

Abstract
PBX1-d is novel splice isoform of pre-B-cell leukemia homeobox 1 (PBX1) that lacks its DNA-binding and Hox-binding domains, and functions as a dominant negative. We have shown that PBX1-d expression in CD4+ T cells is associated with systemic lupus erythematosus (SLE) in a mouse model as well as in human subjects. More specifically, PBX1-d expression leads to the production of autoreactive activated CD4+ T cells, a reduced frequency and function of Foxp3+ regulatory T (Treg) cells and an expansion of follicular helper T (Tfh) cells. Very little is known about the function of PBX1 in T cells, except that it directly regulates the expression of miRNAs associated with Treg and Tfh homeostasis. In the present study, we show that PBX1 directly regulated the expression of CD44, a marker of T cell activation. Two PBX1 binding sites in the promoter directly regulated CD44 expression, with PBX1-d driving a higher expression than the normal isoform PBX1-b. In addition, mutations in each of the two binding sites had different effects of PBX1-b and PBX1-d. Finally, we showed that an enhanced recruitment of co-factor MEIS by PBX1-d over PBX1-b, while there was no difference for co-factor PREP1 recruitment. Therefore, this study demonstrates that the lupus-associated PBX1-d isoform directly transactivates CD44, a marker of CD44 activation and memory, and that it has different DNA binding and co-factor recruitment relative to the normal isoform. Taken together, these results confirm that PBX1 directly regulates genes related to T cell activation and show that the lupus-associated isoform PBX1-d has unique molecular functions.
Keywords: Lupus, PBX1, CD44, T cells
1. Introduction
The pre-B-cell leukemia homeobox 1 (Pbx1) gene has been identified as the lupus susceptibility gene corresponding to the Sle1a1 locus in the MZM2410 mouse model (Cuda et al., 2012). This locus leads to the production of activated autoreactive histone-specific CD4+ T cells in a T-cell intrinsic manner (Chen et al., 2005); Cuda et al., 2012); Cuda et al., 2007). Sle1a1 also leads to a reduction of the frequency and function of Foxp3+ regulatory T cells (Tregs) (Choi et al., 2016); Cuda et al., 2007) as well as an expansion of the follicular helper T cell compartment in the mLN (Tfh) (Choi et al., 2016). These three phenotypes are key features of lupus pathogenesis (Craft, 2012); Mohan et al., 1993); Ohl and Tenbrock, 2015). It is therefore important to understand how Pbx1 is responsible for these events and what molecular interactions drive the observed phenotypes in CD4+ T cells.
Pbx1 is a member of the TALE family of homeodomain-containing transcription factors that modulates the DNA-binding function of Hox proteins (Mann and Chan, 1996). (Berkes et al., 2004) (Longobardi et al., 2014). Pbx1 plays a central role during development and organogenesis by integrating multiple signals through its interaction with numerous partners (Laurent et al., 2008), including Meis and Prep1 TALE proteins that regulate chromatin remodeling and co-activator access (Berkes et al., 2004) (Longobardi et al., 2014). In the development of the immune system, Pbx1 is required to maintain the self-renewal of hematopoietic stem cells (Ficara et al., 2008), restraining myeloid maturation to preserve the differentiation potential of lymphoid progenitors (Ficara et al., 2013). Pbx1-deficient embryonic stem cells fail to generate common lymphoid progenitors, resulting in the absence of B and NK cells, as well as an impaired T cell development (Sanyal et al., 2007). Accordingly, we have shown that mesenchymal stem cells (MSC) from mice expressing Sle1a1 show an accelerated lineage differentiation and expression of innate inflammatory genes, as well as an impaired immunoregulatory capacity (Lu et al., 2015), which corresponds to defects reported in MSCs from lupus patients as well as lupus-prone mice (Collins and Gilkeson, 2013). These complex regulatory networks regulated by reflect the ability of Pbx1 complexed with either Meis or Prep1 to recruit a wide array of Hox and non-Hox co-factors, many of which vary between cell types, and to direct either gene activation or repression (Laurent et al., 2008).
Contrary to B cells (Sanyal et al., 2007), the function of Pbx1 in T cells is unknown, except for the requirement of Prep1/Pbx1 heterodimer DNA binding for thymic development of double negative T cells (Penkov et al., 2008). Out of the seven protein-coding splice isoforms known for Pbx1 in mice, CD4+ T cells express Pbx1-b, which lacks exons 7 and 9 that are present in the full-length Pbx1-a (Cuda et al., 2012). In addition, NZM2410 and Sle1a1 CD4+ T cells express a novel splice isoform, Pbx1-d, which lacks exons 6 and 7 that encode the DNA and a HOX binding domains, respectively (Cuda et al., 2012). Pbx1 amino acid sequence is identical between human and mouse, and we found that PBX1-d is expressed at a significantly higher frequency in the CD4+ T cells of lupus patients as compared to healthy controls, validating the role of this susceptibility allele in lupus (Cuda et al., 2012). Pbx1-d is the only known Pbx1 isoform lacking the DNA binding domain, suggesting that it functions as a dominant negative (DN) binding partner with reduced transcriptional function. We have validated the DN function of Pbx1-d showing that it is equivalent to a Pbx1 knock-down construct in activating MSC differentiation (Sengupta et al., 2012). At the single gene level, the DN function of Pbx1-d was also validated, both as a transcriptional activator of Sox3 in MSCs and a transcriptional repressor of CD44 in Jurkat T cells (Sengupta et al., 2012). We have started to characterize the molecular mechanism by which Pbx1 regulates T cell function, and shown that it directly transactivates the expression of miR-10a, miR-21, and miR-155, three miRNAs that have been implicated in Treg and Tfh homeostasis (Choi et al., 2016). We performed the current study to further define the role of Pbx1 in regulating T-cell specific genes and to assess the ability of the Pbx1-d isoform to recruit the Meis and Prep1 co-factors.
2. Materials and methods
2.1. CHiP-Seq and CHiP-qPCR
Chromatin immunoprecipitation was performed on Jurkat T cells with a CHiP assay kit (Milllipore, Billerica, MA, USA), according to manufacturer’s instructions with an anti-PBX1 polyclonal antibody (P-20, Santa Cruz, CA, USA) or rabbit IgG (Santa Cruz) as negative control in triplicates. The final libraries (average size 350 bp) were quantitated with the Kapa SYBR Fast qPCR reagents (Kapa Biosystems, Wilmington, MA, USA) with an ABI7900HT real-time PCR system (Life Tech., Carlsbad, CA, USA). In preparation for sequencing, barcoded libraries were pooled equimolarly, and diluted to 9 pM for cluster generation on the cBOT (Illumina, San Diego, CA, USA). Samples were sequenced on a single flowcell lane on the HiSeq2000 instrument, using a 2x101 cycle multiplex, paired-end configuration. A typical sequencing run in the HiSeq2000 produced 300–400 million paired-end reads per lane. For ChIP sequencing 50–100 million reads provided sufficient depth for analysis. Peak calling was performed using CisGenome (www.biostat.jhsph.edu/~hji/cisgenome/). Data was averaged between the three replicates precipitated with anti-PBX1 or IgG control and compared by t tests. For CHiP-PCR experiments, 107 Jurkat cells were washed 2 times with cold PBS, and chromatin immunoprecipitation was performed following the cross-linking chromatin immunoprecipitation protocol (http://www.abcam.cn) with the same antibodies as for the CHiP-Seq experiment. Putative PBX1 binding sites in the CD44 intron 1 and promoter were identified using Jaspar3 (http://jaspar.genereg.net/) set at a 75%score threshold. PCR primers are listed in Table S1 as CD44_ChIP P1, P2 and I1. SYBR® Green Supermix (Bio-Rad, Hercules, CA, USA) was used for quantification. Results were expressed as fold enrichment relative to the IgG control. POL2 binding to GAPDH was used as positive control.
2.2. Luciferase assays
The CD44 promoter region containing both P1 and P2 putative PBX1 binding sites (Fig. 1) was amplified with the CD44-PROM primers listed in Table S1, and the 912 bp fragment was ligated into the pGL4.23 luciferase reporter vector (Promega, Madison, WI, USA). HEK 293 cells were transfected with the CD44_PROM-pGL4.23 along with 0–1000 ng DNA of PBX1-b or PBX1-d expression plasmids using Lipofectamine® 2000 (Life Tech.). After 48 h, cells were lysed and the firefly and Renilla luciferase activities were measured using Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instruction. The results were presented as the ratio of Renilla to firefly luciferase activities normalized to the values for cells that were not transfected with PBX1 plasmids.
Fig. 1. PBX1 binds multiple sites in the CD44 gene.

(a) Map of the CD44 gene with the location of the putative PBX1 binding sites in intron 1 (I1) and the promoter (P). (b) Details of two putative promoter PBX1 binding sites (P1 and P2) as well as their corresponding PBX1 binding sequences in upper case. (c) Chromatin from Jurkat T cells was immunoprecipated with antibody against PBX1 or IgG control, and amplified by qPCR with primers flanking each of the 2 promoter sites and the intron 1 site. A positive control (Ctrl) was provided by immunoprecipitation with a POL2 antibody and amplification of the POL2 binding site in the GAPDH promoter. CHiP-qPCR results are expressed in fold enrichment relative to the IgG negative control (N = 3). (d) Representative CHiP-PCR product at the P1 site.
2.3. CD44 message expression
RNA was extracted from HEK 293 cells transfected with 1 ug PBX1-b or PBX1-d expression plasmids using the RNeasy mini kit (Qiagen, Germantown, MD, USA). After cDNA synthesis from 5 ug total RNA using SuperScript First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA), real-time qPCR analysis of CD44 expression was performed using Bio-Rad’ SYBR® Green Supermix and its expression was normalized to GAPDH. Primer sequences are listed in Table S1 as CD44_qRT-PCR. Relative expression was calculated using the 2−ΔΔCt method and normalized to the RQ expression in cells transfected with an empty vector.
2.4. Flow cytometry
Jurkat T cells stably transfected with a lentiviral vector (LV) co-expressing either PBX1-b or PBX1-d and GFP (Sengupta et al., 2012) were stained with an anti-human CD44-Phycoerythrin (PE)-conjugated antibody (IM7, eBioscience, San Diego, CA, USA) or rat IgG2b-PE isotype control. At least 50,000 events were acquired per sample on a FACSCalibur cytometer (BD Biosciences, San Jose, CA, USA). Dead cells were excluded based on forward and side scatter characteristics. Analysis was conducted with the FCSExpress software (De Novo Software, Glendale, CA, USA) comparing CD44 expression in GFP+ and GFP− cells.
2.5. Site directed mutagenesis
The PGL4.23 plasmid containing the two PBX1 P1 and P2 binding sites in the CD44 promoter was amplified by the Q5® high fidelity DNA polymerase (New England Biolabs, Ipswich, MA) with either the Q5CD44M1 primers introducing a BafI restriction site in P1, or the Q5CD44M2 primers introducing a HpyCH4V restriction site in P2. An analysis with Jaspar3 predicted that the introduced mutations abrogated PBX1 binding. The primer sequences are listed in Table S1. The PCR fragments were cloned into pGL4.23 and mutations were confirmed by restriction enzyme analysis and Sanger sequencing.
2.6. Immunoprecipitation and Western blotting
Immunoprecipitation (IP) and Western blotting (WB) was performed on two types of cells: 1) HEK 293 cells transfected with 1 ug PBX1-b or PBX1-d expression plasmids, along with FLAG-tagged MEIS or PREP-1 (a kind gift from Dr. F. Blasi, IFOM, Milano, Italy) and 2) Jurkat T cells transfected with either LV-PBX1-b-GFP or LV-PBX1-d-GFP (Sengupta et al., 2012). Cells were lysed in lysis buffer (150 mm NaCl, 1% NP-40, 50 μm Tris-HCl, pH 8.0) containing a protease inhibitor mixture (Roche, Branford, CT, USA) for 20 min on ice. Lysates were centrifuged at 12,000 rpm for 5 min at 4°C. Supernatants were pre-absorbed with 10% (v/v) of protein A-sepharose beads (Pierce, Waltham, MA, USA) for 1 h, then incubated with 3 μg/ml of IP antibody overnight at 4°C followed by incubation with 10% (v/v) of protein A-sepharose beads for 1 h at 4°C. The beads were then rinsed three times with the lysis buffer with protease inhibitor at 4°C before elution with an equal volume of SDS loading buffer by boiling for 5 min. 15 μl of each sample eluate was run on a 10% SDS-PAGE gel. The gel was then transferred to nitrocellulose membrane. Membranes were blocked with 20 mm Tris, pH 8.0, containing 4% (w/v) milk and 0.05% (v/v) Tween 20. WB antibodies were incubated for 1 h at room temperature. The antibodies used for IP and WB were as follows: anti-PBX1 (P-20), anti-MEIS1/2 (N-17, Santa-Cruz), and anti-FLAG (Sigma, St. Louis, MO, USA).
2.7. Statistical analysis
Statistical analyses were performed using the GraphPad Prism 7 software. Unless indicated, graphs show mean and standard deviation of the mean (SEM) for each group. For comparisons between two groups, paired and unpaired Student’s t-tests were used. 2-way ANOVA with Dunnett’s post-tests were used for multiple comparisons as appropriate. Each experiment was performed at least three times.
3. Results and Discussion
3.1. PBX1 binds to the CD44 promoter
We performed a CHiP-Seq analysis of Jurkat T cells and identified 125 binding sites for which the fold difference (FD) with the anti-PBX1 antibody over the IgG control was at least 2 fold and the p value less than 0.05 (Table S2). Pbx1 expression is low in primary T cells (Cuda et al., 2012) as well as in Jurkat T cells (Sengupta et al., 2012). A much higher number of PBX1 binding sites have been reported in cell lines in which PBX1 expression is high, such as > 20,000 binding sites in MCF7 breast cancer cells (Magnani et al., 2011) and > 2,000 promoter sites reported in OVCAR3 ovarian cancer cells (Thiaville et al., 2012). A high number of binding sites were also identified in mouse embryonic cortex primary cells expressing high levels of PBX (Golonzhka et al., 2015). Fewer binding sites could be expected in cell types such as T lymphocytes that express low levels of PBX1, as this protein may play a regulatory rather than an essential role. The PBX1 binding sites in Jurkat T cells included the PBX1 gene itself (FD = 2.12, p = 0.00519), as well as PAX3 (FD = 2.16, p = 0.012), which is a known target of PBX1 (Chang et al., 2008), and MEF2B (FD = 16.65, p = 0.0158), a gene involved in myogenesis, a process in which PBX1 plays a central role (Sagerstrom, 2004). Interestingly, PBX1 was also bound to the ESRRG locus (FD = 2.55, p = 3.67 × 10−5), and we have found that in the NZM2410 mouse model, reduced expression of Esrrg in CD4+ T cells was associated with an autoimmune phenotype (Perry et al., 2012). None of the genes revealed in this analysis were T cell-specific or associated with a specific T cell function, except for the activation marker CD44 (FD = 2.92, p = 0.015). We have previously shown that transfection of Jurkat T cells with either PBX1-D or PBX1 shRNA transactivated CD44 expression (Sengupta et al., 2012). For these reasons, we focused on characterizing the mechanisms by which PBX1 regulated CD44 expression. The PBX1 binding site identified by CHiP-Seq was located in intron 1 (Fig. 1a). In addition, two PBX1 binding sites, P1 and P2, were identified in the promoter (Fig. 1b). Only a modest binding was detected by CHiP-PCR at the I1 and P2 sites (Fig. 1c), but a strong binding was detected at the P1 site (Fig. 1c and d).
3.2. PBX1-d binding to the CD44 promoter enhances CD44 transcription
We performed luciferase assays to evaluate whether PBX1 binding to the CD44 promoter affected transcription, and to compare the activity of the prevalent PBX1 isoform in T cells, PBX1-b, to that of the SLE-associated DN PBX1-d isoform. The CD44 promoter containing the two PBX1 binding sites directing the expression of a luciferase construct transfected into 293 HEK cells in the presence of PBX1-b or PBX1-b. Both PBX1 isoforms transactivated the CD44 promoter (Fig. 2a). Evidence of the higher activity of PBX1-d was shown in the dose-response analysis of transcriptional yields to increasing amounts of PBX1-b and PBX1-d expression plasmids (Fig. 2b). In addition, PBX1-d transfection significantly enhanced the transcription of the endogenous CD44 gene in HEK 293 cells (Fig. 2c). Finally, we compared CD44 protein expression in Jurkat T cells transfected with PBX1-b-GFP or PBX1-d-GFP. Transfected cells with either isoform expressed more CD44 than their non-transfected GFP-negative counterparts, but cells transfected with PBX1-d expressed significantly more CD44 than cells transfected with PBX1-b (Fig. 2d). These results confirm that PBX1-d transactivates CD44 expression significantly more than the prevalent isoform, and demonstrate that PBX1 binding to the CD44 promoter contributes to this transactivation.
Fig. 2. PBX1-d binding to the CD44 promoter enhances transcription.

(a) Relative luciferase activity units (RU) in 293 HEK cells transfected with CD44_PROM PGL4.23 alone (Ctrl) or with 200 ng of PBX1-b or PBX1-d expression plasmids (N = 3, Dunnett’s multiple comparisons test). (b) Relative luciferase activity units in 293 HEK cells transfected with the CD44_PROM PGL4.23 and 0–1000 ng of PBX1-b or PBX1-d expression plasmids (N = 3, 2-way ANOVA). The same experiment was performed 3 times with similar results. (c) Endogenous CD44 gene expression in the HEK 293 cells transfected with CD44_PROM PGL4.23 alone (Ctrl) or with 1000 ng of PBX1-b or PBX1-d expression plasmids (N = 5, Dunnett’s multiple comparisons test). (d) Representative FACS plots showing CD44 expression in Jurkat T cells transfected with PBX1-b -GFP or PBX1-d-GFP. The graph shows the quantitation for four assays for each cell type. Either PBX1-b or PBX1-d GFP+-transfected cells expressed more CD44 than their non-transfected GFP− counterparts (paired t tests, p < 0.01). Comparison between GFP+ cells was performed with an unpaired t test *: p < 0.05; **: p < 0.01, ***: p < 0.001.
3.3. PBX1-b and PBX1-d have different requirements for the P1 and P2 promoter binding sites
To define the relative requirements of the P1 and P2 binding sites by the PBX1-b and -d isoforms, each binding site was mutated individually (Fig. 3a). Luciferase assays with 200 ng of either PBX1 expression plasmid showed that the mutation in P1 significantly reduced transactivation by PBX1-b but not PBX1-d (Fig. 3b). At this plasmid concentration, the mutation in P2 had no effect with either isoform. A dose-response analysis confirmed that PBX1-b transactivation was significantly reduced in the absence of P1 binding, while it had little effect on PBX1-d transactivation (Fig. 3c). The same dose response analysis confirmed that the abolition of P2 binding has little effect on PBX1-b transactivation, but it significantly increased PBX1-d transactivation in the presence of higher doses of expression plasmid (Fig. 3d). It is possible speculated that the mutation in the P2 promoter prevented PBX1-b binding to fully potentiate transactivation by PBX1-d. It should be noted that at all doses that were tested, transactivation by PBX1-d was significantly higher than transactivation by PBX1-b, for either P1 or P2 mutants as well as for the unmutated WT sequence (Fig. 3c and d). Although PBX1-d does not have a DNA binding domain, it can still bind DNA as a heterodimer complex with MEIS, since MEIS and PBX1 share an identical consensus binding site (Penkov et al., 2013). The PBX1 binding sequence requirement is complex and entirely depends on its co-factors, as the sequence for PBX1 binding alone has not been defined (Longobardi et al., 2014). PBX1-b and PBX1-d share the same sub-optimal HOX-binding ability (Di Rocco et al., 1997), but the reliance of PBX1-d on cofactors for DNA binding may direct PBX1-d complexes to different binding sites than PBX1-b complexes, which is suggested by our results with the CD44 promoter.
Fig. 3. Effects of mutations in the P1 and P2 promoter binding sites on CD44 transactivation by PBX1-b and PBX1-d.

(a) From top to bottom: consensus PBX1 binding site sequence, promoter P1 WT and mutated (M1) sequences, and promoter P2 WT and mutated (M2) sequences. The arrows indicate the location of the restriction sites (BaflI and HpyCH4V) created by the mutations. The predicted binding sequences are in upper case and the mutated nucleotides are in bold. (b) Relative luciferase activity units (RU) in 293 HEK cells transfected with WT, M1 or M2 mutated CD44_PROM PGL4.23 in the presence of 200 ng of PBX1-b or PBX1-d expression plasmids (N = 3, Dunnett’s multiple comparisons test). Relative luciferase activity units in 293 HEK cells transfected with the M1 (c), M2 (d) or the WT CD44_PROM PGL4.23 and 0–1000 ng of PBX1-b or PBX1-d expression plasmids (N = 3, 2-way ANOVA between WT and M1 or M2 for each PBX1-b or PBX1-d transduced cells). *: p < 0.05; **: p < 0.01.
3.4. PBX1-d enhances MEIS recruitment
We compared the interactions of PBX1-b and -d isoforms with their binding partner MEIS and PREP1. Both PBX1-b and PBX1-d were associated with MEIS in Jurkat T cell lines stably transfected with either isoform. However, PBX1-d pulled down more MEIS than PBX1-b did (Fig. 4a) and conversely, MEIS pulled down more PBX1-d than PBX1-b (Fig. 4b). PREP1 was not detected in Jurkat T cells by WB (data not shown). This is consistent with PREP1 being preferentially involved in the transcriptional regulation of house-keeping genes, while MEIS/PBX1 complexes transactivate developmentally regulated genes (Penkov et al., 2013), if we postulate that effector T cell differentiation programs share some features of the embryonic developmental programs.. To better compare recruitment of MEIS and PREP1 to the PBX1-b and -d isoforms, we co-transfected HEK 293 cells with PBX1-b or PBX1-d, and either MEIS-FLAG or PREP-FLAG. Consistent with the results obtained in Jurkat T cells, there was more PBX1-d/MEIS than PBX1-b/MEIS complexes (Fig. 4c). In contrast, there was no difference between the amount of PREP complexed with either PBX1 isoform (Fig. 4d). These results suggest that PBX1-d enhances the recruitment of MEIS as compared to the PBX1-b isoform. Drastically different recruitments of co-activators and co-repressor have been found between the Pbx1a and Pbx1-b isoforms (Asahara et al., 1999). Our results suggest that PBX1-b and PBX1-d not only differ by their binding site preferences but also by the ability to recruit co-factors.
Fig. 4. PBX1-d enhances MEIS recruiting.
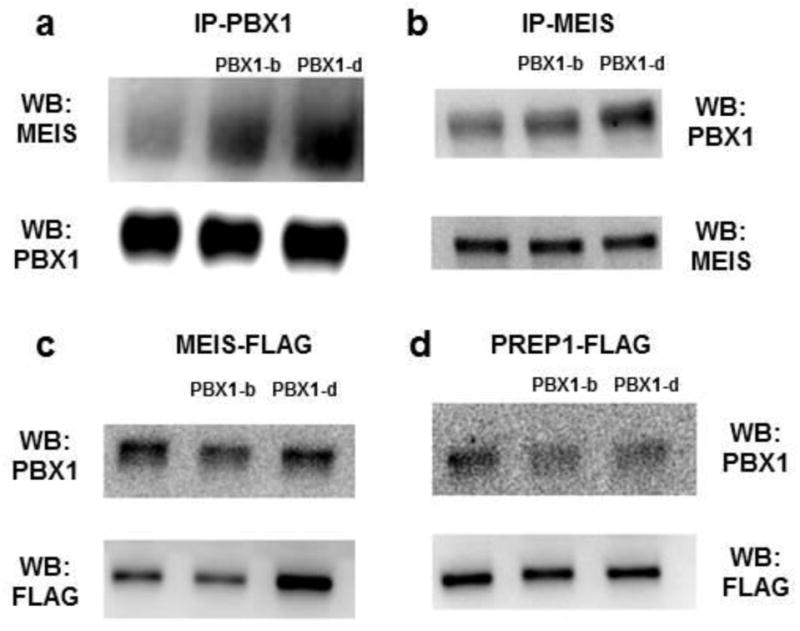
(a) Lysates from Jurkat T cells either untouched, or transfected with PBX1-b or PBX1-b were immunoprecipitated with anti-PBX1, then immunoblotted with anti-MEIS, or anti-PBX1 as control. (b) Lysates from Jurkat T cells either untouched, or transfected with PBX1-b or PBX1-b were immunoprecipitated with anti-MEIS, then immunoblotted with anti-PBX1, or anti-MEIS as control. 293 HEK cells were transduced with MEIS-FLAG (c) or PREP1-FLAG (d), along with either nothing else, PBX1-b or PBX1-d. Lysates were immunoprecipitated with an anti-FLAG antibody, then immunoblotted with either anti-PBX1 or anti-FLAG as control. Each experiment was performed as least three times.
Supplementary Material
Highlights.
PBX1-d is novel splice isoform of PBX1 that is expressed in lupus CD4+ T cells.
PBX1 binds to the CD44 promoter and directly regulates its expression.
PBX1-d enhances CD44 expression relative to PBX1-b, the normal PBX1 isoform
PBX1-d and PBX1-b have different DNA binding and co-factor recruitment requirement.
Acknowledgments
We thank Dr. F. Blasi, Fondazione Istituto FIRC di Oncologia Molecolare (IFOM), Milano, Italy, for the MEIS and PREP expression plasmids. This study was support by the R01 AI045050 grant from the NIH to LM.
Footnotes
Conflicts of interest
The authors have no conflict of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Asahara H, Dutta S, Kao HY, Evans RM, Montminy M. Pbx-Hox heterodimers recruit coactivator-corepressor complexes in an isoform-specific manner. Mol Cell Biol. 1999;19:8219–8225. doi: 10.1128/mcb.19.12.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkes CA, Bergstrom DA, Penn BH, Seaver KJ, Knoepfler PS, Tapscott SJ. Pbx marks genes for activation by MyoD indicating a role for a homeodomain protein in establishing myogenic potential. Mol Cell. 2004;14:465–477. doi: 10.1016/s1097-2765(04)00260-6. [DOI] [PubMed] [Google Scholar]
- Chang CP, Stankunas K, Shang C, Kao SC, Twu KY, Cleary ML. Pbx1 functions in distinct regulatory networks to pattern the great arteries and cardiac outflow tract. Development. 2008;135:3577–3586. doi: 10.1242/dev.022350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Cuda C, Morel L. Genetic determination of T cell help in loss of tolerance to nuclear antigens. J Immunol. 2005;174:7692–7702. doi: 10.4049/jimmunol.174.12.7692. [DOI] [PubMed] [Google Scholar]
- Choi SC, Hutchinson TE, Titov AA, Seay HR, Li S, Brusko TM, Croker BP, Salek-Ardakani S, Morel L. The lupus susceptibility gene Pbx1 regulates the balance between follicular helper T cell and regulatory T cell differentiation. J Immunol. 2016;197:458–469. doi: 10.4049/jimmunol.1502283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins E, Gilkeson G. Hematopoetic and mesenchymal stem cell transplantation in the treatment of refractory systemic lupus erythematosus - Where are we now? Clin Immunol. 2013 doi: 10.1016/j.clim.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol. 2012;8:337–347. doi: 10.1038/nrrheum.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuda CM, Li S, Liang S, Yin Y, Potula HH, Xu Z, Sengupta M, Chen Y, Butfiloski E, Baker H, Chang LJ, Dozmorov I, Sobel ES, Morel L. Pre-B cell leukemia homeobox 1 is associated with lupus susceptibility in mice and humans. J Immunol. 2012;188:604–614. doi: 10.4049/jimmunol.1002362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuda CM, Wan S, Sobel ES, Croker BP, Morel L. Murine lupus susceptibility locus Sle1a controls regulatory T cell number and function through multiple mechanisms. J Immunol. 2007;179:7439–7447. doi: 10.4049/jimmunol.179.11.7439. [DOI] [PubMed] [Google Scholar]
- Di Rocco G, Mavilio F, Zappavigna V. Functional dissection of a transcriptionally active, target-specific Hox-Pbx complex. EMBO J. 1997;16:3644–3654. doi: 10.1093/emboj/16.12.3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficara F, Crisafulli L, Lin C, Iwasaki M, Smith KS, Zammataro L, Cleary ML. Pbx1 restrains myeloid maturation while preserving lymphoid potential in hematopoietic progenitors. J Cell Sci. 2013;126:3181–3191. doi: 10.1242/jcs.125435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficara F, Murphy MJ, Lin M, Cleary ML. Pbx1 regulates self-renewal of long-term hematopoietic stem cells by maintaining their quiescence. Cell Stem Cell. 2008;2:484–496. doi: 10.1016/j.stem.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golonzhka O, Nord A, Tang PL, Lindtner S, Ypsilanti AR, Ferretti E, Visel A, Selleri L, Rubenstein JL. Pbx Regulates Patterning of the Cerebral Cortex in Progenitors and Postmitotic Neurons. Neuron. 2015;88:1192–1207. doi: 10.1016/j.neuron.2015.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent A, Bihan R, Omilli F, Deschamps S, Pellerin I. PBX proteins: much more than Hox cofactors. Int J Dev Biol. 2008;52:9–20. doi: 10.1387/ijdb.072304al. [DOI] [PubMed] [Google Scholar]
- Longobardi E, Penkov D, Mateos D, De Florian G, Torres M, Blasi F. Biochemistry of the tale transcription factors PREP, MEIS, and PBX in vertebrates. Develop Dyn. 2014;243:59–75. doi: 10.1002/dvdy.24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Zeumer L, Sorensen H, Yang H, Ng Y, Yu F, Riva A, Croker B, Wallet S, Morel L. The murine Pbx1-d lupus susceptibility allele accelerates mesenchymal stem cell differentiation and impairs their immunosuppressive function. J Immunol. 2015;194:43–55. doi: 10.4049/jimmunol.1401851. [DOI] [PubMed] [Google Scholar]
- Magnani L, Ballantyne EB, Zhang X, Lupien M. PBX1 genomic pioneer function drives ERalpha signaling underlying progression in breast cancer. PLoS Genet. 2011;7:e1002368. doi: 10.1371/journal.pgen.1002368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann RS, Chan SK. Extra specificity from extradenticle: the partnership between HOX and PBX/EXD homeodomain proteins. Trends Genet. 1996;12:258–262. doi: 10.1016/0168-9525(96)10026-3. [DOI] [PubMed] [Google Scholar]
- Mohan C, Adams S, Stanik V, Datta SK. Nucleosome: a major immunogen for pathogenic autoantibody-inducing T cells of lupus. J Exp Med. 1993;177:1367–1381. doi: 10.1084/jem.177.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohl K, Tenbrock K. Regulatory T cells in systemic lupus erythematosus. Eur J Immunol. 2015;45:344–355. doi: 10.1002/eji.201344280. [DOI] [PubMed] [Google Scholar]
- Penkov D, Mateos San Martin D, Fernandez-Diaz LC, Rossello CA, Torroja C, Sanchez-Cabo F, Warnatz HJ, Sultan M, Yaspo ML, Gabrieli A, Tkachuk V, Brendolan A, Blasi F, Torres M. Analysis of the DNA-binding profile and function of TALE homeoproteins reveals their specialization and specific interactions with Hox genes/proteins. Cell Rep. 2013;3:1321–1333. doi: 10.1016/j.celrep.2013.03.029. [DOI] [PubMed] [Google Scholar]
- Penkov D, Palazzolo M, Mondino A, Blasi F. Cytosolic sequestration of Prep1 influences early stages of T cell development. PloS one. 2008;3:e2424. doi: 10.1371/journal.pone.0002424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry DJ, Yin Y, Telarico T, Baker HV, Dozmorov I, Perl A, Morel L. Murine lupus susceptibility locus Sle1c2 mediates CD4+ T cell activation and maps to estrogen-related receptor gamma. J Immunol. 2012;189:793–803. doi: 10.4049/jimmunol.1200411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagerstrom CG. PbX marks the spot. Dev Cell. 2004;6:737–738. doi: 10.1016/j.devcel.2004.05.015. [DOI] [PubMed] [Google Scholar]
- Sanyal M, Tung JW, Karsunky H, Zeng H, Selleri L, Weissman IL, Herzenberg LA, Cleary ML. B-cell development fails in the absence of the Pbx1 proto-oncogene. Blood. 2007;109:4191–4199. doi: 10.1182/blood-2006-10-054213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta M, Liang S, Potula HHS, Chang LJ, Morel L. The SLE-associated Pbx1-d isoform acts as a dominant-negative transcriptional regulator. Genes and Immunity. 2012;13:653–657. doi: 10.1038/gene.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiaville MM, Stoeck A, Chen L, Wu RC, Magnani L, Oidtman J, Shih Ie M, Lupien M, Wang TL. Identification of PBX1 target genes in cancer cells by global mapping of PBX1 binding sites. PloS one. 2012;7:e36054. doi: 10.1371/journal.pone.0036054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.