10.4049/jimmunol.1100108. [DOI] [PubMed] [Google Scholar]
- 23.Weng M., Ganguli K., Zhu W., Shi H.N., Walker W.A. Conditioned medium from Bifidobacteria infantis protects against Cronobacter sakazakii-induced intestinal inflammation in newborn mice. Am J Physiol Gastrointest Liver Physiol. 2014;306:G779–G787. doi: 10.1152/ajpgi.00183.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunter C.J., Upperman J.S., Ford H.R., Camerini V. Understanding the susceptibility of the premature infant to necrotizing enterocolitis (NEC) Pediatr Res. 2008;63:117–123. doi: 10.1203/PDR.0b013e31815ed64c. [DOI] [PubMed] [Google Scholar]
- 25.Ford H., Watkins S., Reblock K., Rowe M. The role of inflammatory cytokines and nitric oxide in the pathogenesis of necrotizing enterocolitis. J Pediatr Surg. 1997;32:275–282. doi: 10.1016/s0022-3468(97)90194-9. [DOI] [PubMed] [Google Scholar]
- 26.Grassme H., Jendrossek V., Gulbins E. Molecular mechanisms of bacteria induced apoptosis. Apoptosis. 2001;6:441–445. doi: 10.1023/a:1012485506972. [DOI] [PubMed] [Google Scholar]
- 27.Bannerman D.D., Goldblum S.E. Mechanisms of bacterial lipopolysaccharide-induced endothelial apoptosis. Am J Physiol Lung Cell Mol Physiol. 2003;284:L899–L914. doi: 10.1152/ajplung.00338.2002. [DOI] [PubMed] [Google Scholar]
- 28.Rodrigues A., Queiroz D.B., Honda L., Silva E.J., Hall S.H., Avellar M.C. Activation of toll-like receptor 4 (TLR4) by in vivo and in vitro exposure of rat epididymis to lipopolysaccharide from Escherichia Coli. Biol Reprod. 2008;79:1135–1147. doi: 10.1095/biolreprod.108.069930. [DOI] [PubMed] [Google Scholar]
- 29.Kienlen Campard P., Crochemore C., Rene F., Monnier D., Koch B., Loeffler J.P. PACAP type I receptor activation promotes cerebellar neuron survival through the cAMP/PKA signaling pathway. DNA Cell Biol. 1997;16:323–333. doi: 10.1089/dna.1997.16.323. [DOI] [PubMed] [Google Scholar]
- 30.Ouyang X., Ghani A., Malik A., Wilder T., Colegio O.R., Flavell R.A., Cronstein B.N., Mehal W.Z. Adenosine is required for sustained inflammasome activation via the A(2)A receptor and the HIF-1alpha pathway. Nat Commun. 2013;4:2909. doi: 10.1038/ncomms3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hritonenko V., Mun J.J., Tam C., Simon N.C., Barbieri J.T., Evans D.J., Fleiszig S.M. Adenylate cyclase activity of Pseudomonas aeruginosa ExoY can mediate bleb-niche formation in epithelial cells and contributes to virulence. Microb Pathog. 2011;51:305–312. doi: 10.1016/j.micpath.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Munier-Lehmann H., Chenal-Francisque V., Ionescu M., Chrisova P., Foulon J., Carniel E., Barzu O. Relationship between bacterial virulence and nucleotide metabolism: a mutation in the adenylate kinase gene renders Yersinia pestis avirulent. Biochem J. 2003;373:515–522. doi: 10.1042/BJ20030284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shahnazari S., Namolovan A., Mogridge J., Kim P.K., Brumell J.H. Bacterial toxins can inhibit host cell autophagy through cAMP generation. Autophagy. 2011;7:957–965. doi: 10.4161/auto.7.9.16435. [DOI] [PubMed] [Google Scholar]
- 34.Cross T.G., Scheel-Toellner D., Henriquez N.V., Deacon E., Salmon M., Lord J.M. Serine/threonine protein kinases and apoptosis. Exp Cell Res. 2000;256:34–41. doi: 10.1006/excr.2000.4836. [DOI] [PubMed] [Google Scholar]
- 35.Martorana P.A. The role of cyclic AMP in isoprenaline-induced cardiac necroses in the rat. J Pharm Pharmacol. 1971;23:200–203. doi: 10.1111/j.2042-7158.1971.tb08642.x. [DOI] [PubMed] [Google Scholar]
- 36.Basile D.V., Wood H.N., Braun A.C. Programming of cells for death under experimental conditions: relevance to the tumor problem. Proc Natl Acad Sci U S A. 1973;70:3055–3059. doi: 10.1073/pnas.70.11.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Insel P.A., Zhang L., Murray F., Yokouchi H., Zambon A.C. Cyclic AMP is both a pro-apoptotic and anti-apoptotic second messenger. Acta Physiol (Oxf) 2012;204:277–287. doi: 10.1111/j.1748-1716.2011.02273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ugland H., Boquest A.C., Naderi S., Collas P., Blomhoff H.K. cAMP-mediated induction of cyclin E sensitizes growth-arrested adipose stem cells to DNA damage-induced apoptosis. Mol Biol Cell. 2008;19:5082–5092. doi: 10.1091/mbc.E08-01-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kammer G.M. The adenylate cyclase-cAMP-protein kinase A pathway and regulation of the immune response. Immunol Today. 1988;9:222–229. doi: 10.1016/0167-5699(88)91220-0. [DOI] [PubMed] [Google Scholar]
- 40.Zambon A.C., Zhang L., Minovitsky S., Kanter J.R., Prabhakar S., Salomonis N., Vranizan K., Dubchak I., Conklin B.R., Insel P.A. Gene expression patterns define key transcriptional events in cell-cycle regulation by cAMP and protein kinase A. Proc Natl Acad Sci U S A. 2005;102:8561–8566. doi: 10.1073/pnas.0503363102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benz P.M., Feller S.M., Sickmann A., Walter U., Renne T. Prostaglandin-induced VASP phosphorylation controls alpha II-spectrin breakdown in apoptotic cells. Int Immunopharmacol. 2008;8:319–324. doi: 10.1016/j.intimp.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 42.Chen T.C., Hinton D.R., Zidovetzki R., Hofman F.M. Up-regulation of the cAMP/PKA pathway inhibits proliferation, induces differentiation, and leads to apoptosis in malignant gliomas. Lab Invest. 1998;78:165–174. [PubMed] [Google Scholar]
- 43.Rosenberg D., Groussin L., Jullian E., Perlemoine K., Bertagna X., Bertherat J. Role of the PKA-regulated transcription factor CREB in development and tumorigenesis of endocrine tissues. Ann N Y Acad Sci. 2002;968:65–74. doi: 10.1111/j.1749-6632.2002.tb04327.x. [DOI] [PubMed] [Google Scholar]
- 44.Saeki K., Yuo A., Suzuki E., Yazaki Y., Takaku F. Aberrant expression of cAMP-response-element-binding protein (‘CREB’) induces apoptosis. Biochem J. 1999;343 Pt 1:249–255. [PMC free article] [PubMed] [Google Scholar]
- 45.Hunter C.J., Petrosyan M., Ford H.R., Prasadarao N.V. Enterobacter sakazakii: an emerging pathogen in infants and neonates. Surg Infect (Larchmt) 2008;9:533–539. doi: 10.1089/sur.2008.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Committee for the Update of the Guide for the Care and Use of Laboratory Animals; National Research Council . National Academies Press; Washington, DC: 2011. Guide for the Care and Use of Laboratory Animals: Eighth Edition. [Google Scholar]
- 47.Scheifele D.W., Ginter G.L., Olsen E., Fussell S., Pendray M. Comparison of two antibiotic regimens for neonatal necrotizing enterocolitis. J Antimicrob Chemother. 1987;20:421–429. doi: 10.1093/jac/20.3.421. [DOI] [PubMed] [Google Scholar]
- 48.Majumder S., Varadharaj S., Ghoshal K., Monani U., Burghes A.H., Jacob S.T. Identification of a novel cyclic AMP-response element (CRE-II) and the role of CREB-1 in the cAMP-induced expression of the survival motor neuron (SMN) gene. J Biol Chem. 2004;279:14803–14811. doi: 10.1074/jbc.M308225200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Srivastava R.K., Srivastava A.R., Korsmeyer S.J., Nesterova M., Cho-Chung Y.S., Longo D.L. Involvement of microtubules in the regulation of Bcl2 phosphorylation and apoptosis through cyclic AMP-dependent protein kinase. Mol Cell Biol. 1998;18:3509–3517. doi: 10.1128/mcb.18.6.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kwak H.J., Park K.M., Choi H.E., Chung K.S., Lim H.J., Park H.Y. PDE4 inhibitor, roflumilast protects cardiomyocytes against NO-induced apoptosis via activation of PKA and Epac dual pathways. Cell Signal. 2008;20:803–814. doi: 10.1016/j.cellsig.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 51.Caplan M.S., Hedlund E., Adler L., Hsueh W. Role of asphyxia and feeding in a neonatal rat model of necrotizing enterocolitis. Pediatr Pathol. 1994;14:1017–1028. doi: 10.3109/15513819409037698. [DOI] [PubMed] [Google Scholar]
- 52.Feng J., El-Assal O.N., Besner G.E. Heparin-binding epidermal growth factor-like growth factor decreases the incidence of necrotizing enterocolitis in neonatal rats. J Pediatr Surg. 2006;41:144–149. doi: 10.1016/j.jpedsurg.2005.10.018. discussion 144–149. [DOI] [PubMed] [Google Scholar]
- 53.Moon E.Y., Lee Y.S., Choi W.S., Lee M.H. Toll-like receptor 4-mediated cAMP production up-regulates B-cell activating factor expression in Raw264.7 macrophages. Exp Cell Res. 2011;317:2447–2455. doi: 10.1016/j.yexcr.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 54.Johnson A.M., Kaushik R.S., Francis D.H., Fleckenstein J.M., Hardwidge P.R. Heat-labile enterotoxin promotes Escherichia coli adherence to intestinal epithelial cells. J Bacteriol. 2009;191:178–186. doi: 10.1128/JB.00822-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berkes J., Viswanathan V.K., Savkovic S.D., Hecht G. Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut. 2003;52:439–451. doi: 10.1136/gut.52.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Molina N.C., Peterson J.W. Cholera toxin-like toxin released by Salmonella species in the presence of mitomycin C. Infect Immun. 1980;30:224–230. doi: 10.1128/iai.30.1.224-230.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zimmerman N.P., Kumar S.N., Turner J.R., Dwinell M.B. Cyclic AMP dysregulates intestinal epithelial cell restitution through PKA and RhoA. Inflamm Bowel Dis. 2012;18:1081–1091. doi: 10.1002/ibd.21898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schafer P.H., Parton A., Gandhi A.K., Capone L., Adams M., Wu L., Bartlett J.B., Loveland M.A., Gilhar A., Cheung Y.F., Baillie G.S., Houslay M.D., Man H.W., Muller G.W., Stirling D.I. Apremilast, a cAMP phosphodiesterase-4 inhibitor, demonstrates anti-inflammatory activity in vitro and in a model of psoriasis. Br J Pharmacol. 2010;159:842–855. doi: 10.1111/j.1476-5381.2009.00559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bystrom J., Evans I., Newson J., Stables M., Toor I., van Rooijen N., Crawford M., Colville-Nash P., Farrow S., Gilroy D.W. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood. 2008;112:4117–4127. doi: 10.1182/blood-2007-12-129767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bindewald K., Gunduz D., Hartel F., Peters S.C., Rodewald C., Nau S., Schafer M., Neumann J., Piper H.M., Noll T. Opposite effect of cAMP signaling in endothelial barriers of different origin. Am J Physiol Cell Physiol. 2004;287:C1246–C1255. doi: 10.1152/ajpcell.00132.2004. [DOI] [PubMed] [Google Scholar]
- 61.Buxton I.L., Brunton L.L. Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J Biol Chem. 1983;258:10233–10239. [PubMed] [Google Scholar]
- 62.Parnell E., Smith B.O., Yarwood S.J. The cAMP sensors, EPAC1 and EPAC2, display distinct subcellular distributions despite sharing a common nuclear pore localisation signal. Cell Signal. 2015;27:989–996. doi: 10.1016/j.cellsig.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Walsh D.A., Perkins J.P., Krebs E.G. An adenosine 3',5'-monophosphate-dependant protein kinase from rabbit skeletal muscle. J Biol Chem. 1968;243:3763–3765. [PubMed] [Google Scholar]
- 64.Turner J.T., Camden J.M. The influence of vasoactive intestinal peptide receptors in dispersed acini from rat submandibular gland on cyclic AMP production and mucin release. Arch Oral Biol. 1990;35:103–108. doi: 10.1016/0003-9969(90)90170-f. [DOI] [PubMed] [Google Scholar]
- 65.Klingler C., Kniesel U., Bamforth S.D., Wolburg H., Engelhardt B., Risau W. Disruption of epithelial tight junctions is prevented by cyclic nucleotide-dependent protein kinase inhibitors. Histochem Cell Biol. 2000;113:349–361. doi: 10.1007/s004180000143. [DOI] [PubMed] [Google Scholar]
- 66.Gerits N., Kostenko S., Shiryaev A., Johannessen M., Moens U. Relations between the mitogen-activated protein kinase and the cAMP-dependent protein kinase pathways: comradeship and hostility. Cell Signal. 2008;20:1592–1607. doi: 10.1016/j.cellsig.2008.02.022. [DOI] [PubMed] [Google Scholar]
- 67.Kostenko S., Shiryaev A., Dumitriu G., Gerits N., Moens U. Cross-talk between protein kinase A and the MAPK-activated protein kinases RSK1 and MK5. J Recept Signal Transduct Res. 2011;31:1–9. doi: 10.3109/10799893.2010.515593. [DOI] [PubMed] [Google Scholar]
- 68.Zhang L., Zambon A.C., Vranizan K., Pothula K., Conklin B.R., Insel P.A. Gene expression signatures of cAMP/protein kinase A (PKA)-promoted, mitochondrial-dependent apoptosis. Comparative analysis of wild-type and cAMP-deathless S49 lymphoma cells. J Biol Chem. 2008;283:4304–4313. doi: 10.1074/jbc.M708673200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Skalhegg B.S., Landmark B.F., Doskeland S.O., Hansson V., Lea T., Jahnsen T. Cyclic AMP-dependent protein kinase type I mediates the inhibitory effects of 3',5'-cyclic adenosine monophosphate on cell replication in human T lymphocytes. J Biol Chem. 1992;267:15707–15714. [PubMed] [Google Scholar]
- 70.Sugiyama H., Chen P., Hunter M.G., Sitkovsky M.V. Perturbation of the expression of the catalytic subunit C alpha of cyclic AMP-dependent protein kinase inhibits TCR-triggered secretion of IL-2 by T helper hybridoma cells. J Immunol. 1997;158:171–179. [PubMed] [Google Scholar]
- 71.Fan C.F., Mao X.Y., Wang E.H. Elevated p-CREB-2 (ser 245) expression is potentially associated with carcinogenesis and development of breast carcinoma. Mol Med Rep. 2012;5:357–362. doi: 10.3892/mmr.2011.657. [DOI] [PubMed] [Google Scholar]
- 72.Sampurno S., Bijenhof A., Cheasley D., Xu H., Robine S., Hilton D., Alexander W.S., Pereira L., Mantamadiotis T., Malaterre J., Ramsay R.G. The Myb-p300-CREB axis modulates intestine homeostasis, radiosensitivity and tumorigenesis. Cell Death Dis. 2013;4:e605. doi: 10.1038/cddis.2013.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nishihara H., Hwang M., Kizaka-Kondoh S., Eckmann L., Insel P.A. Cyclic AMP promotes cAMP-responsive element-binding protein-dependent induction of cellular inhibitor of apoptosis protein-2 and suppresses apoptosis of colon cancer cells through ERK1/2 and p38 MAPK. J Biol Chem. 2004;279:26176–26183. doi: 10.1074/jbc.M313346200. [DOI] [PubMed] [Google Scholar]