


Abstract
Illumina Infinium DNA Methylation BeadChips represent the most widely used genome-scale DNA methylation assays. Existing strategies for masking Infinium probes overlapping repeats or single nucleotide polymorphisms (SNPs) are based largely on ad hoc assumptions and subjective criteria. In addition, the recently introduced MethylationEPIC (EPIC) array expands on the utility of this platform, but has not yet been well characterized. We present in this paper an extensive characterization of probes on the EPIC and HM450 microarrays, including mappability to the latest genome build, genomic copy number of the 3΄ nested subsequence and influence of polymorphisms including a previously unrecognized color channel switch for Type I probes. We show empirical evidence for exclusion criteria for underperforming probes, providing a sounder basis than current ad hoc criteria for exclusion. In addition, we describe novel probe uses, exemplified by the addition of a total of 1052 SNP probes to the existing 59 explicit SNP probes on the EPIC array and the use of these probes to predict ethnicity. Finally, we present an innovative out-of-band color channel application for the dual use of 62 371 probes as internal bisulfite conversion controls.
INTRODUCTION
DNA methylation is the most studied epigenetic mark in human disease, partly because it can be preserved under most storage conditions and through histological preparations, such as formalin-fixation and paraffin-embedding. DNA methylation states are flexible, yet can be passed on through multiple cell divisions and exert powerful influence on cellular phenotype. It has become increasingly clear that DNA methylation plays an important role in cancer (1), aging (2) and many other physiological or pathological conditions.
The advent of genomic technologies greatly facilitated our understanding of DNA methylation. Three generations of Illumina's DNA methylation microarrays (3), including GoldenGate, Infinium Humanmethylation27 (HM27) and most notably, Infinium HumanMethylation450 (HM450), have been used by many genome-scale studies using human samples, including The Cancer Genome Atlas (TCGA) project (4). The HM450 platform has been used to generate DNA methylation profiles for more than 10 000 samples in TCGA alone. HM450 data on more than 40 000 human samples are publicly available on Gene Expression Omnibus. With these high-throughput technologies, epigenome-wide association studies (EWASs) have become possible to study phenotypic variation attributable to inter-individual epigenomic variations (5).
In 2016, the new HumanMethylationEPIC (EPIC) array, which interrogates a total of 863 904 CpG loci, together with 2932 non-CpG loci and 59 single nucleotide polymorphisms (SNPs), superseded the HM450 array. The EPIC array inherited the HM450 design and >90% of the HM450 probes (Supplementary Data), and included additional probes dedicated to FANTOM5 and ENCODE enhancers, greatly increasing the power of this microarray to study enhancer/regulatory regions.
These DNA methylation microarrays are in essence SNP microarrays (6). Their interrogation of DNA methylation states relies on sodium bisulfite conversion which transforms an epigenetic difference between a modified C (including 5-mC and 5-hmC and other modifications.) and C to a genetic C/T SNP. It has long been recognized that some probes on the Infinium arrays do not perform as expected (7–9) and should be excluded from analyses due to impact of sequence polymorphisms and competing off-target hybridization events. Three groups of probes should generally be filtered out from Infinium microarray analyses: (i) probes with internal SNPs close to the 3΄ end of the probe (Group 1); (ii) probes with non-unique mapping to the bisulfite-converted genome (Group 2); and (iii) probes with off-target hybridization due to partial overlap with non-unique elements (Group 3).
Sequence polymorphisms can affect DNA methylation readouts in the Infinium arrays in three ways: (i) introducing mismatches close enough to the 3΄-end of the probe sequence and interfering with successful extension; (ii) altering the CpG dinucleotide sequence context and hence the ability of target cytosines to be methylated. This also includes the special case where an actual C/T polymorphism is present instead of C/T difference introduced by bisulfite conversion; (iii) causing a switch in the color channel for Type I design by changing the extension base (color-channel-switching, or CCS, SNPs), since the color channels of Type I probes are entirely dependent on the extension base. A SNP in the extension base can cause or not cause a color switch depending on the base change. An A/T SNP, for example, will not cause a color change but an A/G SNP will, as A and T bases are both labeled with red and C (complementary to G) is labeled with green fluorophores (Figure 1A). The first two types of influence are recognized in the literature, and probes with SNPs with a minor allele frequency (MAF) of >1% located within 10 nucleotides (nt) of the target sites are often excluded from analyses (4,7,10,11), although the 10-nt cut-off is largely a rule of thumb. No empirical evidence supports the use of 10 bp as the cut-off. For the third type of influence, one study singled out and excluded all such extension-base SNPs (12). But as discussed, only SNPs that cause a channel switch in this location will be problematic. We describe below how we exploit this information to recover probes that might otherwise be masked.
Figure 1.

Influence of SNPs on probe performance. (A) Illustration of the Infinium probe design. Type I probes utilize a pair of methylated (M) and unmethylated (U) probes, designed against the methylated and unmethylated versions of the target DNA, respectively. Signals representing these two alleles are measured in the same color channel, determined by the base incorporated (nucleotides A and T are labeled red, and C labeled green) complementary to the extension base (D = A, T, G in IUPAC code) (top). Type II probes uses a single probe and the extension occurs at the target CpG, with a red-labeled A measuring the unmethylated allele and green-labeled G measuring the methylated allele (middle). For Type I probes, color-chanel-switching (CCS) SNPs at the extension base can cause signals to come from the alternative color channel (bottom). Green and red signals reflect different alleles of the SNP. (B) Inter-individual variation (calculated as standard deviations; SDs) in beta values measured by Type I probes associated with a SNP located within the probe at a given distance from the 3΄-end of the probe (the target CpG). Normal samples (n = 705) studied in the TCGA project were used to calculate the variation. SD was first calculated within each tissue type (to avoid variance introduced by tissue-specific methylation) and averaged over 13 tissue types; (C) The averaged SD for beta values measured by Type II probes with a SNP present at a given distance from the 3΄ end of the probe, measured in the normal samples studied in the TCGA project; (D) Variation in beta values for Type I probes with CCS SNPs (red), non-channel-switching SNPs (blue) and beta values rescue for CCS SNPs by combining two color channels (green, see ‘Materials and Methods’ section), stratified by minor allele frequency.
The distinctions between Group 2 and 3 probes have not yet been addressed well in the literature. A probe can have unique mapping based on the full 50-nt sequence, and yet a substantial portion can map non-uniquely. Partial hybridization at the 3΄ end with competing molecules may lead to successful extension and subsequent fluorescence while those that do not lead to extension can also compete with the target hybridization and impact signal intensity. Group 3 probes have not been consistently accounted for in different studies, and where they have been, they are defined by overlap with repetitive elements (8). However, this practice is flawed in three ways. First, sequences annotated as repetitive elements may not actually be non-unique or have multiple copies. Some repetitive elements have been annotated based upon knowledge from other species and have been found to be unique or at low copy number in the human genome. Second, some large families of repetitive elements have been co-evolving with the human genome for over 100 million years (13) and have diversified into sequences that can be clearly distinguished from other copies in the same family. More importantly, other sequences not defined as repeats can nevertheless be non-unique as part of retroduplication (14) or pseudogenes (15), particularly in a bisulfite-converted genome and these have been largely ignored in the literature.
The expansion of the EPIC array probe set compared to HM450 was based primarily on enhancer regions whose origins may derive in part from transposable elements (16). Masking based on overlap with repetitive elements could result in a substantial loss of probes to the array. Approximately 22% of the EPIC array probes would be masked if just the last 15 nt are considered for repeat overlap, as suggested in the literature (4). As much as 39% of the probes have been proposed to be excluded from HM450 analyses, largely owing to the overlap with repeats (8). There is a pressing need for a rational approach based on empirical analysis to address artifacts introduced by competing hybridization and extension.
In this manuscript, we present a systematic characterization of how the performance of Illumina Infinium DNA methylation probes is influenced by factors described for each of these potentially problematic categories and provide recommendation for thresholds for probe masking based on empirical evidence obtained by comparison with Whole Genome Bisulfite Sequencing (WGBS) data and mining existing large-scale datasets generated with these platforms. In particular, we discuss SNPs in the extension base of Type I probes. We present a comprehensive annotation and characterization of the latest EPIC array, including accurate mapping to the GRCh38/hg38 genome, characterization of probes influenced by polymorphism based on the latest dbSNP database and probes with non-unique hybridization and extension. In addition, we discuss novel alternative uses of existing probes on the EPIC platforms, as genotyping probes and internal bisulfite control probes.
MATERIALS AND METHODS
SNP
SNP data were obtained from the common SNP definition released with the dbSNP (17) Build 147 (Apr 2016) which include SNPs and INDELs with alternative allele frequency (AAF) of over 1% in any of the 31 population studied in the 1000 Genome Project. We also combined this data with SNPs from the 1000 Genome Project Phase 3 (18) data for genetic variation in sex chromosomes (absent in the common SNP collection from dbSNP) and allele frequencies in each of the 31 human population.
Matched WGBS and HM450 datasets
We downloaded matched WGBS data (as Level 3 beta values) and HM450 data (as Level 1 IDAT files) for 18 samples (Supplementary Data) from the TCGA data portal (https://gdc-portal.nci.nih.gov/). For WGBS data, measurement of retention and conversion from both strands were combined for each CpG dinucleotide. We chose HM450 targets with sums of retention and conversion greater than ten in at least three of the 18 WGBS samples. The HM450 IDATs were processed with the noob background correction (19) and dye bias correction following the TCGA data processing pipeline (4), minus the probe masking (based on detection P-value, SNPs and repeats) performed for Level 3 TCGA data.
Normal HM450 datasets from TCGA
We used HM450 datasets from 705 normal tissue samples to study the impact of sequence polymorphism on beta value readout. This represents data from 13 distinct tissue types (Supplementary Data) each with at least 15 normal samples. The HM450 IDATs were processed in the same way as described above.
Normal colon and HeLa EPIC dataset
We profiled two normal colon samples and cervical cancer cell line HeLa on the Infinium EPIC array. We performed noob background correction (19) and dye bias correction following the TCGA data processing pipeline (4).
Genomic copy number of 3΄ nested subsequence and their overlap with repetitive elements
For each length ranged from 10 to 50 bases (full probe length), we scanned 3΄-subsequence of the probe's source sequence and of the specified length in the bisulfite converted genome (forward, reverse, parent and daughter strands in the GRCh38 assembly) for matches. We convert all the ‘G's in the probe's source sequence to ‘A's during the scanning. Repetitive elements annotation is downloaded from the ISB repeat masker (20). We exclude small repetitive element families with fewer than 30 EPIC probes of which the 15 nt 3΄-subsequences overlap with the family.
Rescued beta value and allele frequency for probes subject to CCS SNPs
To recover the beta value for DNA methylation for probe with CCS SNP, we contrasted the methylated and unmethylated allele after summing signal intensities from red and green channels, i.e.
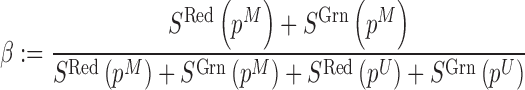 |
where 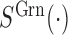 and
and 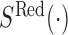 denotes the signal intensity measured the in green and red channel respectively. We can also derive the allele frequency measurement by contrasting signals from the two channels summed from both methylated allele and unmethylated allele. Formally,
denotes the signal intensity measured the in green and red channel respectively. We can also derive the allele frequency measurement by contrasting signals from the two channels summed from both methylated allele and unmethylated allele. Formally,
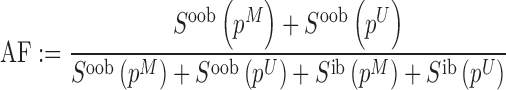 |
where 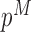 and
and 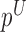 denote the methylated and unmethylated allele of probe
denote the methylated and unmethylated allele of probe 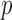 .
. 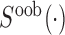 and
and 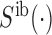 denote the signal intensity measurement in the out-of-band and in-band channels respectively. Signals from both in-band and out-of-band channel were subject to background correction (19) before being used.
denote the signal intensity measurement in the out-of-band and in-band channels respectively. Signals from both in-band and out-of-band channel were subject to background correction (19) before being used.
Constructing predictive model of ethnicity
A random forest classifier is trained using R package randomForest (21) with 200 trees estimated using allele frequencies (obtained above or from designed rs probes separately). A total of 1103 samples from LUAD, ESCA and THCA cancer types are used as test dataset and the remaining 7517 samples are used as training dataset (excluding cell line replica). The self-reported ethnicity labels retrieved from the TCGA synapse data portal. Only samples self-reported as either ‘ASIAN’, ‘BLACK OR AFRICAN AMERICAN’ or ‘WHITE’ are included.
Incomplete bisulfite conversion measurement
Incomplete bisulfite conversion is quantified by the ratio of the mean green signal in CpC probes over the mean green signal in TpC probes, or formally a GCT (Green CpC to TpC) score,
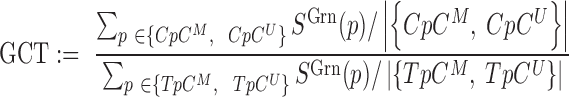 |
where 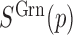 denotes the signal of probe
denotes the signal of probe 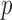 in green channel. GCT is typically greater than one and deviation of GCT from one represent incomplete bisulfite conversion or rare biological methylation of the first cytosine of CpC dinucleotides.
in green channel. GCT is typically greater than one and deviation of GCT from one represent incomplete bisulfite conversion or rare biological methylation of the first cytosine of CpC dinucleotides.
EPIC microarray manifest
MethylationEPIC_v-1-0_B1.csv was downloaded from the Illumina product support website http://support.illumina.com/array/array_kits/infinium-methylationepic-beadchip-kit/downloads.html on 10 May 2016.
Probe mapping
We mapped the probe sequences to bisulfite converted human genome assemblies GRCh37 and GRCh38 using a BWA-mem (22) based aligner specifically designed for bisulfite sequencing reads, included in the BIsulfite Sequencing Comprehensive Utilization and Integration Tools (BISCUIT) (23). BISCUIT mapping uses asymmetrical scoring to ensure that genomic sequence with T/A before bisulfite conversion does not match with C/G in the probe sequence. We exclude the unassembled contigs and haplotypes included in each genomic build. The mapping quality indicates the uniqueness of mapping with 0 presenting poor or equally optimal mapping to multiple positions and 60 representing unique mapping and perfect base pairing. The score in-between represents optimal but imperfect base pairing or with additional suboptimal loci. Ideal probe sequences are with mapping quality 60.
RESULTS
Hybridization quality affected by common polymorphism
We used average within-tissue variation in beta value measurements within 705 normal samples (see ‘Materials and Methods’ section) as a surrogate for SNP-introduced artifacts. It is to be expected that probes influenced by SNPs would have higher measurement variations among individuals. We compared the average within-tissue standard deviation (SD) for probes with SNPs at different positions in relation to the 3΄ end of the probe (marked as position 0), stratified by MAF (Figure 1B and C). The −1 position for Type II probes (the extension base, or C in CpG) was also included. We observed the highest influence in the position that affects the CpG dinucleotide (−1 position for Type II probes and 0 for Type I probes). The influence decreased with increasing distance from the 3΄-end of the probe. When an internal SNP was more than five bases from the 3΄-end of the probe, even with the highest MAF, its effect was negligible. Therefore, the existing practice of masking probes with a polymorphism within 10 bases from the target may be too stringent and could cause over-masking of valuable CpG sites. In addition, a polymorphism at the target CpG dinucleotide with an MAF <1% may still affect the beta value measurement for rare samples. We annotated probes that should be masked based on these criteria, and also in a population-specific manner based on 31 sub-populations studied in the 1000 Genomes Project (Supplementary Data), for users to choose from based on the population under study for optimal usage of the array.
As discussed in the introduction, the influence of CCS SNPs in the extension position for Type I probes had not been previously studied, as neither were they contained within the probe sequence nor part of the target CpG dinucleotides. These switches would cause some or all the signals to come from the unannotated channels, and presumably affect not only the DNA methylation measurement but also normalization methods that rely on the signal intensities of out-of-band (dubbed ‘OOB’) probes (19). The out-of-band channels for probes with a CCS SNP would likely not represent background signals. To find such probes, we overlapped the extension base of Type I probes with common SNPs from dbSNP build 147 and predict 1052 EPIC array probes with a potential color switch (Supplementary Data). When we plotted the in-band versus out-of-band signal for all probes for two normal colon sample and one HeLa cell line sample (Supplementary Data), we can see that most probes were measured in the correct channel and the probes fluorescing in the OOB channel had indeed been predicted to have a CCS SNP. These represented SNPs that these individual carry at the extension positions and causes one or two of the M/U probe pairs (in the case of heterozygous and homozygous loci respectively) to fluoresce from the opposite channel. In different samples, different subsets of these 1,052 probes fluoresced from the OOB channels, depending on the SNPs that these individuals carry (Supplementary Data).
As expected, we also observed an impact on beta value measurements. Again, probes affected by CCS SNPs had a higher average within-tissue variance across the normal samples compared with probes with non-CCS SNPs when the MAF was high (Figure 1D). Re-calculation of beta values by summarizing signals from both channels (Methods) rescued these probes and bring the high variance in the color-switching probes back to normal level (Figure 1D).
We explored the possibility of using of such CCS SNP probes as additional genotyping probes in addition to the explicitly built-in SNP probes (rs probes), as combined M+U signal intensities in the OOB and in-band channels in theory reflected the fraction of the alternative and reference alleles of the SNP in the extension base, respectively. Therefore, the ratio of the out-of-band signal to the sum of in-band and out-of-band signals would represent the AAF (‘Materials and Methods’ section). The density plot of the allele frequency thus derived clearly exhibited three peaks at 0, 0.5 and 1 corresponding to the homozygous reference, heterozygous and homozygous alternative genotypes (Supplementary Data). Both the explicit SNP probes (rs probes) on the array (n = 65 for HM450 and 59 for EPIC) and these implicit SNP probes that we recover (n = 364 for EPIC and 332 for HM450 with >1% MAF) clustered 8620 samples from different ethnicity in TCGA (Figure 2A and B; Supplementary Data). We set aside 1103 samples from LUAD, ESCA and THCA as test datasets and reconstructed random forest classifiers based on 7517 samples from the remaining cancer types. A classifier based on the additional probes resulted in improved concordance (97.6 versus 95.2%) with self-reported ethnicity compared with one based on the explicit SNP probes and trained under the same default parameter (Figure 2C and D). Supplementary Data shows the most predictive probes from the two sets in our classification.
Figure 2.

(A) Distribution of 8620 TCGA samples on the first and the second principal components identified from beta values measured by explicit SNP probes (rs probes designated by the manufacturer). Samples are colored by self-reported ethnicity; (B) Distribution of 8620 TCGA samples on the first and the second principal components identified from allele frequency recovered from CCS SNP probes; (C) Concordance between self-reported ethnicity and predicted ethnicity using explicit SNP probes on the test dataset (n = 1103, methods); (D) Concordance between self-reported ethnicity and predicted ethnicity using the recovered CCS SNP probes on the test dataset.
Hybridization competition with partial probe sequence
Limited overlap with repetitive elements (REs) does not cause measurement deviation
Subsequences of different lengths starting from 3΄ end (termed 3΄ nested subsequence) can be non-unique even though the entire probe is unique. It has been postulated that the overlap of repeat elements (REs) with the 3΄ end of the probe, even minimal, could cause spurious DNA methylation measurements (4), the rationale being that REs could have numerous copies in the genome and impact hybridization. In some studies, any probe overlapping with repeats in the last 15 bases from the 3΄ end was masked (24,25). Based on our mapping, 95.1% of these probes could be mapped uniquely to the human genome, suggesting that most RE-overlapping probes were sufficiently diverse to be discriminated from each other. We compared datasets of 18 samples assayed on both HM450 and WGBS platforms in TCGA. Neither total signal intensities (Supplementary Data) nor the beta values (Supplementary Data) showed a significant difference between probes with REs of different categories and repeat-free probes, even for repeat families with the highest abundance in the human genome including SINEs and LINEs (Supplementary Data). Neither was any difference observed when we restricted our probe set by requiring 40 bases from the 3΄ end of each probe be covered by the repeat family under consideration (Supplementary Data). These again did not support the practice of masking probes based on overlap with all annotated repetitive elements. Low complexity sequences showed significantly lower total intensities than non-repeat class (Mann Whitney U, one tail, P-value < 0.05). This was possibly attributable to the high copy numbers of these sequences in the genome that competed with the true target of the probe but failed to successfully extend, similar to the mechanism discussed below for the 3΄ nested subsequence with limited cross-hybridization. This, however, did not result in a difference in beta values as shown above.
Genomic copy number of 3΄ nested subsequence of sufficient length impacts intensity and beta value
Per the Illumina protocol, hybridization occurs at 37°C followed by a wash at 44°C which likely eliminates some non-specific hybridization events with insufficient length. Therefore, a limited sequence match, like the 15-nt overlap with low-complexity sequences described above, likely introduced competition for hybridization at the target site, but these non-target hybridization events were mostly transient and removed during the washing step. We reasoned that although an overlap of 15 bases did not cause a change in the beta value readout, if hybridization of the competing oligo was strong enough to withstand the washing step and cause an extension, signals would also be generated in the off-target target site(s). This would lead to an increased total signal intensity, and more importantly, a deviated beta value measurement, since in that case the beta value readout would reflect that of more than one locus. Therefore, we investigated the uniqueness of 3΄ nested subsequence of lengths ranging from 10 to 50 bases, independent of their overlap with REs and its impact on the measured signal intensity, as well as beta values.
Sequences under 20 bases long were generally associated with a high copy number due to sequence degeneracy (Figure 3A). Most subsequences became unique in the genome when they reached 22 bases (Figure 3B). We observed a negative correlation between signal intensity and the copy number of 3΄ subsequence when the length is limited, usually under 25 bases, which switched to a positive correlation after the 3΄ subsequence exceeded 25 bases (Figure 3C and D; Supplementary Data). When the 3΄-subsequence was longer than 40 bases, we observed a strong positive correlation between signal intensity in probes with higher copy numbers even though all these probes can be uniquely mapped at their full-length, confirming that hybridization of sufficient length would indeed generate additional signal at off-target sites.
Figure 3.

(A) Box plots showing copy numbers of 3΄ nested subsequence of the EPIC probes in the bisulfite-converted genome, with varying length of the 3΄ subsequence (x axis). (B) The fraction of probes with a unique 3΄ subsequence at a given length from all the probes; (C) Total signal intensities (sum of methylated and unmethylated alleles; y axis) for uniquely mappable Type I probes (see Supplementary Data for Type II probes) with varying copy numbers of the 3΄ subsequence (x axis) of 15, 30 and 40 bases long respectively. The signal intensities are measured in a normal colon sample; (D) Linear regression lines showing the dependence of total signal intensities on the genomic copy number of 3΄-subsequences of different lengths, for Type I probes (left panel) and Type II probes (right panel); (E) Association between the copy number of 3΄ subsequence and measurement accuracy. Averaged absolute differences in beta value measurement between HM450 and WGBS measurements for the same set of samples (n = 18) is plotted against different ranges of the copy number of the 3΄ subsequence of lengths 22, 30 and 40 bases.
We continued to investigate the impact on beta values, in addition to that on signal intensity, by comparing HM450 beta value readouts and WGBS measurements on 18 matched samples studied in TCGA. We indeed observed a higher deviation of HM450 measurement from the WGBS measurement in probes with greater 3΄-subsequence copy numbers (Figure 3E). This trend was increasingly evident when the length of the sub-sequence analyzed exceeded 25 bases, in line with the impact of the copy number to signal intensities observed above. This supported our postulation that non-unique hybridization by the 3΄ subsequence of sufficient length would interfere with the DNA methylation measurement at the target site and should be masked from analyses.
Dual use of probes as internal controls for bisulfite conversion completeness
Assuming equal copy number of the target locus, a Type I probe with a C in the extension base in the reference genome (dubbed a ‘CpC’ probe as it is in the context of CpCpG when combined with the target CpG of the probe on its 3΄ ends) should be indistinguishable in term of fluorescence intensity with a probe having a T in the extension base (‘TpC’ probes) after complete bisulfite conversion (Figure 4A), as C's would be converted to T's after bisulfite conversion and pair with a red-fluorescent A. When there is incomplete bisulfite conversion, the retained C is paired with a green-fluorescent G and any increase in the mean signal in the green channel of CpC probes compared with TpC probes should reflect the level of retention of C after bisulfite conversion. After excluding probes with SNPs, we discover a total of 46 733 CpC probes (41 176 in HM450) and 15 638 TpC probes (13 667 in HM450) in the EPIC array.
Figure 4.

(A) Illustration of the use of CpC and TpC probes as bisulfite conversion controls. Incomplete conversion at C extension sites (CpC probes, 5΄ CYG in template DNA, left) leads to hybridization of a green-fluorescent G with the retained C. Successful bisulfite conversion for CpC probes should be equivalent to a TpC probe (5΄ TYG in template DNA), and lead to hybridization of a red-fluorescent A with T (right panel). In contrast, probes with a T reference allele at the extension site (TpC probes) should not have green signals in the absence of SNPs, and any green signal for these probes should reflect background. (B) Mean green signal across all 46 733 probes for CpC (y axis) versus TpC probes (x axis) in 8652 TCGA samples; (C) Mean CpH beta values vs Green CpC to TpC (GCT) score for TCGA samples (n = 8652). Testicular Germ Cell Tumors (TGCT) within the TCGA datasets are colored by histology, while other tumors are black. Overall Pearson's correlation coefficient = 0.2, P < 1e-10; (D) Top left: density plot of CpG beta value distribution for cell line replicate with the highest GCT score (green) and the lowest GCT score (red); Top right: density plot of CpG beta value distribution for ten samples in Lung adenocarcinoma (LUAD) with the highest GCT score (green) and ten LUAD samples with the lowest GCT score. The same is repeated for Prostate Adenocarcinoma (PRAD, bottom left) and Bladder Urothelial Carcinoma (BLCA, bottom right).
Data production for TCGA samples was performed under tight quality control, including bisulfite conversion tests. Indeed, when we compared the mean green channel signal of CpC probes with the mean green channel signal of TpC probes, we saw that most of the TCGA samples displayed almost equal CpC green signals to TpC green signals (Figure 4B). This is likely also attributable to the fact that incomplete bisulfite conversion within the target sequence would also likely cause mismatches with the probe, disrupting hybridization and subsequent detection and would thus not be well tolerated on this platform. However, we did observe slightly elevated CpC compared to TpC signals, likely representing stochastic incomplete conversion at the extension site without affecting the target sequence. A small number of samples have a CpC/TpC green signal ratio (GCT score, see ‘Materials and Methods’ section) of >1.5. We investigated the overall beta value distribution for such samples with elevated GCT score with samples of the same cancer type but with more balanced GCT score. Samples with an elevated GCT score did exhibit elevated beta values manifested by unmethylated peak being shifted to the higher end (Figure 4C and Supplementary Data). For TCGA, a cell line replicate was assayed together with every batch and repeated 281 times. The same shift is also seen within these technical replicates.
The biggest challenge in determining incomplete conversion has been that bona fide non-CpG (CpH) methylation and incomplete bisulfite conversion are not easily distinguished. However, naturally occurring CpH methylation tends to occur primarily at CpA sites (26) and using CpC probes could help to distinguish these two scenarios. We compare GCT score with CpH methylation level (as measured by the mean beta value from 3091 embedded CpH/ch probes on the HM450 platform), and observed that samples with higher CpH (mostly testicular germ cell tumors (TGCT) of embryonal histology, or mixed histology with embryonal components) did not have higher incomplete conversion estimate while samples with putative incomplete conversion do show slightly higher, likely false positive, CpH measurement (Figure 4D; Pearson's R = 0.20, P < 1e-10). Seminomas within the TGCT cohort did not show such a trend. This also indicated bona fide CpH methylation in the embryonal TGCT samples, consistent with their embryonal origin.
EPIC and HM450 also include ten explicit bisulfite conversion control probes. When measurements from these explicit bisulfite control probes were plotted against CpH methylation levels, we observed a slight positive correlation between the two for the embryonal TGCT cases (Supplementary Data). This suggests that these bisulfite conversion controls probes are actually influenced by real biology and should therefore not be used as bisulfite conversion controls. The design of these probes likely assumed that cytosines in non-CpG contexts were not methylated and could be used as bisulfite conversion controls, without consideration of biological methylation at CpA residues, which can be methylated in embryonic stem cells and neural progenitor cells, and to a lesser extent in other cell types (27). The average measurements from these ten probes displayed a large variance and did not identify the subset of samples with likely incomplete conversion, as shown in Supplementary Data. We therefore recommend that these explicit bisulfite conversion control probes not be used for their designed purposes. Instead, the 46 733 CpC and 15 638 TpC probes provide a more reliable measurement for incomplete bisulfite conversion.
Probe mapping and masking
To fully characterize the EPIC array, we evaluated cross-hybridization by also checking whether the probe sequence could be uniquely and perfectly mapped to the target genomic location as indicated in the array's manifest file based on genome build GRCh37. Table 1 summarizes the number of probes mapped with mapping quality in different ranges (see ‘Materials and Methods’ section). We expected and confirmed (Table 1) that poor mapping quality to appear slightly more in Type I probes than Type II probes, as the unmethylated version (U) for Type I probes has slightly reduced sequence complexity and higher chance of non-unique mapping compared to the methylated version (M) in a bisulfite-converted genome. In cases where one probe of the M and U probes in the Type I design can be mapped to non-target location, the DNA methylation inference can be erroneous. As expected, all probes were mappable to GRCh37. Thirty-eight probes could not be mapped to GRCh38 (Supplementary Data). A higher fraction of probes with low mapping quality were seen for the more recent GRCh38 assembly where more previously unassembled sequences were included. A smaller fraction of Type II probes was subject to mapping issues. Other mapping problems are detailed in the Supplementary Data and corresponding probes are masked accordingly (see probe masking section below). When restricted to the HM450 platform, we compared probes with low mapping quality and other mapping problems identified with ones free of mapping problems. We measured the difference of beta value readouts in 18 datasets assayed on both HM450 and WGBS platforms in TCGA (Methods). Probes with mapping problems exhibited greater deviation from the WGBS measurement (Supplementary Data).
Table 1. Mapping quality of EPIC array probes against the GRCh37 and GRCh38 genomes.
| GRCh38 | ||||||
|---|---|---|---|---|---|---|
| GRCh37 | [0,10] | (10,30] | (30,59] | (59,60] | ||
| CpG | type I A | [0,10] | 7463 | 16 | 3 | 3 |
| (10,30] | 88 | 4580 | 8 | 2 | ||
| (30,59] | 39 | 29 | 7010 | 8 | ||
| (59,60] | 210 | 24 | 65 | 122 714 | ||
| type I B | [0,10] | 5853 | 22 | 1 | 8 | |
| (10,30] | 91 | 4216 | 6 | 4 | ||
| (30,59] | 33 | 23 | 5231 | 10 | ||
| (59,60] | 222 | 25 | 59 | 126 458 | ||
| type II | [0,10] | 18 613 | 44 | 16 | 46 | |
| (10,30] | 373 | 16 321 | 15 | 12 | ||
| (30,59] | 189 | 73 | 35 893 | 41 | ||
| (59,60] | 1048 | 75 | 199 | 648 684 | ||
| CpH | type II | [0,10] | 134 | 1 | 0 | 0 |
| (10,30] | 0 | 153 | 0 | 0 | ||
| (30,59] | 0 | 1 | 327 | 0 | ||
| (59,60] | 1 | 0 | 0 | 2315 | ||
Probes are stratified by the target type (CpG, CpH) and the design type (Type I probe A, Type I probe B and Type II). For SNP probes: 21 Type I-A, 20 Type I-B and 38 Type II probes are with mapping quality 60, 1 Type I-B in (30,59).
Based on all the issues we identified in previous sections, we annotated probes that should be masked or processed differently for the entire EPIC array (Figure 5A showing the size of major categories and Figure 5B showing chromosomal distribution of major categories). We flagged probes that could not be mapped in GRCh38 assembly, or extended to unintended bases in either of the genome assemblies. Probes masked for mapping were enriched in telomeric and peri-centromeric region (Figure 5B). We also masked probes with potential cross-reaction that included (i) Type II probes mapped with a low mapping quality (<10) or with mismatches or INDELs; (ii) Type I probes with a low mapping quality or mismatches or INDELs in either of the M or U allele; (iii) Type I probes with two of its alleles mapped to different genomic locations; and (iv) probes with a non-unique 3΄ nested subsequence of at least 30 bases. This threshold resulted in 12 108 additional unique probes being masked. Only 7639 of these probes were inside repetitive elements annotated in repeat masker.
Figure 5.

(A) Numbers of probes in each masking category, including previous practices that we do not recommend. Black boxes suggest the enclosed masking is recommended; (B) Circos plot of the distribution of (i) number of EPIC probes in each 1Mb bin; (ii) number of NCBI RefGene; (iii) number of masks because of mapping issues; (iv) number of probes masked because of non-unique 30-base or longer 3΄ subsequence; (v) number of masks for SNPs using global allele frequency from the entire 1000 Genome Project population. (C) The resulting population-specific masking with merged population-specific SNP masking and other recommended masking (shown in panel A).
To reduce the influence of SNPs, we provided annotation for probes that should be masked or processed differently based on our exploration: (i) probes with any SNP of global MAF over 1% and within 5 bp from their targets; (ii) Type II probes with SNP of global MAF over 1% affecting the extension base; (iii) Type I probes with putative CCS SNPs. Categories (i) and (ii) should be masked, and Category (iii) could be rescued if in processing signals from both color channels were summarized (‘Materials and Methods’ section). This resulted in the masking of over 8748 probes compared to applying an AAF cut-off of 1% on Illumina's official manifest of EPIC array (Supplementary Data). For subpopulation-specific masking, we suggest SNPs included be of AAF >1% in the specific population and involve more than one individuals (in the 1000 Genome projects) with the alternative allele (Figure 5C). Our investigation also revealed an incorrect exclusion of 4329 probes from the mask based on large (>1%) AAFs provided by the EPIC manifest. This exclusion is based on discrimination of SNPs with AAFs computed from very small numbers of samples (most with allele frequencies being 0.5 based on two samples) and absent from the common SNP collection released with dbSNP Build 147 and 1000 Genomes Project report. We studied these SNPs in the TCGA normal datasets and confirmed little impact on the beta value variation by these SNPs (Supplementary Data). Numbers of probes masked in different categories were summarized in Supplementary Data. Our annotation is downloadable as Supplementary Data and also available at http://research.vai.org/Tools/InfiniumAnnotation/index.html We also include in this site functional annotation of probes including the positions relative to CpG islands, genes, imprinting control regions, chromatin states and binding of CTCF and other transcription factors.
DISCUSSION
On the Illumina Infinium platforms, Type I design utilizes a probe pair while Type II design reduces that to a single probe, and uses the color channels differently. Only signals from the specified color channel (dubbed ‘in-band’) are read in for Type I probes while the ‘out-of-band’ signals have been routinely discarded. Although it appears that Type I design is less efficient in term of chip usage, the out-of-band signals nevertheless contain valuable information. Triche et al. showed that the pooled out-of-band signal could be used to infer the background fluorescence level for signal deconvolution, for their method ‘noob’ (norm-exp deconvolution using out-of-band probes) (19). In this study, we further discover two extra types of usage for subsets of these Type I probes based on the out-of-band signals: as SNP or internal bisulfite conversion control probes. In the former case, the in-band and out-of-band signals represent two different alleles, and in the latter they represent converted and retained non-converted cytosines, respectively (Figure 1A).
There are 59 built in explicit SNP probes (‘rs’ probes) on the EPIC array, a slightly reduced subset compared to the 65 on HM450. These probes have proven useful in identifying potential sample swaps whenever matched samples are included. We show that these probes can be used to predict ethnicity, a useful application for EWAS studies where ethnicity is an important covariate. The additional 1,052 CCS SNP probes that we discover greatly outnumber the explicit SNP probes, and provide additional information that could lead to more powerful applications; for example, to infer finer population composition in an individual. One could propose to use Type II design for similar purposes. However, unlike Type I extension bases, where the summation of the methylated and unmethylated alleles in each color channel yield unambiguous quantification of the SNP allele, the extension base for Type II probes is at the site of measured methylation and the methylation state would influence the beta value readout and yield ambiguous genotype information.
The CCS SNP probes not only expand the utility of the array; when left unnoticed, they can also lead to erroneous DNA methylation measurement and false discoveries in EWAS studies (5), methylation quantitative trait loci (meQTL) studies (28) and any studies where the linkage between genotype and DNA methylation is of interest or can be a confounding factor. We present a method for rescuing the DNA methylation measurement for these probes by simply merging signals from the two color channels.
Understanding the behavior of these probes is important not only for interpreting the final beta value readouts but also for preprocessing and normalization. Some of the current best practices for preprocessing and normalization methods should be modified based on results shown in this paper. For example, the background correction method noob (19) relies on the out-of-band probes. But as we have shown, SNPs in the extension base and incomplete bisulfite conversion could both cause non-background fluorescence in the out-of-band channel. Although these exceptions only affect a small portion of the probes, caution should be taken using signal from these probes to represent background signal. Funnorm (29) uses embedded control probes for HM450 data normalization but we show that the internal bisulfite conversion probes actually are influenced by real biology as they likely contain CpA probes, and can be problematic when samples with high CpH levels are analyzed. We recommend using these new probes that we recover from Type I extension to replace the internal bisulfite controls.
Using CpC and TpC probes, we show that severe incomplete bisulfite conversion is not noticeably observable in the TCGA dataset, and we furthermore suggest that the Infinium platform is inherently relatively impervious to incomplete bisulfite conversion due to resulting interference with probe hybridization. This constitutes one additional advantage of these platforms over WGBS in addition to the low cost and superior quantification. However, small but consistent effects can be picked up in EWAS studies and it is important to include such QC metrics and correct for the low level of incomplete conversion when necessary. Although TpC and ApC probes both fluoresce in red and can be used in the denominator of our incomplete bisulfite conversion QC, TpC has the advantage of sharing the same extension base (‘A’) with CpC probes and could avoid biased cross-channel interference from different base incorporation efficiency.
The artifacts associated with probes affected by polymorphism within the probe sequence have long been recognized. However, the masking of SNP-associated probes has largely been inconsistent and often overdone in the literature. 83% of the EPIC array probes carry SNP annotations and most of them are far away from the target and unique to a certain ethnic group. We performed both in silico mapping and large-scale real data analysis of probe performance to allow sensible choices of probe masking. We show that internal SNPs more than five base pairs from the probes’ 3΄-end do not noticeably affect the DNA methylation measurement. Exclusion based on 15-base overlap at the 3΄ end with annotated repetitive elements is largely unnecessary. Non-unique 3΄-subsequences of 30 or more bases long can impact both signal intensities and beta value calculation. Moreover, a substantial number of probes are pardoned when we adopt a population-specific masking. Our characterization helps minimize unnecessary loss of DNA methylation information from the platform.
We also provide an update to the manufacture's manifest with the latest genome and transcriptome builds, with a correction of erroneous genomic location information. In addition, the EPIC array includes substantial coverage of regulatory elements with half of the probes and all additional probes compared to the HM450 platform designed against such regions. However, the current official manifest does not provide a detailed annotation on these regulatory elements. We annotate these probes with data from REMC and ENCODE TFBS and ChIP-seq experiments, to allow for efficient usage of these probes and assist in the interpretation of data generated.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Timothy J. Triche Jr for insightful discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health/National Cancer Institute [U24 CA143882, R01 CA170550 to P.W.L., in part]; Ovarian Cancer Research Fund Grant [373933 to H.S.]. Funding for open access charge: Van Andel Research Institute (VARI) New Investigator funding (Shen).
Conflict of interest statement. None declared.
REFERENCES
- 1. Shen H., Laird P.W.. Interplay between the cancer genome and epigenome. Cell. 2013; 153:38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013; 14:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bibikova M., Barnes B., Tsan C., Ho V., Klotzle B., Le J.M., Delano D., Zhang L., Schroth G.P., Gunderson K.L. et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011; 98:288–295. [DOI] [PubMed] [Google Scholar]
- 4. TCGA. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013; 368:2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rakyan V.K., Down T.A., Balding D.J., Beck S.. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011; 12:529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bock C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012; 13:705–719. [DOI] [PubMed] [Google Scholar]
- 7. Dedeurwaerder S., Defrance M., Bizet M., Calonne E., Bontempi G., Fuks F.. A comprehensive overview of Infinium HumanMethylation450 data processing. Brief. Bioinform. 2014; 15:929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Naeem H., Wong N.C., Chatterton Z., Hong M.K.H., Pedersen J.S., Corcoran N.M., Hovens C.M., Macintyre G.. Reducing the risk of false discovery enabling identification of biologically significant genome-wide methylation status using the HumanMethylation450 array. BMC Genomics. 2014; 15:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Byun H.M., Siegmund K.D., Pan F., Weisenberger D.J., Kanel G., Laird P.W., Yang A.S.. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum. Mol. Genet. 2009; 18:4808–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morris T.J., Beck S.. Analysis pipelines and packages for Infinium HumanMethylation450 BeadChip (450k) data. Methods. 2015; 72:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Price M.E., Cotton A.M., Lam L.L., Farré P., Emberly E., Brown C.J., Robinson W.P., Kobor M.S.. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin. 2013; 6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen Y.A., Lemire M., Choufani S., Butcher D.T., Grafodatskaya D., Zanke B.W., Gallinger S., Hudson T.J., Weksberg R.. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013; 8:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cordaux R., Batzer M.A.. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009; 10:691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abyzov A., Iskow R., Gokcumen O., Radke D.W., Balasubramanian S., Pei B., Habegger L., Lee C., Gerstein M.. Analysis of variable retroduplications in human populations suggests coupling of retrotransposition to cell division. Genome Res. 2013; 23:2042–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harrison P.M., Zheng D., Zhang Z., Carriero N., Gerstein M.. Transcribed processed pseudogenes in the human genome: An intermediate form of expressed retrosequence lacking protein-coding ability. Nucleic Acids Res. 2005; 33:2374–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sundaram V., Cheng Y., Ma Z., Li D., Xing X., Edge P., Snyder M.P., Wang T.. Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res. 2014; 24:1963–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K.. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001; 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Triche T.J., Weisenberger D.J., Van Den Berg D., Laird P.W., Siegmund K.D.. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013; 41:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smit A., Hubley R., Green P.. Origin of interspersed repeats in the human genome. Curr. Opin. Genet. Devel. 1996; 6:743–749. [DOI] [PubMed] [Google Scholar]
- 21. Liaw A., Wiener M.. Classification and regression by randomForest. R News. 2002; 2:18–22. [Google Scholar]
- 22. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou W. BISCUIT-0.1.3. 2016; 10.5281/zenodo.48262.
- 24. Ling C., Pease M., Shi L., Punj V., Shiroishi M.S., Commins D., Weisenberger D.J., Wang K., Zada G., Ezzat S. et al. A pilot genome-scale profiling of DNA methylation in sporadic pituitary macroadenomas: association with tumor invasion and histopathological subtype. PLoS One. 2014; 9:e96178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun Z., Prodduturi N., Sun S.Y., Thompson E.A., Kocher J.-P.A.. Chromosome X genomic and epigenomic aberrations and clinical implications in breast cancer by base resolution profiling. Epigenomics. 2015; 7:1099–1110. [DOI] [PubMed] [Google Scholar]
- 26. Schultz M.D., He Y., Whitaker J.W., Hariharan M., Mukamel E.A., Leung D., Rajagopal N., Nery J.R., Urich M.A., Chen H.. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature. 2015; 523:212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. He Y., Ecker J.R.. Non-CG methylation in the human genome. Annu. Rev. Genomics Hum. Genet. 2015; 16:55–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rushton M.D., Reynard L.N., Young D.A., Shepherd C., Aubourg G., Darlay R., Gee F., Deehan D., Cordell H.J., Loughlin J.. Methylation quantitative trait locus analysis of osteoarthritis links epigenetics with genetic risk. Hum. Mol. Genet. 2015; 24:7432–7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fortin J.-P., Labbe A., Lemire M., Zanke B.W., Hudson T.J., Fertig E.J., Greenwood C., Hansen K.D.. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014; 15:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.