


Abstract
In vertebrates, the 7SK RNA forms the scaffold of a complex, which regulates transcription pausing of RNA-polymerase II. By binding to the HEXIM protein, the complex comprising proteins LARP7 and MePCE captures the positive transcription elongation factor P-TEFb and prevents phosphorylation of pausing factors. The HEXIM-binding site embedded in the 5΄-hairpin of 7SK (HP1) encompasses a short signature sequence, a GAUC repeat framed by single-stranded uridines. The present crystal structure of HP1 shows a remarkably straight helical stack involving several unexpected triples formed at a central region. Surprisingly, two uridines of the signature sequence make triple interactions in the major groove of the (GAUC)2. The third uridine is turned outwards or inward, wedging between the other uridines, thus filling the major groove. A molecular dynamics simulation indicates that these two conformations of the signature sequence represent stable alternatives. Analyses of the interaction with the HEXIM protein confirm the importance of the triple interactions at the signature sequence. Altogether, the present structural analysis of 7SK HP1 highlights an original mechanism of swapping bases, which could represent a possible ‘7SK signature’ and provides new insight into the functional importance of the plasticity of RNA.
INTRODUCTION
In the nucleus of higher eukaryotes, the non-coding RNA 7SK participates in the regulation of transcription by RNA-polymerase II (1–3). It assists in capturing the positive transcription elongation factor P-TEFb, inhibits its kinase activity and thereby prevents P-TEFb's function, which is to alleviate transcription pauses (4,5). This requires binding to a protein, HEXIM, (HEXIM1, or the minor HEXIM2 in human, which seem to behave similarly with respect to RNA-binding) which in turn binds the cyclin moiety of P-TEFb (6,7). Since P-TEFb is the cellular factor hijacked by human immunodeficiency virus (HIV) to enhance its transcription, 7SK is indirectly linked to the transcription of HIV RNA by monitoring P-TEFb availability (8).
The 7SK RNA belongs to the 7SKsnRNP core particle, together with proteins LARP7 and MePCE, which contribute to its protection against nucleases and its packing into a functional molecule (9–12). Alternative models of the secondary structure of 7SK have been described in the literature. A fold into four domains was proposed after probing the accessibility of the nucleotides of 7SK extracted from human cells (13). Another 2D structure (Figure 1B) was based on the analysis of about a hundred 7SK sequences spanning vertebrates, fly and mollusk species (14). It similarly contains a limited number of domains and linkers, but is closed through base-pairing of conserved sequences. The closure was recently proposed to be facilitated by LARP7 and thus impact 7SKsnRNP assembly (15). The most conserved domains of the 7SK RNA are the 5΄- and the 3΄-hairpins, present in the two 2D structures (16). Both were shown to participate in the function of 7SK (17). The 3΄-hairpin (HP4; 300–331 in human 7SK) was identified as involved in P-TEFb inhibition (17,18) and recently shown to contribute to LARP7 binding (15,18). The 5΄-hairpin (HP1; residues 24–87 in human) was shown to be essential for HEXIM recognition (17,19). It is also able to bind the HIV trans-activator protein Tat (20) which in HIV-infected cells, recruits P-TEFb after anchoring to the TAR domain of the early transcript (21). Both Tat and HEXIM thus bind the cyclin T1 moiety of P-TEFb following interaction with RNA. A major difference is that the HEXIM proteins are dimeric. The dimer interface encompasses a long helix at the C-terminal third of the sequence, which forms an interstrand coiled-coil (22,23). Apart from this domain, HEXIM is an intrinsically disordered protein (24).
Figure 1.

The HP1 5΄ domain of 7SK. (A) Sequence of the 5΄-hairpin (HP1) studied in the present structural work. In red, sequence changes made to facilitate production by enzymatic synthesis while keeping the base-pairing scheme (A25G, C26G, G85C, U86C) or crystallization (loop reduction from the original 11 nucleotides to a tetraloop UUCG). (B) Localization of HP1 (squared) in the full-length 7SK model. The 7SK-motif is shown in red. (C and D) HEXIM-binding assays. (C) Native gel showing the complexes (band noted X) formed by titration with HEXIM of the wild-type RNA HP1 (WT) or after loop reduction to UUCG or GAAA (free RNA band noted R). (D) Binding curves from (C) data.
The HP1 5΄-hairpin comprises a strictly conserved sequence signature, a GAUC repeat framed by single-stranded nucleotides, uridines in most sequences (Figure 1B). Several studies by the group of P. Stadler showed that in a gene with a Polymerase III promoter and terminating by poly-thymidine two GAUC sequences separated by a linker of sufficient length to make a hairpin, is a key feature that characterizes 7SK (14,16,25). It is thus named the 7SK-motif in the following text. In a previous study, we showed that the (GAUC)2 forms a short helix of four base-pairs (26). It was identified as belonging to the HEXIM binding site by monitoring in an NMR experiment the spectral perturbations of the imino-proton signals upon titration with a peptide comprising the RNA-binding sequence of HEXIM proteins. This sequence, an Arginine-Rich Motif (ARM), is the same for HEXIM1 and the minor HEXIM2, and similar to that of Tat (27). We further identified the bulged uridines, together with the (GAUC)2, as specifically required for HEXIM-binding by a mutational analysis monitored by binding assays with the full-length protein (26).
The 7SK-motif is intriguing since it is very small yet nonetheless characterizes 7SK. Moreover, although it is a symmetrical sequence, it binds only one ARM-motif of a HEXIM dimer (26,28). What the second ARM does is still an open issue, but it probably has consequences on the functional mechanism converting HEXIM into a P-TEFb inhibitor (28). It is expected that this mechanism relies on conformational changes of 7SK upon HEXIM binding (29), thus making the highly flexible and modular nature of the 7SK RNA a functional requirement. Interestingly, some of the molecular flexibility may also be expected in the 5΄-domain, which encompasses bulges and internal loops in all 7SK sequences (16,25). Hairpin flexibility was shown to be involved in Tat binding, although another RNA hairpin was considered in that study (30).
In order to uncover the structural characteristics of the 7SK-motif, and with the hope of better understanding its recognition by HEXIM, we launched a structural investigation of the 5΄-domain of 7SK. We present here a crystallographic investigation of the structure of the 5΄-hairpin of 7SK, HP1, modified at the apical loop to facilitate crystal formation. This 57-mer RNA forms a surprisingly straight and compact helical stack with most of the un-paired bases interacting inside the helix. In particular, two un-paired uridines of the 7SK signature fill the groove formed by the (GAUC)2 short helix. However, the structure is flexible, as indicated by the observation of two conformations at the 7SK-motif, with a third uridine either flipped in or out of the molecule. This conformational variability was further investigated by molecular dynamics (MD) simulation. It indicated that the two conformations represent stable alternatives. To further analyze how the structural elements highlighted in the crystal structure impact HEXIM-binding, we performed interaction assays with a series of mutants. These highlighted the importance of the nucleotides framing the (GAUC)2 motif, with a major contribution of the uridine on the 5΄-side of the strand (U40 in human) to recognition by HEXIM. Altogether, the structure of 7SK HP1 provides new insight into the functional importance of the plasticity of RNA molecules.
MATERIALS AND METHODS
The RNA was produced by in vitro transcription with T7 polymerase from a PCR-generated template, purified by denaturing gel-electrophoresis followed by ion exchange chromatography and stored in a buffer containing 10 mM Na cacodylate, 2 mM MgCl2 and 0.25 mM EDTA. Monoclinic crystals grew at 4°C in 30% polyethylene glycol PEG 1000, 50 mM Tris pH 7.5, 50 to 75 mM NaCl, 50 mM MgCl2. The mutational analysis was performed as described previously (26). Data sets were collected from the native crystals and crystals soaked for several hours either with osmium hexamine (2 mM) or with gold (KAu[III]Cl4, 10 mM) in a solution containing 40% PEG 1000 prior to flash-freezing. The structure was solved by molecular replacement using PHASER with a model built using RNAcomposer (31) and the Au-derived crystal data set, exploiting the approach described by Robertson et al. (2010) (32). After refinement with BUSTER (33), the model at 2.3 Å resolution was used to phase the native crystals and osmium-derived crystals, to give three structures, as summarized in Table 1. The molecular dynamics simulation was performed with the Amber force field (34) in a neutral system comprising potassium and sodium ions. More experimental details are provided as Supplementary Text.
Table 1.
| Data collection statistics | Native | Osmium | Gold |
|---|---|---|---|
| Diffraction Source | ESRF BM30 | Soleil Proxima 1 | Soleil Proxima 1 |
| Wavelength (Å) | 0.9797 | 1.13956 | 1.04014 |
| Temperature (K) | 100 | 100 | 100 |
| Detector | ADSC 315 | ADSC 315r | ADSC 315r |
| Crystal-to detector distance (mm) | 395.365 | 318.00 | 354.464 |
| Rotation range per image (°) | 1.0 | 1.0 | 1.0 |
| Total rotation range (°) | 180 | 180 | 370 |
| Exposure time per image (s) | 60 | 0.5 | 0.5 |
| Space group | P21 | P21 | P21 |
| a, b, c (Å) | 45.79, 47.78, 68.67 | 47.26, 47.87, 69.15 | 47.32, 47.83, 69.23 |
| α, β, γ (°) | 90, 99.05, 90 | 90, 105.24, 90 | 90, 105.54, 90 |
| Mosaicity (°) | 0.256 | 0.167 | 0.187 |
| Resolution range (Å) | 30–2.20 (2.33–2.20) | 50–2.35 (2.41–2.35) | 50–2.32 (2.38–2.32) |
| Total No. of reflections | 44 738 (2945) | 41 939 (1943) | 92 102 (4122) |
| No. of unique reflections | 13 144 (1235) | 22 741 (1368) | 23 716 (1157) |
| Completeness (%) | 87.3 (52.0) | 93.4 (75.3) | 93.7 (61.9) |
| Multiplicity | 3.4 (2.38) | 1.8 (1.42) | 3.9 (3.56) |
| {I/σ(I)} | 18.57 (0.97) | 7.37 (1.30) | 9.4 (1.12) |
| CC1/2 | 99.9 (42.5) | 99.5 (77.4) | 99.8 (46.9) |
| R merge 1 (%) | 3.9 (93.9) | 6.2 (45.8) | 9.8 (132.7) |
| R r.i.m. 2 (%) | 4.6 (122.3) | 8.3 (64.4) | 11.3 (154.6 |
| Over all B factor from Wilson plot (Ų) | 59.76 | 54.47 | 54.98 |
| Refinement statistics | |||
| Reflections used in refinement | 13 139 | 12 950.00 | 12 303.00 |
| Reflections used for R-free | 553 | 532.00 | 491.00 |
| R work 3 (%) | 19.24 | 20.27 | 18.63 |
| R free 4 (%) | 23.77 | 23.4 | 22.61 |
| Number of non-hydrogen atoms | |||
| RNA | 2683 | 2660.00 | 2751.00 |
| Ion | 4 | 9.00 | 4.00 |
| Water | 230 | 242.00 | 184.00 |
| R.m.s. Deviations | |||
| Bonds (Å) | 0.01 | 0.01 | 0.01 |
| Angles (°) | 0.79 | 0.93 | 0.83 |
| Average B factors (Ų) | 58.6 | 59.17 | 59.41 |
| RNA | 58.91 | 59.38 | 59.8 |
| Ion | 63.04 | 88.36 | 62.79 |
| Water | 55.48 | 51.63 | 47.76 |
1 R merge = Σhkl Σi |Ii(hkl) – {I(hkl)}| / Σhkl ΣiIi(hkl).
2 R r.i.m. = redundancy independent merging R factor.
3 R work = Σhkl ||Fobs| - |Fcalc|| / Σhkl |Fobs|.
4 R free = Σhkl ||Fobs| - |Fcalc|| / Σhkl |Fobs| calculated using a random set containing 5% of the reflections that were not included throughout refinement.
5The precentage of residues in Ramachandran plot areas were determined using PROCHECK.
RESULTS
The crystallized RNA domain
The HP1 domain (boundaries 24–87) used in the structural work (Figure 1A and B) corresponds to the largest domain common to the two published secondary structures of 7SK (13,14). It was previously shown to fold as a hairpin in the same way as in the full-length 7SK context, and to be sufficient to bind one HEXIM dimer (19,26,28,35).
No crystal was ever obtained with the wild-type hairpin comprising an apical loop of 11 nucleotides. Since this loop is not conserved across species, neither in length or sequence, we exchanged it with a small tetraloop of sequence UUCG, known to fold as a tight stable structure (36–38). The mutant hairpin binds HEXIM with a similar affinity as the wild-type, as shown by electrophoretic mobility shift assays (Figure 1C and D). The shortening of the loop favored formation of monoclinic crystals, which appeared weeks after the crystallization tray was set-up. The crystals used for the structure determination were soaked with osmium or gold salts, but the structure was finally solved by the molecular replacement method since the very low derivation with these salts did not allow exploiting these data sets by the SAD or SIRAS methods. The molecular replacement was achieved using a model constructed with RNAcomposer (31) and the data from the crystal soaked with the osmium salt. The native structure was obtained by phasing the non-isomorphous native data (from crystals obtained in an earlier stage of the project) with the Os model. Altogether, we obtained three independent models at 2.2–2.3 Å resolution (Table 1).
General description of the crystal structures
The RNA molecule is composed of four regions (Figure 2A): the stem (24–33 and 78–87) comprises 10 base-pairs in a stack including three UoG pairs; the central region (34–39 and 68–77) contains one internal loop (34, 75–77) and a bulge (71–72) framed by two stacks of two C-G base-pairs; the 7SK-motif (40–45 and 63–67) comprises four base-pairs and the bulged uridines; the apical region comprises a stem of three C-G base-pairs and the tetraloop UUCG.
Figure 2.
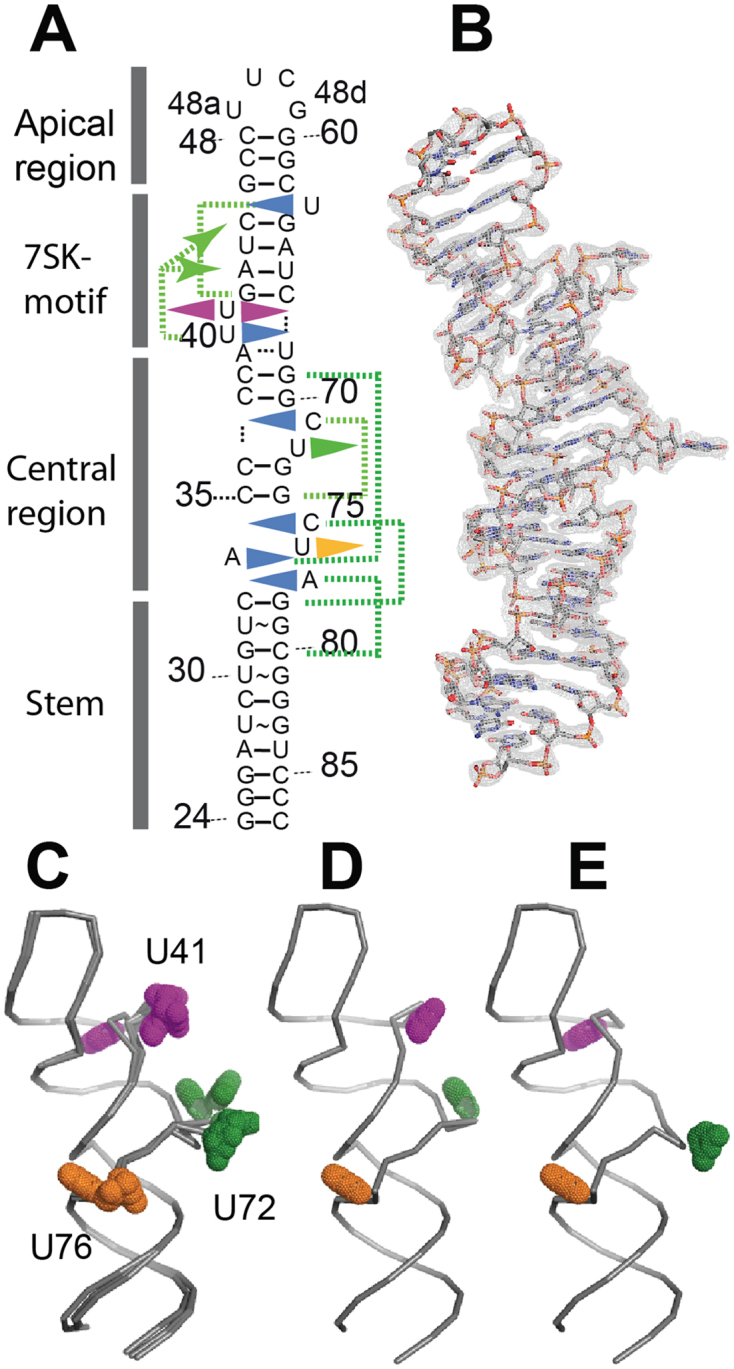
Global view of the molecule highlighting the conformation variability. (A) Sequence of HP1 showing the four regions and highlighting the bulged nucleotides (triangles) forming triple interactions in the helical stack (blue triangle turned inwards) or facing the solvent (triangle outwards) with U41 (magenta), U72 (green) and U76 (orange). Green dotted lines connect the bulged residues with their coplanar base-pairs. (B) Molecule B (grey sticks) in the Au-map contoured at 1.0 σ. (C) Superposition of several molecules (molecule A from Au-, Os- and native maps) showing position variation at U72 or U41. (D) Conformation OUT, as observed in molecule A of Au-map. (E) Conformation IN, as observed in molecule A of Au-map.
All crystals contain two molecules (A and B) in the asymmetric unit, leading to a redundant determination of six RNA structures, which are globally similar (average rmsd = 0.89 Å). However, the structures from the Au and Os data sets differ slightly from that obtained from the native crystals (rmsd 1.35 and 1.23 Å, respectively, for the A molecules, rmsd = 0.38 Å between molecules B of the Au and Os structures).
The global structure (Figure 2B) confirms the 2D base-pairing scheme shown in Figure 1A. It is surprisingly straight, with the four domains piled up in a stack. Of the nine nucleotides represented as bulges in the topology diagram, six are turned inside the helical stack in the crystal structure and form ‘triple’ interactions with base-pairs. Interestingly, these interactions often involve an unexpectedly distant base-pair. This is summarized by green dotted lines in Figure 2A and will be detailed below. As a consequence, the structure has a length similar to a model molecule without bulges or internal loops (Supplementary Figure S1).
The structure is however quite different from a standard helix. The un-paired nucleotides induce a twist, which strongly impacts the orientation of the 7SK-motif. When the basal stem regions of the HP1-RNA and a standard helix are superimposed, the (GAUC)2 are found at a similar distance from the end, but are located on opposite sides, as shown in Supplementary Figure S1A.
The structures vary essentially at position U41
All six structures show extruded bases. In the central region, U72 and U76 face the solvent, where they occupy various positions (Figure 2C). Base U41 is also extruded in most structures but can be found turned into the helical stack (Figure 2C, Supplementary Figure S2A). This variation is drastically different from the conformational flexibility observed in bases U72 and U76, which occupy a more continuous range of positions (Figure 2C, Supplementary Figure S2B). In the following text, the conformation with U41 flipped out, shown in Figure 2D is named OUT, and the other IN, with the base stacked in the helix (Figure 2E). The IN conformation is observed as an alternate conformation with an estimated occupancy of 50% in molecules A in both Os- and Au-derived crystals. The electronic density shows clearly two alternate positions for the phosphate group (Supplementary Figure S3A). The situation is exactly the same in the two maps obtained from the Au and Os data sets. Observation of two alternate conformations rules out the hypothesis that the IN conformation could be constrained by the crystal environment. For the extruded conformation, base U41 can be found either wedged into the minor groove of a symmetry-related molecule (Supplementary Figure S1B), or free in the solvent, due to the different environment of the native crystal. The two conformations thus seem to reflect a flexibility of this part of the molecule, which interestingly is located at the 7SK-motif.
Uridines swap positions at the 7SK-motif
The 7SK-motif comprises four base-pairs (G42-A43-U44-C45 paired with G64-A65-U66-C67) and three uridines (U40-U41 on one strand and U63 on the other strand). Uridines U40 and U63 are nested into the major groove of the regular helix formed by the four base-pairs (GAUC)2. The uridine on the apical side of the 7SK-motif, U63 (in orange in Figure 3) binds at the level of base-pair G42-C67, and forms a trans Watson–Crick/Hoogsteen pair with G42. The imino proton of U63 contributes a H-bond (2.9 Å) with the carbonyl group of G42. Another, more distant, H-bond is possibly made between the O2 oxygen of U63 and the amino group of the base C67 (3.4 Å). In the OUT conformation, U40 lies at the level of base-pair A43-U66 of the (GAUC)2 motif, and interacts with the Hoogsteen face of the adenine (Figure 3A and C). In the IN conformation, U41 occupies this position and makes the same cis Watson–Crick/Hoogsteen interaction (Figure 3D). This induces a displacement of U40 toward the apex of the molecule, at the level of the C45-G64 base-pair, where it is then involved in a trans Watson–Crick/Hoogsteen pair with G64 (Figure 3B). This corresponds to a change of register of two steps in the helical stack. Apart from the exchange of position of U40 and U41, only a few changes are observed for the (GAUC)2 helix and uridine 63 (Supplementary Figure S2). The rmsd between the two conformations is 0.68 Å for U63 and less than 0.3 Å for the AUC sequence. It is slightly higher for guanines G42 and G64 with rmsd values of 1.02 and 0.62 Å, respectively. Interestingly, in the Au-soaked crystal, a low occupancy gold ion is observed at the level of C45-G64, at the position occupied by base U40 in the IN alternate conformation.
Figure 3.

The uridines at the 7SK motif. The (GAUC) repeat and uridines U40 (cyan), U41 (magenta) and U63 (orange). (A) Axial view of conformation OUT in molecule A (Os-map). The black arrow represents the helical axis and shows the direction of the apical loop. (B) Same view for conformation IN. (C) Perpendicular view for conformation OUT. (D) Perpendicular view for conformation IN. H-bonds of the Hoogsteen base-pair are indicated with dotted lines.
Nucleotide A34 from the central region comes to lie near the 7SK-motif
The nucleotide A34 of the 5΄ strand of the internal loop lies between the 7SK-motif and the central region (Figure 4A and C). It inserts into the major groove, in the plane of base-pair G69-C38, resulting in a A34/G69 trans Watson–Crick/Hoogsteen interaction. The amino group of A34 establishes a H-bond with N7 of G69. This special position suggests that A34 could monitor the relative orientation of the blocks formed by the 7SK-motif on one side and the central region on the other. The major groove width is quite small at this level, with a distance of 3.1 Å across the groove, as compared with the typical 4.5 Å. The groove width was obtained with Curves+ (39) and represents the minimal distance between the backbone spline curves passing through the phosphorus atoms.
Figure 4.

The central region. (A) Global view of the central region (from native-map, molecule B), with single-stranded nucleotides A34 (anis), C71 (green), U72 (dark green), C75 (pink), U76 (yellow) and A77 (blue) in stick, showing the magnesium site (Mg, green sphere) surrounded by water molecules (red spheres). (B) Same view turned 90° about the helical axis. (C) Details of C71 interaction with base-pair G74-C35 (green). Dotted lines indicate H-bond, in red if protonation is required. (D) Top view of the triples on either sides of the CUA internal loop involving C71, with G74-C35 (green) and C75, with G78-C33 (pink). (E) Stereo view of the CUA internal loop showing C75 interaction with base-pair G78-C33 (pink) and A77 interaction with base-pair G31-C80 (blue). The intervening base-pair in thin lines is U32oG79.
The central region
The apical part of the central region comprises a bulge of two unpaired nucleotides (C71-U72), among which only U72 is extruded (Figure 4A). C71 is packed in the major groove of the helix, where it makes a cis Watson–Crick/Hoogsteen interaction with the G74 of the C35-G74 base-pair (Figure 4A and CB). One H-bond is formed with a distance of 2.9 Å, between the amino group of C71 and the carbonyl of G74. Another short distance (2.7 Å) is observed between C71(N3) and G74(N7) atoms, but formation of an H-bond requires one nitrogen to be protonated. The insertion of C71 coincides with a widening of the major groove and enlargement of the helix diameter. Interestingly, a magnesium ion was found in the vicinity. It is located in the groove, between bases A34 and C71 (Figure 4A and B). This was observed in molecules B of all three crystals. In the native A molecule, density was also present, but more distorted. It was attributed to a sodium ion, on the basis of larger distances with the coordinated water molecules (around 2.6 Å compared with 2.0 Å for magnesium).
The lower part of the central region comprises nucleotides C75, U76 and A77. While in the sequence, these nucleotides form an internal loop with A34, they behave as a bulge of three in the structure (Figure 4B). Only U76 is extruded. The two other nucleotides pack inside the helix and interact with base-pairs in the stack (Figure 4E). Nucleotide C75 interacts with base-pair C33-G78. The distance (2.8 Å) and angle between the cytidine C75 amino group and the guanine carbonyl group are compatible with H-bond formation. As in the case of C71, a short distance (2.7 Å) is observed with N7 of the guanine, suggesting formation of a C+G Watson–Crick/Hoogsteen pair (40). This requires the cytidine to be protonated at the N3 position. Similar cases have been observed in RNA structures (41), and their importance in RNA folding was stressed (42). The single-stranded adenine A77 lies between the planes of the G31-C80 and U32-G79 base-pairs. It is stabilized by an interaction of its Watson–Crick edge with the Hoogsteen side of G31, with one H-bond formed between its amino group and the N7 nitrogen of G31 (Figure 4D). A distance of 2.7 Å is also observed between the N1 nitrogen atom of A77 and the O6 carbonyl of G31, suggesting the formation of a second H-bond. Such an interaction suggests protonation of the adenine (42). The same situation was observed in the core of yeast tRNAAsp (43). The observation of protonated bases stabilizing triple interaction was a surprise, since the crystals were grown in neutral conditions at pH 7.5. However, crystals grew after several weeks, and the polyethylene glycol used as precipitant is known to acidify upon ageing (44) (https://hamptonresearch.com/documents/growth_101/27.pdf). Together with the formation of two triples, the CUA bulge introduces a surprising twist in the hairpin. This is materialized in Figure 4D, showing that the base-pairs on either sides of the bulge, G74-C35 and C-33-G78, are almost perpendicular. This figure also highlights the symmetry of the central region, in particular the position of bases C71 and C75. On the whole, C71 from the bulge and C75 and A77 from the internal loop form a remarkable stack of triple interactions stabilized by protonation, as schematized in Figure 1A and Supplementary Figure S5. The three triples lock the central region into a tight structure comprising a magnesium ion and introduce a tight twist of the helix.
The stem below the central region contains three UoG base-pairs. They show typical geometries with high shear values and weak stacking interaction with the following base-pair (45,46).
Global information from the MD simulation analysis
The observation of U41 alternating between OUT and IN positions the helical stack, as well as the straightness of helix prompted us to launch an MD simulation to analyze if this reflects the molecular plasticity of this RNA, or was due to some influence of the crystal environment. The MD simulation was performed in a classical fashion using the latest AMBER force field, including all atoms and solvent with magnesium and sodium ions. Two MD simulations were performed in parallel, starting either from structures OUT or IN from the Os crystals. After a first minimization step, dynamics simulation was performed for 80 ns, a time-lapse that allows the evaluation of the stability of the experimental structures. The two structures appeared to be globally stable, as indicated by the variations of rmsd along the trajectory reported in Supplementary Figures S4A and S4B.
The simulation was coupled with an analysis of the helical parameters with the CURVES+ program, which allows the handling of all parameters for the full trajectory (39,47). Analysis of the helical axes bending in the trajectory showed the molecule to be most often more bent in the simulated models than in the crystal (Supplementary Figures S4C and S4D). The angles between the axis of the stem helix (base‐pairs 1 to 10) and the upper stem (base pairs 14 to the loop) observed in the crystal structures, 26° on average, does not correspond to the highest frequency, but is clearly observed in the distribution along the trajectory (Supplementary Figure S4C and D). Interestingly, the bending angle observed in the structure seems to be more frequently observed in the trajectory starting from the IN conformation.
Dynamics at the 7SK-motif
Visual inspection of the trajectories did not reveal conformation exchange in the time lapse of the simulation. In particular, U41 did not move out of the groove in the simulation starting from the IN conformation. Visual inspection of the simulation suggests fluctuations of the U41 base orientation, which turns perpendicularly to the A43-U66 plane and loses the H-bonds. Nonetheless, U41 stays in the groove at the same level (Figure 5A and B). Starting from the OUT conformation, the MD simulation shows that the position of U40 is stable. U41 did not insert into the helical stack in the time lapse of our experiment, as shown in Figure 5C and D.
Figure 5.

Dynamics at the 7SK-motif. (A) Snapshot at 25 ns from the MD simulation started from conformation IN with U41 (pink) and U40 (cyan). (B) Inter-atomic distances, plotted along the trajectory, monitoring the base-pair A43-U66 stability (grey), the U41 motion relative to A43 (dark pink), and the U40 motion relative to C45. (C) Snapshot at 53 ns from the MD simulation started from conformation OUT, showing U41 (pink) and U40 (cyan). (D) Inter-atomic distances, plotted along the trajectory, monitoring the U40 position relative to A43. (E) Same snapshot as in (C) but showing U63 (orange) and U40 (cyan). (F) Variation of the inter-atomic distances monitoring the U63 position relative to G42 (orange) and C67 (red).
The position of U63 at the bottom of the uridines-stack in the major groove of the (GAUC)2 is also remarkably stable when starting from the OUT conformation. This is highlighted by the minimal variations of the distance between the imino proton of U63 and the carbonyl group of G42 along the trajectory (Figure 5E and F). When starting from the IN conformation, U63 moves concomitantly with U41. It adopts a tilted position, breaks its H-bonds, but stays in the major groove.
Thus, both MD simulations confirm that nesting two uridines (U40 and U63) in the major groove at the (GAUC)2 forms a stable structure, and that the stacking of the third uridine (U41) into the same groove is also a viable conformation. The MD experiment does not support the hypothesis that nucleotide U41 might have been trapped inwards or captured outward by contact with another molecule in the crystal. The two positions of U41 observed in the crystal seem thus to represent two alternative structures of the 7SK-motif.
Our MD simulations showed, in contrast, more variations of the structure in the central region below the 7SK-motif.
Dynamics below the 7SK-motif
MD simulation indicates that the nucleotide A34 stays in the major groove, as seen in the crystal structures (Supplementary Figure S5C). It stays extended far in the apical direction although the interaction with the base G69 is lost in favor of an interaction with the backbone. Comparison of Supplementary Figures S5C with S4A suggests that the rotation of the base A34 could impact the major groove width and perhaps the bending of the molecule. This hypothesis was tested by performing a new MD simulation after changing the sequence at A34 for uridine (mutant A34U). However, even after 50 ns simulation, this change did not seem to impact the global bending of the hairpin. The U34 residue stayed in the groove, nevertheless.
Dynamics at the central region
The largest changes observed in the MD simulation were located in the central region. The triple interaction of C71 with the G74-C35 base-pair is lost after about 20 ns in favor of an interaction of C71 with the backbone at G70. C71 thus moves about one step in the apical direction to find a position which seems to relax the backbone, as schematized in Supplementary Figure S5B. The interaction of A77 with base-pair G31-C80 is already lost at the initial minimization step. A77 moves away from the plane of the G31-C80 base-pair, and re-localizes after 10 ns to the level of base-pair C35-G74, three steps away in the apical direction, a considerable change of register (Supplementary Figure S5D). It could be stabilized there by H-bonds with G74 or C35. Similarly, C75 quickly leaves the level of base-pair C33-G78 to reach the upper level of base-pair C36-G73. This corresponds to a +2 change of register. It seems to be stabilized by catching the ribose O2’ and the base O2 oxygens of C36 in a bifurcated H-bond.
These major changes suggested by the MD simulation involve nucleotides C71, C75 and A77, for which the location in the crystal structure depends on protonation. This is not surprising, since the MD was performed at neutral pH.
Further mutational analyses of the HEXIM-binding site
The 7SK-motif is embedded in a larger sequence, named the HEXIM-binding site with reference to our previous analysis by NMR-footprinting (26). Titration with the ARM peptide of the HEXIM RNA-binding sequence showed spectral environmental changes of the imino-protons of several residues at the 7SK-motif and the two stems of three base pairs on either sides. Figure 6A summarizes these previous findings. Besides specific binding, the analysis revealed an increase of solvent accessibility at U44 and G64 and stabilization of the structure at G69, G70 and at the base-pair A39-U68.
Figure 6.

HEXIM-binding study. (A) 2D sequence showing the mutations studied. The two terminal base-pairs (red) were added in the present analysis to ensure efficient production of the folded hairpin. The spheres represent the imino protons, colored according to their sensitivity to titration with the ARM-peptide (Lebars et al. 2010): no change (dark blue), binding (green), increase of solvent accessibility (orange), decrease of solvent accessibility (purple, red for A39-U68); not tested in the NMR experiment (grey). (B) Two views of hairpin HP1 (conformation OUT) with the HEXIM-binding site colored as in A. (C and D) Binding to HEXIM of the HP1 mutants measured with electrophoretic mobility shift assays. The experimental data (average of at least three experiments) were fitted with a model corresponding to one binding site. (C) Binding observed for mutations at the 7SK motif and (D) below the 7SK-motif.
Binding of RNA variants with the purified recombinant human HEXIM1 protein was analyzed by electrophoretic mobility shift assays. Several positions were mutated (Figure 6). Some did not impact HEXIM binding, such as deletion of U72 or change of A34 to uridine (A34U, Figure 6D, dark blue curve). Larger impact was observed for the mutations at the 7SK signature. The mutation of both uridines U40 and U41 into cytidines results in a severe loss of capacity to bind HEXIM, as shown in Figure 6C (dark red curve). These two residues also were shown to be important in our previous study (26), where their simultaneous deletion conduced to a deleterious effect on HEXIM binding. The present subtler change into cytidines indicates that the nature of the bases is important. Furthermore, the individual mutations into cytidine were analyzed. Mutation U41C impacts the binding efficiency (Figure 6C, red curve). The impact is in the range observed previously for the deletion of U63, which was measured again in the present analysis (Figure 6C, orange curve). Mutation U40C shows a much stronger effect, suggesting this residue to be of prominent importance (Figure 6C, light blue curve). On the whole, the contribution to HEXIM-binding of each uridine correlates well with its position in the stack shown in Figure 6B, with a major contribution for the central U40 (cyan in Figure 6B) facing the core base-pairs of the (GAUC)2. The two central base-pairs of the (GAUC)2 were shown previously to be essential for HEXIM recognition, since changing to (GGCC)2 abolished binding (26). Such a modification impacts both grooves of an RNA helix. We thus changed again these two base-pairs, but reversed them in (GUAC)2. The new mutation impacts HEXIM-binding as strongly as observed previously for (GGCC)2 (Figure 6C, dark blue and green curves).
Interestingly, mutant A39G shows a reduced capacity to bind HEXIM, while A39G-U68C restores a full capacity to bind (Figure 6D, red and orange curves). The same result was observed with A39U, which showed less binding, and the double mutant A39U-U68A, which restored binding (Figure 6D, blue and green curves). This suggests that a standard base-pair must be present.
The position of nucleotide A34 just below the 7SK-motif, on the minor groove face, suggested this nucleotide to belong structurally to the HEXIM-binding site. However, the mutation A34U does not impact HEXIM binding.
DISCUSSION
Molecular plasticity of the 5΄-hairpin of 7SK
The straightness of the hairpin conformation rapidly appeared to be a singularity of the crystal structure. During the molecular replacement process, many models were built with available web facilities, such as Assemble (48), RNAcomposer (49), Vfold-3D (50) and MC-fold (51). All those models were more bent than the observed structure in the present crystals, some of them as much as an ‘L’. This is also the case of a solution structure obtained in parallel with NMR (52), which was started when we faced phasing difficulties, and which will be compared in details in a future manuscript. The straightness observed seems to rely essentially on the interactions taking place at the central region. Among them, the insertion of A34 on top of the central region seems to add tension, like a brace restraining the helical flexibility. The extrusion of A34 favors the stacking of the short stem formed by the two base-pairs C35-G74 and C36-G73 on top of the basal stem, thus forming a continuous helix. On the other strand, the ribose–phosphate chain makes a sharp turn allowing C71 of the C71-U72 bulge to insert into the groove. This is stabilized by an interaction between the protonated cytidine C71 with G74, the nucleotide at N+3, reminiscent of a protonated U-turn (53), but with four nucleotides. The nucleotides of the ‘internal loop’, A77 to C75, form another tight loop pinned onto the helical stack. This again maximizes the stacking of bases and maintains the regular spacing of the base-pairs unaffected by the bulged residues. However, this introduces a remarkable twist. As a result, the base-pairs on either sides, C33-G78 and C35-G74 are almost perpendicular (Figure 4D).
The observation of triple interactions formed with base-pairs as distant as three steps in the helical register came as a surprise. The triple interactions at the central region are all formed with G-C base pairs and depend on protonation. They do not hold in the MD simulation, which was performed at neutral pH, closer to physiological conditions. The relaxation observed upon MD simulation corresponds to a change of register of residues of the internal loop (C75, A77) and the bulge (C71) that could be sustained by the presence of the same (G-C)2 sequence on each side of the bulge (Supplementary Figure S5). Toggling between the two situations may be favored by such a symmetrical sequence with five stacked Cs on one strand and Gs on the other, which may sum up as an original source of flexibility.
On the whole, the observed straight hairpin corresponds to a particular conformation favoring stacking of several bases from the bulges into the helix, where they may be trapped upon protonation. Protonation of adenine and cytidines as a potential contribution for hydrogen bond formation and stacking has already been observed in the internal loop of a hairpin ribozyme and proposed to be of biological significance (54,55). The observed straight helix may represent a rare conformation trapped in the crystal, but seems however to depend on peculiar properties of the sequence at the central region.
Interestingly, a magnesium hexahydrate was found in the groove between the bases C71 and A34 (Figure 4A). This suggests that magnesium binding could participate in stabilization of the special conformation observed at the central region. The solution structure realized with the same RNA by NMR at low ionic strength in the absence of magnesium showed a different conformation of the RNA, in particular at the central region (52). Interestingly, the NMR study reported effects following magnesium addition, by monitoring the exchangeable protons, and revealed that the largest changes were obtained in the central region. Although tight, this remarkable packing of the middle bulges still leaves two uridines extruded toward the solvent. They might be involved in protein recognition, or RNA packing into a stable tertiary structure, as suggested by the interaction represented in Supplementary Figure S7. However, none of our experiments sustained that idea, unfortunately.
Structural signature of the 7SK-motif
The (GAUC)2 motif forms a standard helix of four base-pairs. The main surprise was to find the major groove remarkably narrow and filled with uridines. Uridine U63 lies at the level of the bottom base-pair G42-C67. The next base pair of the short helix, A43-U66 is the recipient of a Hoogsteen interaction with one uridine of the lower bulge. This uridine may be either U40 or U41, but when uridine U41 is inserted in the groove, it pushes the other (U40) in the apical direction, where it positions at the level of the top base-pair of the (GAUC)2. Thus, U40 seems always buried in the groove, which imparts to this base a prominent role in the structural motif. Besides the packing of the uridines into the major groove, this dynamic behavior provides another layer of originality. The two positions of U41 represent two snapshots of a peculiar swapping mechanism, which may correspond to a functional distribution. Indeed, in the time-lapse of the MD simulation, both positions of U41, inside or outside of the groove, seem stable.
Uridines at positions equivalent to 40, 41 and 63 are conserved in 7SK sequences from vertebrates. Looking wider in evolution, including arthropods, the U40-U41 bulge may be extended to three residues by U, C or A, but one uridine is always present at the junction with the lower stem (25). This conserved uridine could insert in the groove to interact with the constant AU base-pair of the (GAUC)2 motif. The observation of the prominent contribution of one uridine of the lower bulge (here, U40) as a particular feature of the present structure suggests to expand the sequence for the 7SK-motif to UuGAUC/uGAUC, where capital letters stand for sequences conserved throughout evolution (as far as 7SK has been identified yet) and smaller letters stand for conserved sequences in vertebrates.
The 7SK-motif always tops a central region with bulges and a short stem ending with a Watson-Crick A-U base-pair equivalent to A39-U68. This base-pair was suggested to be dynamic in a previous investigation with NMR, and stabilized upon peptide binding (26). The present crystal structure shows it formed as a standard pair. However, it also reveals a large propeller twist: 19.3° on average as compared with the standard value of 11.6° or the average value of 11.4° for all base-pairs in the six structures. The MD simulation indicates that the A39-U68 base-pair, while keeping the Watson-Crick H-bonds, is susceptible to distortion. Indeed, large variations of the helical parameters (opening, shear and stretch) were observed at the end of the simulation, after ∼70 ns (Supplementary Figure S6). These variations were not observed at the central base-pairs of the (GAUC)2, as e.g. the A43-U65 base-pair (Supplementary Figure S6B, middle panels). The only other variations were observed at the C45-G64 base pair, on the apical side of the (GAUC)2. The global picture suggested by these observation is that the rigid (GAUC)2 recipient for the uridines functions like a knuckle within a more dynamic environment. The internal loop would command the orientation and distance of this knuckle with respect to the core of 7SK, which is maintained by the associated proteins LARP7 and MePCE. The two positions of U41 could then represent a topping conformational signal to be exploited for 7SK functions. In our functional analysis we didn't observe direct effect of point mutations in the central region. However, suppressing both the bulge and the internal loop had an impact on HEXIM-binding, suggesting a need for dynamics at the central region (26).
Is there a biological implication for HEXIM recognition?
What makes the small UuGAUC/uGAUC motif so special thus seems to be the peculiar packing of the single-stranded uridines in the groove, as well as the dynamic nature of the motif and its environment. The importance of these features was analyzed by studying the impact of local mutations on one function of this RNA, namely HEXIM binding. For the 7SK motif, a clear correlation was observed between the position of the mutation in the motif and the strength of the impact with major effects observed for changes at the most internal part of the structural motif, i.e. at the two central base-pairs A43-U66 and U44-A65, as well as at U40. The formation of the U:AU triple fully explains this result, since a cytidine would not be stabilized on the Hoogsteen face of the A43-U65 base-pair. The impact observed with the U41C mutant is clearly smaller than for the U40C mutant. This difference suggests that the extruded position of U41 seems favored. It is indeed observed more often in the different crystals.
Consideration of what is known about how ARM-peptides bind their RNA targets allows to speculate about HEXIM recognition. In most cases observed thus far, the ARM peptide penetrates into the major groove that is consequently widened, thus allowing the basic residues of the peptide to bind the phosphates at the rim of the groove (56,57). A closely related example comes from the protein Tat binding to the TAR RNA from HIV (58,59), BIV (57,60) or EIAV (61). The TAR sequence in EIAV is different from the 7SK sequence bound by Tat. Nevertheless, the ARM peptide of EIAV Tat folds as a helix in the deep major groove of the RNA, where it interacts specifically with several bases and stabilizes a UoG enol conformation, in addition to binding phosphates at the rim of the groove (61). In HIV-TAR the sequence recognized by Tat is similar to that recognized in 7SK by HEXIM and Tat (20). A bulged U between short stems with a base pair equivalent to A43-U66 are essential. Structural studies of HIV-TAR recognition by argininamide or a peptide from Tat showed the bulged U to be pushed into the major groove of the apical stem, in the vicinity of the A-U base-pair. An early work predicted the A-U base pair to form a triple interaction with the U, stabilized by the interaction with argininamide (58,62). This was not observed in later structures of HIV complexes (63), but such triple was formed in BIV-TAR. The Tat peptide stabilizes the TAR conformation around the triple by specific interactions with the bases and backbone of the RNA. In the present structure of the 7SK RNA, a striking difference is that the triples are observed in the free form of the RNA. Moreover, with the major groove almost filled with the uridine bases, the penetration of a peptide, folding in the groove as a β-hairpin in BIV (60) or as a α-helix in EIAV (61), seems prevented. This might suggest recognition on the minor groove, which is more accessible, although poorer in terms of H-bond donor/acceptor variety. However, the experiment where the central AU/UA base-pairs were reversed strongly supports involvement of the major groove. Such mutation does not change much the distribution of H-bonds donors/acceptors in the minor groove, but it clearly does in the major groove (64,65). The strong effect of the mutation indicates thus the major groove as important for HEXIM recognition. In fact, when U41 is extruded, the major groove shows some accessibility, as it opens like a mouth on the apex side (Figure 6D). There are several solvent molecules located in that area (conserved in all molecules) and this is the place where a gold ion was bound in the short soaking process. As often observed, these solvent molecules may indicate the protein binding site. Perhaps the ARM peptide of HEXIM does not fold deep in the groove, but wraps near the rim and around the structure defined by the uridine filling, and is facilitated in the OUT conformation. It cannot be excluded that the external, more hydrophobic, sides of the uridines (C5-C6 atoms) participate to the recognition, as was observed for BIV-TAR (1,57,60). On another hand, the present structure, and more particularly the IN conformation could represent a ‘switched-off’ step of the HEXIM-induced regulation, or another functional state of 7SK, independent of HEXIM-binding and P-TEFb regulation. Indeed, possible involvement of 7SK in other regulation processes, such as translation regulation, was raised (66). Altogether, this merits further investigation, as it provides an interesting case to further explore the slightly different concepts of recognition and binding.
ACCESSION CODES
Atomic coordinates have been deposited in the Protein Data Bank under accession codes 5LYS (Au), 5LYU (native), 5LYV (Os).
Supplementary Material
ACKNOWLEDGEMENTS
Diffraction data were collected at the following synchrotron beam lines: BM30A at the European Synchrotron Radiation Facility (ESRF) (Grenoble, France), and Proxima 1 (proposal ID 20090535) at SOLEIL synchrotron (Saint-Aubin, France). The authors are most grateful to the machine and beam line groups for making these experiments possible.
The authors thank Adam Ben Shem, Pierre Poussin-Courmontagne and Valérie Lamour for their helpful suggestions, Fabrice Jossinet for help with modelling, Isabelle Lebars for discussions and particularly Eric Westhof for invaluable comments on the structure.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Centre National de la Recherche Scientifique; University of Strasbourg; Fondation pour la Recherche Médicale; Sorbonne Universités (UPMC); French National Agency for Research [TrscrREGsnRNP ANR-06-BLAN-0072]; DynamIC [ANR-12-BSV5-0018]; French Infrastructure for Integrated Structural Biology (FRISBI) [ANR-10-INSB-05-01]; INSTRUCT as part of the European Strategy Forum on Research Infrastructures (ESFRI); DMZ benefitted from doctoral fellowships from CONACyT (Mexico) and Association pour la Recherche contre le Cancer. Funding for open access charge: CNRS-UPMC.
Conflict of interest statement. None declared.
REFERENCES
- 1. Peterlin B.M., Brogie J.E., Price D.H.. 7SK snRNA: a noncoding RNA that plays a major role in regulating eukaryotic transcription. Wiley Interdiscip. Rev. RNA. 2012; 3:92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Diribarne G., Bensaude O.. 7SK RNA, a non-coding RNA regulating P-TEFb, a general transcription factor. RNA Biol. 2009; 6:122–128. [DOI] [PubMed] [Google Scholar]
- 3. Barrandon C., Spiluttini B., Bensaude O.. Non-coding RNAs regulating the transcriptional machinery. Biol. Cell. 2008; 100:83–95. [DOI] [PubMed] [Google Scholar]
- 4. Nguyen V.T., Kiss T., Michels A.A., Bensaude O.. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature. 2001; 414:322–325. [DOI] [PubMed] [Google Scholar]
- 5. Yang Z., Zhu Q., Luo K., Zhou Q.. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature. 2001; 414:317–322. [DOI] [PubMed] [Google Scholar]
- 6. Michels A.A., Nguyen V.T., Fraldi A., Labas V., Edwards M., Bonnet F., Lania L., Bensaude O.. MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol. Cell. Biol. 2003; 23:4859–4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Michels A.A., Fraldi A., Li Q., Adamson T.E., Bonnet F., Nguyen V.T., Sedore S.C., Price J.P., Price D.H., Lania L. et al. . Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J. 2004; 23:2608–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sedore S.C., Byers S.A., Biglione S., Price J.P., Maury W.J., Price D.H.. Manipulation of P-TEFb control machinery by HIV: recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res. 2007; 35:4347–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xue Y., Yang Z., Chen R., Zhou Q.. A capping-independent function of MePCE in stabilizing 7SK snRNA and facilitating the assembly of 7SK snRNP. Nucleic Acids Res. 2010; 38:360–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. He N., Jahchan N.S., Hong E., Li Q., Bayfield M.A., Maraia R.J., Luo K., Zhou Q.. A La-related protein modulates 7SK snRNP integrity to suppress P-TEFb-dependent transcriptional elongation and tumorigenesis. Mol. Cell. 2008; 29:588–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krueger B.J., Jeronimo C., Roy B.B., Bouchard A., Barrandon C., Byers S.A., Searcey C.E., Cooper J.J., Bensaude O., Cohen E.A. et al. . LARP7 is a stable component of the 7SK snRNP while P-TEFb, HEXIM1 and hnRNP A1 are reversibly associated. Nucleic Acids Res. 2008; 36:2219–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jeronimo C., Forget D., Bouchard A., Li Q., Chua G., Poitras C., Therien C., Bergeron D., Bourassa S., Greenblatt J. et al. . Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol. Cell. 2007; 27:262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wassarman D.A., Steitz J.A.. Structural analyses of the 7SK ribonucleoprotein (RNP), the most abundant human small RNP of unknown function. Mol. Cell. Biol. 1991; 11:3432–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marz M., Donath A., Verstraete N., Nguyen V.T., Stadler P.F., Bensaude O.. Evolution of 7SK RNA and its protein partners in metazoa. Mol. Biol. Evol. 2009; 26:2821–2830. [DOI] [PubMed] [Google Scholar]
- 15. Uchikawa E., Natchiar K.S., Han X., Proux F., Roblin P., Zhang E., Durand A., Klaholz B.P., Dock-Bregeon A.C.. Structural insight into the mechanism of stabilization of the 7SK small nuclear RNA by LARP7. Nucleic Acids Res. 2015; 43:3373–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gruber A.R., Koper-Emde D., Marz M., Tafer H., Bernhart S., Obernosterer G., Mosig A., Hofacker I.L., Stadler P.F., Benecke B.J.. Invertebrate 7SK snRNAs. J. Mol. Evol. 2008; 66:107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Egloff S., Van Herreweghe E., Kiss T.. Regulation of polymerase II transcription by 7SK snRNA: two distinct RNA elements direct P-TEFb and HEXIM1 binding. Mol. Cell. Biol. 2006; 26:630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Muniz L., Egloff S., Kiss T.. RNA elements directing in vivo assembly of the 7SK/MePCE/Larp7 transcriptional regulatory snRNP. Nucleic Acids Res. 2013; 41:4686–4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Belanger F., Baigude H., Rana T.M.. U30 of 7SK RNA forms a specific photo-cross-link with Hexim1 in the context of both a minimal RNA-binding site and a fully reconstituted 7SK/Hexim1/P-TEFb ribonucleoprotein complex. J. Mol. Biol. 2009; 386:1094–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Muniz L., Egloff S., Ughy B., Jady B.E., Kiss T.. Controlling cellular P-TEFb activity by the HIV-1 transcriptional transactivator Tat. PLoS Pathog. 2010; 6:e1001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dingwall C., Ernberg I., Gait M.J., Green S.M., Heaphy S., Karn J., Lowe A.D., Singh M., Skinner M.A.. HIV-1 tat protein stimulates transcription by binding to a U-rich bulge in the stem of the TAR RNA structure. EMBO J. 1990; 9:4145–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dames S.A., Schonichen A., Schulte A., Barboric M., Peterlin B.M., Grzesiek S., Geyer M.. Structure of the Cyclin T binding domain of Hexim1 and molecular basis for its recognition of P-TEFb. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:14312–14317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schonichen A., Bigalke J.M., Urbanke C., Grzesiek S., Dames S.A., Geyer M.. A flexible bipartite coiled coil structure is required for the interaction of Hexim1 with the P-TEFB subunit cyclin T1. Biochemistry. 2010; 49:3083–3091. [DOI] [PubMed] [Google Scholar]
- 24. Schulte A., Czudnochowski N., Barboric M., Schonichen A., Blazek D., Peterlin B.M., Geyer M.. Identification of a cyclin T-binding domain in Hexim1 and biochemical analysis of its binding competition with HIV-1 Tat. J. Biol. Chem. 2005; 280:24968–24977. [DOI] [PubMed] [Google Scholar]
- 25. Gruber A.R., Kilgus C., Mosig A., Hofacker I.L., Hennig W., Stadler P.F.. Arthropod 7SK RNA. Mol. Biol. Evol. 2008; 25:1923–1930. [DOI] [PubMed] [Google Scholar]
- 26. Lebars I., Martinez-Zapien D., Durand A., Coutant J., Kieffer B., Dock-Bregeon A.C.. HEXIM1 targets a repeated GAUC motif in the riboregulator of transcription 7SK and promotes base pair rearrangements. Nucleic Acids Res. 2010; 38:7749–7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yik J.H., Chen R., Pezda A.C., Samford C.S., Zhou Q.. A human immunodeficiency virus type 1 Tat-like arginine-rich RNA-binding domain is essential for HEXIM1 to inhibit RNA polymerase II transcription through 7SK snRNA-mediated inactivation of P-TEFb. Mol. Cell. Biol. 2004; 24:5094–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martinez-Zapien D., Saliou J.M., Han X., Atmanene C., Proux F., Cianferani S., Dock-Bregeon A.C.. Intermolecular recognition of the non-coding RNA 7SK and HEXIM protein in perspective. Biochimie. 2015; 117:63–71. [DOI] [PubMed] [Google Scholar]
- 29. Krueger B.J., Varzavand K., Cooper J.J., Price D.H.. The mechanism of release of P-TEFb and HEXIM1 from the 7SK snRNP by viral and cellular activators includes a conformational change in 7SK. PloS One. 2010; 5:e12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu J., Wong V., Zhang Y., Tran T., Zhao L., Xia A., Xia T., Qi X.. Distinct conformational transition patterns of noncoding 7SK snRNA and HIV TAR RNAs upon Tat binding. Biochemistry. 2014; 53:675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Purzycka K.J., Popenda M., Szachniuk M., Antczak M., Lukasiak P., Blazewicz J., Adamiak R.W.. Automated 3D RNA structure prediction using the RNAComposer method for riboswitches. Methods Enzymol. 2015; 553:3–34. [DOI] [PubMed] [Google Scholar]
- 32. Robertson M.P., Chi Y.I., Scott W.G.. Solving novel RNA structures using only secondary structural fragments. Methods. 2010; 52:168–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P, Sharff A., Smart O.S., Vonrhein C.. BUSTER version 2.10.2. 2016; Cambridge, United Kingdom: Global Phasing Ltd. [Google Scholar]
- 34. Perez A., Marchan I., Svozil D., Sponer J., Cheatham T.E. 3rd, Laughton C.A., Orozco M.. Refinement of the AMBER force field for nucleic acids: improving the description of alpha/gamma conformers. Biophys. J. 2007; 92:3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Czudnochowski N., Vollmuth F., Baumann S., Vogel-Bachmayr K., Geyer M.. Specificity of Hexim1 and Hexim2 complex formation with cyclin T1/T2, importin alpha and 7SK snRNA. J. Mol. Biol. 2010; 395:28–41. [DOI] [PubMed] [Google Scholar]
- 36. Varani G., Cheong C., Tinoco I. Jr. Structure of an unusually stable RNA hairpin. Biochemistry. 1991; 30:3280–3289. [DOI] [PubMed] [Google Scholar]
- 37. Molinaro M., Tinoco I. Jr. Use of ultra stable UNCG tetraloop hairpins to fold RNA structures: thermodynamic and spectroscopic applications. Nucleic Acids Res. 1995; 23:3056–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ennifar E., Nikulin A., Tishchenko S., Serganov A., Nevskaya N., Garber M., Ehresmann B., Ehresmann C., Nikonov S., Dumas P.. The crystal structure of UUCG tetraloop. J. Mol. Biol. 2000; 304:35–42. [DOI] [PubMed] [Google Scholar]
- 39. Blanchet C., Pasi M., Zakrzewska K., Lavery R.. CURVES+ web server for analyzing and visualizing the helical, backbone and groove parameters of nucleic acid structures. Nucleic Acids Res. 2011; 39:W68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Leontis N.B., Westhof E.. Geometric nomenclature and classification of RNA base pairs. RNA. 2001; 7:499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cate J.H., Gooding A.R., Podell E., Zhou K., Golden B.L., Kundrot C.E., Cech T.R., Doudna J.A.. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science. 1996; 273:1678–1685. [DOI] [PubMed] [Google Scholar]
- 42. Chawla M., Sharma P., Halder S., Bhattacharyya D., Mitra A.. Protonation of base pairs in RNA: context analysis and quantum chemical investigations of their geometries and stabilities. J. Phys. Chem. B. 2011; 115:1469–1484. [DOI] [PubMed] [Google Scholar]
- 43. Westhof E., Dumas P., Moras D.. Restrained refinement of two crystalline forms of yeast aspartic acid and phenylalanine transfer RNA crystals. Acta Crystallogr. A. 1988; 44:112–123. [PubMed] [Google Scholar]
- 44. Jurnak F. Induction of elongation factor Tu-GDP crystal polymorphism by polyethylene glycol contaminants. J. Mol. Biol. 1985; 185:215–217. [DOI] [PubMed] [Google Scholar]
- 45. Masquida B., Westhof E.. On the wobble GoU and related pairs. RNA. 2000; 6:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Varani G., McClain W.H.. The G x U wobble base pair. A fundamental building block of RNA structure crucial to RNA function in diverse biological systems. EMBO Rep. 2000; 1:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lavery R., Moakher M., Maddocks J.H., Petkeviciute D., Zakrzewska K.. Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 2009; 37:5917–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jossinet F., Ludwig T.E., Westhof E.. Assemble: an interactive graphical tool to analyze and build RNA architectures at the 2D and 3D levels. Bioinformatics. 2010; 26:2057–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Popenda M., Szachniuk M., Antczak M., Purzycka K.J., Lukasiak P., Bartol N., Blazewicz J., Adamiak R.W.. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012; 40:e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu X., Zhao P., Chen S.J.. Vfold: a web server for RNA structure and folding thermodynamics prediction. PloS One. 2014; 9:e107504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Parisien M., Major F.. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008; 452:51–55. [DOI] [PubMed] [Google Scholar]
- 52. Bourbigot S., Dock-Bregeon A.-C., Eberling P., Coutant J., Kieffer B., Lebars I.. Solution structure of the 5΄-terminal hairpin of the 7SK small nuclear RNA. RNA. 2016; 22:1844–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gottstein-Schmidtke S.R., Duchardt-Ferner E., Groher F., Weigand J.E., Gottstein D., Suess B., Wohnert J.. Building a stable RNA U-turn with a protonated cytidine. RNA. 2014; 20:1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ravindranathan S., Butcher S.E., Feigon J.. Adenine protonation in domain B of the hairpin ribozyme. Biochemistry. 2000; 39:16026–16032. [DOI] [PubMed] [Google Scholar]
- 55. Tang C.L., Alexov E., Pyle A.M., Honig B.. Calculation of pKas in RNA: on the structural origins and functional roles of protonated nucleotides. J. Mol. Biol. 2007; 366:1475–1496. [DOI] [PubMed] [Google Scholar]
- 56. Ye X., Gorin A., Ellington A.D., Patel D.J.. Deep penetration of an alpha-helix into a widened RNA major groove in the HIV-1 rev peptide-RNA aptamer complex. Nat. Struct. Biol. 1996; 3:1026–1033. [DOI] [PubMed] [Google Scholar]
- 57. Ye X., Kumar R.A., Patel D.J.. Molecular recognition in the bovine immunodeficiency virus Tat peptide-TAR RNA complex. Chem. Biol. 1995; 2:827–840. [DOI] [PubMed] [Google Scholar]
- 58. Puglisi J.D., Tan R., Calnan B.J., Frankel A.D., Williamson J.R.. Conformation of the TAR RNA-arginine complex by NMR spectroscopy. Science. 1992; 257:76–80. [DOI] [PubMed] [Google Scholar]
- 59. Davidson A., Patora-Komisarska K., Robinson J.A., Varani G.. Essential structural requirements for specific recognition of HIV TAR RNA by peptide mimetics of Tat protein. Nucleic Acids Res. 2011; 39:248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Puglisi J.D., Chen L., Blanchard S., Frankel A.D.. Solution structure of a bovine immunodeficiency virus Tat-TAR peptide-RNA complex. Science. 1995; 270:1200–1203. [DOI] [PubMed] [Google Scholar]
- 61. Anand K., Schulte A., Vogel-Bachmayr K., Scheffzek K., Geyer M.. Structural insights into the cyclin T1-Tat-TAR RNA transcription activation complex from EIAV. Nat. Struct. Mol. Biol. 2008; 15:1287–1292. [DOI] [PubMed] [Google Scholar]
- 62. Puglisi J.D., Chen L., Frankel A.D., Williamson J.R.. Role of RNA structure in arginine recognition of TAR RNA. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:3680–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Aboul-ela F., Karn J., Varani G.. The structure of the human immunodeficiency virus type-1 TAR RNA reveals principles of RNA recognition by Tat protein. J. Mol. Biol. 1995; 253:313–332. [DOI] [PubMed] [Google Scholar]
- 64. Weeks K.M., Crothers D.M.. RNA recognition by Tat-derived peptides: interaction in the major groove. Cell. 1991; 66:577–588. [DOI] [PubMed] [Google Scholar]
- 65. Weeks K.M., Crothers D.M.. Major groove accessibility of RNA. Science. 1993; 261:1574–1577. [DOI] [PubMed] [Google Scholar]
- 66. Cosgrove M.S., Ding Y., Rennie W.A., Lane M.J., Hanes S.D.. The Bin3 RNA methyltransferase targets 7SK RNA to control transcription and translation. Wiley Interdiscip. Rev. RNA. 2012; 3:633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.