


Abstract
Promoters and enhancers regulate the initiation of gene expression and maintenance of expression levels in spatial and temporal manner. Recent findings stemming from the Cap Analysis of Gene Expression (CAGE) demonstrate that promoters and enhancers, based on their expression profiles after stimulus, belong to different transcription response subclasses. One of the most promising biological features that might explain the difference in transcriptional response between subclasses is the local chromatin environment. We introduce a novel computational framework, PEDAL, for distinguishing effectively transcriptional profiles of promoters and enhancers using solely histone modification marks, chromatin accessibility and binding sites of transcription factors and co-activators. A case study on data from MCF-7 cell-line reveals that PEDAL can identify successfully the transcription response subclasses of promoters and enhancers from two different stimulations. Moreover, we report subsets of input markers that discriminate with minimized classification error MCF-7 promoter and enhancer transcription response subclasses. Our work provides a general computational approach for identifying effectively cell-specific and stimulation-specific promoter and enhancer transcriptional profiles, and thus, contributes to improve our understanding of transcriptional activation in human.
INTRODUCTION
Spatiotemporal control of gene expression in eukaryotes is coordinated by the interplay of DNA regulatory elements located proximal or distal to the transcription start sites (TSSs) of their target transcripts (1,2). Promoters and enhancers are categories of DNA regulatory elements that have been subjected to extensive studies in recent years. Promoter regions are overlapping with TSSs and sequence motifs that they contain (e.g. TATA box, INR element) are used for anchoring the transcriptional machinery and regulating the initiation of transcription (3). In contrast, enhancers are located few or many thousands base pairs (bp) upstream or downstream from the TSSs and enhance the expression of their target transcripts through interactions with transcription factors (TFs) (or complexes they form) and/or by facilitating chromatin-remodelling activities (4).
It has been recently shown that RNA polymerase II (POL2) -mediated transcription occurs in enhancers on a genome-wide scale, producing a particular class of non-coding RNAs called eRNAs whose functional roles, if any, are elusive (5). This directly implies that enhancers in many cases act as promoters of eRNA transcripts (6). Consequently, promoters and enhancers share biochemical and transcriptional properties that have been reported in recent studies (7–9). Thus, considering their increased similarity, it may be difficult to separate effectively these regulatory classes, since some promoters have also enhancer activity (4,9–11).
Recent findings stemming from CAGE profiling (10) of stimulus-response time courses across multiple cells (12) indicate that transcription of enhancers is the earliest transcriptional event in cells responding to stimulus, followed by a number of coordinated subsequent transcriptional events in mRNA promoters (12). Analysis of CAGE expression profiles from promoters and enhancers suggests that they can be classified into distinct subgroups based on their transcriptional profiles (referred to here as transcription response subclasses) following stimulus, the most prominent being: (i) Rapid short; (ii) Rapid late; (iii) Early standard; (iv) Late standard; (v) Long; and (vi) Late. While this distinction holds across all cells studied, the underlying biology explaining these subclasses remains unclear.
One of the most promising biological features that might explain the difference in transcriptional response between subclasses is the local chromatin environment. A key question is whether it is possible to distinguish different promoter and enhancer transcriptional profiles based solely on local chromatin environment characteristics. Up to now many existing approaches for promoter and enhancer identification, experimental or computational, have been developed and surveyed comprehensively in several review articles (13–17). However, discriminating effectively promoters and enhancers of different transcription response subclasses using information from the local chromatin configuration and identifying subsets of chromatin markers that minimize the classification error between those transcription response subclasses are interesting problems that require further investigation.
Here, we introduce PEDAL (Promoter-Enhancer Discriminative AnaLysis), a computational framework for classifying promoters and enhancers into different transcription response subclasses using histone modification markers, chromatin accessibility and binding sites of transcription factors and co-activators, as input information. As a case study we apply PEDAL to data from human MCF-7 breast cancer cells stimulated by histidine rich glycoprotein (HRG) that triggers differentiation and epidermal growth factor (EGF). PEDAL discriminates effectively almost all promoter and enhancer transcription response subclasses. In addition, results obtained by PEDAL surpass performance of two other state-of-the-art classification algorithms.
To date, our classification algorithm is the first that distinguishes successfully, the transcription response profiles of promoters and enhancers and identifies subsets of chromatin characteristics for categorizing these regions with minimized classification error.
MATERIALS AND METHODS
Available data sets
The primary data for training derive from Arner et al. 2015 (12). In that study, promoters and enhancers were identified using CAGE experiments from a large number of primary cells and tissues. Then, using a rule-based analysis based on hierarchical clustering of individual time courses, promoters and enhancers were categorized into different transcription response subclasses based on their CAGE expression profiles. Here, we focus on promoters and enhancers from human MCF-7 cell-line response to HRG and EGF. More details about the promoter and enhancer identification process (sample collection, library preparation, quality control, differential expression of promoters and enhancers) as well as the categorization procedure into different expression profiles can be found in (12). All promoters and enhancers are classified into the following major transcription response subclasses: (i) Rapid short; (ii) Rapid late; (iii) Early standard; (iv) Late standard; (v) Long; and (vi) Late. Figure 1 panel C shows a stylistic representation (i.e. shape approximation) of each of the major transcription response patterns (i.e. response subclasses) identified in (12).
Figure 1.

Schematic diagram of PEDAL. (A) The first panel shows the data integration and the combination of different replicates for all markers. (B) The second panel shows the preparation of promoter and enhancer regions we use for finding overlaps with input BAM reads. (C) The third panel shows the feature vector generation process and the labeling based on different CAGE expression profiles from (12). (D) The last panel shows an example of discriminating response subclasses using KNN.
From the complete list of MCF-7 promoters and enhancers, we exclude samples that belong simultaneously to two or more subclasses (i.e. we removed multi-labeled cases). The reason is that we formulate the discrimination problem into a multi-class classification problem (should not be confused with the multi-label classification problem), which makes it easier to identify similarities or differences in the chromatin environment, if any, between the considered transcription response subclasses. For the rest of the analysis we do not consider the ‘Unclassified subclass’ containing samples that do not obey the general rules for identifying promoter and enhancer subclasses proposed in (12). Supplementary Figures S1 and S2 present the actual numbers of promoters and enhancers per response subclass for both HRG and EGF stimulations.
Promoter and enhancer samples are represented using numerical values that derived from six input markers. We use the following ENCODE (18) data sets in the BAM format: (i) histone modification H3K4me3 that marks active or poised promoters; (ii) histone modification H3K27ac that marks active or poised enhancers; (iii) Co-activator P300 that is a known enhancer marker; (iv) CTCF which is a marker for insulators; (v) POL2 which transcribes both promoters and enhancers; and (vi) DNase-seq (DHS) data that identify DNA accessible regions. For each marker we retrieved data from two replicates from MCF-7 cells that are pooled together and averaged. All genomic coordinates correspond to the assembly build hg19. The complete list of online sources for the data sets included in this study can be found in Supplementary Table S1.
Feature vectors that describe sample instances are generated as follows: For every promoter and enhancer sample in the data set we identify its ‘centre’. For promoters, the centre is defined as the ‘representative position’, which is the location of the majority of the CAGE signals. For enhancers, the centre corresponds to the middle point of the sequence. Based on that centre, to approximate better promoter and enhancer regions and capture their properties, we generate broader intervals. For enhancers we choose regions 200-bp upstream and 200-bp downstream from the centre since enhancers present symmetrical bidirectional properties. For promoters we choose non-symmetrical regions 300-bp upstream and 100-bp downstream from the centre. Then, using BedTools (19) (intersect command) we map the input data sets to these intervals and we compute the number of reads in the BAM files that overlap promoter and enhancer regions. In this way we quantify the intensity of input signals with respect to promoter and enhancer centres. The values of each feature are normalized by the total number of reads in the data set. The final constructed feature vector includes features corresponding to six different markers and one feature that present the label (integer from 1 to 6).
PEDAL implementation
In PEDAL we reformulate the problem of assigning transcription response subclasses to promoters and enhancers into a multi-class classification task. The underlying computational technique used is the K-nearest neighbors (KNN). KNN is a simple non-linear classifier that runs fast and with optimized K and number of features achieves close to optimal classification performance (20). We decided to use KNN after experimentation and comparison analysis with other classification algorithms (see below the “Comparing PEDAL with other classification algorithms” section). PEDAL framework is implemented in Matlab R2014b using built-in functions for KNN (knnclassify function) and the number of neighbors K is tuned using validation sets completely independent from training and testing sets. The schematic overview of PEDAL is presented in Figure 1. Figure 2 is a graphical representation of the data preparation and learning processes. In our analysis we follow the ‘one versus all’ paradigm and we generate binary classification problems that correspond to different transcription response subclasses. To discriminate promoters of one subclass from all other promoters and similarly enhancers of one subclass versus all other enhancers, we generate six binary classification tasks. In every binary classification task we consider two sets, one that contains the subclass of interest and the “negatives set” that contains all other samples except for the subclass of interest. The positive set for training derives from the subclass of interest and has N data instances (i.e. samples). The negatives for this case contain Q data instances that represent all other samples but not the ones from the subclass of interest. In case N << Q, we select N randomly from the Q negatives to generate the negative set for training (i.e. to be of equal size to the positive). Because we run the training process 1000 times we generate 1000 negative sets of size N that we select randomly with replacement. Exception is the late transcribed promoters response to HRG where N > Q. In this case we generate positive and negative training sets based on all the available samples and the splitting process into training-validation-testing is the same as before. For assessing the classification performance for every individual run we further split the positive and negative sets randomly into training, validation and testing sets. We use 50% of the total size of positive and negative samples for training, 20% for validation and 30% for testing. The validation set is used for tuning the classification model parameters and the testing set for assessing the performance in a completely un-biased way. We finally report the average classification performance of 1000 runs. For assessing classification performance we consider the following performance metrics:
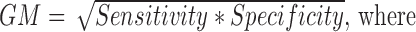
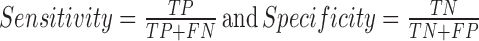
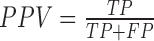
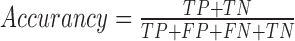
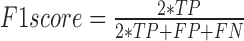
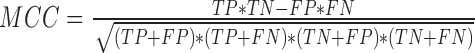
Figure 2.

Flowchart of PEDAL's learning process. The same process was repeated for all transcription response subclasses and all combinations of features.
where GM stands for Geometric mean of Sensitivity and Specificity, PPV stands for Positive Predicted Value, MCC stands for Mathews Correlation Coefficient and TP denotes True Positives, FP denotes False Positives, FN denotes False Negatives and TN denotes True Negatives.
Identifying optimized subsets of input markers
We apply feature selection (FS), to improve the recognition performance of discriminating promoter and enhancer transcription response subclasses and to identify stimulation specific sets of input markers (per subclass) that maximize classification performance by minimizing classification error.
Determination of the relative importance of combinations of input markers using computational techniques is an indirect way to associate important patterns in the input data. Notably, in binary classification tasks, identifying combinations of input variables that minimize classification error imposes that the feature values of these markers for the class of interest (positive) are different from the corresponding values of the other (negative) classes. In our study, this information can be further utilized to generated hypotheses about mechanistic properties and the spatial distribution of chromatin markers of different transcription response subclasses.
The FS problem in different bioinformatics areas is well studied (21,22) and has several applications that span from identification of robust set of chromatin markers for regulatory elements (23,24), characterization of antimicrobial peptide families and subfamilies (25), to the prediction of cancer biomarkers (26). Up to now, several approaches for FS have been proposed based on statistical analysis (27) or search algorithms combined with global optimization techniques (28).
In PEDAL, since the size of feature vector is relatively small we apply a brute force (BF) search algorithm for selecting combinations of input markers that maximize classification performance (or equivalently minimize classification error) based on MCC. In other words, we estimate the performance for every single combination derived from six features resulting in total to 63 combinations. For the rest of the analysis, the term ‘optimized’ refers to the combination of features that maximizes classification performance based on MCC.
To achieve more robust results and to provide a more comprehensive view of the classification performance, we repeat the learning process 1000 times and we compute the average classification performance on the test sets for every combination of features. This process results in training, validating and testing more than 1.5 million individual classification models (63 combinations of features tested 1000 times for 2 stimulations each, for 6 promoter subclasses and for 6 enhancer subclasses). PEDAL source codes and materials are publicly available at https://cloud.kaust.edu.sa/Pages/PEDAL.aspx under an Educational Community Open Source Licence.
RESULTS AND DISCUSSION
Insights on the chromatin environment of MCF-7 transcription response subclasses
To provide insights about similarities or differences of the considered chromatin profiles, we compare first the distributions of the feature values of different input markers for all promoter and enhancer transcription response subclasses (Supplementary Figure S3). To quantify differences in the distributions we perform Wilcoxon rank test under the null-hypothesis that the median of feature values of one input marker for promoters and enhancers of the same subclass does not change. In Table 1 we present the P-values after applying Benjamini–Hochberg correction for multiplicity testing. Considering a level of significance of false discovery rate (FDR) < 0.05, we observe that rapid short transcribed enhancers have different chromatin profiles from rapid short transcribed promoters, except for the DHS marker. In the rapid short and late standard transcribed subclasses only H3K27ac and P300 follow different distributions. Long transcribed promoters and enhancers have different distributions of all input markers except for DHS whereas late transcribed promoters and enhancers have different distributions of all input markers. For visualization purposes, the upper panel of Supplementary Figure S4 (part A for promoters and B for enhancers) shows average profiles of six input markers aligned at the centres of HRG promoters and enhancers regardless of subclass. The lower panel of Supplementary Figure S4 shows the number of input reads that overlap the enhancers and promoter regions from ENCODE data sets replicate 1, visualized as heatmaps. As expected, promoters and enhancers are enriched for POL2 binding, open chromatin (DHS) and H3K4me3 signals, while H3K27ac and P300 enrichment are substantially more prominent in enhancers.
Table 1. FDR scores obtained by Wilcoxon rank test with Benjamini–Hochberg correction for six input markers that belong to promoters and enhancers of the same transcription response subclass.
| Response subclass | Enh H3K4me3 versus Prom H3K4me3 | Enh H3K27ac versus Prom H3K27ac | Enh P300 versus Prom P300 | Enh CTCF versus Prom CTCF | Enh POL2 versus Prom POL2 | Enh DHS versus Enh DHS |
|---|---|---|---|---|---|---|
| Rapid Short | 0.0042 | 2.79E-06 | 2.05E-11 | 0.0003 | 0.0006 | 0.9526 |
| Rapid Long | 0.5771 | 0.0014 | 0.0379 | 0.0892 | 0.1814 | 0.9526 |
| Early Standard | 0.0746 | 0.0016 | 0.0021 | 0.7975 | 0.0746 | 0.9526 |
| Late Standard | 0.8406 | 0.0001 | 0.0379 | 0.7975 | 0.8621 | 0.9526 |
| Long | 0.0216 | 2.64E-07 | 8.64E-06 | 0.0090 | 4.83E-07 | 0.9526 |
| Late | 3.42E-14 | 1.89E-07 | 3.43E-08 | 2.07E-10 | 2.10E-14 | 0.0004 |
Together, all the above findings support the notion that different chromatin characteristics in MCF-7 may be utilized to discriminate different transcription response subclasses of promoters and enhancers. The above issues we will be explored in detail in the next subsections using data from two different MCF-7 stimulations. First, we focus on the stimulation-specific case and we apply PEDAL to distinguish transcription response subclasses of promoters and enhancers stimulated by HRG and EGF. Next, looking the problem in a non-stimulation specific manner, we develop recognition systems for predicting MCF-7 transcription response subclasses using data for training, validation and testing from all the available MCF-7 stimulations.
Applying PEDAL to promoters in MCF-7 stimulated by EGF and HRG
In this section, we explore whether the local chromatin environment of MCF-7 cells, can be used to predict, in a stimulation-specific manner, different promoter transcription response subclasses. To do so, we measure the classification performance of every single promoter subclass versus all other promoter subclasses for both EGF and HRG stimulations.
To get a comprehensive view of the recognition performance, in Supplementary Figure S5 we present the average MCC of 1000 runs for every single combination of input markers for MCF-7 promoters stimulated by EGF. Similarly, in Supplementary Figure S6 we present the average MCC of 1000 runs for all MCF-7 promoters stimulated by HRG. Different combinations of input marker achieve performance that varies in both stimulations and across all transcription response subclasses. Table 2 presents the stimulation-specific optimized combinations of features for both EGF-stimulated and HRG-stimulated MCF-7 cells.
Table 2. Optimized subsets of input markers for all promoter subclasses as derived from the EGF and HRG stimulations-specific PEDAL models.
| Rapid short | Rapid long | Early standard | Late standard | Long | Late | |
|---|---|---|---|---|---|---|
| EGF | H3K4me3 | H3K27ac | H3K4me3 | H3K27ac | CTCF | H3K4me3 |
| H3K27ac | P300 | H3K27ac | P300 | POL2 | H3K27ac | |
| P300 | POL2 | CTCF | POL2 | |||
| CTCF | DHS | |||||
| HRG | P300 | H3K27ac | P300 | H3K4me3 | H3K4me3 | H3K4me3 |
| CTCF | POL2 | H3K27ac | CTCF | POL2 | ||
| POL2 | DHS | P300 | POL2 | DHS | ||
| POL2 | DHS | |||||
| DHS |
An overview of the average classification performance per transcription response subclass for both stimulations using additional performance metrics (GM, PPV, Accuracy and F1-score) is presented in Figure 3A and C. Supplementary Table S4 shows the standard deviation of 1000 runs. In most of the tested subclasses the recognition performance is higher than 70% (by any of the four performance indicators we used). This confirms our hypothesis, that local chromatin environment characteristics can be utilized to discriminate effectively transcription response subclasses. Exceptions are long and late subclasses for EGF stimulation and rapid short and long subclasses for HRG that achieve the lowest performance. This may indicate that, within the computational framework we applied and the considered input data sets, the data samples from these subclasses appear not well separable. The highest performance is achieved for rapid long transcribed promoters for both stimulations. Here, we also wish to highlight that our proposed method is the only one dealing with this particular discrimination problem, and hence, any level of performance achieved provides a first baseline for future studies.
Figure 3.

PEDAL's classification performance for distinguishing promoter and enhancer subclasses using optimized combinations of input markers. All left panels correspond to promoters and all right panels correspond to enhancers: (A and B) correspond to PEDAL models specific to EGF stimulation; (C and D) correspond to PEDAL models specific to HRG stimulation; (E and (F) correspond to the non-stimulation specific PEDAL models trained, validated and tested on all the available EGF and HRG data.
Studying the optimized combinations of input markers presented in Table 2 more closely, we observe that different promoter subclasses are classified optimally using different sets of input markers. Consequently, within our experimentation, promoters of different transcription response subclasses present stimulation-specific chromatin environment fingerprints. Regarding the contribution of individual input markers, it appears that H3K4me3, the typical promoter marker, is part of optimized combinations of input markers for rapid short, early standard, late transcription response subclasses for EGF stimulation and late standard and late for HRG stimulation. In cases such as rapid short transcribed promoters, the P300 marker is selected together with CTCF, and the POL2 marker is selected together with CTCF in long transcribed promoters from both stimulations. This may indicate some stimulation-specific mechanisms of function in MCF-7 cells stimulated by EGF and HRG facilitated by CTCF-mediated DNA looping (29). Notably, H3K27ac, the typical enhancer marker, is selected in seven out of 12 cases in both stimulations (30,31).
Applying PEDAL to enhancers in MCF-7 stimulated by EGF and HRG
Next, we explore whether the local chromatin environment of MCF-7 cells can be utilized to predict, in a stimulation-specific manner, different transcription response subclasses of enhancers. Similar to the case of promoters, we measure the classification performance of discriminating every single enhancer subclass versus all other enhancer subclasses for both EGF and HRG stimulations.
Supplementary Figure S7 presents the average MCC of 1000 runs for every single combination of input markers for MCF-7 enhancers stimulated by EGF. Supplementary Figure S8 presents the average MCC of 1000 runs for all MCF-7 enhancers stimulated by HRG. Table 3 summarizes the stimulation-specific optimized combinations of features for both stimulations. The average classification performance per enhancer transcription response subclass for both stimulations is presented in Figure 3B and D, whereas Supplementary Table S5 shows the standard deviation of 1000 runs.
Table 3. Optimized subsets of input markers for all enhancer subclasses as derived from the EGF and HRG stimulations-specific PEDAL models.
| Rapid short | Rapid long | Early standard | Late standard | Long | Late | |
|---|---|---|---|---|---|---|
| EGF | P300 | H3K4me3 | H3K4me3 | H3K4me3 | CTCF | H3K4me3 |
| CTCF | H3K27ac | P300 | H3K27ac | P300 | ||
| POL2 | CTCF | DHS | CTCF | CTCF | ||
| DHS | POL2 | |||||
| HRG | H3K27ac | H3K4me3 | P300 | H3K27ac | CTCF | H3K4me3 |
| P300 | P300 | DHS | CTCF | H3K27ac | ||
| CTCF | POL2 | CTCF | ||||
| POL2 | DHS | DHS | ||||
| DHS |
Considering a performance threshold of 70% (by any of the four performance indicators we used), we observe that most of the transcription response subclasses from both stimulations can be classified with higher recognition performance. Consequently, within our experimentation, we confirm the hypothesis that different MCF-7 enhancer transcription response subclasses can be distinguished effectively using information from their local chromatin environment. Exceptions are long and late transcription response subclasses from both MCF-7 stimulations, where the performance is lower. Taking all previous results into consideration, we conclude that the utilized input variables for particular subclasses such as late and long transcribed enhancers and promoters in the considered MCF-7 stimulations are not sufficient and additional input information (i.e. different chromatin environment features) is required for the effective prediction of their transcription response subclasses. The highest recognition performance is achieved for the rapid long transcribed subclass for both stimulations.
Regarding the contribution of individual input markers, it appears that long transcribed enhancers from both HRG and EGF stimulations are classified optimally using CTCF. Although the performance in long transcribed enhancers is lower compared to other subclasses, we observe that CTCF is also selected in several other MCF-7 subclasses such as rapid short, rapid long, late standard transcribed enhancers stimulated by EGF or rapid long and late standard transcribed enhancers stimulated by HRG (32). There are also cases where we can discern ‘clear’ enhancer patterns acting as fingerprints for specific MCF-7 subclasses such as H3K27ac-P300 for rapid short transcribed enhancers in HRG stimulation or P300-DHS in early standard transcribed enhancers from the same stimulation. We also observe that H3K4me3 contributes (together with some other markers such as CTCF or P300) to the performance maximization of rapid long, early standard, late standard and late transcribed enhancers from EGF stimulation and rapid long and late transcribed enhancers from HRG stimulation. This may potentially describe specific transcription activation mechanisms for rapid long and late transcribed enhancers in different MCF-7 stimulations via spatial configuration of H3K4me3 (33).
Comparing PEDAL with other classification algorithms
We compare PEDAL's recognition capabilities using KNN with two state-of-the-art classification algorithms, namely Bagged Decision Trees (BDT) and Logistic Regression (LR). For a fair comparison, we follow exactly the same protocols summarized in Figures 1 and 2 and we estimate the classification performance using every single combination of input markers (i.e. in total 63 combinations). We repeat the learning process 1000 times and we select, for every response subclass and algorithm, the combination that achieves the maximum MCC. For all algorithms included in the comparison analysis, we use exactly the same training, validation and testing sets as used for PEDAL with KNN. All implementations held in Matlab R2014b using build-in functions for BDT (TreeBagger function) and LR (glmfit function).
Results obtained by BDT and LR are summarized in Supplementary Figure S9 for all enhancer and promoter transcription response subclasses for both HRG and EGF stimulations. PEDAL with KNN achieves much higher accuracy in all of the tested cases. In most of these cases, the other methods do not achieve performance higher than 70%. This clearly indicates that the PEDAL framework combined with KNN algorithm is the correct choice. Notably, LR, one of the simplest and fastest classification algorithm, achieves comparable and in some cases superior performance to BDT.
A closer look on MCF-7 rapid long transcribed promoters and enhancers stimulated by EGF and HRG
In this subsection, we provide more insights about the optimized combinations of input markers for all MCF-7 promoters and enhancers stimulated by EGF and HRG. As an additional validation process, we focus on the subclass of rapid long transcribed enhancers and promoters since it achieves the highest MCC. The corresponding sets of input markers that maximize MCC for all rapid long subclasses are the following: H3K4me3-H3K27ac-CTCF for EGF-stimulated enhancers, H3K4me3-P300-CTCF-POL2-DHS for HRG-stimulated enhancers; H3K27ac-P300 for EGF-stimulated promoters; and H3K27ac-POL2-DHS for HRG-stimulated promoters.
Figure 4 presents the corresponding chromatin profiles from input data sets replicate 1. All subplots in Figure 4 are based on the actual raw data sets used to generate PEDAL's feature vector (34). The right panels of Figure 4 show the corresponding profiles of all other data samples that do not belong to the rapid long transcribed subclass (i.e. ‘negative’ sets). There are three main observations: (i) the data profiles of the selected input variables distinguish visually the subclasses of interest from all other subclasses (i.e. negative sets) for both stimulations. In a simple way this can explain the maximized classification performance we achieve; (ii) the selected profiles of EGF-stimulated and HRG-stimulated rapid long transcribed promoters are different (number of markers and their spatial distributions) and similarly the profiles of EGF-stimulated and HRG-stimulated rapid long transcribed enhancers are different (number of markers and their spatial distributions); (iii) frequently the profiles of the same markers of the same response subclass of different stimulations follow different spatial distributions. An example is the spatial distribution of H3K4me3 for EGF and HRG rapid long transcribed enhancers or the spatial distribution of H3K27ac for EGF and HRG rapid long transcribed promoters. Based on a limited number of cases examined, our data suggests that the chromatin configuration involved in the studied cases seems to be different, as shown by the set of optimized features and their distributions that generate the best classification results based on MCC. This supports the notion that the local chromatin environment of particular transcription response subclasses follows stimulation-specific spatial organization.
Figure 4.

Optimized input profiles aligned at the ‘center’ for all rapid long transcribed promoters and enhancers for EGF and HRG stimulations. We also show the corresponding profiles for all other promoters and enhancers that do not belong to the considered subclass of interest.
Developing non-stimulation specific models for MCF-7 promoters and enhancers
Finally, we investigate further whether it is possible to develop non-stimulation specific recognition systems for MCF-7 transcription response subclasses for promoters and enhancers. We combine data from HRG and EGF stimulations and we repeat the training, validation and testing processes for every single promoter and enhancer transcription response subclass. Following exactly the same protocols as before, we measure the average classification performance of 1000 executions for every single combination of features and we discover the combinations that maximize MCC. Figure 3E and F summarize the results. In addition, Supplementary Figure S10 compares the overall recognition performance of stimulation-specific models versus non-stimulation specific models. The non-stimulation specific models achieve comparable performance to the ‘best’ stimulation-specific models and always improve the performance of the ‘weaker’ stimulation-specific model. Consequently, they are useful for identifying transcription response subclasses in cells from unknown stimulations, or when the stimulation is known but the existing stimulation-specific models have ‘weak’ recognition capabilities.
CONCLUSION
A novel computational framework for identifying the transcriptional response subclasses of promoters and enhancers using as input, information from their local chromatin environment, is introduced. Our work implicitly links the transcription response subclasses of promoters and enhancers with specific chromatin environment characteristics involving histone modification markers, chromatin accessibility and binding sites of transcription factors and co-activators.
A case study using data from MCF-7 cell-line, reports stimulation specific (HRG-specific and EGF-specific) combinations of input markers that discriminate with maximized MCC, MCF-7 promoters and enhancers of different transcription response subclasses. Looking at the problem in a non-stimulation specific manner, we are further able to develop recognition systems for predicting MCF-7 transcription response subclasses using data from all the available stimulations.
Within the examined cases and based on the data sets used, some markers follow stimulation-specific spatial distributions as shown by the feature values that generate the optimized classification results. All these findings suggest potential mechanisms of function at the chromatin level associated with transcription response subclasses in MCF-7 cells. However, we note that the results obtained from computational methods, although supported by statistical evidence, require further validation steps and targeted wet-lab experiments.
Nonetheless, within the present computational framework, many improvements are possible such as: (i) considering more features that describe the shape of ChIP-seq or DNase-seq signals such as kurtosis, or bimodality or combination of chromatin features with more complex sequence characteristics (e.g. discriminative de novo sequence motifs) may increase the classification performance; (ii) integrating more ChIP-seq data sets form histone marks may shed light to MCF-7 local chromatin configuration rules affecting promoter and enhancer transcription profiles; (iii) performing the same analyses for promoters and enhancers from different cell-lines and tissues may help generalizing findings and inferring more generic chromatin patterns related to promoter and enhancer transcription profiles; (iv) tackling the multi-label classification problem is an independent study that might reveal new insights.
We believe that the results of this analysis will help in better understanding the transcription regulation of mammalian promoters and enhancers. Thus, we anticipate that as more data become available, PEDAL will be readily incorporated into large-scale analyses aiming at identifying more general rules, if any, that can link local chromatin environment characteristics with different expression programs in mammalian cells.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Albin Sandelin and Kristoffer Vitting-Seerup for valuable comments on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
King Abdullah University of Science and Technology (KAUST); Research Grant from MEXT [to the RIKEN Center for Life Science Technologies]. Funding for open access charge: King Abdullah University of Science and Technology (KAUST).
Conflict of interest statement. None declared.
REFERENCES
- 1.Lee T.I., Young R.A.. Transcription of eukaryotic protein-coding genes. Annu. Rev. Genet. 2000; 34:77–137. [DOI] [PubMed] [Google Scholar]
- 2.Heintzman N.D., Ren B.. Finding distal regulatory elements in the human genome. Curr. Opin. Genet. Dev. 2009; 19:541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butler J.E., Kadonaga J.T.. The RNA polymerase II core promoter: a key component in the regulation of gene expression. Genes Dev. 2002; 16:2583–2592. [DOI] [PubMed] [Google Scholar]
- 4.Murakawa Y., Yoshihara M., Kawaji H., Nishikawa M., Zayed H., Suzuki H., Fantom C., Hayashizaki Y.. Enhanced identification of transcriptional enhancers provides mechanistic insights into diseases. Trends Genet. 2016; 32:76–88. [DOI] [PubMed] [Google Scholar]
- 5.Andersson R., Gebhard C., Miguel-Escalada I., Hoof I., Bornholdt J., Boyd M., Chen Y., Zhao X., Schmidl C., Suzuki T. et al. . An atlas of active enhancers across human cell types and tissues. Nature. 2014; 507:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andersson R., Chen Y., Core L., Lis J.T., Sandelin A., Jensen T.H.. Human gene promoters are intrinsically bidirectional. Mol. Cell. 2015; 60:346–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weingarten-Gabbay S., Segal E.. A shared architecture for promoters and enhancers. Nat. Genet. 2014; 46:1253–1254. [DOI] [PubMed] [Google Scholar]
- 8.Core L.J., Martins A.L., Danko C.G., Waters C.T., Siepel A., Lis J.T.. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet. 2014; 46:1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andersson R., Sandelin A., Danko C.G.. A unified architecture of transcriptional regulatory elements. Trends Genet. 2015; 31:426–433. [DOI] [PubMed] [Google Scholar]
- 10.Forrest A.R., Kawaji H., Rehli M., Baillie J.K., de Hoon M.J., Haberle V., Lassmann T., Kulakovskiy I.V., Lizio M., Itoh M. et al. . A promoter-level mammalian expression atlas. Nature. 2014; 507:462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alam T., Medvedeva Y.A., Jia H., Brown J.B., Lipovich L., Bajic V.B.. Promoter analysis reveals globally differential regulation of human long non-coding RNA and protein-coding genes. PloS One. 2014; 9:e109443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arner E., Daub C.O., Vitting-Seerup K., Andersson R., Lilje B., Drablos F., Lennartsson A., Ronnerblad M., Hrydziuszko O., Vitezic M. et al. . Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science. 2015; 347:1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herz H.M., Hu D., Shilatifard A.. Enhancer malfunction in cancer. Mol. Cell. 2014; 53:859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinhold N., Jacobsen A., Schultz N., Sander C., Lee W.. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 2014; 46:1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kleftogiannis D., Kalnis P., Bajic V.B.. Progress and challenges in bioinformatics approaches for enhancer identification. Brief. Bioinform. 2015; 2015:bbv101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shlyueva D., Stampfel G., Stark A.. Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet. 2014; 15:272–286. [DOI] [PubMed] [Google Scholar]
- 17.Narlikar L., Ovcharenko I.. Identifying regulatory elements in eukaryotic genomes. Brief. Funct. Genomics Proteomics. 2009; 8:215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skipper M., Dhand R., Campbell P.. Presenting ENCODE. Nature. 2012; 489:45. [DOI] [PubMed] [Google Scholar]
- 19.Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altman N.S. An introduction to kernel and nearest-neighbor nonparametric regression. Am. Statistician. 1992; 46:175–185. [Google Scholar]
- 21.Saeys Y., Inza I., Larranaga P.. A review of feature selection techniques in bioinformatics. Bioinformatics. 2007; 23:2507–2517. [DOI] [PubMed] [Google Scholar]
- 22.Gola D., Mahachie John J.M., van Steen K., Konig I.R.. A roadmap to multifactor dimensionality reduction methods. Brief. Bioinform. 2015; 2015:bbv038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandez M., Miranda-Saavedra D.. Genome-wide enhancer prediction from epigenetic signatures using genetic algorithm-optimized support vector machines. Nucleic Acids Res. 2012; 40:e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Won K.J., Zhang X., Wang T., Ding B., Raha D., Snyder M., Ren B., Wang W.. Comparative annotation of functional regions in the human genome using epigenomic data. Nucleic Acids Res. 2013; 41:4423–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khamis A.M., Essack M., Gao X., Bajic V.B.. Distinct profiling of antimicrobial peptide families. Bioinformatics. 2015; 31:849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glaab E., Garibaldi J.M., Krasnogor N.. ArrayMining: a modular web-application for microarray analysis combining ensemble and consensus methods with cross-study normalization. BMC Bioinformatics. 2009; 10:358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng T., Wang Y., Bryant S.H.. FSelector: a Ruby gem for feature selection. Bioinformatics. 2012; 28:2851–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soufan O., Kleftogiannis D., Kalnis P., Bajic V.B.. DWFS: a wrapper feature selection tool based on a parallel genetic algorithm. PloS One. 2015; 10:e0117988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Handoko L., Xu H., Li G., Ngan C.Y., Chew E., Schnapp M., Lee C.W., Ye C., Ping J.L., Mulawadi F. et al. . CTCF-mediated functional chromatin interactome in pluripotent cells. Nat. Genet. 2011; 43:630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A. et al. . Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tie F., Banerjee R., Stratton C.A., Prasad-Sinha J., Stepanik V., Zlobin A., Diaz M.O., Scacheri P.C., Harte P.J.. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development. 2009; 136:3131–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holwerda S.J., de Laat W.. CTCF: the protein, the binding partners, the binding sites and their chromatin loops. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013; 368:20120369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pekowska A., Benoukraf T., Zacarias-Cabeza J., Belhocine M., Koch F., Holota H., Imbert J., Andrau J.C., Ferrier P., Spicuglia S.. H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J. 2011; 30:4198–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen L., Shao N., Liu X., Nestler E.. ngs.plot: quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics. 2014; 15:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.