

Summary
Cytomegalovirus (HCMV) contains cholesterol, but how HCMV interacts with host cholesterol metabolism is unknown. We found that in human fibroblasts HCMV infection increased efflux of cellular cholesterol despite reducing the abundance of ABCA1. Mechanistically, viral protein US28 was acting through CDC42, rearranging actin microfilaments, causing association of actin with lipid rafts and leading to a dramatic change in abundance and/or structure of lipid rafts. These changes displaced ABCA1 from cell surface, but created new binding sites for apolipoprotein A-I resulting in enhanced cholesterol efflux. The changes also reduced inflammatory response in macrophages. HCMV infection modified host lipidome profile and expression of several genes and microRNAs involved in cholesterol metabolism. In mice, murine CMV infection elevated plasma triglycerides, but did not affect level and functionality of high-density lipoprotein. Thus, HCMV, through its protein US28, reorganizes lipid rafts and disturbs cell cholesterol metabolism.
Blurb
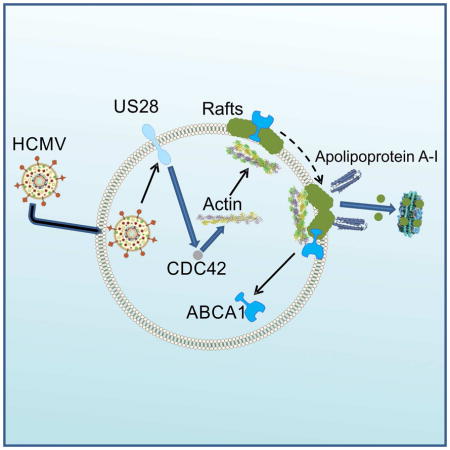
Low et al find that HCMV, through viral protein US28 and cellular Cdc42, rearranges actin microfilaments and modifies lipid rafts to create new binding sites for apolipoprotein A-I on the host plasma membrane. This results in enhanced cellular cholesterol efflux and a reduction in the host inflammatory response.
Introduction
Cytomegalovirus (HCMV) infection is one of the most prevalent viral infections in humans with up to 75% of population carrying the virus. In its latent form, HCMV infection is thought to be largely asymptomatic with only a few viral transcripts expressed. However, when reactivated, HCMV may cause severe disease in immunocompromised individuals. Acute HCMV infection affects many tissues including viral effects on the vascular endothelium leading to vascular dysfunction, bleeding and thrombosis. Whether or not latent HCMV infection contributes to any morbidity is unclear and considering the high prevalence of the infection, causative relationship is difficult to ascertain by traditional epidemiological methods. A number of previous studies have suggested that latent HCMV infection contributes to the risk of atherothrombosis (Zhu et al., 1999) and cancer (Soderberg-Naucler et al., 2013). Given that cellular and systemic cholesterol metabolism plays a central role in the pathogenesis of atherosclerosis and was implicated in pathogenesis of several malignancies (Zhuang et al., 2005), it is possible that a connection between HCMV infection and associated diseases involves an interaction between HCMV and host cholesterol metabolism.
Pathogens of different taxa interact with host cholesterol metabolism (Sviridov and Bukrinsky, 2014). A common purpose of this interaction is, on the one hand, to facilitate formation of plasma membrane lipid rafts or intracellular raft-like membrane structures, which are used by pathogens as entry gates, assembly platforms and budding sites and require the presence of abundant cholesterol. On the other hand, many pathogens disrupt or modify rafts in order to reduce exposure to the immune system which interacts with infected cells via rafts (Triantafilou et al., 2002). HCMV is an enveloped virus and its virions contain host derived cholesterol (Gudleski-O’Regan et al., 2012), but very little is known about the interaction of HCMV with host cholesterol metabolism. Infection of endothelial and epithelial cells and macrophages by HCMV requires receptor mediated endocytosis, a process that is dependent on cholesterol (Haspot et al., 2012; Ryckman et al., 2006), and inhibition of cholesterol biosynthesis in human endothelial cells restrained HCMV replication (Potena et al., 2004). HCMV infection was associated with increased expression of low density lipoprotein related protein (LRP1); silencing of LRP1 concomitantly increased cellular cholesterol content and cholesterol content, yield and infectivity of the virus (Gudleski-O’Regan et al., 2012). HCMV infected cells also have reduced abundance of ABCA1, a key transporter in cholesterol efflux pathway (Sanchez and Dong, 2010). These examples indicate a possible dependence of HCMV replication on host cholesterol metabolism prompting us to investigate mechanisms of interaction between the two. Here, we established that HCMV, through its protein US28, modifies host cholesterol metabolism and restructures lipid rafts enhancing cholesterol efflux from host cells
Results
HCMV infection enhances cholesterol efflux
To investigate the effect of HCMV infection on cholesterol efflux, human fibroblasts were infected with HCMV strain Toledo at a multiplicity of infection of 1 and their ability to release cholesterol to lipid-free apolipoprotein A-I (apoA-I) was tested at 24 and 48 h post infection. The duration of the efflux incubation (2 h) was on a proportional part of the time-dependence curve, and the concentration of apoA-I (30 μg/ml) was close to saturation ensuring that the concentration of the acceptor was not rate-limiting. Labelling of cells with cholesterol (i.e. specific radioactivity of cholesterol) was not affected by the infection (not shown). Figure 1A shows that HCMV infection enhanced the rate of specific cholesterol efflux by 4-fold and 2-fold after 24 and 48 h, respectively. Non-specific efflux (i.e. the efflux in the absence of an acceptor) was unaffected by the infection (not shown).
Figure 1. HCMV enhances cholesterol efflux from human fibroblasts.

A – Time-dependence of the effect of HCMV infection on cholesterol efflux to apoA-I (30 μg/ml); B- The effect of HCMV infection (48h) on cholesterol efflux to apoA-I (30 μg/ml), HDL (40 μg/ml) and methyl-β-cyclodextrin (200 μg/ml); C – The effect of fixing cells with paraformaldehyde (PFA) on the effect of HCMV infection (48h) on cholesterol efflux to apoA-I; D – The effect of HCMV infection (48h) on cell necrosis; E – The effect of HCMV infection (48h) on cell apoptosis; F – The effect of infection with clinical strain of HCMV (Merlin, 48h) on cholesterol efflux to apoA-I. *p<0.05; **p<0.01 (versus uninfected).
Cholesterol efflux to lipid-free apoA-I is solely determined by the activity of the ABCA1 transporter, therefore, we investigated two other acceptors, isolated high density lipoprotein (HDL), an acceptor using ABCG1 and SR-B1 dependent pathways, and methyl-β-cyclodextrin (MβCD), a non-specific acceptor of cholesterol. Both acceptors were used in saturating concentrations. Figure 1B shows that after 48 h of infection efflux to both apoA-I and HDL was significantly enhanced, while the efflux to MβCD was unaffected. To further confirm that the effect of HCMV infection is an active and specific process and is not related to passive diffusion of cholesterol, we tested the efflux of cholesterol from cells fixed with paraformaldehyde prior to the efflux incubation. While the efflux from live cells to apoA-I was enhanced after 48 h of infection, the efflux from fixed cells was similar for infected and uninfected cells (Fig. 1C; note that the conditions of this experiment were different, resulting in higher levels of efflux). To rule out the possibility that changes in cholesterol efflux were related to changes in cell viability, we tested the effect of HCMV infection on necrosis (Fig. 1D) and apoptosis (Fig. 1E) after 24 and 48 h of infection; neither was affected by the infection. Finally, to ensure that the effects were not unique to the laboratory strain of HCMV, we tested the effects of an additional clinical HCMV strain (Merlin). This HCMV strain also caused elevation of cholesterol efflux to apoA-I, although the effect was less profound (Fig. 1F). Thus, HCMV infection enhanced specific cholesterol efflux from host cells.
Involvement of host cholesterol transporters
Increased cholesterol efflux to apoA-I is usually a result of increased abundance of total and/or cell-surface ABCA1. However, when we examined the abundance of ABCA1 in cells infected with HCMV, unexpectedly, we found that it was reduced (Fig. 2A). Densitometry of bands showed that ABCA1/GAPDH ratio was 0.7±0.08 versus 0.4±0.07 (mean±SD) for uninfected versus infected cells, respectively (p<0.003, n=4). Furthermore, the abundance of cell-surface ABCA1 was also reduced in infected cells (Fig. 2A). The abundance of two other transporters responsible for the efflux to HDL, ABCG1 and SR-B1, was unaffected by the infection (Fig. 2B) despite increased efflux to HDL in infected cells (Fig. 1B). We then stimulated expression of ABCA1 and ABCG1 by treating cells with the LXR agonist TO901317. As expected, this treatment increased the rate of cholesterol efflux in both infected and uninfected cells, however the efflux from HCMV infected cells was higher at both conditions and the absolute difference between cholesterol efflux from infected and uninfected cells was similar in cells treated or not treated with the LXR agonist (Fig. 2C). When ABCA1 abundance was reduced by approximately 80% after transfecting cells with siRNAABCA1 (Fig. 2D, inset), the efflux from uninfected cells was significantly reduced, while the efflux from infected cells was unaffected by ABCA1 silencing (Fig. 2D). The pathway of ABCA1-dependent cholesterol efflux also involves the efflux of phospholipids, either simultaneously or prior to the efflux of cholesterol (Vedhachalam et al., 2007). When we tested the effect of HCMV infection on phospholipid efflux, no changes were found (Fig. 2E). Collectively, these findings suggest that the increase of cholesterol efflux after HCMV infection is not mediated by host ABCA1.
Figure 2. Involvement of host cholesterol transporters in the effects of HCMV on cholesterol efflux.

A – The effect of HCMV infection (48h) on the abundance of total (left) and cell-surface (right) ABCA1; B – The effect of HCMV infection (48h) on the abundance of ABCG1 and SR-B1; C – The effect of activation of expression of ABC transporters with LXR agonist TO901317 on cholesterol efflux from HCMV-infected cells (48h infection) to apoA-I; D – The effect of silencing ABCA1 on cholesterol efflux from HCMV-infected cells (48h infection) to apoA-I. Inset – abundance of ABCA1 after silencing with siRNAABCA1; siRNAs, scrambled siRNA; E - The effect of HCMV infection (48h) on phospholipid efflux to apoA-I. *p<0.05; ***p<0.001 (versus uninfected); #p<0.05 (versus untreated).
Involvement of HCMV proteins
An alternative possibility explaining enhanced cholesterol efflux from cells infected with HCMV is that the virus introduces an additional virus-derived efflux pathway into host cell. To test this possibility, we infected cells with HCMV inactivated by UV irradiation. UV irradiation does not affect the capacity of the virus to enter target cells, but blocks viral gene transcription (Zhu et al., 1997). We found that UV inactivation of the virus completely abolished its capacity to stimulate cholesterol efflux (Fig. 3A) and to reduce ABCA1 abundance (Fig. 3B, lanes 1,2,5). Similar effects on cholesterol efflux were demonstrated when UV inactivated Merlin HCMV strain was used (not shown). To further confirm the requirement for active replication, we treated cells with a specific inhibitor of HCMV DNA polymerase, phosphonoacetic acid (PAA) (Stinski, 1977). Treatment with PAA had no effect on cholesterol efflux from uninfected cells, but completely abolished the increase of cholesterol efflux seen in infected cells (Fig. 3C).
Figure 3. Involvement of viral proteins in the effects of HCMV on cholesterol efflux.

A – The effect of UV irradiation on the ability of HCMV (48h infection) to enhance cholesterol efflux to apoA-I; B - The effect of UV irradiation and of deletion of US28 gene on the ability of HCMV (48h infection) to reduce abundance of ABCA1; C – The effect of phosphonoacetic acid (PAA) on the ability of HCMV (48h infection) to enhance cholesterol efflux to apoA-I; D – Cholesterol efflux to apoA-I from cells not infected with HCMV or infected with WT HCMV, HCMV(ΔUS28) and parent virus to HCMVΔUS28 containing US28 (HCMV(+US28)); all infections for 48 h; E –The effect of transient overexpression of tGFP-US28 or hABCA1 on cholesterol efflux. Inset - presence of US28 after transfection with mock (left) or tGFP-US28 (right) plasmid; F – Cholesterol efflux to apoA-I from cells infected with HCMV(+US28), HCMV(ΔUS28) and infected with HCMVΔUS28 (48h infection) in combination with transfection with US28; G - The effect of transient overexpression of tGFP-US28 on the abundance of total (top left) or cell-surface (right) ABCA1; H - The effect of transient overexpression of tGFP-US28 on cholesterol efflux from RAW 264.7 cells. *p<0.01; **p<0.01; ***p<0.001.
HCMV is a complex virus, its genome contains more than 160 open reading frames (ORFs) and the functions of many of the products of these genes are not known. None of the HCMV proteins carries sequences homologous to known sterol binding or sterol sensing domains of mammalian proteins. The HCMV protein US28 is a 41 kDa G-protein-coupled receptor expressed at all stages of the infection and found in perinuclear endosomes and lipid rafts (Droese et al., 2004). It was previously implicated as potentially playing a role in pathogenesis of atherosclerosis by altering smooth muscle cell migration (Streblow et al., 1999). We tested if US28 may be also responsible for the viral effects on cholesterol efflux. We compared uninfected cells with cells infected with the Toledo strain, ΔUS28 viral mutant based on the VR1814 strain and the parental VR1814 strain (+US28). While infection with both Toledo and +US28 viruses similarly increased cholesterol efflux, the ΔUS28 virus failed to do so (Fig. 3D). When the effects on ABCA1 abundance were examined, again +US28 virus reduced abundance of ABCA1, while ΔUS28 virus was less effective (Fig. 3B, lanes 1–4). In a complementary set of experiments, we transiently overexpressed US28 tagged with turboGFP (tGFP) in the host cells in the absence of the virus (Fig. 3E, inset). Overexpression of US28 resulted in an increase of the rate of cholesterol efflux, similar to the positive control, cells overexpressing ABCA1 (Fig. 3E). Furthermore, overexpression of US28 in HCMV(ΔUS28) infected cells elevated cholesterol efflux even above that in cells infected with the parent HCMV(+US28) virus (Fig. 3F). When the effect of US28 overexpression on ABCA1 abundance was examined, the total ABCA1 abundance was unchanged in US28-transfected cells (Fig. 3G). The abundance of cell-surface ABCA1 was, however, lower in US28 versus mock transfected cells (482 versus 662 arbitrary densitometry units) confirming that in cells transfected with US28 ABCA1 is located in a compartment not exposed to the cell surface. A discrepancy between the effects of HCMV infection and US28 transfection on the total abundance of ABCA1 is likely a reflection of different amounts of US28 introduced by viral infection versus transfection or is temporal; however, we cannot exclude a possibility that US28 requires a co-factor for its effects on ABCA1.
Finally, overexpression of US28 in another cell type, mouse macrophage cells RAW 264.7, also elevated cholesterol efflux (Fig. 3H). Thus, it appears that HCMV protein US28 is essential and sufficient for the effects of HCMV on cholesterol efflux; it is also essential, but may not be sufficient for the reduction of ABCA1 abundance in host cells.
Mechanism of the effect of HCMV on cholesterol efflux
To investigate the mechanism of US28-dependent stimulation of cholesterol efflux, we tested the effect of US28 on apoA-I binding to host cells. Infection of cells with +US28 HCMV led to an almost 3-fold elevation of apoA-I binding to cells compared to uninfected cells (Fig. 4A). Infection of cells with US28 deficient HCMV virus led to a reduction of apoA-I binding compared to cells infected with the +US28 virus (Fig. 4A). Next, we tested the effect of overexpression of US28 on apoA-I binding. Transfection of cells with US28 more than doubled the binding of apoA-I compared to mock transfected cells, exceeding the effect of transfection with ABCA1 (Fig. 4B). The effect of US28 on apoA-I binding was further confirmed with two independent methods. First, the amount of apoA-I bound to mock- or US28 transfected cells was analysed by Western blot. Representative images of Western blots of cell-bound [Alexa Fluor 647]apoA-I are shown in Fig 4C. We found that transfection of US28 increased apoA-I/GAPDH ratio by 1.38±0.045 – fold (n=3) in US28 versus mock transfected cells (p<0.05). Second, we used live confocal microscopy to analyse binding of [Alexa Fluor 647]apoA-I to mock or US28 transfected cells. Findings are presented in Fig. 4D (note that for better visualization colours were changed and tGFP is presented as red while Alexa Fluor 647 is shown as green). Left panel in Fig. 4D shows small amount of apoA-I (green dots) bound to mock-transfected cells. Middle panels show apoA-I bound to the surface of the US28-transfected cell (red) but not to the surface of untransfected cell (seen at bright-filed picture on the right panel). Quantitation of the confocal images demonstrated that twice as much apoA-I binds to US28-transfected versus mock-transfected cells (Fig. 4D, right panel).
Figure 4. Mechanism of the effect of HCMV on cholesterol efflux.

A – The effect of HCMV(+US28) and HCMV(ΔUS28) (48h infection) on binding of apoA-I to human fibroblasts. B - The effect of overexpression of US28 or ABCA1 on apoA-I binding (2h) to human fibroblasts; C – Western blot of [Alexa Fluor 647]apoA-I bound to mock or US28 transfected cells; D - Live confocal microscopy images of [Alexa Fluor 647]apoA-I bound to mock or US28 transfected cells. Note that for better visualization colours were changed and GFP is presented as red while Alexa Fluor 647 is shown as green. Left panel – fluorescence image of mock transfected cell, middle panels – florescent (left middle panel) and brightfiled (right middle panel) images of US28 transfected cells, right panel: quantitation of apoA-I binding to US28-transfected and mock-transfected cells; **p<0.01. E – Cross-linking of apoA-I with tGFP-US28 or ABCA1 in cells overexpressing tGFP-US28. The table below the panel explains the conditions: top row specifies if cells were transfected with mock (M) or US28 plasmid; middle row specifies the antibody used for immunoprecipitation; bottom row specifies the antibody used for visualization of the bands. F – The effect of HCMV infection (48h) on the abundance of cell-surface cholesterol; G – The effect of overexpression of US28 on the abundance of cell-surface cholesterol. *p<0.05; ***p<0.001; H – The effect of overexpression of US28 on incorporation of BODIPY-cholesterol into plasma membrane. Left panel: fluorescence of BODIPY-cholesterol bands after separation of membrane lipid extract using TLC; middle panel: abundance of αNa+/K+ATPase in the same samples analysed by western blot; right panel: abundance of flotillin in the same samples analysed by western blot.
We hypothesized that the mechanism of the effect of US28 on apoA-I binding could be a direct binding of apoA-I to US28 on the cell surface leading to a translocation of plasma membrane cholesterol to bound apoA-I. To test this hypothesis, we overexpressed tGFP-tagged US28 in human fibroblasts, incubated cells with human apoA-I, cross-linked with cell-impermeable cross-linker followed by immunoprecipitation with either anti-apoA-I or anti-tGFP antibody. Immunoprecipitation with both anti-tGFP and anti-apoA-I failed to detect a direct binding between US28 and apoA-I (Fig. 4E). At the same time, analysis of complexes immunoprecipitated with anti-apoA-I revealed ample binding of apoA-I to ABCA1 (Fig. 4E, right panel). Thus, it appears unlikely that US28 closely associates with or directly binds to apoA-I. An alternative is that HCMV, through US28 alters the structure of plasma membrane creating sites where apoA-I can bind and solubilise membrane lipids, a mechanism recently suggested for the ABCA1-dependent cholesterol efflux (Vedhachalam et al., 2007).
Binding of apoA-I stimulates transfer of cholesterol to the plasma membrane and increases abundance of cholesterol on the cell surface (Sviridov et al., 2001). We therefore tested if HCMV infection alters the abundance of cholesterol on the surface of plasma membrane (defined as a proportion of cholesterol susceptible to oxidation by cholesterol oxidase). Both HCMV infection and overexpression of US28 significantly increased the abundance of cell-surface cholesterol (Fig. 4F, G). This finding was further confirmed by analysing the amount of BODIPY-cholesterol incorporated into cell membranes after pulse labelling of cells by incubating them for 10 min with BODIPY-cholesterol/cyclodextrin complex. The amount of BODIPY-cholesterol extracted from plasma membranes of US28-transefcted cells was twice that in the mock-transfected cells (Fig. 4H, left panel), while the abundance of the membrane marker αNa+/K+ATPase (“loading control” analysed using western blot in samples from the same experiment) was slightly less (Fig. 4H, middle panel). Enrichment of membranes with cholesterol usually leads to increased abundance of lipid rafts, therefore we also analysed the abundance in the membrane fraction of raft marker, flotillin. Unexpectedly, the abundance of flotillin in cells transfected with US28 decreased when compared to mock-transfected cells (Fig. 4H, right panel) when normalized to loading control (Fig. 4H, middle panel). Thus, HCMV and US28 increased binding of apoA-I and abundance of cholesterol on the cell surface; both factors may contribute to the increased cholesterol efflux.
Lipid rafts are an important structural element of plasma membrane involved in regulation of cholesterol efflux and many other metabolic processes. Therefore, we further investigated the effect of US28 on the abundance of rafts using confocal microscopy. Flotillin and ganglioside GM1 (assessed through binding of cholera toxin subunit B (CT-B)) were used as raft markers and the experiments were conducted in two modifications, in cell suspension, where the plasma membrane can be clearly visualized, and in adherent cells, a more natural state for fibroblasts. In suspended fibroblasts transfected with mock-GFP plasmid, we found a high abundance of flotillin and GM1 positive staining on the plasma membrane, consistent with high abundance of rafts in these cells (Fig. 5A). In cells transfected with US28-tGFP, two types of staining were observed. In some transfected cells US28 was localized to the nucleus, cytoplasm and plasma membrane; in these cells we observed weaker staining with anti-flotillin antibody, but clearly distinguishable staining of the plasma membrane with CT-B (Fig. 5B). In other cells, US28 localized to the perinuclear space and nucleus; in these cells no flotillin or GM1 staining was observed on plasma membrane (Fig. 5C). Quantitation of flotillin abundance derived from confocal microscopy experiments is shown in Fig. 5D. Similar observations were made with adherent cells (Supplemental Figure S1A). Two types of intracellular distribution of US28 were reported previously and are thought to reflect temporal regulation, with nucleus-localized US28 representing later stages of infection (Noriega et al., 2014). We further confirmed these findings in cells infected with +US28 and ΔUS28 HCMV. While in uninfected cells we observed an abundant and even staining of plasma membrane with anti-flotillin antibody and CT-B (Fig. 5E), in cells infected with +US28 virus the abundance of flotillin and GM1 was considerably reduced and became uneven (“clustered”) (Fig. 5F). Abundance and distribution of raft markers in cells infected with ΔUS28 HCMV were similar to uninfected cells (Fig. 5 G), while infection with ΔUS28 virus combined with transfection with US28-tGFP again led to reduced abundance and clustering of lipid raft markers (Fig. 5H; cells transfected with US28-tGFP are identified by tGFP fluorescence). Quantitation of flotillin abundance derived from confocal microscopy experiments is shown in Fig. 5I. Similar observations were made with adherent cells (Supplemental Figure S1B). Thus, HCMV infection and transfection with US28 reduced abundance of rafts and/or severely affected their properties inducing clustering and exclusion of key lipid raft markers.
Figure 5. The effect of HCMV on lipid rafts.

A – The effect of transfection of fibroblasts with mock-GFP on abundance of flotillin and GM1; B – The effect of transfection of fibroblasts with US28-tGFP on abundance of flotillin and GM1 when US28 is distributed to cytoplasm; C – The effect of transfection of fibroblasts with US28-tGFP on abundance of flotillin and GM1 when US28 is distributed to nucleus. D - Quantitation of flotillin abundance derived from confocal microscopy experiments. CTCF – corrected total cell fluorescence; **p<0.01; ***p<0.001 (versus mock GFP or as shown). E – Abundance and distribution of flotillin and GM1 in uninfected cells; F – The effect of infection of fibroblasts with HCMV(+US28) (48h)on abundance and distribution of flotillin and GM1; G – The effect of infection of fibroblasts with HCMV(ΔUS28) (48h) on abundance and distribution of flotillin and GM1. H – The effect of infection of fibroblasts with HCMV(ΔUS28) (48h) and transfection with US28-tGFP on abundance and distribution of flotillin and GM1; Bar -10 μm. I - Quantitation of flotillin abundance derived from confocal microscopy experiments. CTCF – corrected total cell fluorescence; *p<0.05, **p<0.01(versus -HCMV or as shown). See also Figure S1.
To understand a mechanism responsible for re-organization of lipid rafts by HCMV, we investigated status of actin cytoskeleton in infected cells. Actin microfilaments are involved in maintaining the structure of rafts, including their clustering (Chichili and Rodgers, 2007), and are responsible for alteration of raft structure by the influenza virus (Simpson-Holley et al., 2002). Indeed, actin cytoskeleton was altered in HCMV infected cells showing considerable rearrangement; we also observed co-localization of actin with GM1 in the sites of raft clustering (Fig. 6 A, B). Similar changes in actin cytoskeleton and increased co-localization of actin with GM1 was observed in US28 transfected cells when compared to mock-transfected cells (Fig. 6C, D, note that colour of GFP was changed to blue to identify transfected cells).
Figure 6. Mechanism and consequences of the effect of HCMV on lipid rafts.

A, B – The effect of infection of fibroblasts with HCMV on abundance and co-localization of GM1 and β-actin. Bar 20 μm. C, D – The effect of transfection of fibroblasts with US28 on abundance and co-localization of GM1 and β-actin. Note that colour of tGFP was changed to blue to identify transfected cells. Bar 20 μm. E – Western blot of CDC42 with and without siRNA silencing. F –Western blot of [Alexa Fluor 647]apoA-I bound to cells transfected with: mock plasmid and siRNAs (line 1), US28 and siRNAs (line 2), mock plasmid and siRNACDC42 (line 3), US28 and siRNACDC42 (line 4). G – Cholesterol efflux to apoA-I (30 μg/ml) from cells transfected with various combinations of mock and US28 plasmids, siRNAs and siRNACDC42; **p<0.01. H – The effects of transfection with US28 or treatment with methyl-β-cyclodextrin (MβCD) on cytokine secretion from RAW 264.7 macrophages after stimulation with LPS. Ratio of LPS-stimulated changes in US28 transfected versus mock-transfected cells (filled bars) or CD-treated versus untreated cells (dashed bars) are shown (Means ± SD, n=4). *p < 0.05;** p < 0.01; *** p < 0.001 (versus mock-transfected cells); ### p < 0.001 (versus untreated cells). I - The effect of conditioned medium from RAW 264.7 cells transfected with US28 on VCAM1 expression in mouse endothelial cells. *p<0.05. See also Figure S2.
US28 is known to initiate a signalling cascade resulting in activation of CDC42 (Melnychuk et al., 2004; Vischer et al., 2014), a small GTPase of the Rho family implicated in regulation of actin polymerisation (Chadda et al., 2007; Vischer et al., 2014). To test if this mechanism is involved, we silenced CDC42 in human fibroblasts using siRNACDC42. Silencing of CDC42 resulted in approximately 80% reduction of CDC42 abundance (Fig. 6E). When binding of [Alexa Fluor 647]apoA-I was tested in cells transfected with scrambled siRNA, as expected, co-transfection with US28 increased apoA-I binding (Fig. 6F, lanes 1,2). When cells were transfected with siRNACDC42 the increase of apoA-I binding in US28 transfected cells was completely eliminated (Fig. 6F, lanes 3, 4). This finding was confirmed by measuring the effects of US28 on cholesterol efflux from cells after CDC42 silencing: silencing of CDC42 completely eliminated increase of cholesterol efflux caused by US28 transfection (Fig. 6G). Thus, a mechanism of the effects of HCMV on the structure of lipid rafts is through CDC42-mediated remodelling of actin cytoskeleton.
Effect of US28 on inflammation
Changes in lipid rafts may lead to impairment of multiple signalling pathways including inflammatory cytokine production. Therefore we investigated possible physiological implications of the effects of US28 and tested if overexpression of US28 affects elements of inflammation. Mouse macrophage cells RAW 264.7 were transfected with either mock plasmid or with US28, and cytokine secretion was analysed. Transfection of cells with US28 significantly reduced unstimulated secretion of IL-6, IL-27 and IFN-β (Supplemental Figure S2 A). When cells were stimulated with LPS, secretion of IL-6, IL-27 and GM-CSF was reduced in cells transfected with US28 (Fig. 6H, solid bars). To assess the contribution of US28-mediated raft modifications to cytokine secretion, we compared the effects of transfecting cells with US28 with the effects of MβCD, which modifies rafts by extracting cholesterol and does not interact with known chemokine receptors. Treatment of cells with MβCD reduced abundance of rafts (Supplemental Figure S2 B) and significantly reduced LPS-stimulated secretion of IL-6 and GM-CSF, similar to the effects of transfection with US28 (Fig. 6H). However, the effect of MβCD on LPS-stimulated secretion of IL-27 was opposite to that of US28 indicating possible contribution of chemokine scavenging and receptor hijacking previously attributed to US28 (Vischer et al., 2014) to its anti-inflammatory properties. When conditioned medium from US28-transfected macrophages (not stimulated with LPS) was added to the endothelial cells carrying reporter gene under control of VCAM-1 promoter, we observed a 25% reduction in the stimulatory effect on VCAM1 expression, when compared with medium from mock-transfected cells (Fig. 6I).
Effect of HCMV infection on cellular lipid metabolism
Our findings imply that HCMV infection replaces the cell-specific ABCA1-dependent pathway of cholesterol efflux with a virus-specific US28-dependent cholesterol efflux. To elucidate the net effect of such substitution on lipid content, we performed lipidomic analysis of cells infected with HCMV. The findings are summarised in Fig. 7A, with full dataset presented in Supplemental Table S1. The notable effects of HCMV infection on cellular lipids were small, but statistically significant elevation of the abundance of ceramide, dihexosylceramide and GM3 ganglioside. These lipids play an important role in maintaining the structure and functional properties of lipid rafts (Quinn, 2014). Total abundance of two other important components of rafts, cholesterol and sphingomyelin were not affected by the infection; abundance of cholesteryl esters was also unaffected.
Figure 7. The effect of HCMV infection on host lipid metabolism.

A –The effects of HCMV infection (48h) on abundance of cellular lipids (standardized to PC); LPC(O), alkyllysophosphatidylcholine; PG, phosphatidylglycerol; LPE, lysophosphatidylethanolamine; LPI, lysophosphatidylinositol; LPC, lysophosphatidylcholine; DHC, dihexosylceramide; PE(O), alkylphosphatidylethanolamine; Cer, ceramide; THC, trihexosylceramide; MHC, monohexosylceramide; PE(P), alkenylphosphatidylethanolamine; PC(P), alkenylphosphatidylcholine; PC(O), alkylphosphatidylcholine; GM3, GM3 ganglioside; CE, cholesteryl ester; SM, sphingomyelin; PS, phosphatidylserine; PI, phosphatidylinositol; PE, phosphatidylethanolamine; COH, cholesterol; PC, phosphatidylcholine; B- The effect of HCMV infection (48h) on incorporation of [3H]acetate into cholesterol; C- The effect of transfection with US28 on incorporation of [3H]acetate into cholesterol; D – The effect of HCMV infection (48h) on incorporation of [14C]oleic acid into cholesteryl esters; E – The effect of transfection with US28 on incorporation of [14C]oleic acid into cholesteryl esters; F - The effect of transfection of cells with US28 on abundance of total cholesterol and cholesteryl esters. G - The effect of HCMV infection (48h) on the abundance of selected miRNAs related to cholesterol metabolism. *p<0.05; **p<0.01; ***p<0.001 vs Control (Student’s t-test). # p<0.05, ##p<0.01 vs Control (corrected for multiple comparisons using the Benjamini-Hochberg method). See also Table S1 and Figure S3.
Cholesterol efflux is often non-productive: release of cellular cholesterol to extracellular acceptor may be compensated by cholesterol uptake with no net change in overall cellular cholesterol content. We further investigated this issue and measured two surrogate markers of cellular cholesterol content, rates of cholesterol biosynthesis and esterification. The rate of cholesterol biosynthesis in HCMV infected (Fig. 7B) or US28 transfected (Fig. 7C) cells was slightly reduced while the rate of cholesterol esterification was increased (Fig. 7D, E) compared to uninfected cells. These findings are consistent with increased rather than decreased abundance of cholesterol, at least in the regulatory pool in the endoplasmic reticulum. HCMV infection also caused a modest increase in incorporation of [3H]acetate into free fatty acids (1.26±0.04 versus 1.09±0.08 dpm/mg cell protein; p<0.05) and phospholipids (2.89±0.16 versus 2.59±0.08 dpm/mg cell protein; p<0.05) and had no effect on triglyceride biosynthesis. When we measured the abundance of total cellular cholesterol and cholesteryl esters in cells transfected with US28, they were similar to that in mock-transfected cells (Fig. 7F); analogous to the effect of HCMV infection. Thus, elevation of cholesterol efflux by HCMV infection or US28 transfection does not appear to be productive and did not lead to reduction of cellular cholesterol content. It might however lead to a redistribution of cholesterol between cellular pools.
We next investigated the effect of HCMV infection on the expression of lipid metabolism -related genes in host cells. We found that the expression of SR-A, ABCA3, and ABCG1, were increased, while the expression of ABCA12 was reduced and the expression of ABCA1 was unaffected (Supplemental Fig. S3 A). Interestingly, increase in the expression of ABCG1, ABCA3 and SR-A in the infected cells was not translated into increased abundance of these proteins (Fig. 2B, Supplemental Fig. S3 B).
Next we investigated the effect of HCMV infection on the abundance of 10 miRNAs known to target ABCA1 and cholesterol efflux in various cells (for review see (Canfrán-Duque et al., 2014)). Abundance of two miRNAs, miR10b and miR106b, was significantly elevated in infected cells (Fig. 7G) pointing to a possible mechanism of reduction of ABCA1 abundance in cells infected with HCMV. However, the abundance of two other miRNAs known to target ABCA1, miR145 and miR27a was reduced by HCMV infection. Other miRNAs were unaffected. It must be noted that abundance of the affected miRNAs was low compared to that of the main miRNA regulator of ABCA1, miR33a (Supplemental Figure S3 C). The abundance of miR33a and miR33b was unaffected.
Animal studies
Finally, we analysed the effect of CMV infection on lipid metabolism in vivo using a murine model. Balb/c mice were infected intraperitoneally with 1.8 x 106 pfu of murine CMV (MCMV). It should be noted that MCMV does not express US28 (Beisser et al., 2008). At three weeks post infection, uninfected and infected animals were euthanized. Analysis of plasma lipoproteins (Supplemental Figure S4 A–D) revealed that MCMV infection caused modest elevation of total plasma cholesterol, which was mostly due to elevation of non-HDL cholesterol. There was a significant elevation of plasma TG content; HDL cholesterol was unaffected. We also analysed the capacity of mouse plasma to support cholesterol efflux from uninfected mouse macrophages (RAW 264.7). No effect was found (Supplemental Figure S4 E). Further, we analysed the effect of MCMV infection on expression of genes related to cholesterol metabolism in the liver (Supplemental Figure S4 F). Liver of MCMV-infected animals had elevated expression of Abca3, similar to the in vitro findings with HCMV, and also of Hmgcr and Lxra. We next analysed the abundance of Abca1 and Abcg1 in the liver (Supplemental Figure S4 G,H). There was a tendency for lower abundance of Abca1 in the liver of the infected animals, but the difference did not reach statistical significance. However, there was statistically significant elevation of the abundance of Abcg1 in livers of the infected animals.
Discussion
Very little is known about the role of cholesterol in the lifecycle of HCMV, however, the presence of cholesterol in HCMV virions suggests a possible interaction of the virus with cellular cholesterol metabolism. Given the important role such interaction plays in pathogenesis of many viral infections (Sviridov and Bukrinsky, 2014), we comprehensively investigated the interaction between HCMV infection and host cholesterol metabolism in vitro and in vivo.
The main finding of this study is that HCMV restructures lipid rafts in the host cells causing increased cholesterol efflux, at the same time reducing abundance of a key transporter in the host cholesterol efflux pathway, ABCA1. The viral protein responsible for this effect is US28. Mechanistically, the following sequence of events is proposed (Supplemental Figure S5): (i) US28 is acting as chemokine receptor and activates CDC42, (ii) activation of CDC42 leads to rearrangement of actin microfilaments including tight association of actin with rafts, (iii) remodelling of actin affects abundance and/or structure of lipid rafts, causing raft clustering; (iv) changes in lipid rafts lead to creating of plasma membrane regions that are capable of binding of apoA-I and releasing cholesterol; this also leads to displacement of ABCA1 from cell surface, (v) increased binding of apoA-I causes redistribution of intracellular cholesterol to the plasma membrane, (vi) elevated binding of apoA-I and increased amount of accessible cholesterol enhanced cholesterol efflux. Our findings are consistent with the hypothesis that US28 causes a re-distribution of cholesterol between cellular compartments, reorganizes lipid rafts and increases the number of plasma membrane structures capable of binding apoA-I and releasing cholesterol. Current understanding of the mechanism of cholesterol efflux is that ABCA1 changes properties of the plasma membrane domains forming apoA-I bindings sites leading to the efflux (Vedhachalam et al., 2007). It appears that US28 induces similar changes to the plasma membrane without ABCA1.
US28 is a virus-encoded G-protein-coupled receptor; it is expressed in both acute and latent infection (Vischer et al., 2007) and is only found in primate CMVs (Beisser et al., 2008). US28 affects multiple signalling pathways, it is dispensable for viral replication, but plays a key role in immune evasion as well as in inflammatory and proliferative responses of the infected cells ensuring persistence of the infection (for review see (Beisser et al., 2008; Vischer et al., 2007; Vischer et al., 2014)). US28 was also implicated in pathogenesis of atherosclerosis, mainly through the effects on inflammation and migration of smooth muscle cells and macrophages (Streblow et al., 1999; Vischer et al., 2014). Findings of this study may provide a mechanistic explanation for the effects of US28 on inflammatory and proliferative signalling pathways originating from lipid rafts. Dysregulation of cellular cholesterol metabolism inflicted by US28 may contribute to pathogenesis of atherosclerosis through changes in abundance and/or properties of cholesterol-rich plasma membrane domains, rafts and caveolae (Murphy et al., 2008).
Our findings raise the following question: given that expression of US28 is not essential for HCMV replication (Vischer et al., 2007), what advantage is gained by HCMV from enhancing cholesterol efflux, apparently with no net effect on cellular cholesterol content? We propose that this effect is related to the dramatic changes in the abundance and properties of lipid rafts. Lipid rafts are essential for many intracellular pathogens. On the one hand, many viruses use host plasma membrane rafts as an assembly platform, entry gate or binding sites during cell-to-cell transmission (Sviridov and Bukrinsky, 2014); this requires increased abundance of rafts. It is not currently known how and if HCMV interacts with rafts at different stages of its lifecycle. Juckem et al (Juckem et al., 2008) suggested that cholesterol depletion and disruption of lipid rafts result in a block of HCMV infection of fibroblasts. Infection of this cell type takes place by fusion of the virus envelope with the plasma membrane, but whether rafts are also essential for entry into other cell types is unknown. On the other hand, rafts are a structural platform for innate immune response (Fessler and Parks, 2011) and pathogens often target rafts to reduce immune-related signalling originating from rafts and exposure of infection-related factors to the immune system; this requires reduced abundance of rafts. To accommodate these two seemingly contradictory requirements, pathogens modify rafts. Often they do so by modifying lipid fluxes (Sviridov and Bukrinsky, 2014), as even small changes in lipid flux affect raft structural properties severely impairing signalling from TLR receptors (Fessler and Parks, 2011). A frequent form of raft modification is clustering, when rafts converted into more stable and larger structures (Sviridov and Bukrinsky, 2014). We propose that HCMV through the suggested mechanism changes structural and functional properties of lipid rafts, enhancing cholesterol efflux and impairing innate immune responses. This suggestion is supported by our finding that transfection of mouse macrophage cells with US28 reduced secretion of several cytokines measured both directly and by a functional assay assessing the effects of secreted cytokines on VCAM1 expression in endothelial cells. It is also consistent with ability of HCMV to inhibit IFN-α-stimulated responses that are known to rely on intact rafts (Miller et al., 1999). Further, we also found elevated levels of ceramide in HCMV-infected cells; enrichment of rafts with ceramide is known to be associated with altered transmembrane signalling and formation of larger domains (Cremesti et al., 2002). However, the suggestion of physiological role of US28 as a modulator of innate immunity remains speculative and needs to be tested in an animal model, a complex task given that only primate CMVs contain US28. The effects of US28 on host cholesterol metabolism may also be related to a known capacity of HCMV to facilitate co-infection with other viruses, including HIV, which also critically depends on rafts and suppression of ABCA1 for their infectivity (Cui et al., 2012). While our findings are consistent with these hypotheses, critical evidence to support them beyond speculation awaits further experiments.
In a mouse model, MCMV infection did not affect liver abundance of Abca1, while slightly increasing plasma triglyceride and non-HDL cholesterol content. Animal models, however, have limitations in reproducing properties of HCMV infection. Specifically, MCMV does not have a gene for US28 and its chemokine receptor functions are fulfilled by M33 (Beisser et al., 2008); the two proteins have limited homology (64%) and M33 may not share US28 functionality in modifying host cholesterol metabolism.
In conclusion, HCMV through its protein US28 modifies plasma membrane rafts and enhances cholesterol efflux from infected cells. Given the role of cholesterol and lipid rafts in cell metabolism and immunity, findings of this study may have significant implications on understanding of pathogenesis of HCMV infection and its complications and may also identify therapeutic targets for treating HCMV infection.
Experimental procedures
Detailed methodology is provided as Supplemental Experimental Procedures.
Virus and cells
The low passage Toledo strain of HCMV was used unless indicated otherwise. When indicated, HCMV strain VR1814 was used as the basis for deletion of the US28 open reading frame. A clinical strain of HCMV (Merlin) was used in the experiments described in the figure 1F.
Human Foreskin Fibroblast cells (HFF) were used in the experiment described in this study unless indicated otherwise. HFF were infected with HCMV at MOI=1. Unless indicated otherwise, the experiments were conducted 48 h after infection. In the experiments described in Figs. 3H, 5H and Table S1 RAW 264.7 mouse macrophages were used.
Cholesterol and phospholipid efflux
Cholesterol or phospholipids were labelled by incubation with 1μCi of respectively [3H]cholesterol or [3H]-choline, cells equilibrated, and incubated for 2 h with either human apoA-I (30μg/ml) or isolated human HDL (40μg/ml) or methyl-β-cyclodextrin (200 μg/ml). Efflux was expressed as a percentage of cholesterol moved from cell to medium.
Other techniques
Cell-surface ABCA1 was assessed after biotinylation of cell-surface proteins (Cui et al., 2012).
To assess apoA-I binding, apoA-I was fluorescently labelled with Alexa Fluor 350 Carboxyl succinimidyl ester. Cells were incubated with 20μg/ml fluorescent apoA-I for 2 hours, harvested and fluorescence was measured. Alternatively, live cells were observed with confocal microscopy or lysed with RIPA buffer and processed for LiCore Western blot.
Lipid biosynthesis was assessed by the incorporation of [3H]acetate into cholesterol, free fatty acids and triglycerides and phospholipids, and of [14C]oleic acid into cholesteryl esters.
Abundance of cell-surface cholesterol on the plasma membrane was assessed through susceptibility to cholesterol oxidase as described previously (Cui et al., 2012). Alternatively, cells were labelled with BODIPY-cholesterol/methyl-β-cyclodextrin inclusion complex (0.17 mg/ml) at 37°C for 10 min and membranes were isolated as described previously (Cui et al., 2012). Lipids were extracted from the membrane pellet and cholesterol was separated using TLC.
The abundance of lipid rafts was assessed using the Vybrant lipid rafts labelling kit (Life Technologies).
Concentrations of cytokines were determined using the LEGENDplex™ Multi-Analyte Flow Assay kit (BioLegend). SVEC4/VCAM-1 cells were used to assess the effect of secreted cytokines on VCAM-1 expression (D’Souza et al., 2010).
MCMV infection of mice
Animal experiments were carried out in accordance with the Declaration of Helsinki; the experimental protocol was approved by the ethical committee in Warsaw Medical University (#93/2013). Balb/c mice were infected intraperitoneally with 1.8x 106 pfu of MCMV (Smith strain); plasma and liver samples were collected 3 weeks post infection.
Statistics
All data is shown as mean ± SD unless stated otherwise. Statistical significance of the differences was assessed by Student’s t-test when data followed normal distribution or Mann-Whitney U test on ranks.
Supplementary Material
Highlights.
HCMV modifies lipid rafts through viral protein US28 and host Cdc42
Lipid raft modification enhances binding of apoA-I and cholesterol efflux
US28 modifies multiple aspects of host lipid metabolism
Inflammatory response is blunted in US28 transfected macrophages
Acknowledgments
This study was supported by grants from the National Health and Medical Research Council of Australia (GNT1019847, GNT1036352) and NIH (#HL093818) to DS and MB and in part by the Victorian Government’s OIS Program. We acknowledge assistance of Monash micro-imaging at the Alfred Medical Research and Education Precinct.
Footnotes
Author contribution
DS conceived the idea of the study, planned the experiments, oversaw the study and wrote the bulk of the paper, HL, HLC, NM, AH, MD, YF, YL and MBl performed the majority of the experiments, BPM and SA generated and supplied HCMV, PJM performed lipidomic analysis, PR and KP performed animal experiments, WEM supplied ΔUS28 HCMV, CSN, BS, MBu, NM, HL, HLC, participated in designing the study and critically revised the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beisser PS, Lavreysen H, Bruggeman CA, Vink C. Chemokines and chemokine receptors encoded by cytomegalovirus. In: Shenk TE, Stinski MF, editors. Human Cytomegalovirus Current Topics in Microbiology and Immunology. Berlin, Heidelberg: Springer-Verlag; 2008. pp. 221–242. [DOI] [PubMed] [Google Scholar]
- Canfrán-Duque A, Ramírez CM, Goedeke L, Lin CS, Fernández-Hernando C. microRNAs and HDL life cycle. Cardiovasc Res. 2014;103:414–422. doi: 10.1093/cvr/cvu140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadda R, Howes MT, Plowman SJ, Hancock JF, Parton RG, Mayor S. Cholesterol-sensitive Cdc42 activation regulates actin polymerization for endocytosis via the GEEC pathway. Traffic. 2007;8:702–717. doi: 10.1111/j.1600-0854.2007.00565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chichili GR, Rodgers W. Clustering of membrane raft proteins by the actin cytoskeleton. J Biol Chem. 2007;282:36682–36691. doi: 10.1074/jbc.M702959200. [DOI] [PubMed] [Google Scholar]
- Cremesti AE, Goni FM, Kolesnick R. Role of sphingomyelinase and ceramide in modulating rafts: do biophysical properties determine biologic outcome? FEBS Lett. 2002;531:47–53. doi: 10.1016/s0014-5793(02)03489-0. [DOI] [PubMed] [Google Scholar]
- Cui HL, Grant A, Mukhamedova N, Pushkarsky T, Jennelle L, Dubrovsky L, Gaus K, Fitzgerald ML, Sviridov D, Bukrinsky M. HIV-1 Nef mobilizes lipid rafts in macrophages through a pathway that competes with ABCA1-dependent cholesterol efflux. J Lipid Res. 2012;53:696–708. doi: 10.1194/jlr.M023119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza W, Stonik JA, Murphy A, Demosky SJ, Sethi AA, Moore XL, Chin-Dusting J, Remaley AT, Sviridov D. Structure/Function Relationships of Apolipoprotein A-I Mimetic Peptides: Implications for Antiatherogenic Activities of High-Density Lipoprotein. Circ Res. 2010;107:217–227. doi: 10.1161/CIRCRESAHA.110.216507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droese J, Mokros T, Hermosilla R, Schulein R, Lipp M, Hopken UE, Rehm A. HCMV-encoded chemokine receptor US28 employs multiple routes for internalization. Biochem Biophys Res Comm. 2004;322:42–49. doi: 10.1016/j.bbrc.2004.07.076. [DOI] [PubMed] [Google Scholar]
- Fessler MB, Parks JS. Intracellular lipid flux and membrane microdomains as organizing principles in inflammatory cell signaling. J Immunol. 2011;187:1529–1535. doi: 10.4049/jimmunol.1100253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudleski-O’Regan N, Greco TM, Cristea IM, Shenk T. Increased Expression of LDL Receptor-Related Protein 1 during Human Cytomegalovirus Infection Reduces Virion Cholesterol and Infectivity. Cell Host Microbe. 2012;12:86–96. doi: 10.1016/j.chom.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haspot F, Lavault A, Sinzger C, Laib Sampaio K, Stierhof YD, Pilet P, Bressolette-Bodin C, Halary F. Human cytomegalovirus entry into dendritic cells occurs via a macropinocytosis-like pathway in a pH-independent and cholesterol-dependent manner. PLoS One. 2012;7:e34795. doi: 10.1371/journal.pone.0034795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juckem LK, Boehme KW, Feire AL, Compton T. Differential initiation of innate immune responses induced by human cytomegalovirus entry into fibroblast cells. J Immunol. 2008;180:4965–4977. doi: 10.4049/jimmunol.180.7.4965. [DOI] [PubMed] [Google Scholar]
- Melnychuk RM, Streblow DN, Smith PP, Hirsch AJ, Pancheva D, Nelson JA. Human cytomegalovirus-encoded G protein-coupled receptor US28 mediates smooth muscle cell migration through Galpha12. J Virol. 2004;78:8382–8391. doi: 10.1128/JVI.78.15.8382-8391.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DM, Zhang Y, Rahill BM, Waldman WJ, Sedmak DD. Human cytomegalovirus inhibits IFN-alpha-stimulated antiviral and immunoregulatory responses by blocking multiple levels of IFN-alpha signal transduction. J Immunol. 1999;162:6107–6113. [PubMed] [Google Scholar]
- Murphy AJ, Woollard KJ, Hoang A, Mukhamedova N, Stirzaker RA, McCormick SPA, Remaley AT, Sviridov D, Chin-Dusting J. High-Density Lipoprotein Reduces the Human Monocyte Inflammatory Response. Arterioscler Thromb Vasc Biol. 2008;28:2071–2077. doi: 10.1161/ATVBAHA.108.168690. [DOI] [PubMed] [Google Scholar]
- Noriega VM, Gardner TJ, Redmann V, Bongers G, Lira SA, Tortorella D. Human cytomegalovirus US28 facilitates cell-to-cell viral dissemination. Viruses. 2014;6:1202–1218. doi: 10.3390/v6031202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potena L, Frascaroli G, Grigioni F, Lazzarotto T, Magnani G, Tomasi L, Coccolo F, Gabrielli L, Magelli C, Landini MP, et al. Hydroxymethyl-glutaryl coenzyme a reductase inhibition limits cytomegalovirus infection in human endothelial cells. Circulation. 2004;109:532–536. doi: 10.1161/01.CIR.0000109485.79183.81. [DOI] [PubMed] [Google Scholar]
- Quinn PJ. Sphingolipid symmetry governs membrane lipid raft structure. Biochim Biophys Acta. 2014;1838:1922–1930. doi: 10.1016/j.bbamem.2014.02.021. [DOI] [PubMed] [Google Scholar]
- Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J Virol. 2006;80:710–722. doi: 10.1128/JVI.80.2.710-722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez V, Dong JJ. Alteration of lipid metabolism in cells infected with human cytomegalovirus. Virology. 2010;404:71–77. doi: 10.1016/j.virol.2010.04.026. [DOI] [PubMed] [Google Scholar]
- Simpson-Holley M, Ellis D, Fisher D, Elton D, McCauley J, Digard P. A functional link between the actin cytoskeleton and lipid rafts during budding of filamentous influenza virions. Virology. 2002;301:212–225. doi: 10.1006/viro.2002.1595. [DOI] [PubMed] [Google Scholar]
- Soderberg-Naucler C, Rahbar A, Stragliotto G. Survival in patients with glioblastoma receiving valganciclovir. N Engl J Med. 2013;369:985–986. doi: 10.1056/NEJMc1302145. [DOI] [PubMed] [Google Scholar]
- Stinski MF. Synthesis of proteins and glycoproteins in cells infected with human cytomegalovirus. J Virol. 1977;23:751–767. doi: 10.1128/jvi.23.3.751-767.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streblow DN, Soderberg-Naucler C, Vieira J, Smith P, Wakabayashi E, Ruchti F, Mattison K, Altschuler Y, Nelson JA. The human cytomegalovirus chemokine receptor US28 mediates vascular smooth muscle cell migration. Cell. 1999;99:511–520. doi: 10.1016/s0092-8674(00)81539-1. [DOI] [PubMed] [Google Scholar]
- Sviridov D, Bukrinsky M. Interaction of pathogens with host cholesterol metabolism. Curr Opin Lipidol. 2014;25:333–338. doi: 10.1097/MOL.0000000000000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sviridov D, Fidge N, Beaumier-Gallon G, Fielding C. Apolipoprotein A-I stimulates the transport of intracellular cholesterol to cell-surface cholesterol-rich domains (caveolae) Biochem J. 2001;358:79–86. doi: 10.1042/0264-6021:3580079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafilou M, Miyake K, Golenbock DT, Triantafilou K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J Cell Sci. 2002;115:2603–2611. doi: 10.1242/jcs.115.12.2603. [DOI] [PubMed] [Google Scholar]
- Vedhachalam C, Duong PT, Nickel M, Nguyen D, Dhanasekaran P, Saito H, Rothblat GH, Lund-Katz S, Phillips MC. Mechanism of ATP-binding Cassette Transporter A1-mediated Cellular Lipid Efflux to Apolipoprotein A-I and Formation of High Density Lipoprotein Particles. J Biol Chem. 2007;282:25123–25130. doi: 10.1074/jbc.M704590200. [DOI] [PubMed] [Google Scholar]
- Vischer HF, Hulshof JW, de Esch IJP, Smit MJ, Leurs MJ. Virus-encoded G-protein-coupled receptors: constitutively active (dys)regulators of cell function and their potential as drug target. Ernst Schering Foundation Symposiums Proceedings; Springer-Verlag; 2007. pp. 187–209. [DOI] [PubMed] [Google Scholar]
- Vischer HF, Siderius M, Leurs R, Smit MJ. Herpesvirus-encoded GPCRs: neglected players in inflammatory and proliferative diseases? Nat Rev Drug Discov. 2014;13:123–139. doi: 10.1038/nrd4189. [DOI] [PubMed] [Google Scholar]
- Zhu H, Cong JP, Shenk T. Use of differential display analysis to assess the effect of human cytomegalovirus infection on the accumulation of cellular RNAs: induction of interferon-responsive RNAs. Proc Natl Acad Sci U S A. 1997;94:13985–13990. doi: 10.1073/pnas.94.25.13985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Quyyumi AA, Norman JE, Csako G, Epstein SE. Cytomegalovirus in the pathogenesis of atherosclerosis: the role of inflammation as reflected by elevated C-reactive protein levels. J Am Coll Cardiol. 1999;34:1738–1743. doi: 10.1016/s0735-1097(99)00410-6. [DOI] [PubMed] [Google Scholar]
- Zhuang L, Kim J, Adam RM, Solomon KR, Freeman MR. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J Clin Invest. 2005;115:959–968. doi: 10.1172/JCI200519935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.