


Abstract
The majority of genome-wide association study (GWAS) risk variants reside in non-coding DNA sequences. Understanding how these sequence modifications lead to transcriptional alterations and cell-to-cell variability can help unraveling genotype–phenotype relationships. Here, we describe a computational method, dubbed CAPE, which calculates the likelihood of a genetic variant deactivating enhancers by disrupting the binding of transcription factors (TFs) in a given cellular context. CAPE learns sequence signatures associated with putative enhancers originating from large-scale sequencing experiments (such as ChIP-seq or DNase-seq) and models the change in enhancer signature upon a single nucleotide substitution. CAPE accurately identifies causative cis-regulatory variation including expression quantitative trait loci (eQTLs) and DNase I sensitivity quantitative trait loci (dsQTLs) in a tissue-specific manner with precision superior to several currently available methods. The presented method can be trained on any tissue-specific dataset of enhancers and known functional variants and applied to prioritize disease-associated variants in the corresponding tissue.
INTRODUCTION
Regulatory elements tightly orchestrate temporal and spatial patterns of gene expression. Genomic variants of these elements contribute to phenotype change and predisposition to diseases to a large extent (1–5). The recent explosive generation of epigenetic data has made it possible to detect cell-type-specific regulatory regions (6–11). However, the prioritization of regulatory variants remains challenging, partly due to the incomplete understanding of how regulation is achieved at the nucleotide level in different tissues and environmental contexts. Meanwhile, numerous eQTL studies have been performed to determine the regulatory architecture of the human genome (12), however, without revealing causality. This is mainly due to the reason that single nucleotide polymorphisms (SNPs) within a linkage disequilibrium (LD) block are statistically indistinguishable from each other. In spite of that, when eQTLs and SNPs were considered with respect to Deoxyribonuclease I (DNase I) hypersensitive sites (DHSs), ∼50% of eQTLs were found to be dsQTLs (13). Disease- and trait-associated variants identified by GWAS reside predominantly in noncoding regions and were found to perturb TFBSs and local chromatin accessibility (14,15). These observations suggest that causative regulatory SNPs are often associated with focal alterations in chromatin structure through disrupting binding of TFs and lead to deviations from the wild-type gene expression pattern (15–17).
Recent progress on predicting the impact of genetic variants on regulatory element activity has been made by integrating genomic and epigenomic data (18–25), with only a few of them being able to predict causal regulatory eQTLs (22,24,25). For example, by learning a regulatory sequence code from large-scale chromatin-profiling data via a deep-learning approach and integrating evolutionary conservation, DeepSEA (22) outperforms the majority of existing methods in predicting chromatin effects of genetic variants and scoring eQTLs and GWAS SNPs. Nevertheless, this method does not prioritize eQTLs in a tissue-specific manner. Moreover, the ‘black magic’ behind deep learning precludes the users from identifying the underlying mechanism of the sequence variation impact. In addition, some probabilistic frameworks have been developed to fine-map eQTLs in a meta-data fashion. Specifically, RASQUAL (24) utilizes ATAC-seq data of many individuals to identify Quantitative Trait Loci (QTL) by employing iterative genotype correction. Dense genotyping based on the meta-ATAC-seq data used by this method allows accurately identify QTLs. eQTeL (25) incorporates large-scale epigenetic and gene expression data from multiple individuals, expression variance of genes across multiple tissues, and imputed haplotypes to prioritize eQTL SNPs. The requirement of versatile high throughput data limits these methods to be widely applied to different tissues.
We have previously developed a computational approach to systematically dissect the regulatory variants with respect to their potential deleterious effect on essential TF binding in enhancer regions (16,26). These variants are termed candidate killer mutations or deactivating SNPs (deSNPs) due to their ability to deactivate major TF binding sites and to result in abnormal enhancer activity. deSNPs are strongly associated with downstream gene expression and phenotype change. To establish an approach that can identify potential causal regulatory SNPs impacting target gene expression or modulating chromatin states with higher accuracy, we developed a new method aimed to identify CellulAr dePendent dEactivating mutations (CAPE). Our new approach learns regulatory sequence signatures from a large-scale profile of regulatory signal tracks associated with enhancers (including DNase I sensitivity and ChIP-seq of histone marks and major TFs), and models the change of enhancer activity due to a mutation. By integrating two characteristics of a causal regulatory SNP—the variant's disruptive effect on its cognate TF binding and the binding capability of the sequence surrounding the variant—we constructed a set of support vector machine (SVM) models to prioritize genetic variants that deactivate enhancers in a particular cellular context. To test whether these sequence signatures could be adapted to prioritize different functional sequence variants, we trained and tested these models on eQTLs, which affect gene expression, and dsQTLs that modulate chromatin accessibility. To benchmark our method in different cellular contexts, we constructed the eQTL SVM models in two cell lines: the GM12878 lymphoblastoid B cell line (LCL) and the HepG2 hepatocellular carcinoma cell line. We observed that our method is able to accurately prioritize tissue-specific causative regulatory variants, especially eQTLs, and it largely outperforms currently available methods.
MATERIALS AND METHODS
Chromatin signal profiling
The chromatin profiling of DNase I seq, H3K27ac, H3K4me1, H3K4me2, H3K4me3, H2A.Z, P300 and major TFs binding was selected to learn the regulatory sequence code as these signal tracks are strongly associated with cis-regulatory elements.
The DNase I-seq peaks data used in the analysis of local chromatin accessibility for both GM12878 and HepG2 were downloaded from the Encyclopedia of DNA elements (ENCODE) (10) (ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeUwDnase/). The chromatin histone mark ChIP-seq data for both GM12878 and HepG2 were downloaded from the NIH ROADMAP Epigenomics project (27) website (http://egg2.wustl.edu/roadmap/data/byFileType/peaks/consolidated/narrowPeak/). The TF binding data for both GM12878 and HepG2 were downloaded from the ENCODE archive (ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/).
For the signal tracks with two replicates, only peaks that were consistently found in both replicates were chosen. We also excluded peaks with >70% repetitive/retro-transposable elements and regions with <50 bp non-repetitive nucleotides. We used the GRCh37 (hg19) assembly as the reference genome for read mapping and data analysis.
Estimating k-mer weights from different signal tracks
We developed a framework to score k-mers by their ability to capture the binding specificities of major TFs in a given tissue (16). We used a positive set of candidate regulatory DNA sequences, such as peaks of any signal track associated with putative enhancers. We generated a random control set by sampling sequences genome-wide with the same GC content, same repeat content and same length. Then, Fisher's exact test was applied to assess the significance of enrichment of each k-mer in the positive set as compared to the control set. The significance of the Fisher's exact test P-value (–log10(P-value), referred to as binding significance) was used to score the functional constraint of a k-mer, as greater binding significance indicates smaller possibility to detect that k-mer by chance (16) (Figure 1A). The top k-mers significantly enriched in the peaks of the selected signal tracks were likely to be potential binding sites of active TFs (16).
Figure 1.

CAPE framework. (A) The pipeline to calculate binding significance of each k-mer (k = 4 to 12). (B) Binding Significance Change (ΔsigSum) using 10-mers as an example: The pink bars are the 10-mers associated with the wild-type nucleotide T. The light blue bars are the 10-mers associated with the mutated nucleotide G. ΔsigSum is used to quantify the deleterious effect on the binding fitness caused by altering the 10-mer content due to the T/G mutation. (C) Framework explaining the algorithm and introducing the components of CAPE. The SVM takes sequence features estimated based on all signal tracks as input. There are two sequence signatures for each size of k-mer (k = 4, 6, 8, 10, 12): ΔsigSum, and summed binding significance of k-mers in the (−100 bp, 100 bp) flanking window. The output score of the SVM estimates the probability of the genetic variants deactivating a cis-regulatory element in a cellular context.
Binding significance change caused by genetic variant
The basic idea of calculating the binding significance is to quantify the effect of the genetic variant using the score change caused by an altered k-mer content (16). Specifically, given a genetic variant, there are k k-mers associated with each allele. We used the sum of binding significance of the k k-mers overlapping the allele to assess the regulatory activity of the corresponding allele (Figure 1B). Then, the binding significance change (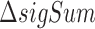 , formula 1, 2) of k k-mers was used to estimate the deleterious impact of the alternative allele on TF binding (Figure 1AB):
, formula 1, 2) of k k-mers was used to estimate the deleterious impact of the alternative allele on TF binding (Figure 1AB):
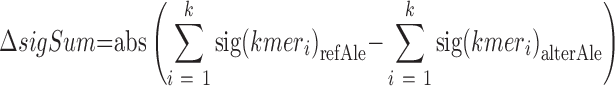 |
(1) |
 |
(2) |
where P(kmer) is the Fisher' exact test P-value of k-mer, k-mer length is an even number ranging from 4 to 12, refAle refers to the reference allele and alterAle refers to its alternative variant.
A k-mer based SVM model to prioritize causal regulatory SNPs
The goal of this study was to develop an approach to accurately identify mutations in enhancers that can disrupt binding of essential TFs and, thus, lead to downstream effects on gene expression or phenotype change. This kind of mutation has been defined as candidate killer mutation or deactivating SNP (deSNP) in our previous studies (16,26).To further enhance the accuracy of deSNPs, we established a classifier by learning the sequence code from large-scale chromatin profiling data of multiple signal tracks, including DNase-seq, H3K27ac, H3K4me1, H3K4me2, H3K4me3, H2A.Z, P300, and major TF binding data of the corresponding tissue (Supplementary Data). Two sequence signatures—the disruptive effect of the mutation on TF binding and co-binding of TFs in its neighborhood—are the basic component of features for each signal.
Specifically, given a set of cis-regulatory elements (promoters or enhancers predicted by a signal track), we used the significance of k-mer enrichment in these cis-regulatory elements to estimate the binding affinity of the k-mers (formula 2). We next used ΔsigSum (formula 1) to assess the binding affinity change of the binding site. The binding significance sum of all k-mers within the flanking (−100 bps, 100 bps) window before mutation ( ) was used to estimate the binding capability of the sequence around the genetic variant. We selected the flanking (−100 bps, 100 bps) window to estimate the overall binding capability of the nearby sequence context due to the fact that dsQTLs typically affect chromatin accessibility in a 200–300 bps region and tend to have substantial correlations with the DNase I sensitivity of their flanking 100 bps window (13). To fine-gauge the binding affinities of the potential cognate TFs and co-factors in the neighborhood, we integrated different sizes of k-mers (k = 4, 6, 8, 10, 12) to represent a binding site (Supplementary Data).
) was used to estimate the binding capability of the sequence around the genetic variant. We selected the flanking (−100 bps, 100 bps) window to estimate the overall binding capability of the nearby sequence context due to the fact that dsQTLs typically affect chromatin accessibility in a 200–300 bps region and tend to have substantial correlations with the DNase I sensitivity of their flanking 100 bps window (13). To fine-gauge the binding affinities of the potential cognate TFs and co-factors in the neighborhood, we integrated different sizes of k-mers (k = 4, 6, 8, 10, 12) to represent a binding site (Supplementary Data).
Next, we built an SVM model with a linear kernel by using (Nk x NkmerSignature x NsignalTrack) features (Figure 1C, formula 3). Nk ( = 5) is the number of k-mer sizes (k = 4, 6, 8, 10, 12). NkmerSignature ( = 2) is the number of signatures including the binding affinity change of the potential binding site due to the mutation and the overall binding capabilities of the nearby sequence context of the genetic variant. NsignalTrack is the number of the chromatin datasets, which equals 19 in GM12878 (Supplementary Data) and 23 in HepG2 (Supplementary Data).
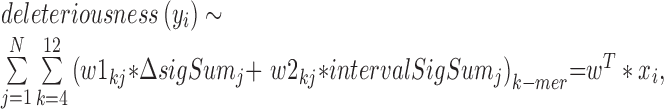 |
(3) |
where N=NsignalTrack, w1 and w2 represent the learned set of feature weights for different chromatin datasets and different k-mers.
To test the accuracy of our classifier, a five-fold cross validation was applied to the positive and control SNPs (we trained the model on every four folds and tested the model on the one remaining fold). We applied the package libsvm (28) to build the classifier. All the features were scaled to z-scores with the mean = 0, and the standard deviation = 1. In addition to DeepSEA (22), the model's performance was also compared to deltaSVM (20), and CATO (21).
Positive and control set of eQTLs and dsQTL SNPs
We trained and tested our method in two different cell lines: GM12878 and HepG2. The k-mer weights were obtained from the ChIP-seq peak regions of each corresponding cell line. To compare our approach to DeepSEA which has been reported as outperforming many currently available methods, the positive SNP set was restricted to a subset of the eQTLs associated with the top 10% highly expressed genes in a given cell line, for which DeepSEA GRASP (29) eQTLs data is also available. Next, we randomly sampled control SNPs from the random negative SNP set provided by DeepSEA and constrained them to any signal region (DNase, H3K27ac, H3K4me1, H2A.Z, P300, and any major TF ChIP-seq peaks) (Supplementary Data) of the corresponding cell line LCL, with similar minor allele frequency distribution. Three-fold negative control SNPs were chosen based on these criteria. Overall, 7949 SNPs including 1948 eQTLs and 3-fold matched control SNPs constitute the training and testing set of eQLs in GM12878 (Supplementary Data); 4176 SNPs comprised of 1044 eQTLs and matched control SNPs constitute the training and testing set of eQTLs in HepG2 (Supplementary Data). We used a 5-fold cross validation to train and test our model. As for the positive set of dsQTL, we used the 574 dsQTLs selected by (20). These dsQTLs are significantly associated with the DNase I sensitivity in a 100 bps window flanking the dsQTL and, therefore, are the most likely causal dsQTL SNPs. As a control set, we conservatively considered only the control SNPs (selected by (20)) overlapping 1% FDR DNase I hotspot peaks in LCL. A 5-fold cross validation was applied to test the accuracy. To compare the performance of our method with CATO, we only considered SNPs that also have a predicted CATO score, resulting in 565 dsQTLs and 2128 control SNPs (Supplementary Data).
k-mer Clustering
To remove the redundancy in the top GM12878 DHS k-mers (k = 10) and validate that the top k-mers correspond to known motifs of major TFs, we clustered the top k-mers (Supplementary Data) using the same clustering strategy as in our previous study (16). Briefly, the clustering included two steps. The first step was to cluster k-mers using a dimer based approach without alignment (16). Next, the motif profiles generated by the first step were further aligned, merged and matched to the known TFBS databases including JASPAR (30) and TRANSFAC (31), using the web-based tool STAMP (32) for similarity, tree-building, and alignment of DNA motifs and profiles.
Enrichment analysis of GWAS traits
The NHGRI GWAS Catalog was downloaded in September 2016 (33). We applied CAPE to prioritize enhancer SNPs (common SNPs located in the active enhancers of GM12878 according to the ROADMAP expanded 18-state model) and to identify deSNPs (FPR ≤ 0.05). To study the enrichment of a set of positive SNPs coinciding with B lymphoblastoid related traits, we generated a null distribution composed of 100× random matched SNP sets with the same size as the tested SNP set. The enrichment of the positive SNPs coinciding with B cell-related trait relative to matched random SNP sets was evaluated as the ratio of the enrichment of tested SNPs on these traits relative to the null distribution. To validate that deSNPs are more likely to be causal SNPs relative to enhancer SNPs, we compared the enrichment of deSNPs in B-cell related GWAS traits to that of the enhancer SNPs. In total, 35 B cell-related traits were kept for the association study (Supplementary Data). The tag SNPs coinciding with the B cell-related GWAS traits were further expanded by LD (r2 > 0.8, minimum distance of 500 bp).
RESULTS
Deactivating mutations in enhancer regions
The basic idea behind this method is to utilize a learned enhancer-associated sequence code to infer the deleterious effect of a potential regulatory SNP on enhancer activity. This approach integrates two characteristics associated with a genomic variation—the ability of a variant to disturb an essential TF binding event and co-binding of other TFs in the neighborhood. Disrupting the binding of an essential TF could lead to a deleterious impact on the enhancer activity and, in some cases, enhancer deactivation (16,34). On the other hand, co-operative binding of multiple TFs often boosts each other's binding affinity (35), whereas co-bound TFs tend to disappear together and are susceptible to genetic knockout of partner TFs (36). In other words, TF binding would be determined not only by the presence and binding affinity of DNA motifs of primary TFs but also by the co-binding of partner TFs (36). We decomposed a potential binding site to a composition of different size k-mers (k = 4, 6, 8, 10, 12). To fine-gauge the k-mer weight, we learned the sequence code at multiple spatial scales and from a diverse set of genome-wide chromatin profiles. The chromatin profiles were selected because of their strong association with the cis-regulatory elements and included DHS, H3K27ac, H3K4me1, H3K4me2, H3K4me3, H2A.Z and major TF binding data from corresponding cell lines (Supplementary Data). Using these features, we established an SVM with a linear kernel to construct a scoring scheme to identify candidate-deactivating mutations and dubbed our algorithm CAPE (Figure 1).
Identified top k-mers correspond to active TFBSs
Prior to identifying genetic variants that could disrupt binding of a TF, one would need to ensure that the k-mers with highest binding significance are indeed able to capture the binding specificities of the essential TFs of a given tissue or cell type. Scoring k-mers based on the DHS data of GM12878, we observed a noticeable sequence similarity among many of the 299 top k-mers from a set of lymphoblastoid B cell DNase I-seq peaks (Materials and Methods), with many of them overlapping each other (Bonferroni-corrected P < 10−3, 494 948 tests, Supplementary Data). To remove the redundancy, we clustered the 299 top k-mers into 64 distinct clusters and mapped them to JASPAR (30) and TRANSFAC (31) using STAMP (32) (Materials and Methods). The k-mer clusters were further merged to 11 clusters. Twenty-eight TFBSs matched these 11 clusters with an E-value cutoff of 5e−3. Nineteen of the TFBSs (68%) were B-cell related. The majority of k-mer clusters were associated with the motif of at least one essential B-cell related TF, such as GABPA, SPI1, NFKB, IRF2, RUNX1 and PAX5 (Figure 2). Many of these TFs play essential roles in development, differentiation, or proliferation of B-lymphocytes (37–44).
Figure 2.

Enriched k-mers in GM12878 enhancers correspond to B-cell TFBSs. The clusters of 299 top k-mers map to known TFBS: 33 k-mer clusters (subclusters) were aligned and merged into 11 motif clusters. STAMP (platform for similarity, tree-building and alignment of DNA motifs and profiles) (32) identified 28 known TFBSs in these clusters, 14 of which are B cell-specific TFs. The inner-circle logos are the motifs for each k-mer subcluster; the matched known TFBSs are labeled on the outer circle. The number within the parentheses indicates the number of k-mers in each k-mer cluster.
CAPE outperforms other classifiers in predicting eQTLs and dsQTLs
Our ultimate goal was to identify causal regulatory SNPs that can impact target gene expression. In order to demonstrate that our method can be applied in a cell-type-specific manner and can be generalized to infer functional regulatory eQTLs, we trained our model in two different cell lines--GM12878 and HepG2--to predict eQTLs (Materials and Methods). Incorporating the sequence features trained from all the selected signal tracks in the corresponding cell line, we built linear SVM models on 1948 eQTLs and matched negative control SNPs in GM12878 and on 1044 eQTLs and matched negative control SNPs in HepG2 (Materials and Methods), respectively. We compared our method to two sequence-base approaches—deltaSVM (20) and DeepSEA (22)—and one more method utilizing both sequence features and DHS data across multiple individuals and multiple tissues (CATO (21)). Our approach outperforms these methods in prioritization of eQTLs. The overall accuracy of our eQTL classifier measured using the area under the curve (AUC) is 78.8% in GM12878 and 73.8% in HepG2, which is higher than all other three methods: DeepSEA, CATO and deltaSVM (Figure 3). This result demonstrated that integration of the two sequence signatures—ability of the variant to disturb the cognate TF binding and the binding capacity of its neighborhood—enables our approach to largely surpass other available methods in prioritizing causative eQTLs, even though only the sequence code learned from the enhancer-associated chromatin profiling were used to build the model.
Figure 3.

CAPE can accurately predict deleterious variants impacting target gene expression. The results of a 5-fold cross validation on GM12878 eQTLs and matched control SNPs (A and B). The results of a 5-fold cross validation on HepG2 eQTLs and matched control SNPs (C and D). (A) ROC curves for CAPE built with sequence features derived from k-mers (k = 4 to 12) learned from 19 signal tracks of GM12878, deltaSVM (GM12878), CATO, and DeepSEA. (B) Precision recall curves for CAPE built with sequence features derived from k-mers (k = 4 to 12) learned from 19 signal tracks of GM12878, deltaSVM (GM12878), CATO, and DeepSEA. (C) ROC curves for CAPE built with sequence features derived from k-mers (k = 4 to 12) learned from 23 signal tracks of HepG2, deltaSVM (HepG2), CATO and DeepSEA. (D) Precision recall curves for CAPE built with sequence features derived from k-mers (k = 4 to 12) learned from 23 signal tracks of HepG2, deltaSVM (HepG2), CATO and DeepSEA. The gray dashed line represents the random expectation.
To build a classifier aimed at prioritizing the genetic variants that disturb TF binding and affect the chromatin state, we utilized a set of dsQTLs which are significantly correlated with the chromatin accessibility of their associated 100 bps DHS in LCLs (Materials and Methods), considering that dsQTLs are enriched in TFBSs and are frequently associated with allele-specific TF binding (13). By incorporating all sequence features (Figure 1), we built an SVM with a linear kernel on the 565 dsQTLs and their controls SNPs (Materials and Methods). Using a 5-fold cross validation, our method has shown the best classification performance (AUC = 72.2%) and was closely followed by deltaSVM and CATO (Figure 4, Supplementary Data). This observation suggests that CAPE is able to capture the sequence code of the active regulatory regions and to distinguish causal from associated variants.
Figure 4.

CAPE can accurately predict deleterious variants impacting chromatin accessibility. The results of a 5-fold cross validation on dsQTLs and control SNPs located in the DHS hotspot region of GM12878. (A) ROC curves for CAPE built with sequence features derived from k-mers (k = 4 to 12) learned from 19 signal tracks, DeepSea, deltaSVM, and CATO. (B) Precision recall curves for CAPE built with sequence features derived from k-mers (k = 4 to 12) learned from 19 signal tracks, deltaSVM, DeepSea, and CATO. The gray dashed line represents the random expectation.
CAPE can be used to pinpoint deleterious regulatory variants
We next tested whether CAPE can identify deleterious regulatory variants associated with tissue-specific disorders or traits. We applied CAPE to prioritize the common SNPs within enhancer regions (GM12878 active enhancers not overlapping the dsQTLs and eQTLs in the training set) (Supplementary Data). SNPs with the FPR ≤ 0.05 were considered as potentially deactivating-enhancer SNPs (deSNPs). To validate that deSNPs are more likely to be causal SNPs compared to regular enhancer SNPs, we compared the enrichment of deSNPs in B-cell related GWAS traits to enhancer SNPs (Figure 5A). We observed that deSNPs were strongly enriched in the B-cell related traits compared to enhancer SNPs (Wilcoxon's test P-value = 3.9e−11). This observation suggests that CAPE is directly applicable to the prioritization and discovery of causal SNPs in GWAS studies. Moreover, the enrichment of B cell-related GWAS traits in deSNPs (identified both by eQTL and dsQTL model) is comparable with that in the top (FPR ≤ 0.05) potential causal SNPs identified by DeepSEA, and higher than deltaSVM and CATO (Supplementary Data).
Figure 5.

CAPE is applicable to the prioritization and discovery of causal SNPs in GWAS studies. (A) Comparison of enrichment of B lympoblastoid-related GWAS traits in deSNPs (identified using the eQTL SVM model) and enhancer SNPs. The y-axis is the ratio of fold enrichment of deSNPs (or enhancer SNPs) as compared with 100 sets of random SNPs. The error bar shows the standard error of the mean value of fold enrichment. One asterisk indicates a Wilcoxon's P-value < 0.01. Two asterisks indicate a P-value < 1e−5. The full list of deSNPs coincided with B-lymphoblastoid related GWAS traits is presented in the Supplementary Data, Supplementary Data. (B) An example of a SNP associated with the trait periodontitis (Mean PAL). rs140900046 is likely to be a causal SNP for periodontitis (Mean PAL). This position associates with the relatively conserved nucleotide T of a GABP binding site and overlaps with a GABP ChIP-seq peak.
One of the deSNPs with the highest CAPE score (≈1) in LCL was rs140900046. This SNP resided within a GABP ChIP-seq peak and is located near two genes, FIZ1 and ZNF524 (Figure 5B). Its alternative allele A is associated with a putative binding site of GABP (Figure 5B), which is a critical regulator of B lymphocyte development (37). In agreement with our prediction that CAPE is capable of identifying causal SNPs from the GWAS studies, rs140900046 coincides with the trait of periodontitis with the target gene FIZ1 (45). Periodontitis is a chronic inflammatory disease which destroys the tissues and bone supporting the teeth and can result in tooth loss. The complex relationship between periodontitis and B cells has been extensively studied (46–49). Based on our analysis, rs140900046 might be one of the causal SNPs for periodontitis and acts via modulating the GABP regulation of the gene FIZ1.
Overall, our results suggest that the regulatory variants with higher CAPE scores are more likely to be detrimental, probably due to disrupting an essential TF binding.
DISCUSSION
We developed a computational approach (dubbed CAPE) to identify causal regulatory SNPs impacting target gene expression and modulating chromatin accessibility. Our k-mer based SVM model incorporates the sequence features learned from enhancer-associated genome-wide sequencing data (including DNase-seq, ChIP-seq of chromatin histone marks and TF binding) to estimate the likelihood of a genetic variant deactivating an enhancer. We took advantage of sets of functional SNPs including eQTLs and dsQTLs to build and test the model. Our approach achieves higher accuracy than existing well-known methods in a cell-type-specific manner. The integration of sequence features of a region hosting a genomic variant and sequence features of the flanking regions (both represented by k-mers) learned from a panel of ChIP-seq and DNase-seq datasets in a machine learning model has resulted in establishing an accurate predictor of deleterious enhancer mutations. The contribution of ChIP-seq data has a non-negligible positive effect on classification accuracy (the AUC of eQTL predictions has increased ∼10% upon inclusion of ChIP-seq data; Supplementary Data). Unlike tools such as DeepSEA (22), which deep learns sequence features without the knowledge of the underlying sequence code, or deltaSVM (20), which learns the sequence features from a single enhancer-associated chromatin profile and considers the k-mer content associated with the genetic variant only, our method decomposes the sequence code of potential binding sites and the binding sites of cofactors from a set of chromatin profiles, and directly quantifies the deactivating effect of a single nucleotide mutation based on the corresponding change in the underlying k-mer profile. Furthermore, different from the tools which require versatile categories of high-throughput sequencing data across multiple individuals such as CATO (21), RASQUAL (24) and eQTeL (25), CAPE prioritizes noncoding variants only by integrating learned sequence signatures based on a single individual, and thus can be widely applied to different tissue-specific datasets.
In addition to sequence features, we also took into account the expression level of potential target genes associated with genetic variants by assigning the nearest gene to the variant. This feature improves the AUC of the dsQTL SVM model by ∼3% when the control SNPs are located in the hotspot DHS region of another cell line or does not improve the AUC otherwise (Supplementary Data). This observation suggests that the expression of nearby genes could be a valuable metric for improving classifier performance but only in selected cases.
The predicted causative regulatory SNPs (FPR ≤ 0.05)—deSNPs—are more likely to be enriched in tissue-specific GWAS traits as compared to enhancer SNPs. This observation suggests that CAPE is capable of pinpointing regulatory variants which disrupt binding of essential TFs (Supplementary Data) and lead to downstream phenotype change in the corresponding cell line. Thus, these inferred causal mutations are prime candidates for disrupting temporal and spatial gene expression programs that define cellular identity.
In summary, our method provides a scoring scheme capable of accurately recognizing deleterious regulatory variants on a genome-wide scale. We constructed a web server (http://cape.dcode.org) and a stand-alone tool to facilitate direct access to the developed algorithm for recognizing and categorizing putative causal enhancer mutations. We expect that our method and tool would contribute to accurate identification of disease-causal mutations in the human and other genomes.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Dorothy L. Buchhagen and Timothy Doerr for critical reading of the manuscript, and grateful to Dr John Spouge for helpful statistical advice.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of the National Institutes of Health; National Library of Medicine. Funding for open access charge: Intramural Research Program of the National Institutes of Health; National Library of Medicine.
Conflict of interest statement. None declared.
REFERENCES
- 1. Visel A., Blow M.J., Li Z., Zhang T., Akiyama J.A., Holt A., Plajzer-Frick I., Shoukry M., Wright C., Chen F. et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009; 457:854–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012; 337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sakabe N.J., Savic D., Nobrega M.A.. Transcriptional enhancers in development and disease. Genome Biol. 2012; 13:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dickel D.E., Visel A., Pennacchio L.A.. Functional anatomy of distant-acting mammalian enhancers. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2013; 368:20120359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Monteiro A.N., Freedman M.L.. Lessons from postgenome-wide association studies: functional analysis of cancer predisposition loci. J. Intern. Med. 2013; 274:414–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ward L.D., Kellis M.. Interpreting noncoding genetic variation in complex traits and human disease. Nat. Biotechnol. 2012; 30:1095–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karczewski K.J., Dudley J.T., Kukurba K.R., Chen R., Butte A.J., Montgomery S.B., Snyder M.. Systematic functional regulatory assessment of disease-associated variants. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:9607–9612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gaffney D.J., Veyrieras J.B., Degner J.F., Pique-Regi R., Pai A.A., Crawford G.E., Stephens M., Gilad Y., Pritchard J.K.. Dissecting the regulatory architecture of gene expression QTLs. Genome Biol. 2012; 13:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thurman R.E., Rynes E., Humbert R., Vierstra J., Maurano M.T., Haugen E., Sheffield N.C., Stergachis A.B., Wang H., Vernot B. et al. The accessible chromatin landscape of the human genome. Nature. 2012; 489:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M.. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kellis M., Wold B., Snyder M.P., Bernstein B.E., Kundaje A., Marinov G.K., Ward L.D., Birney E., Crawford G.E., Dekker J. et al. Defining functional DNA elements in the human genome. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:6131–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Degner J.F., Pai A.A., Pique-Regi R., Veyrieras J.B., Gaffney D.J., Pickrell J.K., De Leon S., Michelini K., Lewellen N., Crawford G.E. et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012; 482:390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A.. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012; 337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li S., Ovcharenko I.. Human enhancers are fragile and prone to deactivating mutations. Mol. Biol. Evol. 2015; 32:2161–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cowper-Sal lari R., Zhang X., Wright J.B., Bailey S.D., Cole M.D., Eeckhoute J., Moore J.H., Lupien M.. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat. Genet. 2012; 44:1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kircher M., Witten D.M., Jain P., O'Roak B.J., Cooper G.M., Shendure J.. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014; 46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ionita-Laza I., McCallum K., Xu B., Buxbaum J.D.. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 2016; 48:214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee D., Gorkin D.U., Baker M., Strober B.J., Asoni A.L., McCallion A.S., Beer M.A.. A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet. 2015; 47:955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maurano M.T., Haugen E., Sandstrom R., Vierstra J., Shafer A., Kaul R., Stamatoyannopoulos J.A.. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat. Genet. 2015; 47:1393–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou J., Troyanskaya O.G.. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods. 2015; 12:931–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alipanahi B., Delong A., Weirauch M.T., Frey B.J.. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015; 33:831–838. [DOI] [PubMed] [Google Scholar]
- 24. Kumasaka N., Knights A.J., Gaffney D.J.. Fine-mapping cellular QTLs with RASQUAL and ATAC-seq. Nat. Genet. 2016; 48:206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Das A., Morley M., Moravec C.S., Tang W.H., Hakonarson H., Margulies K.B., Cappola T.P., Jensen S., Hannenhalli S.. Bayesian integration of genetics and epigenetics detects causal regulatory SNPs underlying expression variability. Nat. Commun. 2015; 6:8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang D., Ovcharenko I.. Identifying causal regulatory SNPs in ChIP-seq enhancers. Nucleic Acids Res. 2015; 43:225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M.J. et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lin C.-J. LIBSVM : a library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011; 27, https://www.csie.ntu.edu.tw/∼cjlin/libsvm/. [Google Scholar]
- 29. Leslie R., O'Donnell C.J., Johnson A.D.. GRASP: analysis of genotype–phenotype results from 1390 genome-wide association studies and corresponding open access database. Bioinformatics. 2014; 30:i185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mathelier A., Zhao X., Zhang A.W., Parcy F., Worsley-Hunt R., Arenillas D.J., Buchman S., Chen C.Y., Chou A., Ienasescu H. et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014; 42:D142–D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matys V., Kel-Margoulis O.V., Fricke E., Liebich I., Land S., Barre-Dirrie A., Reuter I., Chekmenev D., Krull M., Hornischer K. et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006; 34:D108–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mahony S., Benos P.V.. STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res. 2007; 35:W253–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L. et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014; 42:D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heinz S., Romanoski C.E., Benner C., Allison K.A., Kaikkonen M.U., Orozco L.D., Glass C.K.. Effect of natural genetic variation on enhancer selection and function. Nature. 2013; 503:487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spitz F., Furlong E.E.. Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 2012; 13:613–626. [DOI] [PubMed] [Google Scholar]
- 36. Stefflova K., Thybert D., Wilson M.D., Streeter I., Aleksic J., Karagianni P., Brazma A., Adams D.J., Talianidis I., Marioni J.C. et al. Cooperativity and rapid evolution of cobound transcription factors in closely related mammals. Cell. 2013; 154:530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xue H.H., Bollenbacher-Reilley J., Wu Z., Spolski R., Jing X., Zhang Y.C., McCoy J.P., Leonard W.J.. The transcription factor GABP is a critical regulator of B lymphocyte development. Immunity. 2007; 26:421–431. [DOI] [PubMed] [Google Scholar]
- 38. Lloberas J., Soler C., Celada A.. The key role of PU.1/SPI-1 in B cells, myeloid cells and macrophages. Immunol. Today. 1999; 20:184–189. [DOI] [PubMed] [Google Scholar]
- 39. Torlakovic E., Tierens A., Dang H.D., Delabie J.. The transcription factor PU.1, necessary for B-cell development is expressed in lymphocyte predominance, but not classical Hodgkin's disease. Am. J. Pathol. 2001; 159:1807–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gerondakis S., Siebenlist U.. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb. Perspect. Biol. 2010; 2:a000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Minamino K., Takahara K., Adachi T., Nagaoka K., Iyoda T., Taki S., Inaba K.. IRF-2 regulates B-cell proliferation and antibody production through distinct mechanisms. Int. Immunol. 2012; 24:573–581. [DOI] [PubMed] [Google Scholar]
- 42. Blyth K., Slater N., Hanlon L., Bell M., Mackay N., Stewart M., Neil J.C., Cameron E.R.. Runx1 promotes B-cell survival and lymphoma development. Blood Cells Mol. Dis. 2009; 43:12–19. [DOI] [PubMed] [Google Scholar]
- 43. Whiteman H.J., Farrell P.J.. RUNX expression and function in human B cells. Crit. Rev. Eukaryot. Gene Expr. 2006; 16:31–44. [DOI] [PubMed] [Google Scholar]
- 44. Medvedovic J., Ebert A., Tagoh H., Busslinger M.. Pax5: a master regulator of B cell development and leukemogenesis. Adv. Immunol. 2011; 111:179–206. [DOI] [PubMed] [Google Scholar]
- 45. Teumer A., Holtfreter B., Volker U., Petersmann A., Nauck M., Biffar R., Volzke H., Kroemer H.K., Meisel P., Homuth G. et al. Genome-wide association study of chronic periodontitis in a general German population. J. Clin. Periodontol. 2013; 40:977–985. [DOI] [PubMed] [Google Scholar]
- 46. Zhu M., Belkina A.C., DeFuria J., Carr J.D., Van Dyke T.E., Gyurko R., Nikolajczyk B.S.. B cells promote obesity-associated periodontitis and oral pathogen-associated inflammation. J Leukoc. Biol. 2014; 96:349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Berglundh T., Donati M.. Aspects of adaptive host response in periodontitis. J. Clin. Periodontol. 2005; 32(Suppl 6):87–107. [DOI] [PubMed] [Google Scholar]
- 48. Oliver-Bell J., Butcher J.P., Malcolm J., MacLeod M.K., Adrados Planell A., Campbell L., Nibbs R.J., Garside P., McInnes I.B., Culshaw S.. Periodontitis in the absence of B cells and specific anti-bacterial antibody. Mol. Oral Microbiol. 2015; 30:160–169. [DOI] [PubMed] [Google Scholar]
- 49. Tsuruyama T., Imai Y., Takeuchi H., Hiratsuka T., Maruyama Y., Kanaya K., Kaszynski R., Jin G., Okuno T., Ozeki M. et al. Dual retrovirus integration tagging: identification of new signaling molecules Fiz1 and Hipk2 that are involved in the IL-7 signaling pathway in B lymphoblastic lymphomas. J. Leukoc. Biol. 2010; 88:107–116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.