

Abstract
We recently proposed a model for the IκBα-mediated molecular stripping of NFκB from transcription sites. IκBα was shown experimentally to form a transient ternary complex with DNA-bound NFκB, but the mechanism by which the IκBα accelerates dissociation of the NFκB from the DNA was unknown. In this paper we construct and compute free energy profiles for the wild type IκBα-mediated molecular stripping reaction of NFκB from DNA and compare with that for a mutant of IκBα bearing a charge-neutralized PEST. The differences in the free energy profile for stripping originate from the frustrated electrostatic interactions between the negatively charged PEST and the DNA. The PEST occupies two different conformations in the NFκB-IκBα binary complex, one of which occupies the DNA-binding cavity. Specific interactions with positively charged residues in the N-terminal domains of both p50 and p65 apparently draw the domains closer together hindering re-association of DNA. Comparison with the charge-neutralized mutant reveals that all of these functional consequences result from the negative charges in the PEST sequence of IκBα.
Graphical abstract
Energy landscape of molecular stripping of NFκB from DNA by IκBα
Introduction
Members of the NFκB family of transcription factors are held in an inactive state by their bound inhibitors in the cytoplasm until the cell experiences a stress. The nuclear factor κB (NFκB) signaling system regulates cell growth, immune responses, inflammatory viral responses, and apoptotic death 1–5 and is often misregulated in cancer, arthritis, asthma, diabetes, AIDS, and viral infections 6–8. The NFκB family of transcription factors includes many different homodimers or heterodimers of RelA (p65), p50, p52, c-Rel, and RelB, with the RelA/p50 heterodimer being the most abundant form in many cell types 9. The Rel homology domain is over 300 amino acids, and includes an N-terminal domain (NTD), a dimerization domain, and a nuclear localization signal. The dimerization and N-terminal domains bind to DNA while the dimerization domain and the NLS interact with the inhibitor proteins called, IκBs 10–11. In obtaining crystal structures of the multi-domain NFκBs with either DNA or IκB bound it proved necessary to amputate various parts of the proteins. These truncations made it impossible to develop a full mechanistic picture until computer modeling using a coarse grained energy landscape model made it possible to construct a meaningful model of the over 900 amino acid full-length complex of an NFκB (RelA/p50) heterodimer bound with its inhibitor, IκBα (Fig. 1)12.
Figure 1.
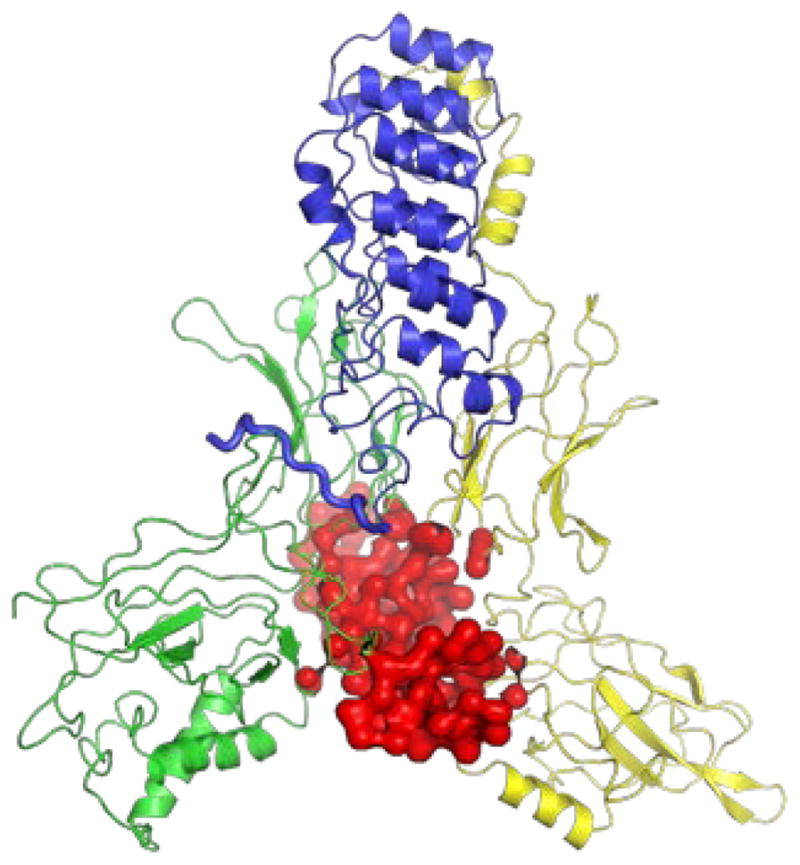
Complete structural model of the NFκB-DNA-IκBα ternary complex 12. The NFκB heterodimer is comprised of p50 (green) and p65 (yellow). The DNA is shown in red and the IκBα in blue. The PEST region is shown in a thicker blue backbone trace line.
The NFκB-IκBα-DNA switch functions in the following way. External signals initiate the degradation of the inhibitors which are already bound to NFκB. This then releases a flood of NFκB molecules. These transcription factors are imported into the nucleus where they turn on hundreds of genes in a temporal pattern set by the duration, timescale and dynamics of the external stimuli. The transcriptional response is then rapidly turned off once the new inhibitor molecules are synthesized, which is initiated when an NFκB molecule binds to an IκBα promoter. Therefore the system presents a classical negative feedback loop which can lead to pulses or oscillations of gene activity 13. Coordinated cessation of NFκB-mediated transcription is desirable due to the large number of activated genes, many of which signal for growth and stress response. In vitro experiments suggest this may be enabled by a kinetic process in which the inhibitor, IκBα, first forms a transient ternary complex with the DNA-bound NFκB and then accelerates the dissociation of the transcription factor from its DNA target 14–15. Such a phenomenon opens up the possibility of molecular kinetic control of binding and release and overturns the classical paradigm of gene regulation based only on thermodynamic control of DNA recognition 16.
We recently proposed a mechanism for this molecular stripping process based on simulations using the Associative memory, Water mediated, Structure and Energy Model (AWSEM). Among other results these studies suggested an important role for the negatively charged PEST sequence at the C-terminus of IκBα in electrostatically repelling the DNA for its release 12. The AWSEM simulations use bioinformatics input based on matching local sequences to sequences for which structures are known. It was natural then to perform these simulations with a model of the PEST sequence based on the crystal structure of the NFκB-IκB complex 10. NMR evidence suggested, however, that the PEST may adopt a wider range of different conformations than is seen in the crystal structure 17. PEST sequences are rich in proline, glutamic acid, serine and threonine, and are predicted to be disordered. Although the C-terminus of IκBα extends to residue 317, previous experimental evidence had suggested that the sequence PESEDEE (residues 281–287) was necessary and sufficient for full NFκB-binding and DNA molecular stripping activity14–15, 18. To more fully explore the role of the PEST sequence in molecular stripping, in this paper we present simulations in which the full distribution of conformers adopted by residues 281–292 in IκBα is explored using the multiple fragment memory approach which takes a somewhat agnostic view of the PEST structure treating it as if there were no direct crystal structure information available 19–20. In addition, we use the same protocol to analyze a charge-neutralized version of the PEST sequence. The results reveal the importance of the negative charges in the PEST in repelling the DNA and reveal a mechanism in which electrostatic frustration between the PEST and DNA accelerates dissociation and then also leads to PEST interaction with specific DNA-binding contacts in NFκB so as to strongly discourage re-association of the DNA to the NFκB-IκBα complex.
Theory and Computational Details
For the interactions within and among the proteins, we carried out enhanced sampling simulations with the AWSEM model20 and modeled the DNA with the 3 Sites Per Nucleotide (3SPN) model21–22. AWSEM is a coarse grained predictive force field whose parametrization is based on the energy landscape formulation of protein folding theory 23. Physically motivated interactions such as hydrogen bonding and solvent interactions are accounted for explicitly in the force field to model tertiary structure relationships in globular proteins. “Evolutionary forces “ are accounted for via bioinformatic terms that act locally in sequence. Both types of interactions cooperate for naturally occurring proteins to sculpt energy landscapes that are highly funneled towards the native state. AWSEM has proven successful in structure prediction of both protein monomers 20 and dimers 24. The 3SPN force field25 is a coarse grained model for DNA that includes physically motivated terms, such as base stacking, base pairing, and electrostatic repulsion of phosphate groups which result in the double helical DNA structure having appropriate thermal and mechanical properties. The range of structural diversity of double helical DNA is well-accounted for by this force field 25. In our simulations the protein and DNA moieties are coupled through steric exclusion and electrostatics which is modeled at the mean field level.
The starting structural models of full-length NFκB bound to DNA, to IκBα, and the NFκB-DNA-IκBα ternary complex were those described previously (Fig. 1) 12. The starting model for the uncharged PEST mutant IκBα was prepared by substituting the charged residues E with Q and D with N, thus effectively neutralizing the PEST sequence residues 281–287 of IκBα. This “PEST-neutralized mutant” was chosen to directly relate to unpublished experimental data. We carried out constant temperature simulations equivalent to more than 100 microseconds for each complex at 300 K which is below the folding temperature of the involved proteins and allows for thorough sampling of the PEST conformations.
To adequately sample states on the DNA dissociation pathway, sampling was enhanced by harmonically biasing the distance between the center of mass of the NFκB binding domains and the DNA in the NFκB-DNA-IκBα complexes, where the DNA orientation was constrained to remain parallel to its orientation in the crystal structure. All of the enhanced sampling simulations were analyzed by weighted histogram analysis method (WHAM) 26 to obtain the 1D free energy profiles. To understand the molecular trajectories, free energies were also mapped onto two-dimensional surfaces calculated from structural coordinates such as the center of mass distance between domains or the end-to-end distances.
Results
Extending the conformational space sampling of the PEST using multiple fragment memories
In our previous study, the energy function governing the structure of the PEST region biased the structure of the PEST towards the structure adopted in the crystal structure 12. To more fully explore the conformations the PEST sequence might adopt in solution, we performed simulations using the multiple fragment memory approach 19–20 and compared the results to simulations using the single fragment memory approach used previously. Figure 2 shows the free energy profiles for DNA dissociation from the NFκB-DNA-IκBα ternary complexes computed with the PEST region modeled with either single memory or with multiple fragment memories. The curves look remarkably similar (Fig. 2). These results gave us confidence that modeling the PEST with multiple fragment memories would yield biologically relevant results. Interestingly, when the DNA dissociates (NFκB-DNA distance of 100 Å), the PEST modeled by the multiple fragment memories apparently adopts a slightly more energetically favorable conformation.
Figure 2.

One dimensional free energy profiles for DNA dissociation from the NFκB-DNA-IκBα ternary complex with the IκBα PEST computed using a single fragment memory (blue) or multiple fragment memories (red).
Conformation of the PEST in the NFκB-IκBα binary complex
In the simulations employing multiple fragment memories the PEST appears to adopt two main conformations, as assessed by the distance between the center of mass of the PEST region and the center of mass of the N-terminal domain of either p50 or p65 (Fig. 3A). One conformation folds back onto the p50 subunit, and is similar to what was found in the single memory simulations. In the other conformation, the PEST extends into the DNA binding cavity and is closer to the p65 (Fig. 4A). In both of these conformations there is a close contact (6 Å) between Arg 52 of the p50 subunit and Glu 282 in the PEST. The folded conformation is farther from p65 than is the extended conformation.
Figure 3.

Two dimensional probability distributions of the PEST-p65 vs the PEST-p50 distance. A) Distribution in the binary complex of wild-type IκBα with NFκB. B) Distribution in the charge-neutralized mutant PEST-containing IκBα with NFκB. C) Distribution in the ternary complex of wild-type IκBα with NFκB and DNA. D) Distribution in the ternary complex of the charge-neutralized mutant PEST-containing IκBα with NFκB and DNA.
Figure 4.

Representative structures of the most probable conformation observed in Fig. 3. All structures in this figure are in the same orientation for ease of comparison. A) Overlay of the two most probable conformations of the PEST in the binary complex between wild type IκBα and NFκB. B) Representative structure of the most probable conformation of the PEST in the binary complex between IκBα with the charge-neutralized mutant PEST and NFκB. C) Overlay of representative structures of the NFκB-DNA-IκBα ternary complexes with wild type IκBα (blue) and with the charge-neutralized mutant of IκBα (purple).
To explore whether the conformations adopted by the PEST were stabilized by electrostatic interactions, we also analyzed simulations with a mutant IκBα in which residues 281–287 (PESEDEE) of IκBα were neutralized to PQSQNQQ. In the binary complex, the PEST-neutralized mutant adopts a broad range of conformations (Fig. 3B) in which the PEST always extends into the DNA-binding cavity (Fig. 4B). Interestingly, in the majority of the conformations, the neutralized PEST was somewhat farther from the p50 subunit, and the distance between Arg 52 of the p50 subunit and Gln 282 in the neutralized PEST was much longer (15 Å). Since all of the observed conformers of the neutralized PEST extended into the DNA-binding cavity, all of them showed the closer distance to both the p50 and p65 subunits.
Conformation of the PEST in the NFκB-DNA-IκBα ternary complex
Simulations of the ternary NFκB-DNA-IκBα ternary complex revealed that the wild type PEST mainly adopted a single conformation (Fig. 3C) with the PEST folded back away from the DNA and interacting more closely with the p50 subunit than with the p65 subunit, (Fig. 4C, blue). Of the several positively charged residues in the p50 NTD, the Arg 55 and 57 provide the major contacts with DNA leaving Lys 75 in the groove between the NTD and the dimerization domain available for interaction with the PEST. The charge-neutralized PEST sequence also adopts a single conformation (Fig. 3D) that is similarly folded-back and extending into the groove between the NTD and the dimerization domain (Fig. 4C, purple).
In an attempt to characterize the two conformations of the PEST, either folded-back on itself, or in the extended form, we measured the end-to-end distance of the PEST in simulations of IκBα alone as well as in simulations of each of the binary and ternary complexes (Fig. 5). The PEST in free wild type IκBα is very extended due to internal electrostatic repulsion of charged residues (solid blue line), while the charge-neutralized PEST in free IκBα is somewhat less extended (dashed cyan line). Consistent with the two main conformations described earlier, two highly probable distances are observed for the PEST in the NFκB-IκBα binary complex with wild type IκBα (solid green line). In contrast, a broad single distribution of end-to-end distances was observed for the charge neutralized PEST in the NFκB-IκBα binary complex. In the NFκB-DNA-IκBα ternary complex, the PEST is folded-back resulting in a shorter end-to-end distance (solid red line). The charge-neutralized PEST adopted a similar folded-back conformation with a similar end-to-end distance (dashed orange line).
Figure 5.

Probability distribution of the end-to-end distance of the PEST region in free IκBα, bound IκBα, and ternary complexes.
Simulations of DNA dissociation from the NFκB-DNA-IκBα ternary complex
To explore the role of the PEST electrostatics in the IκBα-mediated molecular stripping of NFκB from DNA transcription sites, we carried out enhanced sampling simulations of DNA dissociation from the ternary complexes with IκBα bearing the wild type or the charge-neutralized (mutated) PEST. Sampling was enhanced by harmonically biasing the distance between the center of mass of the NFκB binding domains and the DNA and keeping the orientation of the DNA constrained. The free energy profile of the stripping reaction reveals several interesting effects of PEST neutralization (Fig. 6). First, the free energy difference between the bound state (NFκB -DNA distance ~20Å) and the NFκB-IκBα binary complex (NFκB-DNA distance ~100Å) is greater for the neutralized PEST showing that IκBα with the wild type PEST forms a less stable ternary complex than the IκBα with the neutralized PEST. Second, DNA association to the NFκB-IκBα binary complex (reading the free energy profile from right to left) has a barrier for the negatively charged wild type PEST, but is energetically downhill for the charge-neutralized PEST.
Figure 6.

The one dimensional free energy profile for DNA dissociation from the NFκB-DNA-IκBα ternary complex for the wild type PEST (red) as compared to the charge neutralized mutant PEST (grey).The inset shows the same data but with relative free energies measured from the bound state.
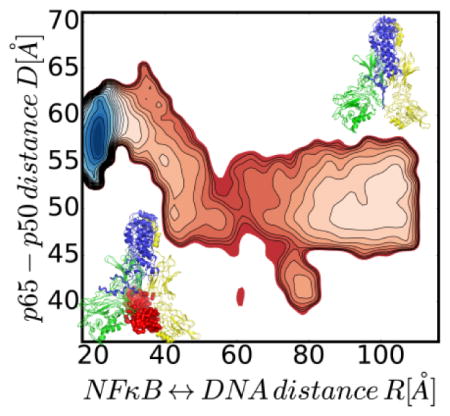
We next generated two dimensional free energy surfaces of DNA dissociation mapped onto the NFκB-DNA and PEST-p65 center of mass distances. These analyses reveal that as molecular stripping occurs, the wild type PEST moves closer to the p65 (Fig. 7A)as compared to p50 (Fig. 7B) and eventually forms distinct clusters which correspond to the NFκB-IκBα binary complex structures shown in Fig. 4A. Similar analyses for the charge-neutralized PEST reveal that it appears to form a relatively stable interaction with the DNA being only partly dissociated (NFκB-DNA distance ~50Å) but forms no specific clusters or stable interactions in the final binary complex( Fig. 7C).
Figure 7.

Two dimensional free energy surface of DNA dissociation mapped onto the NFκB-DNA and PEST-p65 center of mass distances. The wild type PEST moves closer to the p65 and forms distinct clusters (panel A), whereas the mutant PEST is more disordered (panel C). No change was observed in p50 (panels B, D).
Energetic frustration in the NFκB-DNA-IκBα ternary complex
To explore the energy of interaction between the components of the NFκB-DNA-IκBα ternary complex, we took advantage of an algorithm we previously developed for quantifying and visualizing local frustration in biomolecules 27. This algorithm uses the AWSEM Hamiltonians for computing the energetic contributions of contacts of a reference structure relative to the decoy states obtained by residue substitutions in the sequence space. If contacts in the reference structure fall well within the energetically stabilizing region of the decoy energy distribution, they are labeled as minimally frustrated and shown with green dotted lines. If on the other hand the contacts fall well into the energetically destabilizing part of the distribution, then they are labeled as frustrated and shown with red dotted lines. Our previous study, which treated the PEST as a relatively rigid entity, revealed that the NFκB DNA binding cavity is electrostatically frustrated, and binding of either DNA or the negatively charged PEST sequence mitigates this frustration. We further suggested that the ability of the PEST to mitigate the electrostatic frustration contributes to the thermodynamic favorability of molecular stripping 12. In the NFκB-DNA-IκBα ternary complex in which the PEST sequence is folded back away from the DNA as if avoiding electrostatic repulsions, the PEST remains highly frustrated in the presence of DNA (Fig. 8A). Remarkably, for the charge-neutralized PEST, the ternary complex (Fig. 8B) is less frustrated than the binary complex (Fig. 8E). Recalling that the PEST occupies two different conformations in the binary complex, we realized that perhaps the conformation in which the PEST remains folded back against the p50 subdomain may represent the conformation immediately after molecular stripping. We see that this conformation is not frustrated (Fig. 8C). In what we envision is the final binary complex, when the wild type PEST is occupying the DNA-binding cavity, the electrostatic frustration within the cavity is also largely mitigated (Fig. 8D). Even though the charge-neutralized PEST also occupies the DNA-binding cavity, the cavity remains highly frustrated owing to the complete absence of compensating charges(Fig. 8E).
Figure 8.

Local frustration patterns for the PEST-NFκB interactions are highlighted by dimming the rest of the molecule. Highly frustrated interactions are shown in red lines and the arrows indicate the changes in frustration during molecular stripping. A)The wild type IκBα-NFκB-DNA ternary complex. B) The charge -neutralized PEST mutant IκBα-NFκB-DNA ternary complex. C) The alternative conformation of the wild type IκBα-NFκB binary complex with the PEST folded back and interacting with the p50 subunit of NFκB. D) The wild type IκBα-NFκB binary complex with the PEST occupying the DNA-binding cavity of the NFκB. E)The charge-neutralized PEST mutant IκBα-NFκB binary complex with the PEST occupying the DNA-binding cavity of the NFκB.
The wild type PEST sequence prevents DNA rebinding by drawing the N-terminal domains of NFκB close together
To probe domain reorganization during molecular stripping, we also analyzed the change in the center of mass distance between p50 and p65 along the molecular stripping trajectory (Fig. 9). This analysis revealed that the wild type PEST induces significant closure of the distance between p65 and p50 (decreasing from 60 Å down to 40 – 45 Å) whereas this effect was much less pronounced during molecular stripping by IκBα.
Figure 9.

Two dimensional free energy surface of DNA dissociation mapped onto the NFκB-DNA and p65-p50 center of mass distances. The wild type PEST induces significant closure of the distance between p65 and p50 (panel A) whereas this effect is much less in the charge neutralized PEST (panel C).No change was observed for p50 (panels B, D).
Discussion
By comparing the free energy profiles for molecular stripping by wild type IκBα with those obtained for the charge-neutralized PEST-IκBα, we discovered several significant roles played by the electrostatics of the charged PEST sequence in IκBα. First, we see the differences in the free energy profile for stripping (Fig. 6) originate from the frustrated electrostatic interactions between the negatively charged PEST and the DNA. This observation argues that for the wild-type IκBα, electrostatic frustration accelerates the molecular stripping process while for the charge-neutralized PEST, molecular stripping would be less efficient. Conversely, the neutralized PEST stabilizes the ternary complex making the free energy landscape for re-association of DNA to the NFκB-IκBα complex downhill. Thus, charge-neutralization of the PEST is predicted to dramatically impair its ability to remove NFκB transcription factors from their target genesites.
Representative structures of the binary and ternary complexes revealed that the wild type PEST forms specific interactions with the NTD of p50, but also extends into the DNA-binding cavity. The charge-neutralized PEST also extends into the DNA-binding cavity, but does not relieve the frustration of the DNA-binding cavity. In the ternary complexes, the wild type PEST finds an alternative binding site, interacting with a positively charged residue in the NTD of p50 which is not involved in DNA binding. The bound conformation of the PEST is positioned so that its additional negatively charged residues form highly energetically frustrated interactions with the DNA. The frustration analysis reveals that the PEST does not function in a naïve way simply acting to directly repel DNA, but rather it forms specific interactions with the p50 domain of NF κB which fix it in an even more repulsive conformation.
In addition to its positive role in promoting molecular stripping, the wild type PEST also appears to hamper the reverse reaction by preventing DNA re-binding. Upon molecular stripping, the wild type PEST occupies the DNA binding cavity and also interacts with positively charged residues, notably Arg 246, in the p65 NTD. This interaction, which is specifically formed by the wild type PEST residue Glu 287, apparently draws the NTDs of p50 and p65 closer together. This observation explains our previous experimental results that showed that truncation of the PEST even to Glu 285 drastically impaired molecular stripping 14.
Tribute.
We are pleased to dedicate this paper to our wonderful colleague Andy McCammon who first brought us together, and who, with some courage over a long career, reminded biophysicists of the relevance to them of Galileo’s remark, “Eppursi muove”.
Acknowledgments
This work was funded by P01 GM071862 to EAK and PGW and the D.R. Bullard Welch Chair at Rice University, Grant C 00016 (to P.G.W)
ABBREVIATIONS
- NFκB
nuclear factor kappa B
- IκBα
inhibitor of NFκB
- DNA
deoxyribonucleic acid
- NTD
N-terminal domain
Footnotes
Author Contributions: DAP, WZ, DUF, PGW, and EAK planned the work. DAP performed the simulations and wrote some of the manuscript. WZ performed some of the simulations. PGW and EAK wrote the manuscript.
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Hayden MS, Ghosh S. Shared principles in NF-kB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh S, May MJ, Kopp EB. NF-kB and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 3.Martone R, Euskirchen G, Bertone P, Hartman S, Royce TE, Luscombe NM, Rinn JL, Nelson FK, Miller P, Gerstein M, Weissman S, Snyder M. Distribution of NF-kappaB-binding sites across human chromosome 22. Proc Natl Acad Sci U S A. 2003;100(21):12247–12252. doi: 10.1073/pnas.2135255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoffmann A, Natoli G, Ghosh G. Transcriptional regulation via the NF-kB signaling module. Oncogene. 2006;25:6706–6717. doi: 10.1038/sj.onc.1209933. [DOI] [PubMed] [Google Scholar]
- 5.Schreiber J, Jenner RG, Murray HL, Gerber GK, Gifford DK, Young RA. Coordinated binding of NF-kappaB family members in the response of human cells to lipopolysaccharide. Proc Natl Acad Sci U S A. 2006;103(15):5899–5904. doi: 10.1073/pnas.0510996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baldwin AS. Series introduction: the transcription factor NF-kappaB and human disease. J Clin Invest. 2001;107:3–6. doi: 10.1172/JCI11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar A, Takada Y, Boriek AM, Aggarwal BB. Nuclear factor-kappaB: its role in health and disease. J Mol Med. 2004;82:434–448. doi: 10.1007/s00109-004-0555-y. [DOI] [PubMed] [Google Scholar]
- 8.Lee CH, Jeon YT, Kim SH, Song YS. NF-kappaB as a potential molecular target for cancer therapy. Biofactors. 2007;29:19–35. doi: 10.1002/biof.5520290103. [DOI] [PubMed] [Google Scholar]
- 9.Sen R, Baltimore D. Inducibility of k immunoglobulin enhancer-binding protein NF-kB by a posttranslational mechanism. Cell. 1986;47:921–928. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- 10.Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the IkappaBalpha/NF-kappaB complex reveals mechanisms of NF-kappaB inactivation. Cell. 1998;95(6):759–770. doi: 10.1016/s0092-8674(00)81699-2. [DOI] [PubMed] [Google Scholar]
- 11.Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell. 1998;95(6):749–758. doi: 10.1016/s0092-8674(00)81698-0. [DOI] [PubMed] [Google Scholar]
- 12.Potoyan DA, Zheng W, Komives EA, Wolynes PG. Molecular stripping in the NF-κB/IκB/DNA genetic regulatory network. Proc Natl Acad Sci U S A. 2016;113(1):110–115. doi: 10.1073/pnas.1520483112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–1245. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 14.Bergqvist S, Alverdi V, Mengel B, Hoffmann A, Ghosh G, Komives EA. Kinetic enhancement of NF-kB-DNA dissociation by IkBa. Proc Natl Acad Sci U S A. 2009;106(46):19328–19333. doi: 10.1073/pnas.0908797106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alverdi V, Hetrick B, Joseph S, Komives EA. Direct observation of a transient ternary complex during IκBα-mediated dissociation of NF-κB from DNA. Proc Natl Acad Sci U S A. 2014;111(1):225–230. doi: 10.1073/pnas.1318115111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ptashne M. Gene regulation by proteins acting nearby and at a distance. Nature. 1986;322(6081):697–701. doi: 10.1038/322697a0. [DOI] [PubMed] [Google Scholar]
- 17.Sue SC, Dyson HJ. Interaction of the IkappaBalpha C-terminal PEST sequence with NF-kappaB: insights into the inhibition of NF-kappaB DNA binding by IkappaBalpha. J Mol Biol. 2009;388(4):824–838. doi: 10.1016/j.jmb.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergqvist S, Ghosh G, Komives EA. The IκBα/NF-κB complex has two hot spots, on eat either end of the interface. Protein Sci. 2008;17:2051–2058. doi: 10.1110/ps.037481.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hegler JA, Lätzer J, Shehu A, Clementi C, Wolynes PG. Restriction versus guidance in protein structure prediction. Proc Natl Acad Sci U S A. 2009;106(36):15302–15307. doi: 10.1073/pnas.0907002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davtyan A, Schafer NP, Zheng W, Clementi C, Wolynes PG, Papoian GA. AWSEM-MD: protein structure prediction using coarse-grained physical potentials and bioinformatically based local structure biasing. J Phys Chem B. 2012;116(29):8494–8503. doi: 10.1021/jp212541y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinckley DM, Freeman GS, Whitmer JK, de Pablo JJ. An experimentally-informed coarse-grained 3-site-per-nucleotide model of DNA: Structure, thermody-namics, and dynamics of hybridization. J Chem Phys. 2013;139:144903. doi: 10.1063/1.4822042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freeman GS, Hinckley DM, Lequieu JP, Whitmer JK, de Pablo JJ. Coarse-grained modeling of DNA curvature. J Chem Phys. 2014;141(16):165103. doi: 10.1063/1.4897649. [DOI] [PubMed] [Google Scholar]
- 23.Onuchic JN, Luthey-Schulten Z, Wolynes PG. Theory of protein folding: the energy landscape perspective. Ann Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 24.Zheng W, Schafer NP, Davtyan A, Papoian GA, Wolynes PG. Predictive energy landscapes for protein–protein association. Proc Natl Acad Sci U S A. 2012;109:19244–19249. doi: 10.1073/pnas.1216215109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Potoyan DA, Savelyev A, Papoian GAW. Recent successes in coarse-grained modeling of DNA. WIREs Comp Mol Sci. 2013;3:69–83. [Google Scholar]
- 26.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. J Comp Chem. 1992;13(8):1011–1021. [Google Scholar]
- 27.Ferreiro DU, Hegler JA, Komives EA, Wolynes PG. On the role of frustration in the energy landscapes of allosteric proteins. Proc Natl Acad Sci U S A. 2011;108(9):3499–3503. doi: 10.1073/pnas.1018980108. [DOI] [PMC free article] [PubMed] [Google Scholar]