


Abstract
Distinguishing cell states based only on gene expression data remains a challenging task. This is true even for analyses within a species. In cross-species comparisons, the results obtained by different groups have varied widely. Here, we integrate RNA-seq data from more than 40 cell and tissue types of four mammalian species to identify sets of associated genes as indicators for specific cell states in each species. We employ a statistical method, TROM, to identify both protein-coding and non-coding indicators. Next, we map the cell states within each species and also between species using these indicator genes. We recapitulate known phenotypic similarity between related cell and tissue types and reveal molecular basis for their similarity. We also report novel associations between several tissues and cell types with functional support. Moreover, our identified conserved associated genes are found to be a good resource for studying cell differentiation and reprogramming. Lastly, long non-coding RNAs can serve well as associated genes to indicate cell states. We further infer the biological functions of those non-coding associated genes based on their co-expressed protein-coding genes. This study demonstrates that combining statistical modeling with public RNA-seq data can be powerful for improving our understanding of cell identity control.
INTRODUCTION
Cell states or cell identities (e.g. embryonic stem cells (ESCs), heart tissues and the HeLa cell line) are maintained and controlled by a set of key regulators and epigenomic modifications (1–3). Previous studies have revealed crucial roles of some key regulators in controlling gene expression during cell differentiation and developmental processes, including transcription factors (TFs) (3,4), chromatin regulators (5,6), RNA-binding proteins (RBPs) (7,8), microRNAs (9,10) and long non-coding RNAs (lncRNAs) (11,12). In developmental biology and genomics, an important question is to understand how individual biological molecules and their interactions determine cell states, including ESCs, progenitor cells, terminally differentiated tissues and cultured cell lines. Although several regulatory circuits have been found evolutionarily conserved in mammals (13,14), it remains challenging to systematically identify conserved protein-coding genes and lncRNAs that function in various cell states across multiple mammalian species. Given the vast transcriptomic data produced in recent years, comparative analysis of mammalian transcriptomes has become feasible to define the similarities or differences between various mammalian cell states and further identify conserved genes and so reveal molecular mechanisms underlying cell identity control.
More recent high-throughput RNA sequencing (RNA-seq) studies, though, have been able to comprehensively characterize protein-coding genes’ expression patterns in a genome-wide manner across multiple species, providing new insights into the evolution of gene expression (15–19). Recently, it was found that unlike protein-coding genes, the number of non-coding genes increases consistently with the phenotypic complexity of species, suggesting that non-coding RNAs might play critical roles in the evolution of eukaryotes (20,21). However, most RNA-seq-based studies emphasized on the expression patterns of protein-coding genes and did not investigate the expression programs of lncRNAs, though recent studies have revealed that lncRNAs exert critical functions in cell fate regulation (12). Also few efforts have been made to decipher the relationship between expression and evolutionarily conserved programs of lncRNAs (22,23). Nor did previous studies aim to identify conserved key lncRNAs for different cell states.
In addition to characterizing and comparing the transcriptomes within a species and across multiple species, great efforts have been put into the identification of important regulatory factors using high-throughput data. For example, a small set of key TFs that can control cell identity has been identified using a large cohort of existing transcriptomic data sets (24–26). In a recent study, 503 different TFs as candidate core TFs for more than 200 human tissues and cell types have been systematically identified (25). Increasing lines of evidence suggest that RBPs play important and diverse roles in controlling cell states, such as ESCs (2,27). However, post-transcriptional mechanisms of cell identity control by RBPs remain much unexplored.
In this study, we aim to systematically identify conserved genes that could putatively define cell states in multiple mammalian species, as well as to find possible correspondences between cell identities across different species. We collected and processed 307 publicly available polyadenylated RNA-seq data sets for more than 40 tissues and cell types (including ESCs, iPSCs, in vivo tissues and cultured cell lines) from four mammalian species (human, chimpanzee, bonobo and mouse). In order to quantitatively characterize the lineage relationships of those tissues and cell types, we used a statistical method TROM (Transcriptome Overlap Measure) (28,44) to find their correspondence via a comprehensive transcriptome mapping. The results also provide a catalog of protein-coding and long non-coding associated genes, which well capture transcriptome characteristics of various cell identities in different species. Moreover, our analyses revealed that the conserved protein-coding associated genes are highly enriched in biological functions and cellular pathways, which are closely related to the physiology of their associated cell states. Among these conserved protein-coding associated genes, there is also enrichment of master TFs and RBPs, which have been reported to determine cell identities. These results suggest that our identified conserved protein-coding associated genes are good markers of cell identities. In addition to protein-coding genes, we found that lncRNAs also serve well as associated genes for establishing a good correspondence of cell identities across species. We inferred the potential functions of those lncRNAs from the known biological functions of protein-coding genes by constructing a gene co-expression network. We found that the conserved associated lncRNAs exhibit significant enrichment of biological functions related to cell identities, suggesting that these lncRNAs have conserved functions in evolution and are also good markers of cell identities. Our study demonstrates that, by integrating and re-analyzing large-scale public transcriptomic data from multiple species using proper statistical methods, we are able to systematically discover unknown markers of cell identities and provide insights into their molecular characteristics.
MATERIALS AND METHODS
RNA-seq data collection and processing
We compiled a data resource of 307 publicly available poly(A) RNA-seq data sets, which were profiled from four mammalian species: Homo sapiens (human) (183 data sets), Mus musculus (mouse) (77 data sets), Pan troglodytes (chimpanzee) (31 data sets) and Pan paniscus (bonobo) (16 data sets) (Supplementary Data). Our data resource contained ∼19 billion sequencing reads from 307 biological samples, of which seven cell and tissue types (brain, cerebellum, heart, kidney, liver, testis and ESC or iPSC) existed in all four species. In addition, our data resource covered far more cell and tissue types in human (totally 41 tissues and cell types) and mouse (totally 18 tissues and cell types). All RNA-seq data sets were generated from Illumina GAII or HiSeq2000/HiSeq2500 systems. We filtered out low quality reads in each RNA-seq data set using PRINSEQ (29).
We then aligned the RNA-seq data to the mammalian genomes using Tophat v2.0.10 (30,31). Three mammalian genomes were obtained from Ensembl (Homo sapiens hg19, Mus musculus mm10 and Pan troglodytes panTro4). The genome annotation of bonobo (Pan paniscus) was not available in Ensembl, and our analysis for protein-coding genes and lncRNAs requires the annotation. Hence, we followed the analysis strategy of Brawand et al. (16), who used the chimpanzee genome (panTro4) and annotation for bonobo, because of the high similarity (>95%) between bonobo and chimpanzee genomes (32). Our detailed mapping results are summarized in Supplementary Data.
Gene expression estimation
We constructed mammalian reference genome annotations by integrating published annotations from multiple resources. For protein-coding genes, the annotations were from human Gencode v19, mouse Gencode M2 and chimpanzee/bonobo Ensembl_CHIMP2.1.4.73 (33,34). For lncRNAs, the annotations were from a recent publication (23). We then used Cufflinks (30,35) (v2.1.1, supplied with reference annotation, i.e. using ‘-G’ option) to measure the expression levels for both protein-coding genes and lncRNAs in the four mammalian species.
Due to the lack of reference genome annotations for bonobo, following the analysis strategy from Brawand et al. (16), we used the chimpanzee genome (panTro4) as a reference for the RNA-seq data sets from bonobo. To evaluate the potential errors of this strategy, we conducted an additional analysis by mapping the human RNA-seq data to the chimpanzee genome, and compared the resulting gene expression estimates (in fragments per kilobase of transcript per million mapped reads (FPKM) units) with the original gene expression estimates using the human genome. We found that the two sets of gene expression estimates have almost identical distributions (Supplementary Data) and high correlations (Supplementary Data).
Since RNA-seq data sets from public data repositories were generated in different studies by different laboratories, we used a unified procedure to obtain gene expression estimates in FPKM units (30,35). By examining the reproducibility of the FPKM values between replicates within the same tissue or cell type, we found high correlation coefficients between all replicate pairs (see Supplementary Data for examples). In this study, samples from the same cell and tissue type are referred to as ‘(biological) replicates’. Specifically when comparing protein-coding genes with lncRNAs, we observed higher correlations for the former, because protein-coding gene expression levels are generally higher and measured more accurately than those of lncRNAs (36–39). These results show that our processed data lay the basis for further analyses.
Orthologous gene families
To identify conserved cell-state associated genes (including protein-coding genes and lncRNAs), we used the orthologous families of these genes in human, chimpanzee, bonobo and mouse. As an example, suppose gene X is a liver-associated gene in human. If its orthologous genes in the three other species are also liver-associated genes, it is defined as a conserved associated gene of liver. Overall, we identified conserved associated genes in seven cell states: ESC, brain, cerebellum, heart, liver, kidney and testis.
We obtained orthologous families of protein-coding genes from TreeFam v9 (40), a database of tree-based orthology predictions, and orthologous families of lncRNAs from a recent publication (23). The orthologous families are summarized in Supplementary Data. Notably, the conservation analysis of lncRNAs is only based on the three primates, because the number of orthologous families of lncRNAs between primates and mouse is too small.
TF and RBP annotations
For TF analysis in our studies, we downloaded TF annotations of human, mouse, chimpanzee and bonobo from AnimalTFDB 2.0 (41), which provides TF annotations for more than 50 animal genomes. For RBP analysis, we obtained RBP annotations from a recent review, which provides a census of 1542 manually curated RBPs (42).
Identification of cell-state associated genes by TROM
One cell state (i.e. a tissue or cell type, such as ESC, liver, and testis) can be characterized by its ‘associated genes’ that we defined as the relatively highly expressed genes in the cell state as compared with other cell states. We first calculated the relative expression estimate (i.e. Z-scores) of every gene across all cell states: subtracting a gene's FPKM estimates by its mean FPKM estimate of all cell states, and then dividing the differences by its standard deviation of FPKM estimates across all cell states.
 |
where i = 1, 2, …, n (the number of all cell states), ei is the FPKM estimate in cell state i, and
 |
and
 |
are the mean and standard deviation of FPKM estimates across all cell states. Hence, the Z-scores reflect the relative expression of a gene across all cell states.
Because the four species have different numbers of samples, the four species exhibit different Z-score distributions, making it difficult to find a consensus Z-score cutoff to identify associated genes. Hence, we further performed quantile normalization (43) on the Z-scores across the four species, resulting in almost identical normalized Z-score distributions (Supplementary Data). Then for every cell state, we selected its associated genes whose FPKMs are above a threshold and normalized Z-scores are in a top percentile. The FPKM threshold is 1.0 for protein-coding genes and 0 for lncRNAs, and the normalized Z-score percentile is top 5%. These two criteria guarantee that in the given cell state, the expression levels of the associated genes are distinguishable from background noise and are also higher than those in some other cell states.
Another important issue is to evaluate and reduce batch effects in data collected from multiple sources. We used principal component analysis (PCA), a standard analysis method to assess the potential batch effects in our normalized data sets. We performed PCA on the normalized Z-scores of all data sets and examined whether the data sets from the same and/or similar cell and tissue types are clustered together. The data sets (i.e. points) were labeled using their cell and tissue types or batch IDs (Supplementary Data).
Transcriptome mapping by TROM
We then used the TROM algorithm to map two mammalian cell states by their transcriptome similarity (28,44). In the within-species cell state mapping, for every two cell states, we tested the significance of the number of their common associated genes using an ‘overlap test’, whose null hypothesis is that the two cell states’ associated genes are independent samples from the gene population of the species. If significant, two cell states are called mapped. The P-value of the within-species cell state mapping is calculated as
 |
where M is the total number of protein-coding genes or lncRNAs, and m, n and r are the numbers of genes in gene sets A, B and A∩B (Figure 1B left).
Figure 1.

Overview of the RNA-seq data sets and Transcriptome Overlap Measure (TROM) approach. (A) Numbers of RNA-seq data sets and sequencing reads for each mammalian species, including human, chimpanzee, bonobo and mouse. (B) The TROM approach. First, associated genes of each cell state are selected using thresholds on FPKMs and Z-scores (normalized FPKMs across cell states). In the within-species TROM (left panel), the significance of the number of the common associated genes of two cell states is established via an overlap test. In the between-species TROM (right panel), a similar overlap test is carried out, except that orthologous genes are used to connect the two species. Two cell states are called ‘mapped’ if the test is significant. (See Materials and Methods for details.)
In the between-species cell state mapping, for every two cell states from different species, we tested the significance of the number of ortholog pairs between their associated genes using an overlap test. If the test was significant, two cell states were called mapped. The P-value of the between-species cell state mapping is calculated as
 |
where N is the total number of orthologous protein-coding genes or lncRNAs, and m, l and s are the numbers of genes in gene sets A, C and A∩C (Figure 1B right).
The P-values of transcriptome mapping between cell states were adjusted using Bonferroni correction. For both within- and between-species mapping, the TROM scores are calculated as the –log10 transformed Bonferroni corrected P-values.
For details and methodological aspects of the TROM method, please refer to Li et al. (44,45).
To visualize the transcriptome mapping patterns, we plotted the resulting similarity matrices of TROM scores using the R function heatmap(), which performs hierarchical clustering on the matrix rows and columns to generate heatmaps for better visualization. The dendrograms are resulted from the default hierarchical clustering. We did not manually alter or twist the dendrograms.
Enrichment analysis of Gene Ontology and biological pathways
For gene ontology (GO) analysis, we used topGO (46) to estimate the enrichment of biological process terms for different cell states based on their associated genes. We calculated the significance of GO term enrichment in every cell state using a hypergeometric test. The top three most enriched GO terms in every cell state were displayed. For biological pathway analysis, we used KEGGREST (47) to calculate the enrichment of biological pathways, and displayed the results in a similar fashion as in the GO analysis. The P-values were adjusted using Bonferroni correction.
Construction of a co-expression network of protein-coding genes and lncRNAs
We applied a previously published method (48) to reconstructing a co-expression network of protein-coding genes and lncRNAs across human, chimpanzee and bonobo. We did not include mouse because there are few known conserved lncRNAs between mouse and primates. We used the well-established homologous gene families for both protein-coding genes (from TreeFam v9 (40)) and lncRNAs (from (23)) across the three species. We then computed the Pearson correlation coefficients of expression patterns. Given two homologous families, we examined whether the combination of correlation coefficients from the three species was significantly (P-value < 0.01) higher or lower than expected by chance.
Here, we briefly describe the algorithm to compute the P-value of conserved co-expression between two homologous gene families. We defined a homologous gene family as a one-to-one mapping orthologous gene group across multiple species. For example, if gene A in species 1, gene B in species 2 and gene C in species 3 are all orthologous to each other, we call the genes A, B, and C a homologous gene family. Please note that we did not include one-to-many orthologous genes between every two species. For every pair of homologous gene families (m, m'), we computed the probability of observing their Pearson correlation cor(Xm, Xm'), where Xm represents the expression vector of gene family m at all the cell states in all the n species, or even a higher correlation value, relative to those of other gene family pairs. Under the null hypothesis that the Pearson correlations of all gene family pairs are ordered randomly, this probability is the P-value for the pair (m,m’). To calculate this probability, in every species s, we ranked all the homologous gene family pairs based on their Pearson correlations, with larger correlations having smaller ranks. Please note that the ranks could be different in different species, because a gene family may not have a gene in every species. If a species has genes in fewer families, it will have fewer gene family pairs to rank. Then in species s, we divided the ranks of gene family pairs by the total number of gene family pairs in that species, yielding one rank ratio for the (m,m’) gene family pair: rs . Consider all the n species, we obtained n rank ratios for the (m,m’) pair, r1, r2,…, rn. To find out how significant (r1, r2,…, rn) is compared to random observation, we computed the probability of observing a set of n rank ratios such that the ith ordered rank ratio is no greater than r(i), the ith order value of (r1, r2,…, rn), for every i = 1,…, n, by chance. This probability represents the P-value of the observed rank ratios (r1, r2,…, rn) assuming that the order of the species does no matter. If we assume the rs's are drawn independently and uniformly, we can compute the P-value from the joint cumulative distribution of an n-dimensional order statistic:
. Consider all the n species, we obtained n rank ratios for the (m,m’) pair, r1, r2,…, rn. To find out how significant (r1, r2,…, rn) is compared to random observation, we computed the probability of observing a set of n rank ratios such that the ith ordered rank ratio is no greater than r(i), the ith order value of (r1, r2,…, rn), for every i = 1,…, n, by chance. This probability represents the P-value of the observed rank ratios (r1, r2,…, rn) assuming that the order of the species does no matter. If we assume the rs's are drawn independently and uniformly, we can compute the P-value from the joint cumulative distribution of an n-dimensional order statistic:
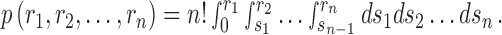 |
We can efficiently compute the above with the recursive formula:
 |
where r0 = 0 and the recursive call to p() supplies all of the original arguments except the (n-i + 1)th argument. Since we included 3 species in the analysis we used n = 3.
We computed correlations for the homologous gene families involving the protein-coding genes and lncRNAs expressed in at least two samples of human, chimpanzee, and bonobo each. Genes were defined as expressed in a cell state if their expression estimates were in the top 90% among all the genes at that cell state. We allowed for both positive and negative correlations in the co-expression network, which is visualized by Cytoscape (49).
GO enrichment in the co-expression network
We identified clusters of highly inter-connected homologous gene families in the co-expression network via the Markov Cluster algorithm (50). Each cluster contains both protein-coding genes and lncRNAs. Then we detected enriched GO (Biological processes) terms in the protein-coding genes of each cluster. We also used the hypergeometric test to evaluate whether the conserved associated lncRNAs of each cell state are enriched in each cluster. The results were visualized using radar plots. The P-values were adjusted using Bonferroni correction.
Code availability
The TROM software is available as an R package with manuals and source codes available at http://www.stat.ucla.edu/∼jingyi.li/software-and-data/trom.html and https://cran.r-project.org/web/packages/TROM/index.html.
RESULTS
Identification of associated genes for different cell states
We first collected and uniformly processed 307 publicly available polyA RNA-seq data sets (∼19 billion sequencing reads) of various tissues and cell types from four mammalian species (mouse and three primates including human, bonobo and chimpanzee) (Figure 1A and Supplementary Data). Among them, the 183 human and 77 mouse data sets span a wide range of developmental stages and lineages. The chimpanzee and bonobo data sets include iPSC and several in vivo tissues (brain/cortex, cerebellum, heart, kidney, liver and testis) (Supplementary Data).
To capture the transcriptome characteristics of different cell states, we define the ‘associated genes’ of a cell state as the protein-coding and long non-coding genes whose expression is relatively high in that cell state and relatively low in some other cell states. To identify the associated genes of different cell states and subsequently use them to compare the cell states from different species, we adapted a statistical method TROM, which were recently developed for comparing developmental stages of D. melanogaster and C. elegans (28,44). We first identified the associated genes of cell states by the following criterion: in a given cell state, its associated genes must have (i) FPKM (35) above a positive constant c (c = 1 for protein-coding genes and c = 0 for lncRNAs, because lncRNAs are generally more lowly expressed than protein-coding genes); (ii) normalized Z-score in the top 5% among its normalized Z-scores in all cell states (see Material and Methods for details, Supplementary Data). The cell state mapping patterns are largely robust to different FPKM cutoffs (Figure 2A and B) and normalized Z-score thresholds (Figure 2C and D). The numbers of associated genes identified by TROM vary across different cell states in each species (Supplementary Data). In addition to our TROM method, we also used PCA to assess the potential batch effects in our normalized data sets. We found that the normalized data sets are better clustered by tissue and cell types rather than by batches in the first three principal component directions (Supplementary Data). Hence, we concluded that the batch effects are not strong in the normalized data sets and would only have negligible effects on our analyses.
Figure 2.

Robust transcriptome mapping patterns. (A) A correspondence map of human cell states by TROM using associated protein-coding genes and lncRNAs under expression cutoff c = 1. (B) A correspondence map of human cell states by TROM using associated protein-coding genes and lncRNAs under expression cutoff c = 0. (C) A correspondence map of human cell states by TROM using associated protein-coding genes under a series of Z-score thresholds. (D) A correspondence map of human cell states by TROM using associated lncRNAs under a series of Z-score thresholds. Columns and rows correspond to biological samples of various cell states. Higher TROM scores (defined as –log10 transformed Bonferroni-corrected P-values from the overlap test) are shown in darker colors.
We then used those identified associated genes to compare the cell states within and between mammalian species (Figure 1B). Specifically, for every pair of cell states, we tested if there is significant overlap between their associated genes. If so, the two cell states are called ‘mapped’. In within-species comparisons, the common associated genes shared by two cell states is considered the overlap; in between-species comparisons, the overlap is defined as the orthologous genes pairs between the associated genes of the two cell states, each from a different species (Supplementary Data). In our study, protein-coding genes were used to compare all the four species, while lncRNAs were only used in the comparisons of the three primate species because orthologous lncRNAs are largely unavailable between primates and mouse. Like other comparative genomic studies, we also used correlation analyses to compare different cell states based on measured gene expression levels. However, in contrast to TROM, these correlation analyses failed to find clear or informative correspondence patterns among the mammalian cell states (Supplementary Data). This was expected and consistent with our previous findings (28,44), because correlation values depend heavily on the accuracy of gene expression estimates. On the other hand, TROM finds correspondence between cell states based on their associated genes and is more robust to noise and biases in gene expression estimates. More detailed comparison and power analysis of TROM and Pearson and Spearman correlation coefficients can be found in Li et al. (44).
Within-species cell state mapping
Lineage relationships rediscovered by within-species cell state correspondence maps
We first applied TROM to map transcriptomes of human cell states (i.e. tissues and cell types) using their associated protein-coding genes, resulting in a clear correspondence map (Figure 3A). As expected, transcriptomes of the same or similar tissue types form prominent mapping blocks, including (i) ESCs and iPSCs, (ii) cultured cell lines, (iii) immune cells and tissues, (iv) neural tissues, (v) liver tissues and (vi) testis tissues. These blocks are consistent with the known physiology of tissues and cells. For example, similar to ESCs, iPSCs are capable of generating all types of differentiated tissues and cells.
Figure 3.

Cell states encoded by associated genes in human. (A) A correspondence map of human cell states by TROM using associated protein-coding genes. Columns and rows correspond to biological samples of various cell states. Higher TROM scores (defined as –log10 transformed Bonferroni-corrected P-values from the overlap test) are shown in darker colors. Axis colors represent cell states, and colored boxes mark the prominent mapping patterns. (B) The number of protein-coding genes (top) and lncRNAs (bottom) associated with different number of human cell states. For associated protein-coding genes, the proportion of housekeeping genes in each group are shown. (C) Enriched gene ontology (GO) (biological processes) terms of 19 human tissues. More significant enrichment scores (defined as –log10 transformed Bonferroni-corrected P-values) are shown in darker colors. (D) Enriched KEGG cellular pathways of 19 human tissues. More significant enrichment scores (defined as –log10 transformed Bonferroni-corrected P-values) are shown in darker colors.
Next we compared those cell states again by TROM using only associated TFs or associated lncRNAs (Supplementary Data). Interestingly, the resulting mapping patterns are similar to Figure 3A but have smaller mapping blocks, confirming the higher tissue specificity of TFs (51) and lncRNAs (36) than protein-coding genes. For example, placenta, trophoblast-like cells and mesenchymal stem cells form a clear mapping block when compared using TFs. This observation can be explained by the fact that trophoblasts develop into a large part of the placenta, and placenta-derived cells have mesenchymal stem cell potentials (52). These results suggest that the identified associated TFs and lncRNAs serve as good representatives of cell identities, consistent with previous findings on their context specific activations and regulatory roles (36,51).
In addition to human, we compared the transcriptomes of cell states within every other mammalian species using (i) all the associated protein-coding genes, (ii) the associated TFs only and (iii) the associated lncRNAs only (Supplementary Data). The mapping patterns are largely consistent with what we observed in human. These within-species mapping results demonstrate that our approach is powerful and robust in revealing cell state similarity in multiple mammalian species, and verify our identified associated genes as good markers of cell identities.
We further examined the number of associated cell states for the associated genes we identified in human and mouse (Figure 3B and Supplementary Data). Among 41 human cell states, the highest proportion (about 28%) of the union of associated protein-coding genes are associated with three cell states. By contrast, a great proportion (about 33%) of the associated lncRNAs are only associated with one cell state, confirming the stronger cell type-specificity of lncRNAs (36). In addition, we investigated the expression broadness and dynamics of housekeeping genes. Housekeeping genes are defined as the genes that are expressed in all cell types and are required for the maintenance of basic cellular functions. However, previous studies suggest that expression levels of some housekeeping genes vary across tissue types and experimental conditions (53,54). We found that housekeeping genes (55) are more enriched in the protein-coding genes associated with more cell states (Figure 3B). This observation is consistent with the definition of housekeeping gene and also suggests that our TROM method can identify the housekeeping genes expressed at higher levels in specific cell types.
Biological functions of cell-state associated protein-coding genes
To investigate the biological functions of our identified associated genes, we started with the associated protein-coding genes, which have more complete functional annotations than lncRNAs have. We first identified the GO terms enriched in the associated protein-coding genes for various cell states in human (Figure 3C) and mouse (Supplementary Data). The results revealed that the associated protein-coding genes are enriched with biological processes that largely define the identities of the respective cell states. For example, kidney and liver are enriched with metabolic processes (including organic acid metabolic process, carboxylic acid metabolic process, oxoacid metabolic process and small molecule metabolic process), and testis is enriched with spermatogenesis and sexual reproduction. Interestingly, although kidney and liver share some metabolic processes, liver is additionally enriched with processes related to lipid and fatty acid metabolism. In addition, we found great similarity between the GO terms enriched in associated genes and the ‘tissue-specifically enriched GO terms’ identified using super-enhancer-associated genes found by Hnisz et al. (56). For example, GO terms such as ‘muscle contraction’, ‘muscle system process’ and ‘heart contraction’ are enriched in the associated genes of heart and skeletal muscle in our study and the super-enhancer-associated genes found by Hnisz et al. (56). This observation suggests that our identified associated genes have a strong association with the super-enhancers and possibly other cis-regulatory elements.
We next identified the KEGG cellular pathways enriched in the associated protein-coding genes of different cell states in human (Figure 3D) and mouse (Supplementary Data). The resulting enriched cellular pathways are biologically meaningful. For example, heart-associated genes are highly enriched with pathways related to oxidative phosphorylation and hypertrophic cardiomyopathy. Interestingly, we also found that these genes have obvious enrichment with pathways related to neurodegenerative diseases (e.g. Alzheimer's disease, Parkinson's disease and Huntington's disease). One reason for this phenomenon could be that a number of heart-associated genes encode NADH dehydrogenase subunits and Cytochrome C oxidase subunits, which are involved in Alzheimer's disease (57), Parkinson's disease (58,59) and Huntington's disease (60) based on previous biochemical evidence. This observation is also supported by previous studies, which suggest that heart failure is linked to neurodegenerative diseases, such as Alzheimer's disease (61,62).
Given those identified enriched GO terms and cellular pathways, we further characterized their dynamics during the differentiation process from stem cells to differentiated neural cells. We selected the following tissues and cell types to represent this differentiation progress: (i) ESC/iPSC, (ii) embryonic germ layers (including mesendoderm, ectoderm, mesoderm and endoderm), (iii) neuroectodermal spheres and neural progenitor cells, (iv) differentiated neural tissues (including brain and cerebellum). By investigating the enriched biological functions at each of these cell states, we observed a clear shift in biological processes as the differentiation goes on: cell states closer to the stem cell end, such as the embryonic germ layers, are enriched with basic metabolic processes; on the other hand, fully differentiated neural tissues contain enriched functions of neuronal development and signaling (Supplementary Data), consistent with previous studies (63).
Between-species cell state mapping
Conserved expression patterns revealed by between-species cell state correspondence maps
As the within-species results have shown the effectiveness of our approach in mapping cell states and finding cell state markers, we further extended this approach to comparing cell states between different mammalian species. We evaluated the similarity of two cell states from different species by adopting TROM, which is based on the number of orthologous gene pairs in the associated genes of two cell states, each from a different species (see Materials and Methods for details). We first performed a comprehensive mapping between human and mouse cell states based on their associated protein-coding genes (Figure 4A). Several prominent mapping blocks emerged between corresponding tissues in human and mouse, including neural tissues, heart and skeletal muscle tissues, and testis tissues.
Figure 4.

Cell state correspondence maps between human and other mammalian species. (A) A correspondence map of various cell states between human and mouse by TROM using associated protein-coding genes. Rows correspond to human cell states, and columns correspond to mouse cell states. (B) A correspondence map of various cell states between human and mouse by TROM using associated TFs. Rows correspond to human cell states, and columns correspond to mouse cell states. (C) A correspondence map of various cell states between human and chimpanzee by TROM using associated lncRNAs. Rows correspond to human cell states, and columns correspond to chimpanzee cell states. In (A–C), higher TROM scores (defined as –log10 transformed Bonferroni-corrected P-values from the overlap test) are shown in darker colors. Axis colors represent cell states, and colored boxes mark the prominent mapping patterns.
In addition to these expected mapping patterns, other interesting mappings between tissues and cells of different types are also observed. The two most prominent mapping patterns are (i) human ESCs, iPSCs and cancer cell lines versus mouse ESCs, iPSCs, germ layers and neural progenitor cells and (ii) human liver and kidney tissues versus mouse liver and kidney tissues. Such mapping patterns are consistent with the known physiology of those tissues and cell lines. For (i), ESCs and cultured cancer cell lines are both characterized by rapid cell proliferation, short cell cycles and blocks in differentiation (64,65), which can explain our observed transcriptome similarity between human ESCs, iPSCs and cancer cell lines versus mouse ESCs and iPSCs. For (ii), we observed similarity between human and mouse liver and kidney transcriptomes, which has been reported by previous studies using microarray (66,67), RNA-seq (16,68) and DNase-seq data (69). These mapping patterns show that that orthologous genes are conserved not only at the sequence level but also at the transcription level in terms of gene expression patterns (70,71). They also confirm that the associated protein-coding genes are good cell state indicators.
We next performed a similar human-mouse cell state mapping using only the associated TFs (Figure 4B). Similar to the within-species results, the mapping patterns obtained using TFs are more sparse with smaller mapping blocks than their counterparts observed using protein-coding genes. Different mapping patterns have emerged. For example, almost no mapping is found between human cultured cancer cell lines and mouse ESCs/iPSCs (Figure 4B). This result is also in line with our previous observations in the within-species mapping using TFs, indicating that ESCs/iPSCs are distinct from cancer cell lines in terms of TF expression.
We also compared the cell states of human and those of bonobo and chimpanzee using the associated protein-coding genes, TFs or lncRNAs (Figure 4C and Supplementary Data). Since most lncRNAs are more conserved between closely related species, we restricted our study to primates when performing between-species cell state mapping using the associated lncRNAs. Expectedly, using the associated lncRNAs between primates leads to clear mapping patterns, which are highly similar to the mapping results based on the associated TFs. In those mapping results, tissues of the same type are mapped across primate species.
Conserved cell-state associated protein-coding genes are potentially key regulators
We hypothesized that among the associated protein-coding genes of each cell state, the conserved genes, which were recurrently identified across different species, may represent more important cell-state associated functionality and higher cell-state specificity than the non-conserved ones. In fact, we found that the conserved associated protein-coding genes (Supplementary Data) are significantly enriched with known tissue-specific genes from the TiGER database (72) (Supplementary Data). Note that our cell-state associated genes are not restricted to the specific genes of that cell state but may include the genes that are also associated with other cell states. Cross-species strategy can be used to refine these complex associations to identify the genes with stronger cell-state specificity. In addition, we found that small portions of the TiGER tissue-specific genes were not identified as the associated genes. A possible explanation is that the TiGER tissue-specific genes were identified using fewer tissue types than our study has (Supplementary Data).
Gene expression programs are regulated in large part by TFs through recognizing and binding to specific sequences in the genome. Previous ChIP-seq-based studies revealed that a remarkably small number of key TFs could mainly define tissue-specific gene expression programs (3,56,73–76). We identified conserved associated TFs for seven cell states (Table 1 and Supplementary Data), and reasoned that these TFs should be functionally important based on our above analyses and previous literature. In addition, most of these conserved associated TFs received top specificity ranking in a recent study by Young et al. that defined core TFs for over 200 human cell types and tissues (25) (Supplementary Data). The conserved associated TFs have a large overlap with the core TFs, suggesting that they are putative transcriptional regulatory factors for controlling cell identities.
Table 1. Conserved cell-state associated TFs, RBPs and lncRNAs.
| Cell state | Total # of conserved associated TFs | TFs supported by other evidence | Total # of conserved associated RBPs | RBPs supported by other evidence | Total # of conserved associated lncRNAs | lncRNAs supported by other evidencea |
|---|---|---|---|---|---|---|
| ESC | 33 | APEX1, BLM, CARM1, FOXD3, HCFC1, JARID2, LIN28A, MYBBP1A, MYBL2, MYCN, NANOG, POU5F1, POU5F1B, PRDM14, PRDM5, RARG, SALL4, TERF1, TP53, ZFP42, ZNF281, ZSCAN10 | 22 | EIF5A, LIN28A, PARP1, PTBP1, RPL12, TRIM71, YTHDF2 | 26 | — |
| Brain | 7 | ARNT1, BCL11A, FEZF2, KCTD1, MAP3K10, NEUROD6 | 2 | — | 34 | — |
| Cerebellum | 17 | BARHL1, BARHL2, LHX1, NEUROD1, ST18 | 4 | ADARB1 | 58 | TMEM161B-AS1 (ENSG00000247828, blastnOrangutan.Locus_265345) |
| Heart | 12 | ANKRD1, EBF2, GATA4, HAND2, HEYL, MEF2A, MYOCD, NKX2-5, SMYD1, TBX5, TBX20 | 4 | RBM20, RBM24 | 63 | H19 (ENSG00000130600) |
| Liver | 9 | CREB3L3, FOXA3, LPIN2, MLXIPL, NR1H4, NR1l2, NR1l3 | 3 | ANG, AZGP1 | 48 | — |
| Kidney | 12 | GLIS2, HOXA9, HOXD8, HOXD10, PAX8, SIM1 | 2 | BICC1 | 69 | LINC00853 (ENSG00000224805), CYP4A22-AS1 (ENSG00000225506) |
| Testis | 41 | BRDT, CREM, FHL5, GTF2A1L, MAK, OVOL1, OVOL2, RFX2, SFMBT1, SOX30, TCFL5, YBX2, ZFY | 48 | ADAD1, AKAP1, BOLL, CALR3, CPEB2, DAZL, DDX25, DDX4, DZIP1, MEX3B, NSUN7, NXF3, PIH1D3, PIWIL1, PIWIL2, PLD6, RANBP17, RBM44, RPL10L, RPL39L, TDRD5, YBX2 | 477 | TUSC7 (ENSG00000243197) |
This table summarizes the conserved associated TFs, RBPs and lncRNAs of seven cell states. Conservation definition: human-chimpanzee-bonobo-mouse for TFs and RBPs; human-chimpanzee-bonobo for lncRNAs. Reported tissue-specific biological functions of some TFs, RBPs and lncRNAS are listed as ‘other evidence’ in the 3rd, 5th and 7th columns of this table. The complete list of conserved associated TFs, RBPs and lncRNAs are provided in Supplementary Data, respectively. Reference supports for conserved associated TFs and RBPs are summarized in Supplementary Data, respectively.
aAnnotated in lncRNAdb (http://www.lncrnadb.org).
Most of these conserved associated TFs have been reported essential for tissue development and physiology (see the references of related literatures in Supplementary Data). For example, conserved associated TFs in ESCs include known self-renewal and pluripotency factors such as Oct4 (Pou5f1), Nanog, Sall4, Jarid2 and Myc (64) (Table 1). A recent study found that, compared to the commonly used ‘OSKM factors’ (Oct4, Sox2, Klf4 and Myc), the ‘SNEL factors’ (Sall4, Nanog, Esrrb and Lin28) can generate iPSCs more efficiently (77). Sall4, Nanog and Lin28 were successfully identified as conserved associated TFs in our analysis. Additionally, our conserved associated TFs in heart tissues significantly overlap with known lineage reprogramming factors that directly aid the conversion of fibroblasts to cardiomyocytes in human and mouse (Supplementary Data) (78): out of the six common reprogramming factors (GATA4/Gata4, HAND2/Hand2, MEF2C/Mef2C, MESP1/Mesp1, MYOCD/Myocd and TBX5/Tbx5) for human/mouse cardiomyocytes, we identified GATA4/Gata4, HAND2/Hand2, MEF2C/Mef2C, MYOCD/Myocd and TBX5/Tbx5 as conserved associated TFs. More importantly, some of these conserved associated TFs have not been well studied and could serve as interesting candidates for further functional studies on tissue development, physiology and cell state conversion. For example, it still remains unclear how these conserved testis-associated TFs function in the development and physiology of testis tissues (Supplementary Data).
Recent studies highlight that some RBPs might play key roles in regulating cell homeostasis and differentiation post-transcriptionally (2,79). We identified conserved associated RBPs for seven cell states (Table 1 and Supplementary Data). The biological functions of some of these conserved associated RBPs in regulating tissue development and physiology have been reported (see the references of related literatures in Supplementary Data). For example, we successfully identified PTBP1 and YTHDF2 as the conserved associated RBPs of ESCs; they are well-known RBPs in regulating ESC differentiation and development (80,81). More importantly, we have little knowledge about most of the conserved associated RBPs' molecular functions in controlling cell states, and these RBPs could be putative post-transcriptional regulators. For example, we identified CPSF4, an essential component of the 3΄ end processing machinery (82), as one of the conserved associated RBPs of ESCs. However, it remains to be verified whether CPSF4 is involved in the regulation of self-renewal and pluripotency properties of ESCs.
Inferring lncRNA functions using co-expression modules
Although we have identified more conserved associated lncRNAs than TFs and RBPs (Table 1 and Supplementary Data), fewer lncRNAs have known biological or molecular functions. Hence, our results will provide important new insights into the biological functions of lncRNAs. We exemplified three conserved associated lncRNAs in Figure 5A, accompanied with their relative expression levels across seven cell states. We identified LINC01108 as a conserved associated lncRNA of ESCs and iPSCs, indicating that it is a pluripotency lncRNA (83). This result is supported by a report that knockdown of LINC01108 resulted in human ESC differentiation (83). We also identified CYP4A22-AS1 (also known as ncRNA-a3) as a conserved associated lncRNA of kidney. Although CYP4A22-AS1 was known to function as an enhancer to stimulate TAL1 gene expression in MCF7 cells (84), its regulatory mechanism in kidney remains unclear. Moreover, NTM-IT was discovered as a conserved associated lncRNA of cerebellum, but its molecular functions are largely unknown. Same as the conserved associated TFs, we also expect those conserved associated lncRNAs to be a valuable resource for future functional studies.
Figure 5.

Inferring functions of conserved associated long non-coding RNAs (lncRNAs). (A) Three examples of conserved associated lncRNAs in embryonic stem cells (ESCs) and iPSCs (LINC01108, ENSG00000226673) (top), kidney (CYP4A22-AS1, ENSG00000225506) (middle) and cerebellum (NTM-IT, ENSG00000238262) (bottom). The expression estimates of the three lncRNAs across seven cell states in human, chimpanzee and bonobo are shown. (B) The 19 largest clusters in the co-expression network of protein-coding genes and lncRNAs. Colors of dots distinguish protein-coding genes (orange) and lncRNAs (blue). (C) Enriched GO terms (biological processes) of the protein-coding genes in the 19 largest clusters. Higher enrichment scores (defined as –log10 transformed Bonferroni-corrected P-values) are shown in darker colors. (D) Radar plots illustrate the extents to which the conserved associated lncRNAs of different cell states are enriched in different clusters. The cell states include cerebellum (top left), heart (top right), kidney (bottom left) and testis (bottom right).
Although we have identified the associated cell types for lncRNAs, we still do not clearly know their biological functions, because every cell type has multiple biological functions and we do not know which functions belong to each lncRNA. Hence, we constructed a co-expression network of lncRNAs and protein-coding genes to investigate the enriched biological functions of the lncRNAs. We analyzed a set of 16 354 protein-coding gene families and 13 404 lncRNA families across the three primates (human, chimpanzee and bonobo). We calculated Pearson correlations of expression levels for all gene family pairs and ranked the correlations in each species. Then we tested if the cross-species combination of ranks was significantly (P-value < 0.01) better than expected by chance (see Materials and Methods for details). Finally, the significant gene family pairs formed a network with 15 333 nodes (14 369 protein-coding and 964 lncRNAs) and 1 387 285 edges.
We then inferred the potential biological functions of the 964 lncRNAs based on the functions of their co-expressed protein-coding genes. Using the Markov clustering algorithm (50), we identified 19 clusters of highly inter-connected genes (Figure 5B). The protein-coding genes in these clusters are enriched with tissue-specific functions (Figure 5C), such as sperm chromatin condensation, male gamete generation and spermatogenesis in clusters 3 and 15 (testis), nervous system development and neurogenesis in cluster 8 (neural tissues), cardiac muscle tissue development and muscle system process in cluster 2 (heart and skeletal muscle) and immune response and immune system process in cluster 19 (immune cells and tissues). Another interesting finding is that the conserved associated lncRNAs of various cell states are consistently more enriched in the clusters that have corresponding tissue-specific functions (Figure 5D). For example, the conserved associated lncRNAs in cerebellum are most enriched with the protein-coding genes in cluster 14, whose most enriched biological functions are regulation of neurogenesis and heparin metabolic process. Notably, the clusters with highest lncRNA proportions (clusters 1 and 3) are enriched with spermatogenesis functions, in agreement with the predominant lncRNA specificity in testis (23).
DISCUSSION
A central goal of developmental biology is to understand the molecular mechanisms underlying the differentiation of ESCs or progenitor cells into various terminally differentiated tissues and cell types. Comparison of gene expression profiles from different cell states shed new light on deciphering such mechanisms. Here, we applied a statistical method, TROM, to perform a comprehensive transcriptome mapping for diverse tissues and cell types within and across four mammalian species. We identified associated protein-coding genes as good cell state markers, which capture transcriptome characteristics and lead to reasonable correspondence maps of cell states both with-species and between-species. We also identified associated lncRNAs, which are also found as good cell state markers and even more cell state specific than protein-coding genes. Furthermore, we compared the transcriptomes of various cell states across multiple mammalian species using TROM, and identified conserved associated protein-coding genes and lncRNAs. Finally, we confirmed known and discovered potential biological functions for these conserved associated genes. In addition to uncovering novel insights into mammalian transcriptomes, this study also provides a useful resource of conserved cell-state associated TFs, RBPs and lncRNAs, which characterize transcriptomes of various cell states and enable researchers to explore new hypotheses in developmental biology.
We realize that our current cell state mapping results are limited in two aspects. First, our tissue samples are heterogeneous populations of cells, such as the brain tissues containing multiple types of neurons. Because of that, our mapping results cannot well detect neuronal subtypes, though we partially resolved this issue by using lncRNAs or TFs, which exhibit higher tissue expression specificity than protein-coding genes. In addition, we found that some samples have obvious within-tissue heterogeneity. For example, some chimpanzee liver samples have a stronger mapping to mouse spleen and kidney than to mouse liver tissues. This result is probably due to the fact that liver tissues include fibroblasts, vascular tissues and many other cell types in addition to hepatocytes. We anticipate that this heterogeneity issue could be resolved with the availability of single-cell RNA-seq data. Despite this limitation, our mapping strategy performs well in assessing transcriptome similarities across a wide range of mammalian tissues and cell lines, as demonstrated by our results.
Second, lncRNA expression estimates are not as accurate as the protein-coding gene expression estimates, because lncRNAs have much lower expression levels than those of protein-coding genes. Many previous studies have revealed that the median expression level of lncRNAs is only about a tenth of that of mRNAs (36,38,85–89). This makes accurate estimation of lncRNA expression a generally difficult task. Our curated RNA-seq data sets were generated over a span of more than five years, resulting in a great variety in their sequencing depths and read lengths. Notably, the data we used from early studies are short read RNA-seq (<50 base pair single-end reads) of moderate sequencing depths (∼10–20 million reads). Advances in RNA-seq library preparation and sequencing technologies now enable the generation of hundreds of millions of paired-end reads with longer than 100 base pairs in length. Greater sequencing depths and increased read lengths allow more accurate abundance assessment of lowly expression genes (90), and thus could improve our cell state mapping results. In addition, we used polyadenylated RNA-seq data sets in our analysis. Please note that compared with mRNAs, lncRNAs are significantly enriched in non-polyadenylated RNA-seq samples (88). Since our study is based on polyadenylated RNA-seq data sets, we did not include the non-polyadenylated lncRNAs in our expression analyses. However, given that most transcribed transcripts, including both mRNAs and lncRNAs, are poly(A)+ or bimorphic (91), we still expect our lncRNA results to be biologically meaningful.
Past years have seen great progress in studying cell-state transitions. For example, fibroblasts (92) and neural progenitor cells (93) have been reprogrammed into pluripotent stem cells, which can again differentiate into specific lineages under defined growth conditions and/or through gene expression perturbation (94). Some previous studies revealed that knocking down and/or overexpression of key regulatory factors in the initial cell state could aid successful cell state conversion (7,95–96). However, most of those studies only focused on transcription factors. Our study for the first time provides the repertoires of various cell states’ associated protein-coding and non-coding genes identified based on RNA-seq technologies. We anticipate that these genes are valuable candidates for further functional studies to improve the efficiency and fidelity of cell-state conversion and reprogramming (78).
In addition to the increasingly accumulation of RNA-seq data, a large amount of microarray data sets have been produced and deposited during the past decade. To this end, our cell state mapping approach allows us to integrate RNA-seq and microarray data for studying more cell states in more species. Our method can also be extended to incorporate additional epigenomic data types, such as DNA methylation, histone modification, DNA accessibility and genome-wide transcription factor binding data. Some recent studies demonstrated that cell and tissue lineages could be well clustered based on their DNA accessibility and histone modification patterns (69,97–98). Ongoing efforts to further the knowledge of cell-state associated regulatory genes will greatly advance our understanding of ESC state maintenance, as well as our capability in cell reprogramming and directed differentiation.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank other members from Lu Lab and Li Lab for their comments and suggestions. Especially, the authors would like to thank Dr. Mark D. Biggin at Lawrence Berkeley National Laboratory for his insightful comments and detailed edits on this work.
Authors’ contributions: J.J.L. and Z.J.L. conceived and designed the project; Y.Y. and Y.T.Y. performed all the analyses; J.Y. helped in the co-expression network analysis; Y.Y. and Y.T.Y. prepared the figures and tables; J.J.L., Z.J.L., Y.T.Y. and Y.Y. wrote the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
To Y.Y., Y.T.Y., J.Y. and Z.J.L.:National Key Research and Development Plan of China [2016YFA0500803]; National High-Tech Research and Development Program of China [2014AA021103]; National Natural Science Foundation of China [31522030, 31271402]; Tsinghua University Initiative Scientific Research Program [2014z21045]; Computing Platform of National Protein Facilities (Tsinghua University). To J.J.L.: Department of Statistics at UCLA; Hellman Fellowship from the Hellman Foundation; National Science Foundation [DMS 1557727 and DMS 1613338]; NIH/NIGMS [R01GM120507]. Funding for open access charge: NIH /NIGMS [R01 GM120507].
Conflict of interest statement. None declared.
REFERENCES
- 1. Furusawa C., Kaneko K.. A dynamical-systems view of stem cell biology. Science. 2012; 338:215–217. [DOI] [PubMed] [Google Scholar]
- 2. Ye J., Blelloch R.. Regulation of pluripotency by RNA binding proteins. Cell Stem Cell. 2014; 15:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Young R.A. Control of the embryonic stem cell state. Cell. 2011; 144:940–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Spitz F., Furlong E.E.. Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 2012; 13:613–626. [DOI] [PubMed] [Google Scholar]
- 5. Chen T., Dent S.Y.. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat. Rev. Genet. 2014; 15:93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liang G., Zhang Y.. Genetic and epigenetic variations in iPSCs: potential causes and implications for application. Cell Stem Cell. 2013; 13:149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Han H., Irimia M., Ross P.J., Sung H.K., Alipanahi B., David L., Golipour A., Gabut M., Michael I.P., Nachman E.N. et al. MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature. 2013; 498:241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mallinjoud P., Villemin J.P., Mortada H., Polay Espinoza M., Desmet F.O., Samaan S., Chautard E., Tranchevent L.C., Auboeuf D.. Endothelial, epithelial, and fibroblast cells exhibit specific splicing programs independently of their tissue of origin. Genome Res. 2014; 24:511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shenoy A., Blelloch R.H.. Regulation of microRNA function in somatic stem cell proliferation and differentiation. Nat. Rev. Mol. Cell Biol. 2014; 15:565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun K., Lai E.C.. Adult-specific functions of animal microRNAs. Nat. Rev. Genet. 2013; 14:535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fatica A., Bozzoni I.. Long non-coding RNAs: new players in cell differentiation and development. Nat. Rev. Genet. 2014; 15:7–21. [DOI] [PubMed] [Google Scholar]
- 12. Flynn R.A., Chang H.Y.. Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell. 2014; 14:752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de la Pompa J.L., Wakeham A., Correia K.M., Samper E., Brown S., Aguilera R.J., Nakano T., Honjo T., Mak T.W., Rossant J. et al. Conservation of the Notch signalling pathway in mammalian neurogenesis. Development. 1997; 124:1139–1148. [DOI] [PubMed] [Google Scholar]
- 14. Farh K.K., Grimson A., Jan C., Lewis B.P., Johnston W.K., Lim L.P., Burge C.B., Bartel D.P.. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005; 310:1817–1821. [DOI] [PubMed] [Google Scholar]
- 15. Barbosa-Morais N.L., Irimia M., Pan Q., Xiong H.Y., Gueroussov S., Lee L.J., Slobodeniuc V., Kutter C., Watt S., Colak R. et al. The evolutionary landscape of alternative splicing in vertebrate species. Science. 2012; 338:1587–1593. [DOI] [PubMed] [Google Scholar]
- 16. Brawand D., Soumillon M., Necsulea A., Julien P., Csardi G., Harrigan P., Weier M., Liechti A., Aximu-Petri A., Kircher M. et al. The evolution of gene expression levels in mammalian organs. Nature. 2011; 478:343–348. [DOI] [PubMed] [Google Scholar]
- 17. Gilad Y., Mizrahi-Man O.. A reanalysis of mouse ENCODE comparative gene expression data. F1000Research. 2015; 4:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin S., Lin Y., Nery J.R., Urich M.A., Breschi A., Davis C.A., Dobin A., Zaleski C., Beer M.A., Chapman W.C. et al. Comparison of the transcriptional landscapes between human and mouse tissues. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:17224–17229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Breschi A., Djebali S., Gillis J., Pervouchine D.D., Dobin A., Davis C.A., Gingeras T.R., Guigo R.. Gene-specific patterns of expression variation across organs and species. Genome Biol. 2016; 17:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mattick J.S. Non-coding RNAs: the architects of eukaryotic complexity. EMBO Rep. 2001; 2:986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Necsulea A., Kaessmann H.. Evolutionary dynamics of coding and non-coding transcriptomes. Nat. Rev. Genet. 2014; 15:734–748. [DOI] [PubMed] [Google Scholar]
- 22. Washietl S., Kellis M., Garber M.. Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals. Genome Res. 2014; 24:616–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Necsulea A., Soumillon M., Warnefors M., Liechti A., Daish T., Zeller U., Baker J.C., Grutzner F., Kaessmann H.. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014; 505:635–640. [DOI] [PubMed] [Google Scholar]
- 24. Cahan P., Li H., Morris S.A., Lummertz da Rocha E., Daley G.Q., Collins J.J.. CellNet: network biology applied to stem cell engineering. Cell. 2014; 158:903–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. D'Alessio A.C., Fan Z.P., Wert K.J., Baranov P., Cohen M.A., Saini J.S., Cohick E., Charniga C., Dadon D., Hannett N.M. et al. A systematic approach to identify candidate transcription factors that control cell identity. Stem Cell Reports. 2015; 5:763–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rackham O.J., Firas J., Fang H., Oates M.E., Holmes M.L., Knaupp A.S., Consortium F., Suzuki H., Nefzger C.M., Daub C.O. et al. A predictive computational framework for direct reprogramming between human cell types. Nat. Genet. 2016; 48:331–335. [DOI] [PubMed] [Google Scholar]
- 27. Emani M.R., Narva E., Stubb A., Chakroborty D., Viitala M., Rokka A., Rahkonen N., Moulder R., Denessiouk K., Trokovic R. et al. The L1TD1 protein interactome reveals the importance of post-transcriptional regulation in human pluripotency. Stem Cell Reports. 2015; 4:519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li J.J., Huang H., Bickel P.J., Brenner S.E.. Comparison of D. melanogaster and C. elegans developmental stages, tissues, and cells by modENCODE RNA-seq data. Genome Res. 2014; 24:1086–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schmieder R., Edwards R.. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011; 27:863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L.. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012; 7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trapnell C., Pachter L., Salzberg S.L.. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Prufer K., Munch K., Hellmann I., Akagi K., Miller J.R., Walenz B., Koren S., Sutton G., Kodira C., Winer R. et al. The bonobo genome compared with the chimpanzee and human genomes. Nature. 2012; 486:527–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Flicek P., Ahmed I., Amode M.R., Barrell D., Beal K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fairley S. et al. Ensembl 2013. Nucleic Acids Res. 2013; 41:D48–D55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L.. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010; 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cabili M.N., Trapnell C., Goff L., Koziol M., Tazon-Vega B., Regev A., Rinn J.L.. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011; 25:1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dinger M.E., Amaral P.P., Mercer T.R., Pang K.C., Bruce S.J., Gardiner B.B., Askarian-Amiri M.E., Ru K., Solda G., Simons C. et al. Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res. 2008; 18:1433–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guttman M., Amit I., Garber M., French C., Lin M.F., Feldser D., Huarte M., Zuk O., Carey B.W., Cassady J.P. et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009; 458:223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakaya H.I., Amaral P.P., Louro R., Lopes A., Fachel A.A., Moreira Y.B., El-Jundi T.A., da Silva A.M., Reis E.M., Verjovski-Almeida S.. Genome mapping and expression analyses of human intronic noncoding RNAs reveal tissue-specific patterns and enrichment in genes related to regulation of transcription. Genome Biol. 2007; 8:R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schreiber F., Patricio M., Muffato M., Pignatelli M., Bateman A.. TreeFam v9: a new website, more species and orthology-on-the-fly. Nucleic Acids Res. 2014; 42:D922–D925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang H.M., Liu T., Liu C.J., Song S., Zhang X., Liu W., Jia H., Xue Y., Guo A.Y.. AnimalTFDB 2.0: a resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res. 2014; 43:D76–D81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gerstberger S., Hafner M., Tuschl T.. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014; 15:829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Amaratunga D., Cabrera J.. Analysis of data from viral DNA microchips. J. Am. Stat. Assoc. 2001; 96:1161–1170. [Google Scholar]
- 44. Li W.V., Chen Y., Li J.J.. TROM: A Testing-Based Method for Finding Transcriptomic Similarity of Biological Samples. Stat. Biosci. 2016; doi:10.1007/s12561-016-9163-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. R package. TROM: Transcriptome Overlap Measure. 2016; https://cran.r-project.org/web/packages/TROM/index.html.
- 46. Alexa A., Rahnenfuhrer J., Lengauer T.. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006; 22:1600–1607. [DOI] [PubMed] [Google Scholar]
- 47. Tenenbaum D. KEGGREST: Client-side REST access to KEGG. R Package Version 1.6.4.
- 48. Stuart J.M., Segal E., Koller D., Kim S.K.. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003; 302:249–255. [DOI] [PubMed] [Google Scholar]
- 49. Smoot M.E., Ono K., Ruscheinski J., Wang P.L., Ideker T.. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011; 27:431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. van Dongen S., Abreu-Goodger C.. Using MCL to extract clusters from networks. Methods Mol. Biol. 2012; 804:281–295. [DOI] [PubMed] [Google Scholar]
- 51. Vaquerizas J.M., Kummerfeld S.K., Teichmann S.A., Luscombe N.M.. A census of human transcription factors: function, expression and evolution. Nat. Rev. Genetics. 2009; 10:252–263. [DOI] [PubMed] [Google Scholar]
- 52. Fukuchi Y., Nakajima H., Sugiyama D., Hirose I., Kitamura T., Tsuji K.. Human placenta-derived cells have mesenchymal stem/progenitor cell potential. Stem Cells. 2004; 22:649–658. [DOI] [PubMed] [Google Scholar]
- 53. Barber R.D., Harmer D.W., Coleman R.A., Clark B.J.. GAPDH as a housekeeping gene: analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiol. Genomics. 2005; 21:389–395. [DOI] [PubMed] [Google Scholar]
- 54. Greer S., Honeywell R., Geletu M., Arulanandam R., Raptis L.. Housekeeping genes; expression levels may change with density of cultured cells. J. Immunol. Methods. 2010; 355:76–79. [DOI] [PubMed] [Google Scholar]
- 55. Eisenberg E., Levanon E.Y.. Human housekeeping genes, revisited. Trends Genet. 2013; 29:569–574. [DOI] [PubMed] [Google Scholar]
- 56. Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-Andre V., Sigova A.A., Hoke H.A., Young R.A.. Super-enhancers in the control of cell identity and disease. Cell. 2013; 155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aksenov M.Y., Tucker H.M., Nair P., Aksenova M.V., Butterfield D.A., Estus S., Markesbery W.R.. The expression of several mitochondrial and nuclear genes encoding the subunits of electron transport chain enzyme complexes, cytochrome c oxidase, and NADH dehydrogenase, in different brain regions in Alzheimer's disease. Neurochem. Res. 1999; 24:767–774. [DOI] [PubMed] [Google Scholar]
- 58. Varghese M., Pandey M., Samanta A., Gangopadhyay P.K., Mohanakumar K.P.. Reduced NADH coenzyme Q dehydrogenase activity in platelets of Parkinson's disease, but not Parkinson plus patients, from an Indian population. J. Neurol. Sci. 2009; 279:39–42. [DOI] [PubMed] [Google Scholar]
- 59. Mizuno Y., Ohta S., Tanaka M., Takamiya S., Suzuki K., Sato T., Oya H., Ozawa T., Kagawa Y.. Deficiencies in complex I subunits of the respiratory chain in Parkinson's disease. Biochem. Biophys. Res. Commun. 1989; 163:1450–1455. [DOI] [PubMed] [Google Scholar]
- 60. Arenas J., Campos Y., Ribacoba R., Martin M.A., Rubio J.C., Ablanedo P., Cabello A.. Complex I defect in muscle from patients with Huntington's disease. Ann. Neurol. 1998; 43:397–400. [DOI] [PubMed] [Google Scholar]
- 61. Willis M.S., Patterson C.. Proteotoxicity and cardiac dysfunction–Alzheimer's disease of the heart. N. Engl. J. Med. 2013; 368:455–464. [DOI] [PubMed] [Google Scholar]
- 62. Ramanan V.K., Saykin A.J.. Pathways to neurodegeneration: mechanistic insights from GWAS in Alzheimer's disease, Parkinson's disease, and related disorders. Am. J. Neurodegenerative Dis. 2013; 2:145–175. [PMC free article] [PubMed] [Google Scholar]
- 63. Wu J.Q., Habegger L., Noisa P., Szekely A., Qiu C., Hutchison S., Raha D., Egholm M., Lin H., Weissman S. et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:5254–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kim J., Orkin S.H.. Embryonic stem cell-specific signatures in cancer: insights into genomic regulatory networks and implications for medicine. Genome Med. 2011; 3:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Reya T., Morrison S.J., Clarke M.F., Weissman I.L.. Stem cells, cancer, and cancer stem cells. Nature. 2001; 414:105–111. [DOI] [PubMed] [Google Scholar]
- 66. Chan E.T., Quon G.T., Chua G., Babak T., Trochesset M., Zirngibl R.A., Aubin J., Ratcliffe M.J., Wilde A., Brudno M. et al. Conservation of core gene expression in vertebrate tissues. J. Biol. 2009; 8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xing Y., Ouyang Z., Kapur K., Scott M.P., Wong W.H.. Assessing the conservation of mammalian gene expression using high-density exon arrays. Mol. Biol. Evol. 2007; 24:1283–1285. [DOI] [PubMed] [Google Scholar]
- 68. Merkin J., Russell C., Chen P., Burge C.B.. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science. 2012; 338:1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Vierstra J., Rynes E., Sandstrom R., Zhang M., Canfield T., Hansen R.S., Stehling-Sun S., Sabo P.J., Byron R., Humbert R. et al. Mouse regulatory DNA landscapes reveal global principles of cis-regulatory evolution. Science. 2014; 346:1007–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zheng-Bradley X., Rung J., Parkinson H., Brazma A.. Large scale comparison of global gene expression patterns in human and mouse. Genome Biol. 2010; 11:R124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Su A.I., Cooke M.P., Ching K.A., Hakak Y., Walker J.R., Wiltshire T., Orth A.P., Vega R.G., Sapinoso L.M., Moqrich A. et al. Large-scale analysis of the human and mouse transcriptomes. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:4465–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu X., Yu X., Zack D.J., Zhu H., Qian J.. TiGER: a database for tissue-specific gene expression and regulation. BMC Bioinformatics. 2008; 9:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A.. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013; 153:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Buganim Y., Faddah D.A., Jaenisch R.. Mechanisms and models of somatic cell reprogramming. Nat. Rev. Genet. 2013; 14:427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Morris S.A., Daley G.Q.. A blueprint for engineering cell fate: current technologies to reprogram cell identity. Cell Res. 2013; 23:33–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yamanaka S. Induced pluripotent stem cells: past, present, and future. Cell Stem Cell. 2012; 10:678–684. [DOI] [PubMed] [Google Scholar]
- 77. Buganim Y., Markoulaki S., van Wietmarschen N., Hoke H., Wu T., Ganz K., Akhtar-Zaidi B., He Y., Abraham B.J., Porubsky D. et al. The developmental potential of iPSCs is greatly influenced by reprogramming factor selection. Cell Stem Cell. 2014; 15:295–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Xu J., Du Y., Deng H.. Direct lineage reprogramming: strategies, mechanisms, and applications. Cell Stem Cell. 2015; 16:119–134. [DOI] [PubMed] [Google Scholar]
- 79. Jangi M., Sharp P.A.. Building robust transcriptomes with master splicing factors. Cell. 2014; 159:487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Shibayama M., Ohno S., Osaka T., Sakamoto R., Tokunaga A., Nakatake Y., Sato M., Yoshida N.. Polypyrimidine tract-binding protein is essential for early mouse development and embryonic stem cell proliferation. FEBS J. 2009; 276:6658–6668. [DOI] [PubMed] [Google Scholar]
- 81. Wang Y., Li Y., Toth J.I., Petroski M.D., Zhang Z., Zhao J.C.. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014; 16:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Martin G., Gruber A.R., Keller W., Zavolan M.. Genome-wide analysis of pre-mRNA 3΄ end processing reveals a decisive role of human cleavage factor I in the regulation of 3΄ UTR length. Cell Rep. 2012; 1:753–763. [DOI] [PubMed] [Google Scholar]
- 83. Ng S.Y., Johnson R., Stanton L.W.. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 2012; 31:522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Orom U.A., Derrien T., Beringer M., Gumireddy K., Gardini A., Bussotti G., Lai F., Zytnicki M., Notredame C., Huang Q. et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010; 143:46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pauli A., Valen E., Lin M.F., Garber M., Vastenhouw N.L., Levin J.Z., Fan L., Sandelin A., Rinn J.L., Regev A. et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012; 22:577–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sigova A.A., Mullen A.C., Molinie B., Gupta S., Orlando D.A., Guenther M.G., Almada A.E., Lin C., Sharp P.A., Giallourakis C.C. et al. Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc. Natl. Acad. Sc. U.S.A. 2013; 110:2876–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ulitsky I., Shkumatava A., Jan C.H., Sive H., Bartel D.P.. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011; 147:1537–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., Guernec G., Martin D., Merkel A., Knowles D.G. et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012; 22:1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Guttman M., Garber M., Levin J.Z., Donaghey J., Robinson J., Adiconis X., Fan L., Koziol M.J., Gnirke A., Nusbaum C. et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010; 28:503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Toung J.M., Morley M., Li M., Cheung V.G.. RNA-sequence analysis of human B-cells. Genome Res. 2011; 21:991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yang L., Duff M.O., Graveley B.R., Carmichael G.G., Chen L.L.. Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 2011; 12:R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Takahashi K., Yamanaka S.. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126:663–676. [DOI] [PubMed] [Google Scholar]
- 93. Kim J.B., Sebastiano V., Wu G., Arauzo-Bravo M.J., Sasse P., Gentile L., Ko K., Ruau D., Ehrich M., van den Boom D. et al. Oct4-induced pluripotency in adult neural stem cells. Cell. 2009; 136:411–419. [DOI] [PubMed] [Google Scholar]
- 94. Murry C.E., Keller G.. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008; 132:661–680. [DOI] [PubMed] [Google Scholar]
- 95. Morris S.A., Cahan P., Li H., Zhao A.M., San Roman A.K., Shivdasani R.A., Collins J.J., Daley G.Q.. Dissecting engineered cell types and enhancing cell fate conversion via CellNet. Cell. 2014; 158:889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Rackham O.J., Firas J., Fang H., Oates M.E., Holmes M.L., Knaupp A.S., Consortium F., Suzuki H., Nefzger C.M., Daub C.O. et al. A predictive computational framework for direct reprogramming between human cell types. Nat. Genet. 2016; 48:331–335. [DOI] [PubMed] [Google Scholar]
- 97. Amin V., Harris R.A., Onuchic V., Jackson A.R., Charnecki T., Paithankar S., Lakshmi Subramanian S., Riehle K., Coarfa C., Milosavljevic A.. Epigenomic footprints across 111 reference epigenomes reveal tissue-specific epigenetic regulation of lincRNAs. Nat. Commun. 2015; 6:6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Li W.V., Razaee Z.S., Li J.J.. Epigenome overlap measure (EPOM) for comparing tissue/cell types based on chromatin states. BMC Genomics. 2016; 17:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.