


Abstract
The evolution in next-generation sequencing (NGS) technology has led to the development of many different assembly algorithms, but few of them focus on assembling the organelle genomes. These genomes are used in phylogenetic studies, food identification and are the most deposited eukaryotic genomes in GenBank. Producing organelle genome assembly from whole genome sequencing (WGS) data would be the most accurate and least laborious approach, but a tool specifically designed for this task is lacking. We developed a seed-and-extend algorithm that assembles organelle genomes from whole genome sequencing (WGS) data, starting from a related or distant single seed sequence. The algorithm has been tested on several new (Gonioctena intermedia and Avicennia marina) and public (Arabidopsis thaliana and Oryza sativa) whole genome Illumina data sets where it outperforms known assemblers in assembly accuracy and coverage. In our benchmark, NOVOPlasty assembled all tested circular genomes in less than 30 min with a maximum memory requirement of 16 GB and an accuracy over 99.99%. In conclusion, NOVOPlasty is the sole de novo assembler that provides a fast and straightforward extraction of the extranuclear genomes from WGS data in one circular high quality contig. The software is open source and can be downloaded at https://github.com/ndierckx/NOVOPlasty.
INTRODUCTION
The circular genomes of chloroplasts and mitochondria are frequently targeted for de novo assembly. Both genomes are usually maternally inherited, have a conserved gene organization and are often used in phylogenetic and phylogeographic studies, or as a barcode in plant and food identification (1). Different in vitro strategies to isolate these genomes from the much larger nuclear chromosomes have been developed, but this task has proven to be particularly challenging. Before the development of NGS technology, organelle genome assembly was based on conventional primer walking strategies, using long range PCR and cloning of PCR products, which are laborious and costly (2–4). NGS made it possible to develop novel strategies to construct the entire chloroplast or mitochondrial genome, thereby dramatically reducing time and costs compared to the more conventional methods. It is now affordable to obtain whole genome data in a short timespan by using genomic DNA extracted from whole cells (5). Besides nuclear sequences, a high copy number of extranuclear sequences will be present in the sample, usually around 5 to 10% of chloroplast DNA (6) and around 1–2% of mitochondrial DNA (7), allowing to assemble both nuclear and extranuclear genomes from one simple experiment. Shallow sequencing of genomic DNA will result in comparatively deep sequencing of the high-copy fraction of the genome; this approach is called genome skimming. Although assembling the complete data set will generate contigs for the organelle genomes, it is also possible to first isolate the chloroplast or mitochondrial reads, and then assemble this subset. The best strategy depends on the data set, computational power and reference genome availability.
When a reference genome sequence is available from a closely related organism, a genome sequence can easily be assembled by mapping the reads to the reference (5). However, when the reference is too distant, the assembly will contain numerous mismatches. Reference assemblies generally require less computational time and virtual memory, but can only handle a limited amount of variances between the targeted and reference genome to stay accurate. In many cases, a de novo assembly is the preferred strategy for an accurate assembly.
When sequence reads are obtained from a total DNA extract, there will be a large excess of reads from the nuclear genome. To reduce the runtime and computational resources needed for the assembly of the several order of magnitudes smaller organelle genomes, it is suggested to work with a relatively low total number of reads (6). The copy number of organelle genomes being much higher than the copy number of the nuclear genome, working with a whole genome data set of low coverage is largely sufficient (8). One strategy often used to reduce the ratio of nuclear to organelle reads prior to the assembly consists in filtering the extra-nuclear sequences, either by keeping only regions of higher coverage or by mapping reads to a reference genome. Filtering by differential coverage will often result in the undesirable exclusion of regions of low or high GC content (see Figure 1), as many NGS systems will perform less efficiently in these regions (7). Another option is to isolate plastid or mitochondrial DNA, prior to sequencing, by capturing these molecules using specific probes. However, many specific probes need to be designed to cover the complete organelle genome, such that this approach is only recommended when many samples must be sequenced in parallel.
Figure 1.

Coverage depth for a 12 000 bp long region of the mitochondrial genome of Gonioctena intermedia. There are several regions with a low GC content, resulting in a reduced read coverage.
With recent technological advances and cost reduction of shotgun sequencing, the most reliable and straightforward method to a complete assembly of an extranuclear genome would be to sequence a whole genome extract and utilize the complete data set for the assembly, a bioinformatic procedure that is not always straightforward with the tools currently available. Here, we present a novel algorithm, NOVOPlasty, specifically developed for the de novo assembly of mitochondrial and chloroplast genomes from whole genome data. We compared its performance with available software commonly used for organelle genome assembly, through the benchmarked assembly of new and reference mitochondrial and chloroplast genomes from multiple organisms.
MATERIALS AND METHODS
Sequencing
All in-house non-human samples were sequenced on the Illumina HiSeq platform (101 bp or 126 bp paired-end reads). The human mitochondria samples (PCR-free) were sequenced on the Illumina HiSeqX platform (150 bp paired-end reads).
Two public data sets of Arabidopsis thaliana and of Oryza sativa were downloaded from the European Nucleotide Archive (http://www.ebi.ac.uk). Data sets SRR1174256 (A. thaliana), SRR1810277 (A. thaliana) and ERR477442 (O. sativa) were sequenced on the Illumina HiSeq 2000 platform and consists out of paired end reads with a read length of respectively 90 bp, 101 bp and 96 bp. Data sets DRX021298 (A. thaliana) and SRR1328237 (O. sativa) were sequenced on the Illumina HiSeq 2500 platform and consisted of paired end reads with a read length of respectively 150 bp and 151 bp. A total of 20% of data sets SRR1174256 and SRR1810277, and 8% of SRR1328237 were sub-sampled for the benchmarking study.
De novo assembly
All assemblies were executed on a Intel Xeon CPU machine containing 24 cores of 2.93 GHz and a total of 96.8 GB of RAM. Our program NOVOPlasty is written in Perl. In addition, four open-source assemblers (MITObim (9), MIRA (10), ARC (https://github.com/ibest/ARC) and SOAPdenvo2 (11)) and the pay-for-use CLC assembler (CLCbio, Aarhus, Denmark) were used on the same data, for comparison.
Quality assessment
To obtain a reliable quality assessment, we chose to benchmark the different tools with well known model organisms. A human data set was used for a mitochondrial assembly and Arabidopsis thaliana and Oryza sativa for the chloroplast assembly. The latter two were executed in duplicate, their accession numbers in the European Nucleotide Archive are listed in the ‘Sequencing’ section. In addition to these data sets, one unknown mitochondrial genome (Gonioctena intermedia) and one unknown chloroplast genome (Avicennia marina) were included in the comparison. The mitochondrial genome of G. intermedia contains a highly repetitive section, which was useful to assess the performance on long repetitive regions. In this case a reference genome was obtained by assembling long PacBio (Pacific Biosciences) reads together with short Illumina reads using the MIRA assembler. Some of the PacBio long reads cover the complete mitochondrial genome, which made it possible to assemble a reliable reference genome for the benchmark study. The NOVOPlasty assembly of the mitochondrial genome of G. intermedia was submitted to GenBank (KX922881). The other reference genomes were retrieved from the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov). GenBank entry AP000423.1 was selected for the A. thaliana assemblies, KM103369.1 for data set SRR1328237 of O. sativa, KM088022.1 for data set ERR477442 of O. sativa and X93334.1 for H. sapiens. Even though these references are very accurate, some variances between individual samples is expected. To detect these putative variances between our data set and the used reference, we realigned all reads to each assembly with bowtie2 (12) for visual inspection with Tablet (13). Each data set contained a small number of single nucleotide polymorphisms (SNPs), which were corrected in the respective reference genomes to acquire a perfect reference for our benchmark study. Visual proof of the SNPs justifies these corrections and can be examined in the Supplementary Data.
The different data sets were used for a benchmarking study comparing six assemblers, namely, MIRA, MITObim, SOAPdenovo2, CLC, NOVOPlasty and ARC. ARC was only used for the mitochondrial assemblies since it still relies on reference genomes and when a very close reference was lacking, the assemblies resulted in a lower genome coverage than with the de novo approach. All tested assemblers were evaluated for speed, memory efficiency, disk usage, genome coverage, assembly accuracy and number of contigs. Comparing speed and system requirements was straightforward, since each assembler ran on the same machine and made use of the same input data set. The quality indicators were measured relative to the corresponding reference as mentioned above. The genome coverage represents the percentage of the reference genome that was assembled minus ambiguous nucleotides. The accuracy represents the percentage of correctly assembled nucleotides relative to the ‘perfect’ validated alignments. The highest possible score (100%) for speed, memory efficiency, disk space, genome coverage, assembly accuracy and number of contigs were set to respectively 0 min, O GB of RAM, 0 GB, 100%, 100% and 1 contig. The lowest score (0%) was always chosen close to the average of the assembler that performed the worst to get a clear difference between the assemblers. All percentages were rounded off to two decimal digits. The absolute values for each assembly can be examined in the Supplementary Data.
NOVOPlasty
NOVOPlasty is a seed-extend based assembler similar to string overlap algorithms like SSAKE (14) and VCAKE (15). It starts with storing the sequences into a hash table, which allows quick accessibility of the reads (Figure 2). The assembly has to be initiated by a seed, which is iteratively extended bidirectionally. This seed sequence is not used for initiating the assembly, but to retrieve one sequence read of the targeted genome from the NGS data set. This strategy can handle a wider range of seed inputs without incorporating mismatches into the assembly. The seed sequence can be one sequence read, a conserved gene or even a complete organelle genome from a distant species. The end and start of the seed are scanned for overlapping reads in the hash table and stored separately. All putative extensions are identified and subsequently cross-checked with the paired reads to verify if they are positioned correctly. Relatively similar sequences are grouped together and every base extension is resolved by a consensus between the overlapping reads. When there is more than one possible consensus extension (i.e. more than one group of sufficient size), the assembly splits and two new contigs will be created. Unlike most assemblers, NOVOPlasty does not try to assemble every read, but will extend the given seed until the circular genome is formed. The assembly will circularize when the length is in the expected range and both ends overlap by at least 200 bp. When a repetitive region is detected, the circularization will be postponed until the assembly exits the repetitive region. Since whole genome data usually contain a high coverage of extranuclear sequences, the algorithm is capable of extending one read into a complete circular genome (Figure 2).
Figure 2.

Work flow of NOVOPlasty. For simplicity the work flow was limited to unidirectional extension. (A) All reads are stored in a hash table with a unique id. A second hash table contains the ids for the read start = k-mer parameter (default = 38) of the corresponding read. (B) Scope of search 1 is the region where a match of the ‘read start’ indicates a extension of the sequence. All these matching reads are stored separately. (C) The position of the paired reads are verified by aligning each paired read to a previous assembled area, which is determined by the library insert size (scope of search 2). (D) A consensus sequence of the different extensions is determined.
Besides a new linear approach to assembling, NOVOPlasty achieves higher quality assemblies by incorporating case based adjustments. Problematic regions caused by sequencing errors or inclusion of genomic elements are automatically detected and will be resolved as best as possible by tuning the parameters and by initiating appropriate strategies. Current Illumina sequencing technology (Illumina HiSeq and MiSeq) has for example a very high error rate after a long single nucleotide repeat (SNR) stretch, which makes the subsequent sequence unreliable (16). Those error-prone regions may thus cause interruptions in contig assembly, which are particularly problematic for our linear assembly strategy. An essential strategy of NOVOPlasty to ensure continuity lies in the early detection of these SNR stretches and the disposal of the most erroneous reads prior to building the consensus. Defining the exact length of the SNR stretch is not straightforward due to a strong variance of SNR length between the individual reads and an overall low quality score. The most frequent recurrent length will be selected as the correct one, as this option is observed as the most accurate. When the consensus cannot be resolved, NOVOPlasty ‘jumps’ over the problematic region using paired end information and restarts the assembly from there. Because the SNR region will be approached from both directions, it is possible to avoid the highly erroneous parts of the reads. One of the other major issues of short read assembly is complex repetitive sequences. Mitochondrial genomes of beetles often contain a long highly repetitive section (17), while chloroplast genomes can contain shorter dispersed repetitive DNA (18). When NOVOPlasty encounters a repetitive region, it determines the repeated sequence and the region adjacent upstream. All reads that start with the sequence prior to the repetitive DNA are filtered out for further analysis. Should the length of the repetitive region be less than the length of the reads, the assembly of the region can be resolved directly. Otherwise, the algorithm will temporary tune the parameters very stringently to search for small variations between the repetitive sequences that could serve as a reference point. When the region can not be resolved, the assembly will be terminated and reinitiated as a new contig at the sequence following the repetitive region.
RESULTS
Chloroplast assembly
Seventeen unpublished chloroplast genomes and four public data sets were successfully assembled with NOVOPlasty. Since chloroplast genomes contain a large inverted repeat, two versions of the assembly are generated, that vary only in the orientation of the region between the two repeats. The correct orientation needs to be resolved manually. One data set of Avicennia marina and two of A. thaliana and O. sativa each were used for a benchmarking study between different assemblers (Table 1 and Supplementary Data). As for the chloroplasts of A. thaliana and O. sativa, only NOVOPlasty and CLC were successful in assembling the complete genome. Since all of the other tested tools were unsuccessful in producing a comparable amount of genome coverage, the discussion will be restricted to the assemblies of those two algorithms. NOVOPlasty assembled entirely the five genomes in one circular contig each with a coverage of 100% (Figure 3). The assembly of SRR1174256 contained one deletion of 1 bp against the reference from GenBank, located in a long SNR. This is likely caused by systematic errors inherent to Illumina technology. The assemblies from the other three public data sets did not contain any mismatch. CLC generally assembles a chloroplast genome in three contigs, which represent the long single copy section, the short single copy section and the inverted repeat (Figure 3). However, each of these contigs can in fact be a scaffold of several contigs merged by strings of ambiguous nucleotides (‘N’). These ambiguous stretches can represent gaps of unknown length, or can be subsequently resolved manually if both ends overlap each other. The CLC assembly of SRR1174256 comprises 3 scaffolds (8 contigs), including a deletion of 31 bp and 4 mismatches. Besides a few short fragments previously sequenced using classic Sanger technology, there was no reference genome available for A. marina. Since NOVOPlasty achieved the highest coverage and quality for the four public data sets, and its assembly, that consists of one circular contig, showed a 100% identity with previously known fragments, we manually verified the NOVOPlasty assembly by realigning all reads and visually inspected it with Tablet (14). Because we found no dubious sequence, we used our assembly as a reference for the quality assessment of the other tools. The assembly by MITObim resulted in one contig with a coverage of 99.63% and an accuracy of 99.8%. CLC obtained a coverage of 99.3% in 8 scaffolds (22 contigs) with an accuracy of 99.99% (Table 1). Given that we used the NOVOPlasty assembly as a reference, additional proof of mismatches for the CLC and MITObim assemblies were visualized by realigning all reads to each assembly, which can be found along with more elaborated results and statistics in Supplementary Data.
Table 1. Benchmarking results for the assembly of the A. marina chloroplast.
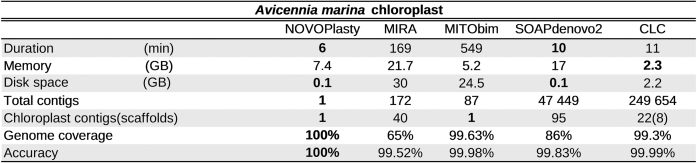
|
Figure 3.

Comparison between the NOVOPlasty and CLC alignments of three different chloroplast assemblies against their respective reference. (A) CLC and NOVOPlasty assemblies of SRR1174256 (A. thaliana) against GenBank entry AP000423.1. (B) CLC and NOVOPlasty assemblies of ERR477442 (O. sativa) against GenBank entry KM088022.1. (C) CLC assembly of A. marina against the manually inspected NOVOPlasty assembly.
Mitochondrial assembly
NOVOPlasty has currently been tested for the assembly of seven mitochondrial genomes from three different species. Besides the mitochondrion of the leaf beetle Gonioctena intermedia, all assemblies resulted in a complete circular genome. Detailed results and statistics can be found in Supplementary Data.
All assemblers besides ARC successfully constructed the complete human mitochondrion in a single contig. ARC was not successful which was unexpected since the reference was almost identical. According to the developers this problem might be resolved by removing the adapters before the assembly. Quality assessment showed that the MIRA assembly contained one mismatch and four unidentified nucleotides, while the other assemblies were identical to the reference. MITObim performed best in memory consumption, 1.5 GB and NOVOPlasty had the shortest runtime, 4 m 04 s (see Supplementary Data).
More interesting was the assembly of the G. intermedia mitochondrion. Like other beetle mitochondrial genomes, it contains a large AT-rich region (control region), including a highly repetitive section (17), resulting in a more complex de novo assembly. The mitochondrial genome was first assembled with long PacBio reads that covered the control region entirely to build a reliable reference. The resulting genome includes a repetitive region of 1820 bp, consisting in tandem repeats of variable length (∼120 bp). Assembling this region with short reads will be infeasible, regardless of which software is used. Nevertheless we were interested which software was able to deliver the most complete and accurate assembly. An assembly generated by mapping reads to a reference genome, using ARC, was used as mapping software reference to evaluate the possible benefits of a de novo assembly. We chose ARC because it is able to use multiple references and has been successfully tested for mitochondrial genome assembly (http://biorxiv.org/content/early/2015/01/31/014662). The nine closest mitochondrial genomes available at GenBank were selected as references. The resulting ARC assembly was 99.99% accurate but comprised only 85.39% of the genome. Except SOAPdenovo2, all de novo assemblers were able to assemble a larger fraction of the genome (Table 2). Since it is not possible to assemble the tandem repeats accurately with short reads, we also calculated the coverage and accuracy against the mitochondrion reference without the repetitive section of the control region. This significantly increased the accuracy for the assemblies that partially covered the tandem repeats. The MIRA and MITObim assemblies have the highest genome coverage, but the lowest accuracy. If we only look at the repetitive region, the accuracies are 95.44% and 90.62% for respectively MIRA and MITObim. This shows that these assemblers are not reliable for problematic regions and lose their advantage of higher genome coverage. From the four assemblers with an accuracy above 99.97%, NOVOPlasty has the highest genome coverage (Table 2).
Table 2. Benchmarking results for the assembly of the G. intermedia mitochondrion.
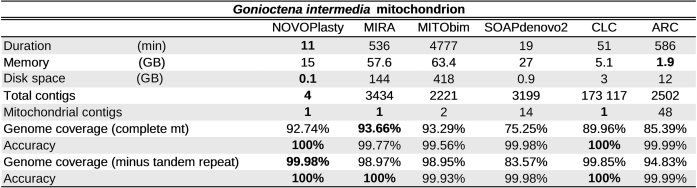
|
A second data set of G. intermedia, with an average coverage depth of 301 and a read length of 126 bp (in comparison to a read length of 101 bp and average coverage depth of 157 for the data set used above), was excluded from the benchmarking study as a result of insufficient memory capacity during the MIRA and MITObim assemblies. Both assemblies were automatically terminated after they exceeded 91 GB of RAM. NOVOPlasty, CLC and SOAPdenovo2 successfully executed the assembly (see Supplementary Data). The increase in depth of coverage and read length resulted in a higher genome coverage for the NOVOPlasty assembly. It rose from 92.74% to 94.66%. A second assembly of this data set with an average coverage depth of 892 improved the genome coverage further to 95.18%. This shows that increased read length with deeper coverage can help NOVOPlasty to resolve problematic regions, since the gain was from the highly repetitive and AT-rich region. More remarkable were the results of CLC and SOAPdenovo2, both assemblies resulted in a reduced genome coverage and accuracy. The genome coverage of SOAPdenovo2 and CLC were reduced by respectively 64% and 1.4%. The reduction of CLC could be explained by differences in the sample preparation or in the sequencing run (resulting in reads of reduced quality or underrepresented region), but this can not explain the large reduction for SOAPdenovo2. One explanation could be the high coverage depth, which can cause problems with some assembly tools. This was tested by repeating the assembly with a sub-sample of 50% of the previous data set. The results showed an increase in genome coverage from 30.8% to 46.8% for the SOAPdenovo2 assembly, showing a reverse effect of an increased coverage depth on the quality of the assembly for SOAPdenovo2.
Overall performance
The average performance of speed, memory efficiency, disk space, genome coverage, accuracy and contig count were calculated based on seven assemblies and presented in a score graph (Figure 4). NOVOPlasty scores slightly higher on speed and disk space than CLC, although the difference is negligible. CLC consistently performed better on memory usage, while NOVOPlasty has nevertheless the lowest memory usage of the four open-source assemblers. Regarding contig counts, CLC and NOVOPlasty have a similar high score, caused by the average high contig count of SOAPdenovo2 (125 contigs). The small difference in score is still significant since NOVOPlasty was able to achieve a 100% coverage in a single contig for all assemblies besides the G. intermedia mitochondrion, including the 16 chloroplast genomes that were not included in the benchmark study. On the other hand, CLC generated a minimum of 3 scaffolds for chloroplast genomes and even 8 scaffolds (comprising 22 contigs) for A. marina (see Figure 3). NOVOPlasty offers yet another significant advantage, as all the other tested assemblers output a pool of contigs, originating from the nuclear, mitochondrial and chloroplast genomes. This pool can contain in some cases more than 1 700 000 contigs (see Supplementary Data), which can make it problematic to isolate the organelle genome. Finally, NOVOPlasty scores best on genome coverage and accuracy, the two most important indicators for an accurate assembly.
Figure 4.

Score graph derived from the benchmark study. Each property of each assembler was given a score proportional to the other assemblers. Each score was based on the average results of seven assemblies and expressed in percentage. A score of 100% is always seen as most favorable, more detailed explanation can be found in the ‘Quality assessment’ section of Materials and Methods. (*) Highest score for the corresponding property.
Seed compatibility
One of the possible criticisms of de novo assembly with seed sequences is that you need previous knowledge regarding the organism of interest. Therefore, we developed a very flexible seed input that accepts sequences from more distantly related species. The main difference with traditional seed dependent assemblers, is that NOVOPlasty does not use the seed sequence to initiate the assembly, but uses it to retrieve one sequence read of the targeted genome from the data set, that subsequently will be elongated until the genome is circularized. This new strategy was tested with a variety of seed sequences, originating from closely to relatively distantly related species. Twelve different mitochondrial (Figure 5) and chloroplast (Figure 6) genomes were tested as a seed sequence for the assembly of respectively the mitochondrial genome of Homo sapiens and the chloroplast genome of Arabidopsis thaliana.
Figure 5.

Seed compatibility test for the de novo assembly of the human mitochondrium with 12 different mitochondrial genomes as seed sequence. A green dot means that the mitochondrial genome of that species can be used as a seed for the mitochondrial assembly of H. sapiens. Red X means unsuccessful. Phylogenetic tree based on information extracted from the NCBI taxonomy database (20), using phyloT (http://phylot.biobyte.de/).
Figure 6.

Seed compatibility test for the de novo assembly of the chloroplast from Arabidopsis thaliana with 12 different chloroplast genomes and 12 subunits (RuBP) as a seed sequence. A green dot means that the chloroplast genome of that species can be used as a seed for the chloroplast assembly of A. thaliana. Red M indicates that NOVOPlasty assembled the mitochondrial genome instead of the chloroplast genome. Same color indications for the RuBP unit. Phylogenetic tree based on information extracted from the NCBI taxonomy database (20), using phyloT (http://phylot.biobyte.de/).
Regarding the mitochondrial assembly, all seed sequences derived from vertebrates were sufficient to result in a successful assembly. This shows that this new strategy greatly simplifies the seed selection. For the chloroplast genomes, the assembly was successful in only half of the cases, and initiating instead the assembly of the mitochondrial genome in the other cases. This is caused by the occurrence of similar sequences in the chloroplast and mitochondrial genomes, likely the result of intergenomic transfer between them (19). To prevent this, it is recommended to use, as seed, short regions specific to the chloroplast genome, that have no equivalent in the mitochondrial genome (instead of the complete genome sequence). This was empirically confirmed by selecting the Rubisco-bis-phosphate oxygenase (RuBP) subunit as a seed sequence (Figure 6), which resulted in a successful assembly of the chloroplast genome in 10 out of 12 seed sequences. We were unable to initiate the assembly with the RuBP subunits of Pyropia perforata (RuBP subunit not present) and Lobosphaera incisa as a seed sequence. While these two algae species are evolutionarily very distant from Arabidopsis, we were able to assemble the chloroplast genome of Arabidopsis using the complete chloroplast genomes of both algae as seed. We would therefore recommend to try short specific portions of the genome as seed at first, but then to try the complete chloroplast genome as seed if unsuccessful.
DISCUSSION
Mitochondrial and chloroplast genomes represent a significant portion of the total deposited genomes in GenBank (21) (see Supplementary Data) and the annual deposition is growing each year. Many peer-reviewed articles are describing the assembly of these genomes, making organelles the most sequenced eukaryotic genome (22). Expanding the repository of organelle genomes can be beneficiary for a wide range of scientific fields, from forensics to evolutionary studies and food industry. With the continuous advancements in sequencing technologies, sequencing costs and data availability will not be a limiting factor in the future. High quality reads can be obtained by different platforms, but the outcome can vary greatly depending on the assembly software.
Due to a lack of reliable and user-friendly open-source software for the assembly of mitochondrial and, especially, plastid genomes, many researchers select the CLC pay-for-use assembler (1,9,23,24). We present NOVOPlasty, an open-source alternative software, specifically designed for assembling organelle genomes that is capable of delivering the complete genome sequence within 30 min. The algorithm takes full advantage of the high coverage available for organelle genomes in NGS data, which makes it even capable of assembling reads from problematic regions, like AT-rich stretches. No reference genome is needed and the assembly can be initiated by a wide range of seed sequences. When the final assembly delivered by NOVOPlasty is made of several contigs, those are automatically arranged sequentially, which facilitates finishing the assembly using complementary methods.
In our benchmark study, the average performance of speed, memory efficiency, disk space, genome coverage, accuracy and contig count were calculated based on seven assemblies, two mitochondria and five chloroplasts (see Figure 4). NOVOPlasty was the best on all criteria except for memory usage in respect to CLC. Note that all our mitochondria and chloroplasts assemblies done for the present work needed less than 16 GB of RAM. Most importantly, NOVOPlasty was the software achieving by far the most assemblies in a unique contig, and produced the best accuracy and genome coverage. We obtained reliable quality scores by validating each assembly by mapping original reads on to the assembly and visually inspect the complete genome. By its simple seed-and-extend design, NOVOPlasty seems also to be the best software to make use of longer Illumina reads (comparison available for the G. intermedia mitochondrion). Finally, by assembling only the genome requested by the user, NOVOPlasty simplifies the post-hoc tasks required by other assemblers. At the time of acceptance, NOVOPlasty has been tested by 19 beta-users across four continents, producing with success assemblies for more than 90 organelle genomes (mostly chloroplasts). All but six, characterized by particularly complex repeats, displayed a single contig.
The software is open source and can be downloaded at https://github.com/ndierckx/NOVOPlasty. Besides a standard Perl installation, there are no software or module requirements to run the script. All paired-end Illumina whole genome data sets are compatible with NOVOPlasty. It is recommended to have sufficient coverage (30X for the organelle genome) and to use untrimmed reads to assemble a complete circular genome. Incomplete assemblies caused by low coverage regions (low GC) could be resolved by using higher coverage (up to 1000X or more), but be cautious that higher coverage will slow down the assembly and will require more virtual memory. A manual and an example of the configuration file can also be found on the github page.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Prof. Ludwig Triest from the VUB Plant Biology and Nature Management laboratory for providing the A. marina chloroplast sequences. The authors thank all the beta users for testing NOVOPlasty and especially Dr Matthew Barrett from Kings Park and Botanic Garden, Australia for his constructive comments. The authors would also like to thank Chedly Kastally from Evolutionary Biology & Ecology department, ULB for his work on the the G. intermedia mitochondrion.
Authors’ contributions: N.D. conceived, designed and scripted NOVOPlasty. N.D. and G.S. analyzed the results. P.M. and G.S. initiated and funded the research project. PM provided the G. intermedia sequences. N.D. wrote the manuscript. P.M. and G.S. reviewed the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
N.D. was supported through a ULB-VUB PhD seed funding given to the Interuniversity Institute of Bioinformatics in Brussels and a PhD bursary of the Belgian Kids Fund. Funding for open access charge: Hôpital Universitaire des Enfants Reine Fabiola through a support from the Fonds iris-Recherche to G.S.
Conflict of interest statement. None declared.
REFERENCES
- 1. Brozynska M., Furtado A., Henry R.J.. Direct chloroplast sequencing: Comparison of sequencing platforms and analysis tools for whole chloroplast barcoding. PLoS One. 2014; 9:e110387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bignell G.R., Miller A.R., Evans I.H.. Isolation of mitochondrial DNA. Methods Mol. Biol. 1996; 53:109–106. [DOI] [PubMed] [Google Scholar]
- 3. Jansen R.K., Raubeson L.A., Boore J.L., dePamphilis C.W., Chumley T.W., Haberle R.C., Wyman S.K., Alverson A.J., Peery R., Herman S.J.. Elizabeth AZ, Eric HR. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods in Enzymology. 2005; Academic Press; 348–384. [DOI] [PubMed] [Google Scholar]
- 4. Khan A., Khan I.A, Asif H., Azim M.K.. Current trends in chloroplast genome research. Afr. J. Biotechnol. 2010; 9:3494–3500. [Google Scholar]
- 5. Wu J., Liu B., Cheng F., Ramchiary N., Choi S.R., Lim Y.P., Wang X-W.. Sequencing of chloroplast genome using whole cellular DNA and Solexa sequencing technology. Front. Plant Sci. 2012; 3:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ahmed I. Chloroplast genome sequencing: some reflections. Next Gen. Seq. Appl. 2015; 2:2. [Google Scholar]
- 7. Ekblom R., Smeds L., Ellegren H.. Patterns of sequencing coverage bias revealed by ultra-deep sequencing of vertebrate mitochondria. BMC Genomics. 2014; 15:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McPherson H., van der Merwe M., Delaney S.K., Edwards M.A., Henry R.J.. Capturing chloroplast variation for molecular ecology studies: a simple next generation sequencing approach applied to a rainforest tree. BMC Ecol. 2013; 13:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hahn C., Bachmann L., Chevreux B.. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads–a baiting and iterative mapping approach. Nucleic Acids Res. 2013; 41:e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chevreux B., Wetter T., Suhai S.. Genome sequence assembly using trace signals and additional sequence information. Comput. Sci. Biol. 1999; 99:45–56. [Google Scholar]
- 11. Luo R., Liu B., Xie Y., Li Z., Huang W., Yuan J., He G., Chen Y., Pan Q., Liu Y. et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012; 1:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Langmead B., Salzberg S.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Milne I., Bayer M., Stephen G., Cardle L., Marshall D.. Tablet: visualizing next-generation sequence assemblies and mappings. Methods Mol. Biol. 2016; 1374:253–268. [DOI] [PubMed] [Google Scholar]
- 14. Warren R.L., Sutton G.G., Jones S.J., Holt R.A.. Assembling millions of short DNA sequences using SSAKE. Bioinformatics. 2007; 23:500–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jeck W.R., Reinhardt J.A., Baltrus D.A., Hickenbotham M.T., Magrini V., Mardis E.R., Dangl J.L., Jones C.D.. Extending assembly of short DNA sequences to handle error. Bioinformatics. 2007; 23:2942–2944. [DOI] [PubMed] [Google Scholar]
- 16. Ross M.G., Ross M.G., Russ C., Costello M., Hollinger A., Lennon N.J., Hegarty R., Nusbaum C., Jaffe D.B.. Characterizing and measuring bias in sequence data. Genome Biol. 2013; 14:R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mardulyn P., Termonia A., Milinkovitch M.C.. Structure and evolution of the mitochondrial control region of leaf beetles (Coleoptera: Chrysomelidae): a hierarchical analysis of nucleotide sequence variation. J. Mol. Evol. 2003; 56:38–45. [DOI] [PubMed] [Google Scholar]
- 18. Tsai C.H., Strauss S.H.. Dispersed repetitive sequences in the chloroplast genome of Douglas-fir. Curr. Genet. 1989; 16:211–218. [DOI] [PubMed] [Google Scholar]
- 19. Alverson A.J., Wei X.X., Rice D.W., Stern D.B., Barry K., Palmer J.D.. Insights into the evolution of mitochondrial genome size from complete sequences of citrullus lanatus and cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 2010; 27:1436–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Federhen S. The NCBI taxonomy database. Nucleic Acids Res. 2012; 40:D136–D143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Benson D.A., Cavanaugh M., Clark K., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W.. GenBank. Nucleic Acids Res. 2013; 41:D36–D42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smith D.R. The past, present and future of mitochondrial genomics: have we sequenced enough mtDNAs?. Brief. Funct. Genomics. 2015; 15:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Naito K., Kaga A., Tomooka N., Kawase M.. De novo assembly of the complete organelle genome sequences of azuki bean (Vigna angularis) using next-generation sequencers. Breed. Sci. 2013; 63:176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ye C.-Y., Lin Z., Li G., Wang Y.-Y., Qiu J., Fu F., Zhang H., Chen L., Ye S., Song W. et al. Echinochloa chloroplast genomes: insights into the evolution and taxonomic identification of two weedy species. PLoS One. 2014; 9:e113657. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.