

Abstract
Amyloid-β (Aβ) is a 39–42 residue protein produced by the cleavage of the amyloid precursor protein (APP), which subsequently aggregates to form cross-β amyloid fibrils that are a hallmark of Alzheimer’s disease (AD). The most prominent forms of Aβ are Aβ1–40 and Aβ1–42, which differ by two amino acids (I and A) at the C-terminus. However, Aβ42 is more neurotoxic and essential to the etiology of AD. Here, we present an atomic resolution structure of a monomorphic form of AβM01–42 amyloid fibrils derived from over 500 13C−13C, 13C−15N distance and backbone angle structural constraints obtained from high field magic angle spinning NMR spectra. The structure (PDB ID: 5KK3) shows that the fibril core consists of a dimer of Aβ42 molecules, each containing four β-strands in a S-shaped amyloid fold, and arranged in a manner that generates two hydrophobic cores that are capped at the end of the chain by a salt bridge. The outer surface of the monomers presents hydrophilic side chains to the solvent. The interface between the monomers of the dimer shows clear contacts between M35 of one molecule and L17 and Q15 of the second. Intermolecular 13C−15N constraints demonstrate that the amyloid fibrils are parallel in register. The RMSD of the backbone structure (Q15–A42) is 0.71 ± 0.12 Å and of all heavy atoms is 1.07 ± 0.08 Å. The structure provides a point of departure for the design of drugs that bind to the fibril surface and therefore interfere with secondary nucleation and for other therapeutic approaches to mitigate Aβ42 aggregation.
Graphical abstract
INTRODUCTION
Amyloid fibrils are filamentous structures formed by an extensive menu of peptides and proteins. The molecules vary in length from a few to a few hundred amino acids, and both hydrophobic and hydrophilic residues are present in the protein sequences. These fibrils are of considerable medical importance because they are associated with more than 40 different diseases1 including Parkinson’s disease, type 2 diabetes, dialysis related amyloidosis, Huntington’s disease, prion diseases, and importantly Alzheimer’s disease (AD). At present, 5.4 M Americans are living with AD, and, in addition to the enormous personal cost associated with this devastating disease, there is a projected annual cost for patient care in 2016 of $236 billion.2 There are multiple studies of natural and model peptides that have revealed factors that favor amyloid formation, including the amino acid sequence, and the effective charge and patterning of hydrophobic and hydrophilic groups.3–6 Nevertheless, there is a paucity of basic experimental information about the details of the molecular structures present in amyloids, and therefore there are many fundamental physical and chemical questions regarding the molecular mechanism of amyloid formation, and the nature of the intermolecular interactions that lead to the remarkable stability of these macromolecular structures.
Although amyloid fibrils are microscopically well ordered (vide infra), they are macroscopically disordered and have low solubility. As a consequence, their molecular structures cannot be determined to high resolution with X-ray diffraction or with solution state nuclear magnetic resonance (NMR), the two primary tools of structural biology. Although fiber diffraction does show that fibrils contain extended β-sheets with the β-strands in the sheets oriented perpendicular to the fibril direction, there are many other features of the structure of fibrils that are presently unknown: for example, the protein fold and the orientation of the amino acid side chains and how they are packed into a fibril structure.
In situations such as this, magic angle spinning (MAS) NMR spectroscopy has proven to be a powerful technique to elucidate the atomic resolution structural details.7–9 In particular, MAS NMR has provided information on backbone conformations, supramolecular organization, and registry of interstrand arrangements of amyloid fibrils. This has led to an atomic resolution structure of amyloid fibrils formed by a small peptide derived from transthyretin (TTR105–115), which was determined utilizing a combination of MAS NMR spectra and cryo-electron microscopy.10–12 In addition, similar approaches have been used to determine a partial structure of the prion Het-s and more recently elegant complete structures of E22Δ-Aβ1–40, the Osaka mutant of Aβ1–40,13–17 the protein associated with a familial form of AD, and α-synuclein,18 associated with Parkinson’s disease.
In the case of Aβ, the predominant proteins present in fibrils range from 39 to 43 residues in length in vivo, and are produced from cleavage of the amyloid precursor protein (APP) by β- and γ-secretases.19,20 The most prevalent alloforms are peptides with 40 (Aβ1–40) and 42 (Aβ1–42) amino acid residues, with the latter identified as the more toxic species that possesses a significantly higher aggregation propensity and as a result nucleates fibril formation.21,22 While a great deal of attention has been devoted to modeling structures of Aβ1–40,23–39 very little is known about the structure of Aβ1–42,8 and how it forms reactive surfaces for secondary nucleation, which in turn generate toxic species from monomers in a fibril-catalyzed reaction.40–43 Thus, elucidating the structural details of Aβ1–42 fibrils is an important first step toward understanding this autocatalytic process. Subsequently, detailed atomic resolution structures can guide the rational design of therapeutic tools with which to diagnose and treat AD.
MAS NMR structural studies are based on dipolar recoupling7–9 and require isotopic labeling with 13C and 15N44–47 to measure 13C–13C and 13C–15N distances and torsion angles. Because of its short length (39–42 residues), Aβ labeling can be accomplished using peptide synthesis, an approach used in many previously published investigations. However, peptide synthesis has three significant drawbacks. First, it requires expensive quantities of a 13C/15N amino acid for each position labeled, and consequently only about four or five residues per peptide are labeled in each of the many published studies.24–27,31–38,48 Second, because of the small number of labeled residues, the number of structural constraints available from a specifically labeled Aβ is correspondingly reduced. Thus, in a recent tour-deforce study of Aβ1–42, Xiao et al.48 prepared 17 different labeled Aβ1–42’s, but obtained only 11 and 9 long-range intra- and intermolecular constraints, respectively. Thus, with specific labels it is possible to miss important contacts. For example, Xiao et al.48 did not simultaneously label Q15, L17, and M35 and therefore did not observe cross-peaks corresponding to the intermolecular dimer interface presented here. Third, peptide synthesis does not always produce samples that have the proper chirality and sequence homogeneity. Thus, multiple seeding steps are often required to purify the sample and obtain a single thermodynamically stable conformation.48 As will be seen below, the conformational heterogeneity (or homogeneity) of Aβ is best accessed by looking for multiple cross-peaks in MAS NMR spectra.
In this Article, we describe an atomic resolution structure of AβM01–42 fibrils, based on ∼490 unique 13C−13C and 13C−15N intra- and intermoleclar distance constraints obtained from MAS NMR spectra recorded from samples that are produced recombinantly and 13C labeled using as a carbon source U−13C6-glucose, 1,6−13C2-glucose, 1,3−13C2-glycerol, or 2−13C-glycerol and 15N labeled using 15NH4Cl. Furthermore, the biosynthetic samples can be purified to high homogeneity, and, in contrast to other recent investigations, the fibrils formed do not require repeated seeding steps to obtain a monomorphic sample.49,50 The fibril dimensions and mass-per-length were obtained from scanning tunneling electron microscopy (STEM) measurements.51 The fibril structure is derived by incorporation of the experimental constraints into an energy minimization procedure that reveals the manner in which amyloid fibrils disperse electrostatic and hydrophobic interactions to fold the full-length Aβ protein sequence. The Aβ42 monomer is an S-shaped (or mirror image S-shaped) structure and the fibril subunit is a dimer with 2-fold symmetry, and therefore we observe a single set of well-defined cross-peaks. In addition, there are well-defined intramolecular contacts that determine the fold of the monomer. These include a salt bridge between the A42−13COO− and the K2815NH3+, and contacts between I41–G29, I41–K28, F19–I32, F20–V24, and F19–A30 that form two hydrophobic pockets that define the S-shape. Thus, the salt bridge and these hydrophobic pockets delineate the core of the monomer that consists of residues 15–42. In contrast, the outer surface of the monomers presents to the solvent hydrophilic side chains from K28, S26, D23, and E22 and two hydrophobic patches including residues V18 and A21 and V40 and A42. The first 15 residues M0–14 are dynamic and are observed in TOBSY spectra as we reported previously.49 The remaining salient structural feature is the interface between the two members of the dimer consisting of contacts between M35 on one molecule and Q15 and L17 on the second so that two molecules of Aβ42 are arranged back-to-back as the primary structural unit of the AD fibril. The 13C/15N chemical shift assignments and the structure can be accessed via the BMRB code 30121 and PDB ID 5KK3, respectively.
RESULTS AND DISCUSSION
Obtaining monomorphic amyloid fibrils is an essential condition to generate high-resolution MAS NMR spectra that in turn permit atomic resolution structural characterization. Previously, we have described the preparation of such Aβ42 amyloid fibrils obtained without employing repeated seeding steps that showed a consistent set of chemical shifts from sample to sample.49,50 We initially characterized a U−13C/15N sample, assigning the vast majority of the residues, both backbone and side chains, with secondary chemical shifts predicting four β-strands between residues 16 and 42. We have subsequently prepared additional samples with several biosynthetic labeling schemes. Spectra of these samples permitted us to generate a list of constraints that we used to produce an atomic resolution structure with a heavy atom backbone RMSD of ∼0.7 Å and overall RMSD of ∼1.1 Å discussed below. The presence of only one set of chemical shifts implies that only one conformation is present within mature amyloid fibrils. These spectra could be accounted for by having an amyloid fibril that consists of a single monomer, or possessing symmetry if multiple monomers are present within the mature fibril. As we have stated above, our data are consistent with a dimeric structure that forms the core of the fibril.
NMR Spectroscopy of AβM01–42 Fibrils
Traditionally, assignments and torsion angles are obtained on a uniformly labeled 13C/15N sample, which has the optimal signal-to-noise for a given amount of sample, but cross-peaks observed from such a sample can arise from both intramolecular or intermolecular contacts. The former is essential to determining the structure of the monomer, which in turn is the first step in the structure determination once the spectra are assigned and the location of the β-strands specified. In the case of AβM01–42, the list of contacts observed in the 100% labeled sample does not converge to a single structure, and contains both intra- and intermolecular contacts. To conclusively distinguish intramolecular from intermolecular contacts and in turn generate a monomeric structure, we used a sample consisting of 30% uniformly labeled material and 70% natural abundance material that was formed from separately isolated monomers that were efficiently mixed prior to fibrillization. The isotopic dilution attenuates the contributions from intermolecular contacts, leaving only cross-peaks from intramolecular interactions. We use both the 100% uniformly labeled and the 30% uniformly labeled samples extensively to produce a substantial percentage of the contacts we used in the dimer structure shown below.
Proton-assisted recoupling (PAR)52,53 and dipolar-assisted rotational resonance (DARR)54 are established as the methods of choice for observing 13C−13C correlations corresponding to long distance constraints. In our previous publication,49 we reported DARR spectra for a sample of U−13C/15N-AβM01–42, from which we obtained a large number of contacts between amino acid residues distant from one another. More recently, we recorded 13C−13C PAR spectra and observed many additional structural restraints that are reported herein.
In particular, we illustrate in Figure 1 the spectrum obtained with a PAR52,53 experiment using τmix = 20 ms from the 100% U−13C/15N sample, and Figure S1 shows the spectrum obtained from the 30% labeled sample. The excellent resolution present in both of these spectra allowed us to extract a total of 239 sequential, medium, and long-range distance constraints for the 100% labeled sample and 111 sequential, medium, and long-range distance constraints for the 30% labeled sample. Some of the contacts important for determining the fold of the monomer structure are between F19–I32, F19–A30, F20–V24, V24–G29, I31–V36, G33–V36, G29–I41, and K28–A42, while important intermolecular contacts that specify the structure of the Aβ42 dimer are between L17–M35 and Q15–M35. Many of the cross-peaks that correspond to these contacts are visible in the expanded portion of the spectrum shown at the top of Figure 1. The effect of reducing the concentration of uniformly labeled monomers to 30% on a PAR spectrum is shown in Figure 2. It shows slices from the 20 ms PAR spectrum of a 100% labeled sample (see Figure 1) and from the 20 ms PAR spectrum of a 30% labeled sample (Figure S1). In the 100% labeled sample, M35Cε shows 13 contacts to Q15, L17, and L34, while 10 of these contacts are absent in the 30% labeled sample. Hence, these 10 “disappearing” contacts between M35 and Q15, L17, and L34 can be classified as intermolecular.
Figure 1.
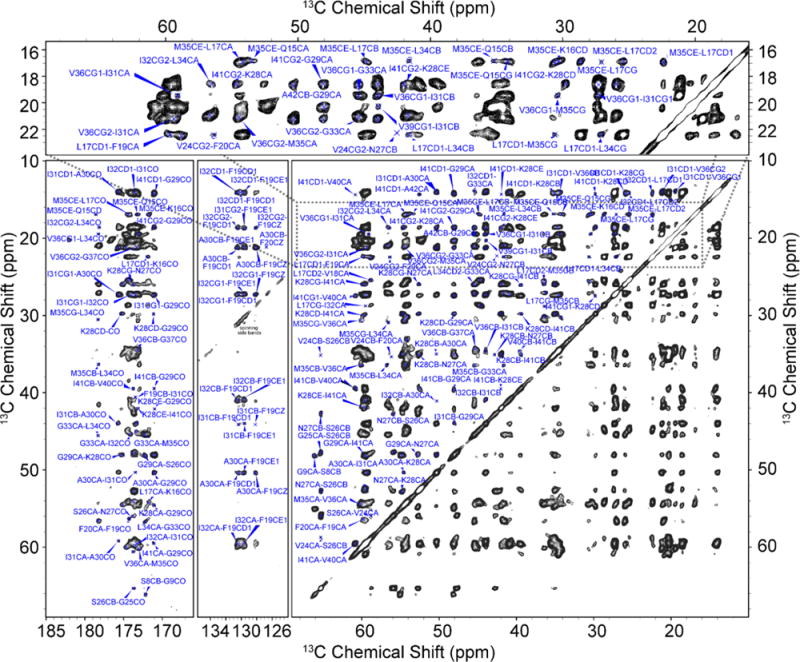
2D 13C−13C MAS PAR spectrum of U−13C/15N-AβM01–42 fibrils recorded at ω0H/2π = 800 MHz, T = 277 K, ωr/2π = 20 kHz. τmix = 20 ms, and ω1H/2π = 83 kHz decoupling field. For optimal PAR mixing, the radio frequency (RF) fields were set to ω1C/2π = 62.5 kHz and ω1H/2π = 55 kHz on the 13C and 1H channels, respectively. Several important inter-residue cross-peaks are denoted with red labels in the expanded region of the spectrum. The inset shows several important intermolecular contacts including Q15–M35 and L17–M35, while the main panel shows numerous intramolecular contacts used in calculating the structure, including F19–I32, F19–A30, V24–F20, V24–G29, I41–G29, and K28–A42.
Figure 2.
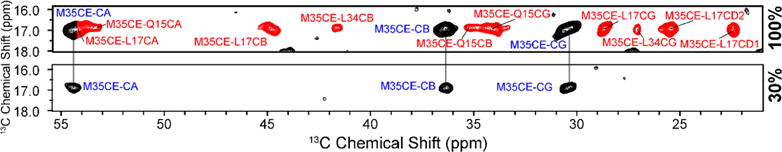
Slices from two 20 ms PAR spectra illustrating the presence of intermolecular contacts at the interface between the two members of the Aβ42 dimer. The top slice shows a total of 13 inter- and intramolecular cross-peaks involving M35CE from the 100% U−13C/15N labeled sample. By diluting the sample to 30% with natural abundance material (lower slice), 10 of these cross-peaks, shown in red in the top slice and assigned to contacts between M35CE and Q15 and L17, are no longer present, confirming that they are intermolecular in origin.
In addition, we recorded a PAIN55 spectrum illustrated in Figure 3a, which revealed long-range 13C−15N contacts and provided information about the backbone to side chain interactions. Some particularly important cross-peaks correspond to F20–G25, V18–L34, L17–L34, G29–I41, V24–A30, and I31–V36. A complete list of these contacts is provided in Table S1.
Figure 3.
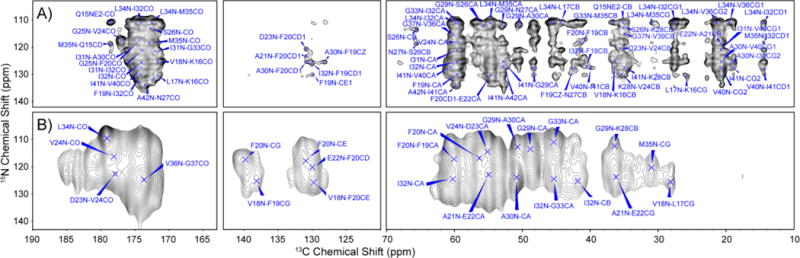
(A) 2D 13C−15N MAS PAIN spectrum of U−13C/15N-AβM01–42 fibrils recorded at ω0H/2π = 750 MHz, T = 277 K, ωr/2π = 20 kHz, τmix = 30 ms, with ω1H/2π = 83 kHz 1H decoupling field. Particularly relevant intramolecular contacts include F20–G25, G29–I41, V24–A30, and I31–V36, and intermolecular contacts include V18–L34 and L17–L34. (B) 2D 13C−15N ZF-TEDOR (τmix = 16 ms) spectrum of 2−13C1-glycerol/15N mixed sample recorded at 600 MHz, ωr/2π = 12.5 kHz, VT gas regulated to 105 K with 83 kHz TPPM during acquisition. The cross-peaks observed in this spectrum confirm that the fibrils are PIR. A total of 24 cross-peaks are observed, with the most relevant cross-peaks observed being I32N–I32CA, F20N–F20CA, G29N–G29CA, G33N–G33CA, L34N–L34CO, V24N–V24CO, and V36N–G37CO.
Although there are substantial differences in published models and structures of Aβ amyloid fibrils, including those of Aβ40, Aβ42, and the Osaka mutant E22Δ-Aβ39, one commonality that is largely shared is the presence of a salt bridge between the K28NζH3+ and a carboxylic acid group (an exception to this statement is ref 56). In the case of Aβ40, the salt bridge was assigned to be paired with D23Cγ,24,27,34,35 and in E22Δ-Aβ3913,14 the bridge connects K28 and E3Cδ. Although it may be an indirect correlation, we find it interesting that of the fibril structures reported to date, the ones obtained for peptides that are more toxic as compared to Aβ40 have the salt bridge located in different regions. The main driving force for fibril formation are interactions involving hydrophobic groups, the structure is governed by constraints imposed by the detailed sequence, and salt bridges are formed if compatible with the rest of the structure. Recently, Ishii’s group reported observation of a salt bridge formed between K28 and the C-terminus of A42, and speculated that this interaction may account for some of the pronounced differences in the aggregation rates between Aβ40 and Aβ42.48 Given the very similar chemical shifts between the fibrils we prepared and those prepared by Ishii’s group, we looked for a similar salt bridge. In particular, we recorded FS-REDOR57 build-up curves for both the 100% uniformly labeled sample and also the 30% uniformly labeled sample (Figure 4).
Figure 4.
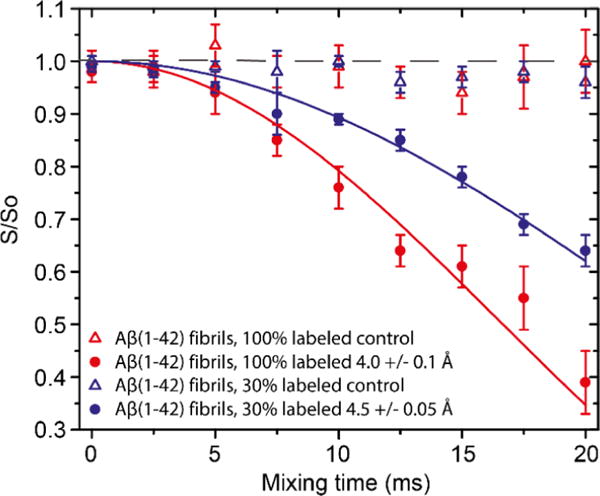
FS-REDOR of AβM01–42 fibrils recorded at 750 MHz, T = 277 K, and ωr/2π = 8 kHz with ω1H/2π = 83 kHz 1H decoupling field applied during acquisition. The Gaussian selective π pulse on 13C was 0.6 ms long and set on the resonance of A42−13COO−. For 15N, ω1S/2π = 33 kHz during REDOR and set to the resonance of the Nζ of K28. The S and S0 signals were measured with and without the 15N selective pulse, respectively. The curve fits show that a salt bridge exists between K28 and A42 with a distance of 4.0 Å in the 100% labeled sample and 4.5 Å in the 30% sample. Intermolecular contacts between the PIR fibrils are responsible for the discrepancy in the dephasing observed.
We note that the K2815Nζ resonance is well separated and can be easily selectively excited. On the other hand, the 13C Gaussian pulse excites not only the A4213CO but also its neighboring resonances. The dephasing curve of these resonances, which actually showed no dephasing, was conveniently used as a control. The dephasing curves of A4213CO were simulated using an analytic approach described elsewhere57,58 with a scaling factor of 0.9 to account for the imperfections of the pulse sequence and hardware. We obtained A4213CO to K2815Nζ distances of 4.0 and 4.5 Å on the samples with 100% [U−13C,15N]AβM01−42 and 30% [U−13C,15N]AβM01−42, respectively. The FS-REDOR57 experiment confirms that a salt bridge exists between K28−15NζH3+ and the 13COO− of A42. We attribute the discrepancy in the two distances to the fact that the intermolecular contacts in the 30% sample are attenuated and can be neglected. On the other hand, contributions from these contacts in the 100% [U−13C,15N]AβM01–42 sample lead to a more rapid dephasing, and thus a shorter apparent distance. Therefore, for the structure calculation, we used 4.5 Å as the intramolecular distance between A4213CO and K2815Nζ, implying a relatively weak interaction or an exchange between free groups and salt-bridged groups.
To determine the intermolecular registry of molecules within the fibrils, we utilized a mixed sample, consisting of 50% 2−13C-glycerol labeled protein where the Cα positions are preferentially labeled with 13C and 50% 15N labeled material. Thus, a 13C−15N correlation experiment (PAIN55 or ZF-TEDOR59) will exhibit 13Cα−15NH cross-peaks exclusively from intermolecular contacts. If these cross-peaks correspond to the same positions as found in U−13C/15N spectra, then the molecules are arranged in a parallel-in-register (PIR) array. This strategy has been successfully employed in a variety of systems, PI3-SH3, β2m, ΔN6, etc.,60–62 all of which show PIR β-strands. However, the ∼4.5 Å 13Cα−15NH distance requires τmix ≈ 15–20 ms, and dynamic processes lead to significant attenuation of signal intensities. Thus, the experiment is most successful at low temperatures (∼100 K) where many dynamic processes are frozen out, yielding more intense cross-peaks in the ZF-TEDOR59 spectrum. The temperature leads to broader spectra particularly in the 15N dimension, yet as shown in Figure 3b several intense cross-peaks are observed that are entirely intermolecular in origin. Assignment of these cross-peaks dictates that the interstrand arrangement in AβM01–42 is PIR.
To further probe for unambiguous intermolecular constraints, we prepared a 2−13C-glycerol/1,3−13C2-glycerol mixed sample (Figure S4), which should provide us with intermolecular contacts and aid in generating a global structure. Unlike the 2-glycerol sample/15N mixed sample, not all cross-peaks are intermolecular, but rather have unambiguous intramolecular contacts, ambiguous intramolecular or intermolecular contacts, and finally unambiguous intermolecular contacts. The unambiguous intermolecular contacts can further be broken into ones that report about interstrand arrangement (i.e., PIR), while others report about intermolecular contacts between adjacent monomers. We observe several additional contacts, which are consistent with a parallel in register arrangement including S8Cα−Cβ, G9Cα−S8Cβ, L17Cδ1−Cγ, L17Cα−Cγ, G25Cα–V24CO, S26Cα−Cβ, G29Cα−CO, A30Cα−G29CO, A30Cα−I31Cγ1, A30CO−I31Cγ1, I32Cα−Cβ, G33Cα−CO, G33Cα−L34Cα, V36Cα−G37Cα, V40Cγ2−Cβ, and I41Cβ−V40CO. In contrast to the ZF-TEDOR59 experiment that requires cryogenic temperatures for the long mixing times necessary to observe long distances, the PAR experiment performs well at 277 K, enabling us to retain the excellent spectral resolution and obtain well-resolved cross-peaks.
A common technique used in structural characterization of samples by MAS NMR is sparse 13C labeling of samples, which yields narrower line widths by eliminating dipolar and secular coupling from adjacent 13C atoms. Additionally, it minimizes dipolar truncation from homonuclear dipolar couplings, resulting in improved efficiency for dipolar recoupling for inter-residue contacts. Accordingly, we prepared a sample using 1,6−13C2-glucose, which typically labeled –CH3 groups. 13CH3−13CH3 contacts can be very helpful in elucidating structures as they are likely to provide long distance information. Accordingly, we recorded a 13C−13C 200 ms DARR spectrum, along with 12 ms mixing ZF-TEDOR55 spectra (shown in Figure S3), and observed several contacts from these spectra, but importantly I31–N27 contacts. Figure 5 serves to summarize the 487 distance constraints obtained from the various dipolar recoupling experiments that were used to calculate the structure of Aβ42.
Figure 5.
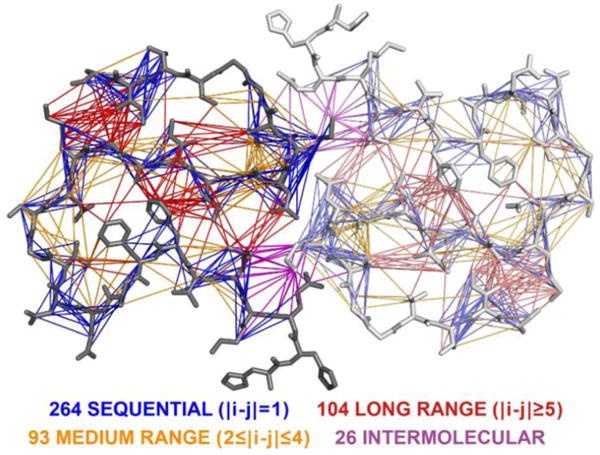
Schematic representation of unique constraints used for structure calculation. There were a total of 487 unique distance constraints of which 264 were sequential contacts, 93 medium range, 104 long-range, and 26 intermolecular distance constraints.
Mass per Unit Length STEM Measurements
Although MAS dipolar recoupling NMR experiments provide detailed atomic resolution structural data (0.1 Å), they are limited to a length scale <10 Å. It is therefore necessary to rely on STEM and cryoEM measurements to assemble the atomic resolution NMR structures into the cryo EM electron density and a composite fibril with dimensions up to 2000 Å. This is the procedure followed in our recent study of TTR105–115.11,12 The initial step in this process is to perform STEM measurements of the mass per unit length (MPL) that determines the number of AβM01–42 molecules present in the fibrils. Figure 6 (left) illustrates a typical STEM micrograph that shows the two varieties of fibrils present in our samples, a dominant component with an MPL of 4.880 kDa/Å and a second with 2.474 kDa/Å. The number distribution of these fibril types is shown on the right where we have fitted Gaussian curves to the distributions that are centered at MPL values above with widths of 273 and 449 kDa/nm, respectively. The vertical lines indicate the MPL expected for 2–7 molecules/fibril. Using an interstrand spacing of 4.5 Å, measured with 13C−13C dipole recoupling experiments on TTR105–11563 and a molecular weight for U−13C/15N-AβM01–42 of 4909.2 Da, we find that the peaks in the MPL curves correspond to 2.26 and 4.47 molecules/fibril. Thus, the STEM measurements are consistent with the dimeric structure that emerges from the NMR structure calculations and for a tetrameric fibril with 2-fold symmetry. The latter corresponds to a fibril in which two filaments wind around each other, as seen by cryo-EM,40,64,65 and each filament contains multiple planes of the dimeric structure determined here. If we use the 4.8 Å for the β-strand spacing, then we obtain 2.41 and 4.38 for the number of molecules/fibril. Note that the STEM measurements were performed on samples that were used previously for the MAS NMR spectra, so that we are sure that they correspond to monomorphic fibrils.
Figure 6.
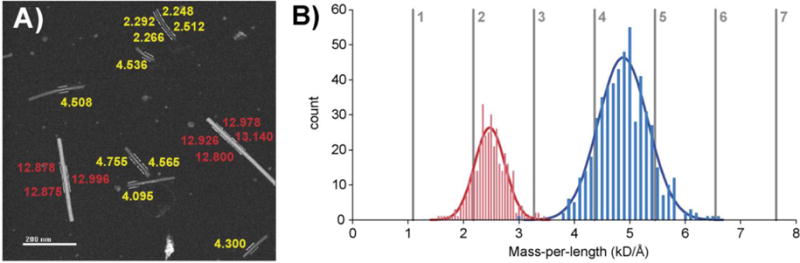
(A) STEM micrograph showing the ∼2.5 kDa/Å and ∼4.5 kDa/Å fibrils present in the AβM01–42 samples. The numbers adjacent to the particle indicate the molecular weight in kD observed for that segment. In addition, there are two particles present that are the standard TMV (13.1 kDa/Å) used in STEM measurements. Note that the length of the AβM01–42 fibrils is ∼50–200 nm, which is shorter than found for Aβ1–40. In other micrographs (Figure S7), we observe similar fibril masses and lengths. (B) Distribution of fibril masses determined from the STEM measurements on 894 different segments and Gaussian fits to the distributions. The distributions are centered at 2.474 and 4.880 kDa/Å with widths of 0.273 and 0.449 kDa/Å, respectively. The lower and higher molecular weights we associate with dimeric and tetrameric fibrils, respectively. The vertical lines indicate the theoretical MPL for integer numbers of molecules of MW = 4909.2 Da, which is weight expected for U-13C/15N-AβM01–42.
Examination of higher magnification STEM images showed bright spots adjacent to many fibers but not near the TMV standard. The images with the least salt had smaller SD for both TMV and fibers and a fiber M/L closer to 2.3 kDa/Å. Therefore, we conclude that phosphate buffer (used in preparation of the fibrils) was causing the higher M/L value. This is a well-known effect observed in previous studies using phosphate.66
Structure Determination
We assigned a total of 1866 peaks from a total of 12 spectra, including PAR,52,53 DARR,54 PAIN,55 TEDOR,59 and FS-REDOR57 of differently labeled samples (see Table S1). From those, 870 were intraresidue contacts, 615 sequential, 119 medium range, 220 long-range, and 42 intermolecular constraints as illustrated in Figure 5. For each cross-peak in the 12 spectra, we set an upper limit of visibility (in Å); that is, we chose a distance that is certain to satisfy each contact we observe in the particular spectrum. We chose the final distance for each constraint based on the spectra in which it was observed and then chose the one with the lowest upper limit (see Table S2). With this procedure we arrived at a list of 487 unique distance constraints, of which 264 were sequential contacts, 93 medium range, 104 long-range, and 26 intermolecular distance constraints. Additionally, we used 28 dihedral angle constraints for residues Q15–V18, S26–K28, A30–I32, and V39–A42. Dihedral angles for these residues are involved in β-strand formation as determined by secondary chemical shift (see Figure S6). For these residues, we also included 13 hydrogen bonds per monomer. Because the spectra only show one set of peaks with sharp lines, the fibril is expected to be a quasi-crystal. Hence, the backbone heavy atoms were set to be between 4.5 and 5.1 Å. We used CYANA 3.9767 to select the 10 lowest energy structures from 500 calculations that converged to a heavy atom backbone RMSD of 0.71 ± 0.12 Å and an RMSD of 1.07 ± 0.08 Å for all heavy atoms (Table 1).
Table 1.
Structural Statistics
| Conformational Constraints | |
|---|---|
| distance constraints | 487 |
| intra residual | 0 |
| sequential (|i − j| = 1) | 264 |
| medium range (2 ≤ |i − j| ≤ 4) | 93 |
| long range (|i − j| ≥ 5) | 104 |
| intermolecular | 26 |
| hydrogen bonds | 13 |
| dihedral angle restraints (ϕ/ψ) | 28 |
| Restraint Violations | |
|---|---|
| CYANA target function value (Å2) | 1.81 ± 0.4 |
| RMS distance restraint violation (Å) | 0.0035 ± 0.0007 |
| maximal distance restraint violation (Å) | 0.0048 |
| RMS dihedral angle restraint violation (deg) | 0.075 ± 0.037 |
| maximal dihedral angle restraint violation (deg) | 0.146 |
| RMSD of the Central Dimer | |
|---|---|
| heavy atom backhone (Q15–A42) (Å) | 0.71 ± 0.12 |
| all heavy atoms {Q15–A42) (Å) | 1.07 ± 0.08 |
The resulting structure illustrated in Figure 7A–C is a dimer formed from two S-shaped monomers with most hydrophobic residues hidden in the fibril core. Notably, we observe two distinct hydrophobic cores, one containing residues I31, V36, V39, and I41, and the other containing L17, F19, F20, V24, A30, and I32. The latter is bridged by M35 and L34 of both monomers to form one continuous hydrophobic interior across the monomer interface. Moreover, the stacking of monomer pairs along the fibril means the hydrophobic cores will be repeated along the fibril axis and run as continuous hydrophobic cores inside the fibril. Meanwhile, the hydrophilic residues, K16, E22, D23, S26, and K28, are exposed to the solvent and form the main turn/loop between the three main β-strands. We note that the local RMSD is slightly elevated around E22 and D23 (see Figure S9). While it is not clear if this is caused by missing restraints, this could also be caused by a strain around this loop region. It is interesting to note that numerous familial mutations related to early onset Alzheimer’s disease68–70 are found within this loop region, again, particularly surrounding E22 and D23. The interface between the two monomers appears to serve to hide the hydrophobic residues that are present in the other main turn present in these fibrils, L34, M35, and V36, which have intermolecular contacts with V18, L17, K16, and Q15. The first 14 residues are more dynamic than the residues present within the fibril core, which we have reported previously.49 These first 14 residues possess mainly hydrophilic residues and are likely disordered and immersed in the solvent. We note that while several of these residues are observed by CP-based experiments, S8, G9, and Y10, no contacts are observed between these residues and any residues within the fibril core. Similar to the results published by Ishii et al., we observe a salt bridge between K28 and the C-terminus of A42. This feature is absent in Aβ40 fibrils where instead K28 pairs with D23 (see above); this variation results from the absence of V41–A42 and the very different hydrophobic core packing in Aβ40 relative to Aβ42 fibrils. Matching K28 with A42 enables E22 and D23 to face the solvent, as discussed above.
Figure 7.
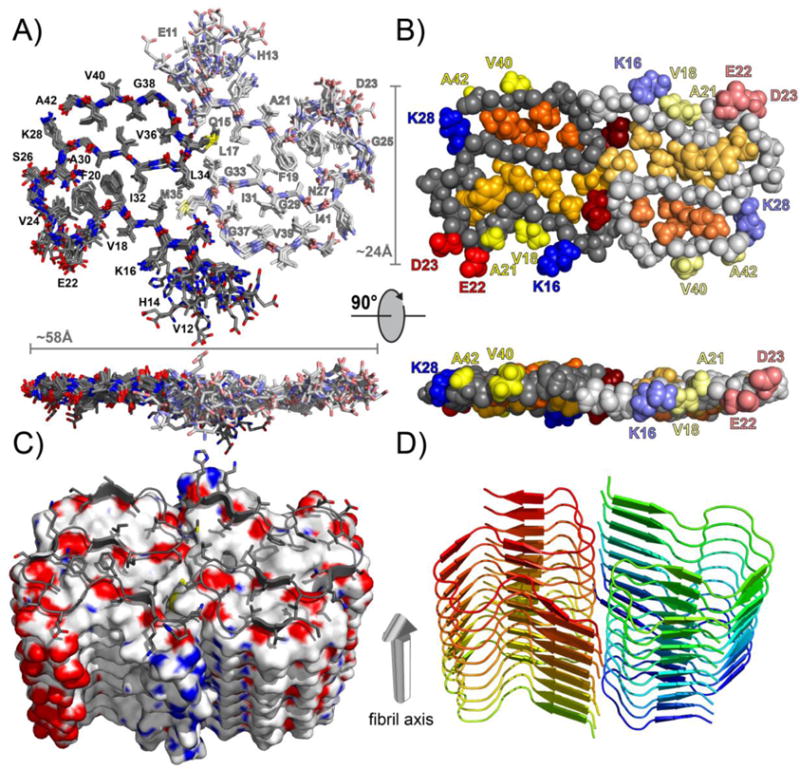
(A) Stick model representation of the 10 lowest energy structures. Shown is the central dimer; one monomer is in bright and one in pale colors. The structures were aligned using the backbone heavy atoms of residues Q15–A42. The structures converged to a heavy atom backbone RMSD of 0.77 ± 0.17 Å and an RMSD of 1.11 ± 0.14 Å for all heavy atoms. (B) CPK model showing the two center monomers from the top (top image) and from the side of a fibril (bottom image). One monomer is in bright and one in pale colors. Only residues Q15–A42 are shown. The hydrophobic side chain of one buried cluster (I31, V36, V39, I41) is shown in orange, and of the other buried cluster (L17, F19, F20, V24, A30, I32, L34) in gold. M35 is shown in dark red. E22 and D23 are shown in red and K16 and K28 in blue. Hydrophobic side chains facing the solvent are shown in yellow (V18, A21 and V40, A42). (C) Surface representation of the lowest energy structure. Carbon atoms are shown in gray, oxygen atoms in red, nitrogen atoms in blue, and sulfur atoms in yellow. Only residues Q15–A42 are shown. (D) Ribbon representation of the lowest energy structure showing the alignment of the dimers along the fibril axis. Only residues Q15–A42 are shown. All figures were generated using the PyMOL software package.
Our structure predicts the intermolecular contacts between L34 and L′34 on different monomers within the dimer. We note that glycerol efficiently labels leucine. In the 2−13C-glycerol/1,3−13C2-glycerol 13C−13C mixed sample, we observe very strong contacts between all atoms within the side chain of L34; however, contacts between the CO and the side chain are absent. Because of the labeling scheme employed along with the fact that leucine is efficiently selectively labeled using this labeling scheme, the contacts observed are conclusively deemed to be intermolecular. However, a question remains whether these contacts are between monomers within the same plane of the dimer, or if they are parallel in register contacts between β-strands above or below one another. We suspect that a combination of both is likely, the latter of which is supported by the absence of cross-peaks between L34N and the side chain atoms within the 2−13C-glycerol/15N mixed sample (Figure 3b).
Finally, we note that the structure shown in Figure 7 is a dimer in contrast to that reported by Xiao et al.,48 who reported a monomer. In particular, the structure is consistent with our STEM data discussed above and with the observation of cross-peaks between M35 and Q15 and L17 (Figure 2) corresponding to the interface. As pointed out above, these cross-peaks were not present in the spectra of Xiao et al. because none of the 17 samples they examine simultaneously contained 13C/15N labels at the three crucial residues Q15, L17, and M35.
Molecular Determinants of Fibril Formation
Although several intermediate forms such as nuclei, oligomers, and protofibrils of various sizes, forms, and structures may exist, the fact that thermodynamic properties are independent of path allows us to discuss the driving forces for fibril formation based on the structures of the end states: the fibril structure presented here and the largely unstructured monomers.65 Twelve hydrophobic side chains (L17, F19, F20, V24, A30, I31, I32, L34, M35, V36, V39, and I41) are largely exposed to solvent in the unstructured monomer and buried in the fibril. This suggests that the self-assembly reaction is governed by the hydrophobic effect and increased entropy of water upon desolvation.71 This is a very strong driving force for self-assembly, which overcomes the loss in entropy upon packing of monomers into fibrils, which limits the number of configurations, and the stronger electrostatic repulsion between negatively charged monomers in the fibrils as compared to monomers. The repulsion in the fibril is diminished through ion pairing (e.g., the C-terminal carboxyl to K28 side chain) and charge modulation,72 yet the free energy difference between intramolecular salt bridges and the free side chains interacting with water may be small. The specificity of the fibril fold, on the other hand, may be governed by neat packing of hydrophobic side chains and avoidance of steric clashes.73,74 Ion pairing and repulsive electrostatic interactions may contribute to the specificity and disfavor alternative structures in which the repulsion would be higher.73–75
Comparison with Other Aβ Alloforms
Multiple structural models for Aβ40 amyloid fibrils have been reported by several groups.26,36–38,76–78 While there are subtle differences among these models, they do share some features. These include the presence of only a single turn and extended β-strands, which include residues 10–40, with the first 10 residues either being observed by J-based transfer experiments or invisible to both CP and J-based transfer experiments. It has been reported that the first 10 residues of the N-terminus are dynamic and are either observed only with J-based transfers or are “invisible” to both J-based and CP-based transfers. In contrast, Reif and co-workers have reported that they have seeded an Aβ40 sample and attained a single form, which possessed two sets of chemical shifts, leading them to conclude that the basic subunit for these fibrils is an asymmetric dimer.23 This dimer has a single set of resonances visible from 12 to 40, while the other is from 21 to 38. In contrast, the Osaka mutant, which has 39 residues, has several turns. This amyloid fibril was reported to have all residues incorporated into the fibril core, with the N-terminus forming contacts with the central part of the peptide.
The structural differences between Aβ40 and Aβ42 may hold valuable insights into why the two alloforms have such different aggregation kinetics. Using detailed kinetic analyses, it was recently found that all three underlying microscopic steps (primary nucleation, secondary nucleation, and elongation) have lower rate constants for Aβ40 as compared to Aβ42.43 While the large difference in primary nucleation rate may be explained by Aβ42 monomers being more hydrophobic than Aβ40 monomers, the latter two steps are fibril dependent, and their higher rate in the case of Aβ42 may at least in part be explained by distinct features of the fibril structures. One striking difference between the models proposed for Aβ40 and structure of the Aβ42 fibrils is found in the exposed hydrophobic patches that can be inferred to run along the sides of the fibrils (Figure 7). The Aβ42 structure presented here reveals one hydrophobic groove composed of V18 and A21, which is gated by E22 and K16, and one hydrophobic patch composed of V40 and A42. The former groove is seen in several Aβ40 models,31 but the latter patch is a unique feature for Aβ42 fibrils. While A42 is absent in Aβ40, V40 is buried in several Aβ40 models. Although the catalytic surface for secondary nucleation can in principle be anywhere on the fibril, it is likely to involve a major fraction of the fibril surface. The reaction order for secondary nucleation with respect to monomer is two,79 and it is likely that the nucleus contains at least two monomers and has at least similar dimensions as the dimer shown here. It is therefore possible that the presence of this additional hydrophobic stretch (V40, A42) that lines the Aβ42 fibrils can explain at least in part its higher rate constant for secondary nucleation as compared to Aβ40. Moreover, in mixtures, Aβ40 and Aβ42 fail to form mixed fibrils; they interact only at the level of primary nucleation, while the fibril-specific processes are highly specific events.80 The two residues I41–A42 cannot be accommodated in Aβ40 structures for steric reasons, as there is no room to fit two more residues next to V40. However, removal of the same residues from the Aβ42 fibril structure presented here would lead to very significant destabilization because one of the hydrophobic cores would be disrupted by removal of V41.
We note that two sites for several of the familial mutations, E22 and D23, are located in a loop region, and exposed to the surrounding solution, while A21 is part of one hydrophobic groove, and D7 and A2 are located in the flexible N-terminus. The structure presented here may serve as a starting point for modeling of mutant fibrils81 and interpretation of changes in aggregation properties due to familial and designed mutations.69,82,83 It may also serve as a starting point for designing additional substitutions to study the molecular determinants of surface-catalyzed secondary nucleation.64 The structure presented here may also shed some light on the A21C/A30C-Aβ42 variant, in which the introduced cysteines form a disulfide bond within the monomer, which is compatible with protofibril but not fibril formation.84 In the Aβ42 fibril model, the A30 side chain is located in one of the hydrophobic cores, while the A21 side chain is exposed in one hydrophobic groove on the surface; thus a cross-link between positions 21 and 30 would not be possible.
The chemical shifts of the assigned residues we reported recently,85 and those simultaneously reported by Ishii86 (and the very recent chemical shifts of Ravotti et al.87), are essentially identical, which suggests that the Xiao et al. model of the monomer and the structure reported here are highly similar. Our structure has similar structure elements and long-range contacts, including F19–I32, K28–A42, etc., which are all present in our spectra. However, we also observe numerous additional contacts and cross-peaks, allowing us to calculate a structure to superior resolution. The model of Xiao et al. had 11 long-range contacts, and 2 intermolecular contacts,48 while we have 105 long-range contacts, and 15 intermolecular contacts. We note the similarity between the two structures shown in Figure S9, which is in contrast to the multitude of Aβ40 amyloid fibril models and literature that abounds with discussions of polymorphism. Thus, it appears that the structure presented in Figure 7 may represent the thermodynamically most stable form given that two groups have independently produced nearly identical fibrils by different methods (nonseeding vs repeated seeding steps), using material prepared differently (recombinant vs peptide synthesized). Moreover, the variation between the two studies in terms of pH, peptide concentration, and salt concentration suggests that the Aβ42 fibril structure is relatively invariant to changes in solution conditions.
In addition, we note that even though we have an N-terminal methionine (M0) present in our samples, we still observe chemical shifts essentially identical to those of Aβ without M0. This together with the fact that we find that residues M0–14 are dynamic indicate that the presence of this residue does not affect our structural results. As a further confirmation of this fact, we have prepared Aβ1–42 via a SUMO expression system and find the spectra essentially identical to those reported here.88
Finally, there are several structural studies of Aβ42 employing cryo-EM to characterize the Aβ42 amyloid fibrils.89–91 While this work was performed carefully, it appears difficult to reconcile the structure reported here, or the model of Xiao et al., with the published electron density profiles. Furthermore, no MAS NMR spectra of the samples were reported, so it is not possible to determine if the samples were mono- or polymorphic. Thus, at the moment we do not have a satisfactory explanation for differences between the cryoEM and the MAS NMR results. In addition, a structure of Aβ42 was reported92 based on hydrogen exchange and mutation experiments combined with the structural model for Aβ40 proposed by Tycko and co-workers. This structure is very different from that reported here.
CONCLUSIONS
We have utilized a variety of MAS NMR dipolar recoupling sequences to generate ∼490 intermolecular and intramolecular contacts for Aβ42 amyloid fibrils, which yielded an atomic resolution structure. The monomer structure possesses an S-shape with residues 15–42 comprising the amyloid fibril core and residues 1–14 appearing to be unstructured. We observe intermolecular contacts between Q15, L17, L34, and M35, which imply that the basic subunit of the mature fibrils exists as a dimer, consistent with data from STEM experiments. The structure has a heavy atom backbone RMSD of ∼0.7 Å. We find that two hydrophobic clusters are buried in the interior of the fibril, while two hydrophobic patches are exposed on the outside and line the fibrils surface. One of these contains V18 and A21, and the other V40 and A42. The structure presented here provides a valuable resource for understanding the aggregation of Aβ42 and its variants, and will serve as a starting point for the design of molecules with high affinity for the surface of the fibril and the potential to interfere with secondary nucleation. The structure may thus enhance our understanding of molecular events underlying Alzheimer’s disease and improve the possibilities for therapeutic invention.
EXPERIMENTAL SECTION
Sample Preparation
The biosynthetic 13C/15N labeling and preparation of Aβ42 fibrils were described in detail previously.49 As in our previous investigation,49 we utilized a 100% uniformly 13C/15N labeled sample to obtain intermolecular and intramolecular constraints. To detect −13CH3’s interacting with other sites, we prepared a sample with 1,6−13C2-glucose93,94 as the 13C source, which sparsely labels mostly −13CH3 groups.94 To differentiate intermolecular from intramolecular contacts, we used a sample that consisted of 30% U−13C/15N material in a matrix of 70% natural abundance material. The peptides were first separately purified and isolated as monomers that were mixed in a 3:7 molar ratio on ice and subsequently allowed to fibrillize by incubating the mixed solution at room temperature for at least 16 h, as described previously.49 Similarly, we prepared a sample consisting of 50% carbon labeled material (derived from growth with 2-glycerol) and 50% 15N labeled material. For this sample, we expressed Aβ42 in M9 media, with 2 g/L of 2−13C-glycerol substituted for glucose, and separately prepared material grown from M9 media containing natural abundance glucose and 15NH4Cl. Finally, we employed a similar process to produce a 13C 2−13C-glycerol/1,3−13C2-glycerol mixed sample, which was used in PAR experiments to detect intermolecular contacts for example between M35 and Q15 and L17. All samples were lyophilized and rehydrated followed by ultra-centrifugation to pellet the fibrils and subsequently packed into a rotor. Previously, we reported that lyophilization of 13C/15N and rehydration does not alter the chemical shifts of the MAS NMR spectra.49 Typically, ∼20–30 mg of hydrated AβM01–42 fibrils was packed into a 3.2 mm Bruker rotor (Bruker Biospin, Billerica, MA) using a home-built centrifugal packing tool.
MAS NMR Experiments
13C−13C and 13C−15N spectra were acquired on a Bruker 800 MHz AVANCE III spectrometer equipped with a 3.2 mm triple channel HCN Bruker probe (Bruker Biospin, Billerica, MA). For the DARR,54 RFDR,95,96 and ZF-TEDOR59 experiments, we used ωr/2π = 20 ± 0.01 kHz and T = 277 K. FS-REDOR57 spectra were recorded at ω0H/2π = 750 MHz ωr/2π = 8 ± 0.01 kHz and T = 277 K. Spectra recorded at ω0H/2π = 800 and 750 MHz were processed using TopSpin 3.1 and the NMRPipe software package,97 respectively, and all spectra were analyzed in Sparky.98 The ZF-TEDOR59 spectra used to establish the parallel-in-register arrangement of the fibrils were recorded at 105 K on a Bruker 600 MHz AVANCE III spectrometer equipped with a triple channel HCN Bruker probe at ωr/2π = 12.5 ± 0.01 kHz.
2D DARR spectra were recorded using τCP99 = 1.2 ms, ω1/2π = 62.5 kHz, τmix = 80 ms, ω1H/2π = 20 kHz, and a recycle delay of 2.5 s. During acquisition, the TPPM 1H decoupling field was ω1H/2π = 83.3 kHz. The FID matrix containing 2048 × 1024 points was averaged over 16 transients. Subsequently, a squared sine window function with a sine bell shift of 3.5 was used, and the FID matrix was zero filled to 4096 × 2048 points and Fourier transformed.
Ambient temperature Z-filtered transferred echo double resonance (ZF-TEDOR)59 was acquired using τmix = 1.6 ms, 13C and 15N π-pulses with ω1/2π = 50 kHz, ω1H/2π = 83 kHz TPPM decoupling during acquisition, and a 3 s recycle delay. The FID matrix of 1024 × 512 points was averaged for 32 scans.
Measurements of the distance between A4213COO− and K2815Nζ were performed using the frequency selective REDOR (FS-REDOR)57 sequence on a Cambridge Instruments NMR spectrometer (courtesy of Dr. D.J. Ruben) operating at ω0H/2π = 750 MHz. The experiments used a 1H/13C/15N 3.2 mm E-free probe (Bruker BioSpin, Billerica, MA) and ωr/2π = 8 kHz. During cross-polarization, ω1H/2π = 50 kHz and ω1H/2π = 83 kHz otherwise. The 180° selective Gaussian pulse on 13C was τ180 = 0.6 ms long and set on resonance for A42 13COO−. On the 15N channel, the selective Gaussian pulse of τ180 = 1.25 ms, selectively inverting the K2815Nζ, sandwiched between two REDOR periods consisting of rotor synchronized 15N π pulses. The RF power level on 15N was ω1S/2π = 33 kHz during REDOR. The S and S0 signals were measured with and without the 15N selective pulse, respectively.
Low temperature ZF-TEDOR59 spectra were acquired using τmix = 16 ms mixing with ω1S/2π = 50 kHz 13C and 15N π-pulses and ω1H/2π = 125 kHz decoupling during evolution and ω1H/2π = 83 kHz 1H TPPM decoupling during acquisition and a 3 s recycle delay.
The PAR spectrum shown in Figure 1 was acquired with 2048 complex data points in the direct dimension and 1048 complex data points in the indirect dimension using the protocol of Ruben and coworkers.100 Prior to indirect evolution and PAR mixing, transverse 13C magnetization was prepared using CP99,101 with τmix = 1.2 ms. For optimal PAR mixing, the radio frequency (RF) fields were set to ω1C/2π = 62.5 kHz and ω1H/2π = 55 kHz to optimize transfer to side-chain resonances according to a thorough and rigorous search of available PAR conditions.
The PAIN55 spectrum shown in Figure 3 was acquired with 1024 complex data points in the direct dimension and 512 data points in the indirect dimension. Prior to indirect evolution and PAIN mixing, transverse 15N magnetization was prepared using CP from the 1H channel with τmix = 1 ms contact time before the indirect evolution period. The PAIN RF fields for the 13C and 15N channels were chosen to be ω1/2π = 50 kHz, and for the 1H channel ω1/2π = 55 kHz.
Structure Calculation
Structure calculations were performed using CYANA 3.9767 as previously described for other fibril structure determinations with some variations.14,68 The first 10 residues expected to be dynamic were omitted for the structure calculation. The structure was set up to be comprised of 14 monomers linked by LL2 linkers with the following scheme, (Aβ11–42–30×LL2-)6-Aβ11–42–50×LL2-(Aβ11–42–30×LL2-)6-Aβ11–42. Dihedral angles for residues involved in β-strand formation as determined by secondary chemical shift were set to −200.0° ≤ ϕ ≤ −80.0° and 40.0° ≤ ψ ≤ 220.0° (i.e., residues 15–18, 26–28, 30–32, and 39–42). Additionally, hydrogen bonds were defined for these residues using H–O distance restraints of 1.8–2.0 Å and N–O distance restraints of 2.7–3.0 Å. Hydrogen restraints were weighted 10 times as compared to other constraints. To reflect the semicrystalline nature of the fibril, the backbone heavy atoms along the fibril axis were kept between 4.5 and 5.1 Å for residues 15–42. These restraints were weighted 10 times as compared to other constraints. The salt bridge between K28 and A42 was defined as follows: the distance of K28Nζ and A42CO was set to be between 4.0 and 4.7 Å, and the distance between K28Nζ and A42O was set to be below 4.0 Å. Salt bridge restraints were weighted 10 times as compared to other constraints. Sequential, medium range, and long-range contacts were used as intramolecular restraints. Used spectra and distance constraints can be found in Table S2. As a constraint, the distance of the spectrum was used in which the specific contact was observed. Intermolecular constraints were implemented as constraints between two monomers on the same level along the fibril axis. 500 conformers were generated with 35 000 torsion angle dynamics steps for each conformer. The final structure bundle is comprised of the 10 conformers with the lowest CYANA target function.
Supplementary Material
Acknowledgments
The research was supported by grants from the National Institutes of Biomedical Imaging and Bioengineering (EB-003151, EB-002026, and EB-002804) to R.G.G. and by the Swedish Research Council (VR) and a European Research Council (ERC) Advanced Grant to S.L. R.S. is funded by a DFG research fellowship (SI2105/1-1). We thank Salima Bahri for discussion and proof reading.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b05129.
Additional spectra utilized in the structure calculation, along with tables detailing the constraints obtained (PDF)
Notes
The authors declare no competing financial interest.
NOTE ADDED IN PROOF
After this manuscript was accepted we learned of another paper (ref 102) describing the structure of Aβ1–42 fibrils formed at pH=7.4 in the presence of 100 mM NaCl and 100 μM Zn2+. The structure is essentially identical to the structure reported here in the ordered region (residues 15–42) and shows disorder in the N-terminal tail (residues 1–14).
References
- 1.Chiti F, Dobson C. Annu Rev Biochem. 2006;75:333. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Alzheimer’s Assn. 2016 http://www.alz.org/facts/overview.asp.
- 3.Lopez, De La Paz M, Goldie K, Zurdo J, Lacroix E, Dobson CM, Hoenger A, Serrano L. Proc Natl Acad Sci U S A. 2002;99:16052. doi: 10.1073/pnas.252340199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tartaglia G, Vendruscolo M. Chem Soc Rev. 2008;37:1395. doi: 10.1039/b706784b. [DOI] [PubMed] [Google Scholar]
- 5.Goldschmidt L, Teng P, Riek R, Eisenberg D. Proc Natl Acad Sci U S A. 2010;107:3487. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ventura S, Zurdo J, Narayanan S, Parreno M, Mangues R, Reif B, Chiti F, Giannoni E, Dobson CM, Aviles FX, Serrano L. Proc Natl Acad Sci U S A. 2004;101:7258. doi: 10.1073/pnas.0308249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Griffin RG. Nat Struct Biol. 1998;5:508. doi: 10.1038/749. [DOI] [PubMed] [Google Scholar]
- 8.Lansbury PT, Costa PR, Griffiths JM, Simon EJ, Auger M, Halverson KJ, Kocisko DA, Hendsch ZS, Ashburn TT, Spencer RGS, Tidor B, Griffin RG. Nat Struct Biol. 1995;2:990. doi: 10.1038/nsb1195-990. [DOI] [PubMed] [Google Scholar]
- 9.Su Y, Andreas L, Griffin RG. Annu Rev Biochem. 2015;35:1. doi: 10.1146/annurev-biochem-060614-034206. [DOI] [PubMed] [Google Scholar]
- 10.Jaroniec CP, MacPhee CE, Bajaj VS, McMahon MT, Dobson CM, Griffin RG. Proc Natl Acad Sci U S A. 2004;101:711. doi: 10.1073/pnas.0304849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Debelouchina GT, Bayro MJ, Fitzpatrick AW, Ladizhansky V, Colvin MT, Caporini MA, Jaroniec CP, Bajaj VS, Rosay M, MacPhee CE, Vendruscolo M, Maas WE, Dobson CM, Griffin RG. J Am Chem Soc. 2013;135:19237. doi: 10.1021/ja409050a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fitzpatrick AWP, Debelouchina GT, Bayro MJ, Clare DK, Caporini MA, Bajaj VS, Jaroniec CP, Wang LC, Ladizhansky V, Muller SA, MacPhee CE, Waudby CA, Mott HR, De Simone A, Knowles TPJ, Saibil HR, Vendruscolo M, Orlova EV, Griffin RG, Dobson CM. Proc Natl Acad Sci U S A. 2013;110:5468. doi: 10.1073/pnas.1219476110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huber M, Ovchinnikova OY, Schutz AK, Glockshuber R, Meier BH, Bockmann A. Biomol NMR Assignments. 2015;9:7. doi: 10.1007/s12104-013-9535-x. [DOI] [PubMed] [Google Scholar]
- 14.Schutz AK, Vagt T, Huber M, Ovchinnikova OY, Cadalbert R, Wall J, Guntert P, Bockmann A, Glockshuber R, Meier BH. Angew Chem, Int Ed. 2015;54:331. doi: 10.1002/anie.201408598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siemer AB, Ritter C, Ernst M, Riek R, Meier BH. Angew Chem, Int Ed. 2005;44:2441. doi: 10.1002/anie.200462952. [DOI] [PubMed] [Google Scholar]
- 16.Van Melckebeke H, Wasmer C, Lange A, AB E, Loquet A, Bockmann A, Meier BH. J Am Chem Soc. 2010;132:13765. doi: 10.1021/ja104213j. [DOI] [PubMed] [Google Scholar]
- 17.Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH. Science. 2008;319:1523. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 18.Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, Wan W, Stubbs G, Schwieters CD, Lee VMY, George JM, Rienstra CM. Nat Struct Mol Biol. 2016;23:409–415. doi: 10.1038/nsmb.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Nature. 1998;391:387. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 20.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller L, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Science. 1999;286:735. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 21.Jarrett JT, Berger EP, Lansbury PT. Ann N Y Acad Sci. 1993;695:144. doi: 10.1111/j.1749-6632.1993.tb23043.x. [DOI] [PubMed] [Google Scholar]
- 22.Berger EP, Jarrett JT, Lansbury PT. J Am Chem Soc. 1993;115:76. [Google Scholar]
- 23.Amo JMLd, Schmidt M, Fink U, Dasari M, Fandrich M, Reif B. Angew Chem, Int Ed. 2012;51:6136. doi: 10.1002/anie.201200965. [DOI] [PubMed] [Google Scholar]
- 24.Antzutkin ON, Balbach JJ, Leapman RD, Rizzo NW, Reed J, Tycko R. Proc Natl Acad Sci U S A. 2000;97:13045. doi: 10.1073/pnas.230315097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antzutkin ON, Balbach JJ, Tycko R. Biophys J. 2003;84:3326. doi: 10.1016/S0006-3495(03)70057-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antzutkin ON, Leapman RD, Balbach JJ, Tycko R. Biochemistry. 2002;41:15436. doi: 10.1021/bi0204185. [DOI] [PubMed] [Google Scholar]
- 27.Balbach JJ, Petkova AT, Oyler NA, Antzutkin ON, Gordon DJ, Meredith SC, Tycko R. Biophys J. 2002;83:1205. doi: 10.1016/S0006-3495(02)75244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bertini I, Gonnelli L, Luchinat C, Mao JF, Nesi A. J Am Chem Soc. 2011;133:16013. doi: 10.1021/ja2035859. [DOI] [PubMed] [Google Scholar]
- 29.Dasari M, Espargaro A, Sabate R, del Amo JML, Fink U, Grelle G, Bieschke J, Ventura S, Reif B. ChemBioChem. 2011;12:407. doi: 10.1002/cbic.201000602. [DOI] [PubMed] [Google Scholar]
- 30.del Amo JML, Schneider D, Loquet A, Lange A, Reif B. J Biomol NMR. 2013;56:359. doi: 10.1007/s10858-013-9755-5. [DOI] [PubMed] [Google Scholar]
- 31.Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC. Cell. 2013;154:1257. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paravastu AK, Leapman RD, Yau WM, Tycko R. Proc Natl Acad Sci U S A. 2008;105:18349. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paravastu AK, Qahwash I, Leapman RD, Meredith SC, Tycko R. Proc Natl Acad Sci U S A. 2009;106:7443. doi: 10.1073/pnas.0812033106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. Proc Natl Acad Sci U S A. 2002;99:16742. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petkova AT, Ishii Y, Tycko R. Biophys J. 2002;82:320. [Google Scholar]
- 36.Petkova aT, Leapman RD, Guo ZH, Yau WM, Mattson MP, Tycko R. Science. 2005;307:262. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 37.Petkova AT, Leapman RD, Yau WM, Tycko R. Biophys J. 2004;86:506. [Google Scholar]
- 38.Petkova aT, Yau WM, Tycko R. Biochemistry. 2006;45:498. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tycko R, Ishii Y. J Am Chem Soc. 2003;125:6606. doi: 10.1021/ja0342042. [DOI] [PubMed] [Google Scholar]
- 40.Arosio P, Cukalevski R, Frohm B, Knowles TPJ, Linse S. J Am Chem Soc. 2014;136:219. doi: 10.1021/ja408765u. [DOI] [PubMed] [Google Scholar]
- 41.Cohen SIA, Arosio P, Presto J, Kurudenkandy FR, Biverstål H, Dolfe L, Dunning C, Yang X, Frohm B, Vendruscolo M, Johansson J, Dobson CM, Fisahn A, Knowles TPJ, Linse S. Nat Struct Mol Biol. 2015;22:207. doi: 10.1038/nsmb.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen SIA, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TPJ. Proc Natl Acad Sci U S A. 2013;110:9758. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meisl G, Yang XT, Hellstrand E, Frohm B, Kirkegaard JB, Cohen SIA, Dobson CM, Linse S, Knowles TPJ. Proc Natl Acad Sci U S A. 2014;111:9384. doi: 10.1073/pnas.1401564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hong M. J Magn Reson. 1999;139:389. doi: 10.1006/jmre.1999.1805. [DOI] [PubMed] [Google Scholar]
- 45.Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein K, Oschkinat H. Nature. 2002;420:98. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- 46.Pauli J, Baldus M, Rossum Bv, de Groot H, Oschkinat H. ChemBioChem. 2001;2:272. doi: 10.1002/1439-7633(20010401)2:4<272::AID-CBIC272>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 47.LeMaster DM, Kushlan DM. J Am Chem Soc. 1996;118:9255. [Google Scholar]
- 48.Xiao YL, Ma BY, McElheny D, Parthasarathy S, Long F, Hoshi M, Nussinov R, Ishii Y. Nat Struct Mol Biol. 2015;22:499. doi: 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Colvin MT, Silvers R, Frohm B, Su Y, Linse S, Griffin RG. J Am Chem Soc. 2015;137:7509. doi: 10.1021/jacs.5b03997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walsh DM, Thulin E, Minogue A, Gustavsson N, Pang E, Teplow D, Linse S. FEBS J. 2009;276:1266. doi: 10.1111/j.1742-4658.2008.06862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wall JS, Hainfeld JF. Annu Rev Biophys Biophys Chem. 1986;15:355. doi: 10.1146/annurev.bb.15.060186.002035. [DOI] [PubMed] [Google Scholar]
- 52.De Paepe G, Lewandowski JR, Loquet A, Bockmann A, Griffin RG. J Chem Phys. 2008;129 doi: 10.1063/1.3036928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lewandowski JR, De Paepe G, Eddy MT, Struppe J, Maas W, Griffin RG. J Phys Chem B. 2009;113:9062. doi: 10.1021/jp810280t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takegoshi K, Nakamura S, Terao T. Chem Phys Lett. 2001;344:631. [Google Scholar]
- 55.Lewandowski JR, De Paepe G, Griffin RG. J Am Chem Soc. 2007;129:728. doi: 10.1021/ja0650394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tay WM, Huang DT, Rosenberry TL, Paravastu AK. J Mol Biol. 2013;425:2494. doi: 10.1016/j.jmb.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jaroniec CP, Tounge BA, Herzfeld J, Griffin RG. J Am Chem Soc. 2001;123:3507. doi: 10.1021/ja003266e. [DOI] [PubMed] [Google Scholar]
- 58.Mueller KT. J Magn Reson, Ser A. 1995;113:81. [Google Scholar]
- 59.Jaroniec CP, Filip C, Griffin RG. J Am Chem Soc. 2002;124:10728. doi: 10.1021/ja026385y. [DOI] [PubMed] [Google Scholar]
- 60.Bayro MJ, Debelouchina GT, Eddy MT, Birkett NR, MacPhee CE, Rosay M, Maas WE, Dobson CM, Griffin RG. J Am Chem Soc. 2011;133:13967. doi: 10.1021/ja203756x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Debelouchina GT, Platt GW, Bayro MJ, Radford SE, Griffin RG. J Am Chem Soc. 2010;132:17077. doi: 10.1021/ja107987f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Su YC, Sarell CJ, Eddy MT, Debelouchina GT, Andreas LB, Pashley CL, Radford SE, Griffin RG. J Am Chem Soc. 2014;136:6313. doi: 10.1021/ja4126092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caporini MA, Bajaj VS, Veshtort M, Fitzpatrick A, MacPhee CE, Vendruscolo M, Dobson CM, Griffin RG. J Phys Chem B. 2010;114:13555. doi: 10.1021/jp106675h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cohen SI, Arosio P, Presto J, Kurudenkandy FR, Biverstal H, Dolfe L, Dunning C, Yang X, Frohm B, Vendruscolo M, Johansson J, Dobson CM, Fisahn A, Knowles TP, Linse S. Nat Struct Mol Biol. 2015;22:207. doi: 10.1038/nsmb.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cukalevski R, Yang X, Meisl G, Weininger U, Bernfur K, Birgitta, Frohm, Knowles TPJ, Linse S. Chem Sci. 2015;6:4215. doi: 10.1039/c4sc02517b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wall JS, Simon MN, Lin BY, Vinogradov SN. In: Globins and Other Nitric Oxide-Reactive Proteins. Pt A, Poole RK, editors. Vol. 436. Elsevier Academic Press Inc; San Diego, CA: 2008. p. 487. [Google Scholar]
- 67.Güntert P, Mumenthaler C, Wüthrich K. J Mol Biol. 1997;273:283. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 68.Huber M, Ovchinnikova OY, Schutz AK, Glockshuber R, Meier BH, Bockmann A. Biomol NMR Assignments. 2015;9:7. doi: 10.1007/s12104-013-9535-x. [DOI] [PubMed] [Google Scholar]
- 69.Basun H, Almkvist O, Axelman K, Wahlund LO, Lannfelt L. Neurobiol Aging. 2002;23:S154. doi: 10.1007/978-3-7091-6139-5_12. [DOI] [PubMed] [Google Scholar]
- 70.Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Naslund J, Lannfelt L. Nat Neurosci. 2001;4:887. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 71.Berne B, Weeks J, Zhou R. Annu Rev Phys Chem. 2009;60:85. doi: 10.1146/annurev.physchem.58.032806.104445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lund M, Jonsson B. Q Rev Biophys. 2013;46:265. doi: 10.1017/S003358351300005X. [DOI] [PubMed] [Google Scholar]
- 73.Eisenberg D, Jucker M. Cell. 2012;148:1188. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Linse S, Voorhies M, Norstrom E, Schultz D. J Mol Biol. 2000;296:473. doi: 10.1006/jmbi.1999.3452. [DOI] [PubMed] [Google Scholar]
- 75.Kurnik M, Hedberg L, Danielsson J, Oliveberg M. Proc Natl Acad Sci U S A. 2012;109:5705. doi: 10.1073/pnas.1118640109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Balbach JJ, Petkova AT, Oyler NA, Antzutkin ON, Gordon DJ, Meredith SC, Tycko R. Biophys J. 2002;83:1205. doi: 10.1016/S0006-3495(02)75244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. Proc Natl Acad Sci U S A. 2002;99:16742. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bateman DA, Tycko R, Wickner RB. Biophys J. 2011;101:2485. doi: 10.1016/j.bpj.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TP. Proc Natl Acad Sci U S A. 2013;110:9758. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cukalevski R, Yang XT, Meisl G, Weininger U, Bernfur K, Frohm B, Knowles TPJ, Linse S. Chem Sci. 2015;6:4215. doi: 10.1039/c4sc02517b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schledorn M, Meier BH, Bockmann A. Frontiers in molecular biosciences. 2015;2:14. doi: 10.3389/fmolb.2015.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Meisl G, Kirkegaard JB, Arosio P, Michaels TCT, Vendruscolo M, Dobson CM, Linse S, Knowles TPJ. Nat Protoc. 2016;11:252. doi: 10.1038/nprot.2016.010. [DOI] [PubMed] [Google Scholar]
- 83.Cukalevski R, Yang X, Cohen SI, Boland B, Frohm B, Thulin E, WalsXh DM, Knowles TP, Linse S. J Neurochem. 2013;125:235. [Google Scholar]
- 84.Lendel C, Bjerring M, Dubnovitsky A, Kelly R, Filippov A, Antzutkin O, Nielsen N, Hard T. Angew Chem, Int Ed. 2014;53:12756. doi: 10.1002/anie.201406357. [DOI] [PubMed] [Google Scholar]
- 85.Colvin MT, Silvers R, Frohm B, Su Y, Linse S, Griffin RG. J Am Chem Soc. 2015;137:7509. doi: 10.1021/jacs.5b03997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xiao Y, Ma B, McElheny D, Parthasarathy S, Long F, Hoshi M, Nussinov R, Ishii Y. Nat Struct Mol Biol. 2015;22:499. doi: 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ravotti F, Wälti MA, Güntert P, Riek R, Böckmann A, Meier BH. Biomol NMR Assignments. 2016 doi: 10.1007/s12104-016-9682-y. [DOI] [PubMed] [Google Scholar]
- 88.Colvin, M. T.; Fredrick, K. K.; Jacavone, A. C.; Lindquist, S. L.; Linse, S.; Griffin, R. G., in prep.
- 89.Schmidt M, Rohou A, Lasker K, Yadav JK, Schiene-Fischer C, Fandrich M, Grigorieff N. Proc Natl Acad Sci U S A. 2015;112:11858. doi: 10.1073/pnas.1503455112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Grigorieff N, Ceska TA, Downing KH, Baldwin JM, Henderson R. J Mol Biol. 1996;259:393. doi: 10.1006/jmbi.1996.0328. [DOI] [PubMed] [Google Scholar]
- 91.Fandrich M, Schmidt M, Grigorieff N. Trends Biochem Sci. 2011;36:338. doi: 10.1016/j.tibs.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Doeli H, Schubert D, Riek R. Proc Natl Acad Sci U S A. 2005;102:17342. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Eddy MT, Andreas L, Teijido O, Su YC, Clark L, Noskov SY, Wagner G, Rostovtseva TK, Griffin RG. Biochemistry. 2015;54:994. doi: 10.1021/bi501260r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Korzhnev DM, Religa TL, Lundstrom P, Fersht AR, Kay LE. J Mol Biol. 2007;372:497. doi: 10.1016/j.jmb.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 95.Bennett AE, Ok JH, Griffin RG, Vega S. J Chem Phys. 1992;96:8624. [Google Scholar]
- 96.Bennett AE, Rienstra CM, Griffiths JM, Zhen WG, Lansbury PT, Griffin RG. J Chem Phys. 1998;108:9463. [Google Scholar]
- 97.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 98.Goddard, T. D.; Kneller, D. G. University of California, San Francisco.
- 99.Pines A, Gibby MG, Waugh JS. J Chem Phys. 1973;59:569. [Google Scholar]
- 100.States DJ, Haberkorn RA, Ruben DJ. J Magn Reson (1969–1992) 1982;48:286. [Google Scholar]
- 101.Hartmann SR, Hahn EL. Phys Rev. 1962;128:2042. [Google Scholar]
- 102.Wälti MA, et al. Proc Natl Acad Sci USA. in press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.