


Abstract
DNA polymerases catalyze a metal-dependent nucleotidyl transferase reaction during extension of a DNA strand using the complementary strand as a template. The reaction has long been considered to require two magnesium ions. Recently, a third active site magnesium ion was identified in some DNA polymerase product crystallographic structures, but its role is not known. Using quantum mechanical/ molecular mechanical calculations of polymerase β, we find that a third magnesium ion positioned near the newly identified product metal site does not alter the activation barrier for the chemical reaction indicating that it does not have a role in the forward reaction. This is consistent with time-lapse crystallographic structures following insertion of Sp-dCTPαS. Although sulfur substitution deters product metal binding, this has only a minimal effect on the rate of the forward reaction. Surprisingly, monovalent sodium or ammonium ions, positioned in the product metal site, lowered the activation barrier. These calculations highlight the impact that an active site water network can have on the energetics of the forward reaction and how metals or enzyme side chains may interact with the network to modulate the reaction barrier. These results also are discussed in the context of earlier findings indicating that magnesium at the product metal position blocks the reverse pyrophosphorolysis reaction.
INTRODUCTION
The multiple isoforms of DNA polymerase (pol) found in nature are categorized among several families (1), but are believed to utilize a common two-metal ion mediated nucleotidyl transferase reaction to insert or remove nucleotides at the 3΄-end of a DNA strand (2). These reactions are template base directed, but occur with varying efficiencies depending on the specialized biological function of each DNA polymerase (3). The replicative DNA polymerases, for example, exhibit rapid nucleotide insertion for correct incoming nucleotides, whereas repair and translesion bypass DNA polymerases are significantly slower. This range in insertion rates for DNA synthesis among DNA polymerases presumably reflects differences in the respective interactions with substrates, cofactors and products. Another important factor is the microscopic equilibrium between the forward and reverse reactions of nucleotidyl transfer, since the observed DNA synthesis rate reflects both the forward and reverse reactions around the transition state as well as enzyme strategies (i.e. conformational changes) to facilitate the forward reaction.
The reaction path of the nucleotidyl transfer reaction by pol β was characterized in previous work (4,5). A bimetallic reaction scaffold was identified in the active site involving two closely positioned magnesium ions. One of these metals, the catalytic metal (Me(c)) or metal A, lowers the pKa of the primer terminus O3΄ while the other metal, the nucleotide-binding metal (Me(n)) or metal B, facilitates incoming nucleoside 5΄-triphosphate binding (5). In the beginning of the reaction path, deprotonation of O3΄ occurs with a low energy cost of ∼3 kcal/mol (6). Since this proton transfer step leads to a local energy minimum within the reaction path, the overall reaction proceeds through a two-phase pathway where the O3΄ proton transfer precedes the second phase of chemistry. The activation barrier for the overall reaction is ∼18 kcal/mol, and the experimentally measured rate constant that is in the range of 1–10 s−1 corresponds to an activation barrier of ∼16–17 kcal/mol (6). Both experimentally and computationally, Asp256 functions as the Lewis base in the proton transfer from O3΄ (7). Once the proton moves from O3΄ to Asp256, chemistry occurs through an in-line attack by the newly formed primer O3΄ oxyanion on Pα of the incoming nucleotide involving a penta-coordinated transition state. This transition state is unstable, and the reaction proceeds to completion with formation of the new O3΄–Pα bond and breaking of the substrate Pα–Oαβ bond, generating pyrophosphate (PPi) and the DNA strand extended by one deoxyribonucleotide monophosphate (dNMP) residue.
Recently, time-lapse crystallography studies of the Y-family pol η (8) and X-family pol β (9) have identified a transient product-associated magnesium ion in the active site (Figure 1B). This product metal (Me(p)) was not observed at the beginning of the reaction and dissociated from the enzyme prior to product dissociation. Importantly, the observed product metal coordinates oxygens of the reaction products, the inserted dNMP and PPi. Accordingly, there is a renewed interest in roles of divalent metal ions in the DNA polymerase nucleotidyl transfer reaction, since the reaction has long been thought to be mediated by two closely spaced magnesium ions (2) (Figure 1A). The product-associated metal has been proposed to play an essential chemical role for the forward reaction in pol η (8,10). In contrast, computational analyses suggested that the role of this product metal in pol β is to deter the reverse pyrophosphorolysis reaction, thereby driving the net reaction forward (11). Since these prior computational studies (6,7,12) did not consider the influence of this metal ion on the forward reaction, the first part of the present study evaluates the possibility of a direct catalytic role of the product-associated metal in the pol β forward reaction.
Figure 1.
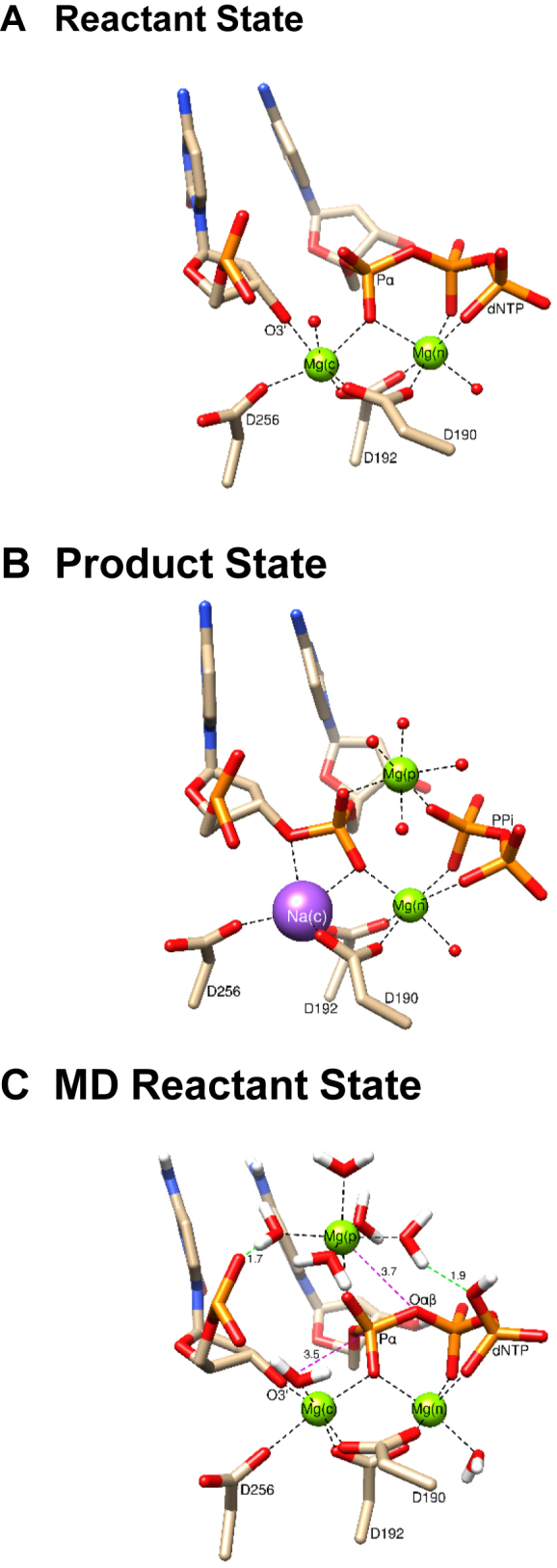
Active site structure of the ternary complexes of pol β. (A) Reactant state from crystallographic structure (PDB ID: 4KLE) having magnesium ions in the catalytic and nucleotide binding sites; (B) product state from crystallographic structure (PDB ID: 4KLG) having magnesium ions in the nucleotide and product metal sites and a sodium ion in the catalytic metal site; (C) reactant state from the final point of a lengthy well equilibrated MD trajectory calculation based on the structure in panel (A) where the product metal ion was initially introduced to the system based on the location of this metal in a crystallographic product structure (PDB ID: 4KLG). Important distances (Å) are indicated (red dashed lines) as are metal coordination (black dashed lines). Two water molecules solvating the product metal make hydrogen bonds with phosphate oxygens of the primer terminal nucleotide and Pγ of the incoming dCTP (green dashed lines).
MATERIALS AND METHODS
Computational procedure for QM/MM calculations
Initial structures for theoretical calculations were prepared using a high-resolution crystallographic structure of the ternary reaction complex (PDB ID: 4KLE) where both catalytic and nucleotide (dCTP) binding metal sites are occupied by magnesium ions (13). This structure was obtained by first crystallizing the ternary substrate complex with Ca2+ which does not support catalysis and then flash freezing the system after immersing the crystal in a solution containing Mg2+ ions for 10 s. Since the reaction was allowed to proceed for only a short time, 70% of the system remains in a reactive substrate state and the rest is converted to product with PPi occupying the positions occupied by the corresponding atoms in dCTP. Using the product metal position from the crystal structure after 40 s of reaction (PDB ID: 4KLE), the product metal was introduced into the initial configuration of the system. Molecular dynamics (MD) simulations were carried out in a completely solvated aqueous medium after hydrogens and neutralizing sodium ions were added. Positions of all crystallographic water molecules were preserved initially. The total charge on dCTP was taken to be −3 with only one oxygen on the γ-phosphate protonated and this choice was due to the fact that the pKa value of H(dTTP)3− in solution is 6.52 (14). All MD trajectory calculations were carried out with the Amber12SB force-field using the PMEMD module of Amber.12 (15). Water molecules were represented by the TIP3P model (16). Long range interactions were treated with the particle mesh Ewald method (17). The ground metal ion (i.e. Mg2+ introduced at the potential product metal site in the initial setup) was not part of the constraint atomic assembly. After initial NPT trajectory at 10 K, to adjust the density of the system near 1.0 g/cm3, a 20 ns constant volume/constant temperature (T = 300 K) equilibrium simulation with the sequentially decreasing harmonic constraint force constants (from 50 to 0.1 kcal/mol/nm) applied to the protein, DNA and metal ions in the crystallographic structure ensures that the system coordinates represent a pre-catalytic state. Prior to QM/MM calculations, we optimized several configurations selected from MD simulations and used the lowest energy system as the starting configuration for the reaction path calculation; other configurations are within 3 kcal/mol to this lowest energy conformation. A two metal system originated from this system where the ground metal and a distal water molecule exchange their positions and the geometry re-optimized.
The QM/MM systems were prepared from the final optimized structures of the above systems. The quantum region included parts of Asp190, Asp192 and Asp256, the primer terminal nucleotide including the 5΄-phosphate group (excluding the base), the incoming nucleotide triphosphate (excluding the base), and the three metal ions in the catalytic, nucleotide and product metal positions. In addition, there were eight water molecules solvating the metal ions included in the QM region. This system contained 100 QM atoms; in the two metal system, there were 99 QM atoms. Atoms within 10 Å of the quantum atoms were treated using the Amber force field and allowed to move. The rest of the atoms remained frozen during optimizations. Each system contained over 10 000 atoms in the MM region.
The reaction scheme for bond formation (and bond breaking) was studied using the hybrid QM/MM potential with the ONIOM(MO:MM) framework (18) implemented in the Gaussian-09-D1 (19). The QM region was treated using B3LYP exchange-correlation function and 6-31g* basis set. The classical region was handled using the Amber ff12SB force-field (15). Calculations were performed within the electronic embedding scheme (18,19) to accommodate the polarization of the QM region by the partial charges in the MM region. The QM/MM boundary is treated with the pseudo-atom approach (18) when the boundary involves a covalent bond and in the present work, each system consisted with such such pseudo-atoms. The only reaction coordinate present in the current protocol was the distance between the α-phosphorous atom of the incoming nucleotide triphosphate and the primer terminus 3΄-oxygen atom, since the path calculation was started with the O3΄-proton transferred to Asp256 in the optimized structures.
Protein purification and crystallization
Human wild-type pol β was overexpressed in Escherichia coli and purified as previously described (20). Binary complex crystals with a templating guanine in a one-nucleotide gapped DNA were grown as previously described (21). The time-lapse crystallography approach was performed as described previously to generate the inserted Sp-dCTPαS product complex and briefly summarized here (9). A binary pol β:DNA complex crystal was soaked with Sp-dCTPαS (Trilink) in the presence of calcium for 30 min and then transferred to a cryo-solution containing 200 mM MnCl2 to initiate the reaction. After 120 s, the reaction was stopped by flash freezing and data collected at the home source, 1.54 Å to observe the anomalous signal after phasing. Data were processed and scaled using the HKL2000 software (22). An initial model was determined using 2FMS (PDB ID), refinement using PHENIX, and model building using Coot (23,24). Figure 3 was prepared in PyMol and all density maps were generated after performing simulated annealing (25).
Figure 3.
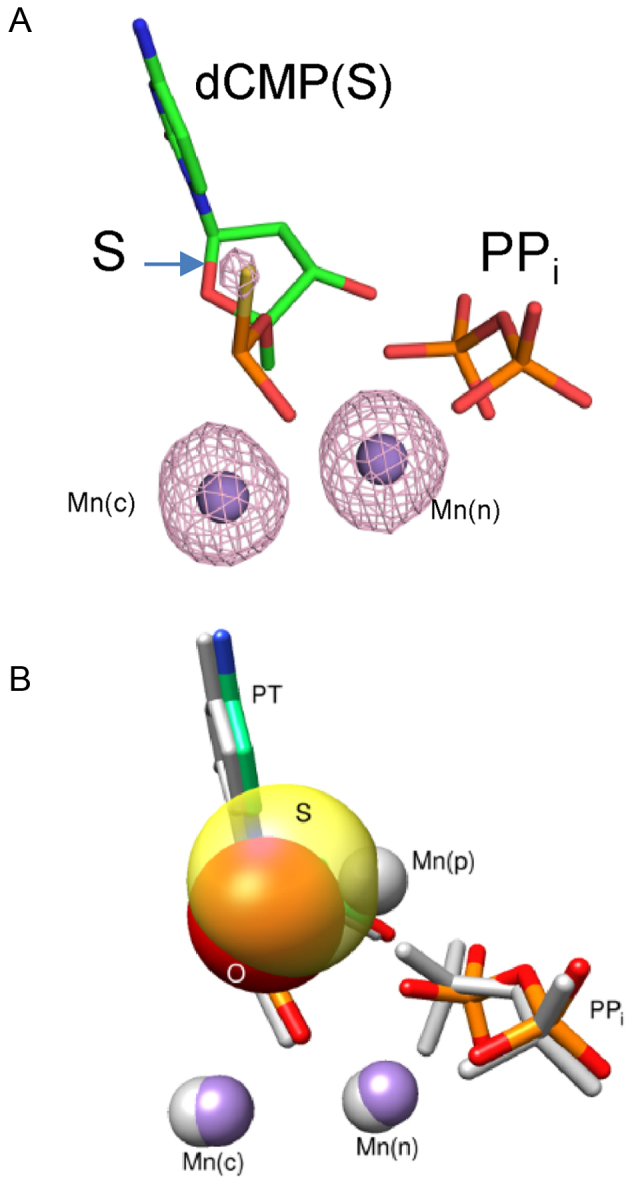
Crystallographic structure of a product complex formed after insertion of the Sp-isomer of dCTPαS. (A) Active site after insertion of the Sp-isomer of dCTPαS. An anomalous density map contoured to 5σ is shown indicating that Mn2+ occupies the catalytic and nucleotide metal binding sites. (B) Pol β active site structural comparison of the products of dCTP (PDB ID 4KLH) and Sp-dCTPαS (PDB ID 5U9H) insertion. A product metal is observed only during the incorporation of a natural nucleotide. Sulfur (transparent yellow) appears to exclude product metal binding. The equivalent oxygen of dCTP (i.e. pro-Sp) is red (appears orange in the overlap with the sulfur van der Waals radius). Three Mn2+ (gray) are observed during insertion of dCTP whereas only two manganese ions (purple) are detected during the insertion of the Sp-isomer of dCTPαS.
RESULTS AND DISCUSSION
Since a third magnesium ion observed during in crystallo structural analysis has been suggested to lower the activation barrier for the forward reaction (10), we undertook a computational approach to investigate the influence of this adjunct metal on the forward reaction. Previously, we showed that this metal deters pyrophosphorolysis (i.e. the reverse reaction) (11). The location of the product magnesium does not permit it to be present prior to breaking the Pα and Oαβ bond. In the current study, to locate the position for the third metal at the beginning of the reaction, we employed unconstrained molecular dynamics to obtain a set of coordinates with Mg2+ bound in a region proximal to the product metal site. This magnesium will be denoted as Mg(p) (i.e. product metal). By this process, the Mg2+ finds a stable position that is ∼2.0 Å from the observed product metal site and 3.7 Å from the bridging oxygen between Pα and Pβ of the incoming dNTP (Figure 1C). In this position, the pro-Sp oxygen of Pα coordinates Mg(p) along with a shell of five water molecules. One of these water molecules makes a strong hydrogen bond with a phosphate oxygen of the primer terminal nucleotide (1.7 Å) while another water molecule from this coordination shell makes a strong hydrogen bond with an oxygen on Pγ of the incoming nucleotide (1.9 Å). We initiated the forward reaction path calculations with the O3΄ proton located on Asp256, and in this optimized conformation, the O3΄–Pα distance is ∼3.5 Å.
Part A
QM/MM calculations with the three-magnesium ion system
Previous pol β QM/MM reaction path calculation (6,7,12) had shown that the bond to be broken (i.e. Pα–Oαβ bond) begins to stretch early in the reaction path and when the O3΄–Pα distance is ∼2.3 Å, the bond has been stretched by 0.2 Å (to 1.8 Å), and the penta-coordinated transition state begins to appear. With three magnesium ions, the reaction path yields an activation barrier between 16.6 and 18.1 kcal/mol (Figure 2A). A comparison of the reactant structure with the product structure fails to display differences in most atomic positions, except near PPi (Figure 2B). The new adjunct magnesium ion maintains its coordination with the five water molecules of the starting conformation (2.10–2.15 Å). The Mg(p)–Oαβ distance has shortened slightly in the product structure (from 3.7 to 3.5 Å) while two of the water molecules in the coordinating shell of Mg(p) establish hydrogen bonds with the newly created oxyanion of PPi (1.7–1.8 Å); the protons participating in these hydrogen bonds were 2.9 and 2.7 Å from Oαβ in the starting configuration. Finally, the QM/MM product structure (Figure 2B), indicates a breaking of several water shell hydrogen bonds in order to attain the conformation observed in the crystallographic product structure (Figure 1B) where the product magnesium directly interacts with the newly formed primer terminus and PPi.
Figure 2.

Energy profiles for the pol β nucleotidyl transfer reaction. (A) Profile obtained from QM/MM calculations with either two (blue) or three (red) magnesium ions. The reaction coordinate is chosen to be the distance between the primer terminal nucleophile O3΄ and Pα of the incoming dCTP; the distance is reduced from right to left. In the designated transition state region (TS), the dissociating Pα–Oαβ bond has been stretched by 0.2 Å over its equilibrium distance. Also, the reactant (RS) and product states (PS) are indicated. (B) The starting (gray) and final (color) configurations of the QM sub-system (100 atoms) in the three magnesium calculation. Magnesium ions are represented by solid spheres (gray or green). All quantum waters are shown. Key water and Oαβ interactions in the final state are indicated with red dashed lines. (C) The starting and final configurations of the QM sub-system (99 atoms) with two magnesium ions. A water interaction with Oαβ in the final state is highlighted (red dashed line).
The use of B3LYP/6-31G* facilitates direct comparison of the results from the present calculations with the reported data in the literature for similar systems with varying environments (6,7,12). It should be noted that single point calculations performed with diffuse and larger basis sets (ONIOM/(B3LYP/6-31+g*/Amber) and ONIOM(B3LYP/6-311++G**/Amber) resulted in quite comparable energy barriers of about ∼20 kcal/mol for this system (results not shown).
Comparative QM/MM calculations with the two-magnesium ion system
To further evaluate whether the presence of the Mg(p) alters the forward reaction, we preformed similar calculations with a corresponding two-metal system, i.e. containing the catalytic and nucleotide-binding magnesium ions. QM/MM calculations of the forward reaction had previously been performed for a two-metal system (6,7,12), and this earlier work included the catalytic and nucleotide-binding magnesium ions, but was with a restricted number of QM atoms. Those calculations did not, for instance, include the phosphate group of the primer terminus. Accordingly, we re-evaluated the pol β two-metal ion system with a larger number of atoms in the QM sub-system, so as to enable direct comparison with the corresponding three-metal system.
These calculations revealed a similar activation barrier as that found for the three-metal system, i.e. 17.5–18.6 kcal/mol (Figure 2A). Compared to the results with the three-metal system, the reaction path and product positions in the two-metal system were similar, except for a small number of atoms around PPi (Figure 2C). Several additional water molecules were included in the QM sub-system in this calculation to allow compatibility with the three-metal system and to maintain the hydrogen-bonding network that extends from the primer terminus phosphate and triphosphate of the incoming nucleotide. These added water molecules mimic the water molecules solvating Mg(p) in the three-metal system. Notably, none of these water molecules is in contact with Oαβ in the reactive state. After the reaction, a water molecule moves to make a strong hydrogen bond (1.6 Å) with the oxyanion on PPi. As in the three-metal system, the Pα–Oαβ bond distance (1.7 Å) expands to 1.8 Å as the O3΄–Pα distance contracts from 2.2 to 2.0 Å. Thus, a very similar transition region is observed for the two- and three-metal systems.
As the reaction path approaches the transition state (O3΄–Pα distance closes from 2.7 to 2.4 Å), the presence of Mg(p) assists the reaction (2–3 kcal/mol, Figure 2A). However, this advantage disappears as the system achieves the transition state. There are competitive events in the three-metal system, as Mg(p) struggles to maintain its octahedral coordination shell while adjustments are required in the atomic positions around the newly formed PPi due to its new electronic structure. In other words, there is a competition between the product metal and the PPi oxyanion for specific water molecules. This indicates that the energy gains seen at the earlier part of the reaction path are neutralized by these competing forces around the oxyanion site of PPi. Overall, the two- and three-metal systems give rise to similar activation barriers, and the adjunct product Mg2+ remains locked at its binding site and does not hasten nucleotidyl transfer.
Effect of an incoming phosphorothioate on the nucleotidyl transfer reaction
To experimentally evaluate a potential role of a divalent metal ion in the product metal position, we made use of an incoming dNTP phosphorothioate analog. Phosphorothioate nucleotide analogs, where a non-bridging oxygen on Pα is substituted with sulfur, have been used to determine reaction stereochemistry (26), metal coordination (26), and whether enzyme conformational steps limit the observed nucleotide insertion rate (26). In the second case, it is recognized that Mg2+ is thio-phobic preferring binding to oxygen rather than sulfur (27). In addition, the larger van der Waals radius of sulfur (1.89 Å) compared to that of oxygen (1.50 Å) (28) and the 30–35% longer phosphate–sulfur bond (29) physically limits metal ligand binding to the Sp-sulfur on Pα (Figure 3).
The observed rate of insertion of the Sp-isomer of dCTPαS is only 3-fold slower (<1 kcal/mol in activation energy) than dCTP (30) suggesting that the product metal is not essential for the forward reaction. Additionally, time-lapse crystallography fails to observe a product metal during insertion of Sp-dCTPαS (Table 1; Figure 3A). Figure 3 illustrates a structure of pol β incorporating dCTP (PDB ID: 4KLH) (9) superimposed with one that has Sp-dCMPαS inserted immediately after complete nucleotide insertion (i.e. the point at which product metal occupancy is the greatest) (9). From the anomalous electron density for manganese (Figure 3A), the product metal site is empty when dCTP(αS) is incorporated. Although a product metal is clearly observed (Figure 3B) during the incorporation of a natural nucleotide, sulfur appears to exclude product metal binding. The kinetic and structural results suggest that binding of a divalent product metal near the product metal site was not required for the forward reaction, and the results were consistent with the calculated activation barriers for the natural nucleotide (Figure 2A), indicating a lack of significant effect of the adjunct product metal on the forward reaction.
Table 1. Data collection and refinement statistics of a ternary pol β:DNA co-complex with the inserted Sp-isomer of dCTP(αS).
| Data collection | |
|---|---|
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 50.9, 80.0, 55.1 |
| α, β, γ (°) | 90, 109.1, 90 |
| Resolution (Å) | 50–1.85 |
| R sym or Rmergea (%) | 6.7 (54.1) |
| I/σI | 20.7 (2.1) |
| Completeness (%) | 99.7 (99.9) |
| Redundancy | 4.5 (3.0) |
| Refinement | |
| Resolution (Å) | 1.85 |
| No. of reflections | 67 715 |
| R work/ R free | 19.2/22.9 |
| No. of atoms | |
| Protein | 2673 |
| DNA | 649 |
| Water | 356 |
| B-factors (Å2) | |
| Protein | 33.2 |
| DNA/dCαS/PPV | 36.2/19.7/35.3 |
| Water | 39.7 |
| R.m.s deviations | |
| Bond length (Å) | 0.007 |
| Bond angles (°) | 0.9 |
| PDB ID | 5U9H |
aHighest resolution shell is shown in parentheses.
Part B
Effect of a sodium ion in the product metal site
Time-lapse crystallographic snapshots of pol β revealed that the product magnesium ion appeared at the end of the nucleotidyl transfer reaction path and strictly correlated with product formation (9), and as described above, computational analysis indicated that a magnesium ion in the nearby site plays no role in the forward reaction. Nevertheless, we further explored the influence of occupancy of the adjunct product metal site on the nucleotidyl transfer reaction. Previously, while analyzing the reverse reaction (pyrophosphorolysis) by pol β, we examined Na+ instead of Mg2+ at the product metal site (11). This use of Na+ softened the coordination sphere around the metal, capturing features of its potentially dynamic water network. As noted above, calculations in the presence of the tightly coordinated Mg2+, in fact, failed to yield a reverse reaction; however, changing the product metal from Mg2+ to Na+ resulted in a biologically viable reaction barrier. Thus, employing Na+ permitted coordinating water molecules to have greater freedom as the reaction proceeded, whereas Mg2+ resulted in structural rigidity at this position that retains solvating waters.
In this situation, when Na+ occupies the ground state product metal site, rather than Mg2+, the reaction path displays a significantly lower activation barrier (11.6–13.2 kcal/mol) (Figure 4A). The transition state in this case occurs with the O3΄-Pα distance between 2.3 and 2.1 Å. In the early part of the reaction path, the energy profile follows that of the three-magnesium system until the reaction approaches the transition state region. Around the transition state, the paths diverge with the Na+ case yielding a 5 kcal/mol lower energy barrier. Once again, there are minimal deviations in most QM atoms during the path except for water molecules near PPi (Figure 4B). The Na+ coordination distances of the water molecules are 2.3–2.5 Å, significantly longer than those of the three-magnesium system (∼2.1 Å) (Figure 5C). Only one water molecule coordinating Na+ makes a strong hydrogen bond (1.8 Å) with the oxyanion on PPi (Figure 4B); prior to product formation, this water was the nearest sodium-coordinating ligand.
Figure 4.

Energy profiles for the pol β nucleotidyl transfer reaction. (A) Profile obtained from QM/MM calculations with a sodium ion in the product metal site (green). The two- (blue) and three-metal (red) site profiles are also shown for comparison (see Figure 2). This system is used to account for an ion near the product metal binding site that could potentially exhibit the transient behavior during catalysis. The transition (TS, described in the text), reactant (RS), and product states (PS) are indicated. (B) The starting (gray) and final (color) QM sub-system configurations (100 atoms) of the three-metal system with a sodium ion at the product metal site are shown. Metal ions are represented by solid spheres (gray or green). The red dashed line represents a newly formed interaction between a Na(p)-bound water and Oαβ in the final state.
Figure 5.

Metal coordination distance variations along the reaction coordinate. (A) Coordination distances for the catalytic, (B) nucleotide binding, and (C) product metal sites. (D) Metal–metal distances along the reaction coordinates (top panel, Mg(c)–(Mg(n) for all cases; bottom panel, Mg(c)–M(p) and Mg(n)–M(p) with M being magnesium or sodium). In (A) and (B), the Mg(c)Mg(n), Mg(p) system is in the top panel, the Mg(c)Mg(n) system is in the middle panel, and the Mg(c)Mg(p)Na(p) system is in the bottom panel. The same color scheme for the top panel is followed for the other panels in (A–C).
Key coordination distances to the various metals along the reaction path are given in Figure 5 for all three systems. All three systems display the largest range in coordinating distances with Mg(c) even at the beginning of the reaction (1.9–2.5 Å), where the distance between Asp256(OD2) and Mg(c) is the greatest. Also, progression of the reaction results in the atoms coordinated with Mg(c) moving nearer to it, except O3΄ that moves away from Mg(c) during product formation. The deviation in coordinating distances is much narrower for Mg(n) (2.0–2.2 Å). Once products are formed, the coordinating ligand distances undergo dramatic changes for both Mg(c) and Mg(n) (Figure 5A–B). In contrast, the product Mg2+ displays the smallest variations in coordination distances (Figure 5C), and the water coordination shell remains rigid throughout the reaction path. When product metal site is occupied by Na+, variations and fluctuations are found for water molecules around this site. A shortening of the Mg(c)–Mg(n) distance (∼0.2 Å) is observed during the approach leading to the transition state of all three systems (Figure 5D). Assuming a 2+ charge on each magnesium ion, a simple application of Coulomb's law predicts a ∼20 kcal/mol increase in repulsion energy between the two magnesium ions, i.e. very small changes in the Mg(c)–Mg(n) distance results in large energy changes. For the system with Na+, variations in the distances for Mg(n)–Na(p) (decrease) and Mg(c)–Na(p) (increase) are observed, whereas the corresponding distances in the three-magnesium case do not change appreciably (Figure 5D).
Since Mg2+- and Mn2+-dependent nucleotide insertion rates are similar for pol β (31,32), the effect of replacing the product metal ion with manganese on the reaction barrier was evaluated. This was tested by replacing the magnesium at the product metal site with a manganese ion and then re-optimizing each point along the reaction path. No appreciable change in the reaction barrier was observed when a manganese ion occupied the product metal site when compared the three-magnesium profile (results not shown).
A water network around the product metal site modulates the reaction barrier
The calculated activation barriers for the cases considered are between 13 and 18 kcal/mol, and the reaction leads to the expected products, i.e. extended primer and PPi (Figure 4A). The two- and three-metal systems where the metal sites are occupied by magnesium ions generate nearly identical activation barriers (Figure 2A). However, when a monovalent metal ion with relaxed coordination properties is introduced into the ground state product metal site, a decrease in the activation barrier of ∼5 kcal/mol is observed (Figure 4A). This lower barrier provides support for the possible role of a metal in the product metal site in modulating DNA polymerase catalyzed nucleotidyl transfer reactions. An examination of the molecular architecture around the product metal site is useful in finding clues as to how the enzyme might modulate charge transfer near the leaving group.
In Figure 6, we focus on the initial reactant and final product metal coordination with an extended hydrogen bond and water network for each system. The five water molecules coordinating Mg(p) in the initial state are shown (Figure 6A, ∼2.1 Å). The remaining coordinating oxygen (pro-Sp of Pα, dNTP) is 2.0 Å away from this metal. Two of these water molecules also make hydrogen bonds with the oxygens of the primer terminus phosphate group. One of these oxygens also makes a hydrogen bond to a water solvating Mg(c). Another water, coordinating Mg(p), makes a hydrogen bond with a non-bridging oxygen of Pγ (dNTP). The nearest Mg(p)-coordinating water hydrogen bonds to Oαβ, that becomes the PPi oxyanion, is between 2.7 and 2.9 Å from the metal. A closer look at the product structure reveals that the coordination of Mg(p) is not altered and includes two water molecules strongly interacting with the primer terminal phosphate. The major difference between the reactant and product states is that two waters from the Mg(p) coordination shell now participate in hydrogen bonds with the PPi oxyanion. Importantly, one of these two water molecules that had coordinated Pγ (1.9 Å), now hydrogen bonds with an oxygen on Pβ giving rise to one of the two hydrogen bonds with the oxyanion. Since this represents an initial phase of product formation, there still exists a strong hydrogen bond network that bridges phosphate groups at the primer terminus and PPi via product state metal coordination.
Figure 6.

Hydrogen-bond network that connects the phosphate group on the primer terminus with the dNTP (or PPi). Involvement of the product metal in strengthening the network is apparent. (A) Mg(c)Mg(n)Mg(p), (B) Mg(c)Mg(n), and (C) Mg(c)Mg(n)Na(p). Hydrogen bonds are indicated in purple dashed lines and the product metal coordination is shown in blue.
In the two-magnesium scenario (Figure 6B), a network of water molecules links the primer terminus phosphate group with the incoming dNTP. This network becomes stronger during product formation. A water molecule within this network makes a stronger hydrogen bond (1.6 Å) during product formation with the oxyanion on PPi, neutralizing its charge. A similar interaction network between the primer terminus phosphate and dNTP is observed for the Mg(c)–Mg(n)–Na(p) system in the early stage of the reaction path (Figure 6C). In contrast, after the reaction is complete with Na(p), the water coordination distances surrounding sodium vary, suggesting a weaker interacting network compared to the other two systems; only a single water molecule makes a strong hydrogen bond with the oxyanion of PPi and this water has the shortest metal–ligand distance in the product state (2.3 Å).
A product metal site in other DNA polymerases
In ternary substrate complex crystallographic structures of A- and B-family DNA polymerases, a conserved lysine residue is located in the vicinity of the triphosphate of dNTP. The positively charged Nζ+ is found in the position occupied by the product metal identified in X- and Y-family polymerases (9). The distances from Lys(Nζ) to Oαβ of the incoming nucleoside triphosphate in crystallographic structures of A- and B-family enzymes are ∼2.9–4.1 Å (Supporting Information, Supplementary Table S1). In the optimized configurations of pol β, the Mg(p) and Na(p), are 3.7 and 4.0 Å to Oαβ, respectively. To examine the potential consequences of the presence of this lysine residue located in the vicinity of the incoming dNTP, we performed a separate QM/MM calculation with NH4+ at the product metal site as a reasonable approximation to model the influence of a lysine side chain. The resultant energy profile for the reaction path is shown in Figure 7. It is evident that the reaction occurs at a relatively low energy barrier, and the barrier occurs at an earlier point (2.4–2.5 Å) along the reaction path (Figure 7). In a comparison of the reactant and product state structures, this system has a more relaxed water structure solvating NH4+ than in the case of Mg(p) (Supporting Information, Supplementary Figure S1); the waters also exhibit larger coordination distances from the nitrogen (2.7–3.2 Å, Supporting Information, Supplementary Figure S2). Meanwhile, the coordination distances of the catalytic and nucleotide-binding metals remain similar to those in the other systems (Supporting Information, Supplementary Figure S3). As described in Supporting Information, charge variations are also quite comparable with the Na+ counterpart (Supplementary Figures S4–S6).
Figure 7.
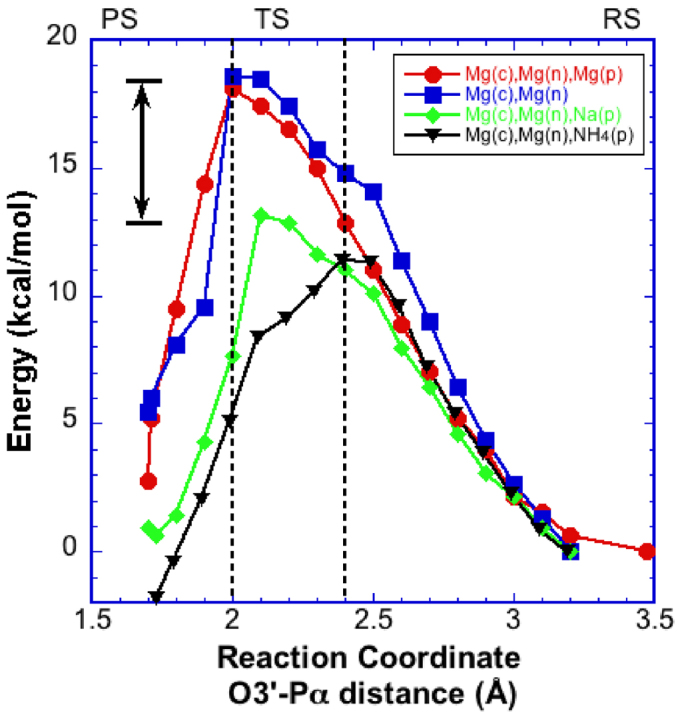
Energy profiles for the pol β nucleotidyl transfer reaction. The profile (black) calculated with an ammonium ion is used to mimic a charged lysine residue found in the position of Mg(p) (13). This lysine residue is conserved in A- and B-family DNA polymerases. The energy profiles of the other three systems are included to facilitate comparison.
As noted in Figure 7, reaction energies are also altered at different magnitudes, when different metals are present at the product metal site. The reaction energy is exoergic in the absence of the product metal, while the system with a magnesium ion at the product metal site, the reaction energy is decreased by ∼2 kcal/mol. In the system with a sodium ion at the product metal site, the reaction energy is neither endoergic nor exoergic, but in the model system with the product metal site occupied by an ammonium ion, the reaction energy is found to be endoergic, by ∼2 kcal/mol. However, these reaction energies correspond to the final stage of only the bond formation and further research is needed to fully estimate the reaction energy differences.
SUMMARY
QM/MM calculations reveal that the pol β nucleotidyl transfer forward reaction in a two-Mg2+ system proceeds with an energy barrier approximating the observed forward rate of insertion in solution. Interestingly, a virtually identical energy barrier, and reaction path, was observed with a three-Mg2+ system, including a Mg2+ bound near the product metal site. Thus, the product metal was neither required for nor able to facilitate the forward reaction. This observation was consistent with crystallography and kinetic experiments carried out with a phosphorothioate dCTP that deters Mg2+ and Mn2+ binding at the product metal site. On the other hand, the presence of a monovalent cation (e.g. Na+ or NH4+) at the product metal site resulted in a significant reduction in the energy barrier. This effect involved changes in a network of water molecules linking phosphates of the primer terminus and incoming dNTP. Taken together with previous QM/MM calculations for the reverse reaction, the product metal does not hasten the forward reaction, but it influences product formation by deterring the reverse reaction. Thus, the net effect of the product metal is to increase the chemical equilibrium constant by decreasing the rate of the reverse reaction.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Research Project [Z01-ES043010 to L.P., Z01-ES050158 and Z01-ES050159 to S.H.W] in the Intramural Research Program of the US National Institutes of Health, US National Institute of Environmental Health Sciences and in association with the US National Institutes of Health Grant [U19CA105010]. Funding for open access charge: Intramural Research Program of the US National Institutes of Health, US National Institute of Environmental Health Sciences.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bebenek K., Kunkel T.A.. Functions of DNA polymerases. Adv Protein Chem. 2004; 69:137–165. [DOI] [PubMed] [Google Scholar]
- 2. Beese L.S., Steitz T.A.. Structural basis for the 3΄-5΄ exonuclease activity of Escherichia-coli DNA-polymerase-I – a 2 metal-ion mechanism. EMBO J. 1991; 10:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beard W.A., Shock D.D., Vande Berg B.J., Wilson S.H.. Efficiency of correct nucleotide insertion governs DNA polymerase fidelity. J. Biol. Chem. 2002; 277:47393–47398. [DOI] [PubMed] [Google Scholar]
- 4. Pelletier H., Sawaya M.R., Kumar A., Wilson S.H., Kraut J.. Structures of ternary complexes of rat DNA polymerase beta, a DNA template-primer, and ddCTP. Science. 1994; 264:1891–1903. [PubMed] [Google Scholar]
- 5. Steitz T.A., Steitz J.A.. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:6498–6502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin P., Pedersen L.C., Batra V.K., Beard W.A., Wilson S.H., Pedersen L.G.. Energy analysis of chemistry for correct insertion by DNA polymerase beta. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:13294–13299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Batra V.K., Perera L., Lin P., Shock D.D., Beard W.A., Pedersen L.C., Pedersen L.G., Wilson S.H.. Amino acid substitution in the active site of DNA Polymerase beta explains the energy barrier of the nucleotidyl transfer reaction. J. Am. Chem. Soc. 2013; 135:8078–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakamura T., Zhao Y., Yamagata Y., Hua Y.J., Yang W.. Watching DNA polymerase eta make a phosphodiester bond. Nature. 2012; 487:196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Freudenthal B.D., Beard W.A., Shock D.D., Wilson S.H.. Observing a DNA polymerase choose right from wrong. Cell. 2013; 154:157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gao Y., Yang W.. Capture of a third Mg (2) (+) is essential for catalyzing DNA synthesis. Science. 2016; 352:1334–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Perera L., Freudenthal B.D., Beard W.A., Shock D.D., Pedersen L.G., Wilson S.H.. Requirement for transient metal ions revealed through computational analysis for DNA polymerase going in reverse. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E5228–E5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lin P., Batra V.K., Pedersen L.C., Beard W.A., Wilson S.H., Pedersen L.G.. Incorrect nucleotide insertion at the active site of a G:A mismatch catalyzed by DNA polymerase beta. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:5670–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Freudenthal B.D., Beard W.A., Shock D.D., Wilson S.H.. Observing a DNA polymerase choose right from wrong. Cell. 2013; 154:157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sigel H., Griesser R.. Nucleoside 5΄-triphosphates: self-association, acid-base, and metal ion-binding properties in solution. Chem. Soc. Rev. 2005; 34:875–900. [DOI] [PubMed] [Google Scholar]
- 15. Case D.A., Darden T.A., Cheatham T.E. III, Simmerling C.L., Wang J., Duke R.E., Luo R., Walker R.C., Zhang W., Merz K.M. et al. . 2012; San Francisco: University of California. [Google Scholar]
- 16. Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L.. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983; 79:926–935. [Google Scholar]
- 17. Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G.. A smooth particle mesh ewald method. J. Chem. Phys. 1995; 103:8577–8593. [Google Scholar]
- 18. Vreven T., Byun K.S., Komaromi I., Dapprich S., Montgomery J.A., Morokuma K., Frisch M.J.. Combining quantum mechanics methods with molecular mechanics methods in ONIOM. J. Chem. Theory Comput. 2006; 2:815–826. [DOI] [PubMed] [Google Scholar]
- 19. Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A. et al. . 2009; Wallingford, CT: Gaussian, Inc. [Google Scholar]
- 20. Beard W.A., Wilson S.H.. Purification and domain-mapping of mammalian DNA polymerase beta. DNA Replic. 1995; 262:98–107. [DOI] [PubMed] [Google Scholar]
- 21. Batra V.K., Beard W.A., Shock D.D., Krahn J.M., Pedersen L.C., Wilson S.H.. Magnesium-induced assembly of a complete DNA polymerase catalytic complex. Structure. 2006; 14:757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Otwinowski Z., Minor W.. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997; 276:307–326. [DOI] [PubMed] [Google Scholar]
- 23. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al. . PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D, Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D, Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 25. Schrödinger LLC. The AxPyMOL Molecular Graphics Plugin for Microsoft Powerpoint. 2010; Version 1.0, Schrodinger LLC. [Google Scholar]
- 26. Burgers P.M., Eckstein F.. A study of the mechanism of DNA polymerase I from Escherichia coli with diastereomeric phosphorothioate analogs of deoxyadenosine triphosphate. J. Biol. Chem. 1979; 254:6889–6893. [PubMed] [Google Scholar]
- 27. Pecoraro V.L., Hermes J.D., Cleland W.W.. Stability constants of Mg2+ and Cd2+ complexes of adenine nucleotides and thionucleotides and rate constants for formation and dissociation of MgATP and MgADP. Biochemistry. 1984; 23:5262–5271. [DOI] [PubMed] [Google Scholar]
- 28. Alvarez S. A cartography of the van der Waals territories. Dalton Trans. 2013; 42:8617–8636. [DOI] [PubMed] [Google Scholar]
- 29. Liang C.X., Allen L.C.. Sulfur does not form double-bonds in phosphorothioate anions. J. Am. Chem. Soc. 1987; 109:6449–6453. [Google Scholar]
- 30. Berg B.J.V., Beard W.A., Wilson S.H.. DNA structure and aspartate 276 influence nucleotide binding to human DNA polymerase beta – Implication for the identity of the rate-limiting conformational change. J. Biol. Chem. 2001; 276:3408–3416. [DOI] [PubMed] [Google Scholar]
- 31. Liu J., Tsai M.D.. DNA polymerase beta: pre-steady-state kinetic analyses of dATP alpha S stereoselectivity and alteration of the stereoselectivity by various metal ions and by site-directed mutagenesis. Biochemistry. 2001; 40:9014–9022. [DOI] [PubMed] [Google Scholar]
- 32. Batra V.K., Beard W.A., Shock D.D., Pedersen L.C., Wilson S.H.. Structures of DNA polymerase beta with active-site mismatches suggest a transient abasic site intermediate during misincorporation. Mol. Cell. 2008; 30:315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.