


Abstract
We have developed a new λ Red recombineering methodology for generating transient selection markers that can be used to transfer mutations between bacterial strains of both Escherichia coli and Salmonella enterica. The method is fast, simple and allows for the construction of strains with several mutations without any unwanted sequence changes (scar-free). The method uses λ Red recombineering to generate a marker-held tandem duplication, termed Duplication-Insertion (Dup-In). The Dup-Ins can easily be transferred between strains by generalized transduction and are subsequently rapidly lost by homologous recombination between the two copies of the duplicated sequence, leaving no scar sequence or antibiotic resistance cassette behind. We demonstrate the utility of the method by generating several Dup-Ins in E. coli and S. enterica to transfer genetically linked mutations in both essential and non-essential genes. We have successfully used this methodology to re-construct mutants found after various types of selections, and to introduce foreign genes into the two species. Furthermore, recombineering with two overlapping fragments was as efficient as recombineering with the corresponding single large fragment, allowing more complicated constructions without the need for overlap extension PCR.
INTRODUCTION
The need to introduce a specific mutation present in one strain into another bacterial strain is common in bacterial genetics, molecular biology and biochemistry. Except for the rare cases when the mutation itself confers a selectable phenotype, the process is often tedious and time-consuming. Usually it involves placing an antibiotic resistance (AbR) determinant close to the mutation, either by random transposon-hops or using λ Red mediated genetic engineering (recombineering) to generate precisely targeted insertions (1–3). Once the AbR marker is introduced, it can be transferred to other strains by bacteriophage mediated generalized transduction, which results in a fraction of the transductants co-inheriting the linked mutation from the donor strain.
It is often undesirable to leave an antibiotic resistance cassette in the resulting strain as the marker itself could affect the phenotype of the bacterium (e.g. by affecting expression of nearby genes or by the potential cost of expression of the antibiotic resistance protein itself). Furthermore, if there is a need to combine several mutations into one strain, there is a risk that an insufficient number of usable selection markers are available. One way to minimize these concerns is to remove the antibiotic resistance marker in a later step, e.g. by using resistance cassettes flanked by Flp-recombinase target sites (FRT) and removing the cassette by expression of Flp (1). However, there are limitations to the use of FRT-flanked resistance markers. As Flp leaves one copy of the FRT sequence behind there can be unwanted and unpredictable effects even after excision of the marker. For instance, we have noticed that an FRT ‘scar’ at the end of the rpsT transcript has a measurable deleterious impact on growth rate (4). Furthermore, there are concerns that upon expression of Flp in a strain with multiple FRT sequences in its genome, large-scale genomic rearrangements could occur (1). Another reason to avoid leaving resistance cassettes or scar sequences behind in the final strains is that they could cause unwanted phenotypes. Thus, every mutation that is present in a strain in addition to the mutations of interest for the particular experiment necessitates another control for the experiment. Isogenic strains that differ only in the absence or presence of the mutation of interest but carry all the same resistance cassette insertions or scar sequences need to be constructed and tested to ensure that the additional sequences do not influence the results of the experiment. However, if no cassette or scar is left behind, the recipient strain (often the wild-type) from the previous step in a strain construction is the only control necessary.
Selection markers can also be seamlessly excised. Markers that are targets for endonuclease I-SceI can be removed by co-expression of λ Red and I-SceI, and counter-selectable markers can also be removed by a second round of λ Red recombineering (5–7). These methods can be used to remove cassettes without leaving any scar sequence, but both require a second transformation step.
We wanted to find a more efficient strategy for introducing a selectable marker in the genome. The marker should be transferable through generalized transduction to enable co-transduction of nearby mutations and the marker should be easily lost without leaving any scar sequence. Finally, the method for losing the marker should not require any additional transformation or any specific genetic background.
Before the introduction of recombineering for gene replacements, suicide plasmids were used to move constructed mutations to the chromosome (8–10). In principle, the mutation of interest was first cloned or constructed on a suicide plasmid vector. The plasmid was then introduced into a recipient strain under conditions where it could not replicate. Selection for an antibiotic resistance marker carried on the plasmid allowed for the isolation of recombinants where the plasmid had undergone homologous recombination with the chromosome. This left a duplication with the plasmid sequence at the junction between two copies of the locus of interest, usually a wild-type copy derived from the chromosome and a mutant copy from the plasmid. Through selection against a counter-selectable marker on the plasmid, segregants that had lost the integrated plasmid and one copy of the duplicated sequence were found at a high frequency.
Here, we have developed a fast and easy variation of this methodology. It entails creating an engineered duplication (in a single step using λ Red recombineering) in a strain that already contains the mutation of interest. At the junction between the two copies of the duplicated sequence, a selectable and counter selectable marker is placed to allow for positive and negative selection. As the generated duplications contain an insertion at the junction between the two copies, we refer to them as duplication-insertions (Dup-Ins). Once the strain containing the Dup-In is constructed, the Dup-In and the linked mutation can be transferred by generalized transduction into other strains, selecting for the resistance marker at the duplication junction. Dup-Ins are easily lost through homologous recombination, and Dup-In free clones are easily isolated by selection against the inserted cassette. This method eliminates the risk of running out of usable selectable markers in multi-step strain constructions.
We demonstrate the Dup-In methodology in the model organisms Salmonella enterica and Escherichia coli, where much of today's detailed studies of bacterial biochemistry, molecular biology and genetics are done. Although we have not tested in other organisms, the methodology should be extendable to other bacteria with developed genetic methods and where λ Red recombineering or analogous methods work.
MATERIALS AND METHODS
Strains and growth conditions
All bacterial strains (Supplementary Data) are derivatives of S. enterica serovar Typhimurium strain LT2 or E. coli K12 strain MG1655. Generalized transductions in S. enterica were performed with P22 HT int (11) and in E. coli with P1 vir (12). For rich medium, we used either SOC (20 g/l tryptone [oxoid], 5 g/l yeast extract [oxoid], 0.5 g/l NaCl, 0.25 mM KCl, 10 mM MgCl2, 4 g/l glucose) or LB (10 g/l NaCl, 10 g/l tryptone [oxoid] and 5 g/l yeast extract [oxoid]). The LB was supplemented with 15 g/l agar (oxoid) to make LB agar (LA) plates. Sucrose selection plates (LB agar without NaCl and with 50 g/l sucrose [Sigma]) were used for counter-selection against sacB. When needed, antibiotics (Sigma) were used at the following concentrations: Tetracycline, 7.5 mg/l; Chloramphenicol, 12.5 mg/l; Kanamycin, 100 mg/l.
Selectable and counter selectable cassettes
The cat-sacB-T0 cassette (GenBank KM018298) and its derivatives are all surrounded by the same primer-binding sites: ‘P1’ (5΄-GTGTAGGCTGGAGCTGCTTC-3΄) and ‘P2’ (5΄-CATATGAATATCCTCCTTAGTTCC-3΄). Variants of the cat-sacB-T0 cassette provide an additional colorimetric or fluorometric screen: Acatsac1, Ycatsac1 and Tomcatsac1, containing a gene encoding a chromoprotein (AmilCP from Acropora millepora) or a fluorescent protein (sYFP2 or dTomato), transcribed from a strong synthetic promoter, CP25 (13) and terminated by the phage P22 late transcriptional terminator (TP22late). The additional elements are placed between the ‘P1’ primer binding site and the dual promoters of the cat-sacB operon (Supplementary Data). The cat gene in all cassettes was also exchanged by a kan gene to allow for selection in strains already carrying a cat gene. Those cassettes were named Akansac1, Ykansac1 or Tomkansac1.
λ Red recombineering
PCR reactions to generate DNA for recombineering were performed with Phusion DNA polymerase (Thermo Fisher) according to the manufacturers instructions. All primer sequences are listed in Supplementary Data, and PCR programs are described in Supplementary Data. When making cells competent for λ Red transformation through electroporation, cultures of cells containing the pSIM5-Tet plasmid (14) were grown over night at 30°C in ‘No Salt LB’ (LB with no NaCl) with 2 g/l glucose and 7.5 mg/l tetracycline. Cultures were diluted 1:100 in the same medium (pre-warmed to 30°C), and grown at 30°C until OD600 ≈ 0.2 (measured with 1 cm light path in a Shimadzu UV mini 1240 spectrophotometer). Once the target OD was reached, the culture flasks were moved to a 42°C shaking (185 rpm) water bath to induce expression of the temperature-controlled λ Red genes. After 15 min at 42°C (OD600 ≈ 0.3), the cultures were cooled in an ice-water bath for at least 10 min. The cells were pelleted by centrifugation at 4°C (4000 × g, 6–10 min) and all the medium was removed. The cells were washed once in ice-cold 10% glycerol (∼1/4 of the culture volume), pelleted (4000 × g, 6 min) and re-suspended in ice-cold 10% glycerol (200–400 μl per 25 ml initial culture volume).
Prior to electroporation, the electrocompetent cells (20–40 μl) and DNA (up to about 50 fmol PCR product in 1–2 μl H2O) were mixed on ice in electroporation cuvettes (1 mm gap, Bio-Rad), and electroporated in a Gene Pulser Xcell or Gene Pulser (Bio-Rad) at 2.5 kV, 400 Ω and 25 μF. After electroporation, cells were immediately moved to 200 μl pre-warmed (42°C) SOC in a 42°C waterbath and incubated without shaking for at least 15 min before plating on LA plates with 12.5 mg/l chloramphenicol at 37°C. Sometimes this incubation at 42°C was followed by, or replaced by, shaking incubation at 37°C for several hours or over night. In cases where we wanted to keep the pSIM5-Tet plasmid in the transformants, the additional incubation at 42°C was replaced by shaking at 30°C for 2–5 h, and the plates were incubated at 30°C.
Segregation of Dup-Ins
To isolate segregants (Dup-In free, SucR clones), colonies were picked from plates (with or without antibiotic) and streaked on sucrose selection plates. Using the Acatsac1, Ycatsac1 or Tomcatsac1 cassettes (or their KanR derivatives) typically only resulted in occasional false positives (blue or fluorescent SucR colonies) among thousands of white, non-fluorescent colonies.
For segregation assays in liquid media, overnight cultures grown in LB + chloramphenicol at 37°C were diluted 1:1000 in No Salt LB without chloramphenicol. After growth at 37°C overnight, the cultures were serially diluted and plated on sucrose selection plates to determine the number of SucR cells per ml (corresponding to ∼5 × 109 cfu). All Dup-Ins used for this experiment are listed in Table 1.
Table 1. Segregation pattern after transfer of insertion-duplications and linked mutations by transduction.
| Segregation pattern (number of transductants)d | |||||||
|---|---|---|---|---|---|---|---|
| Duplicated region | Dupl. size (bp)a | Mutation | Distance (bp)b | Phenotypec | Homozygous mute | Homozygous wild-typef | Heterozygousg |
| hisBHAF h | 2497 | ΔhisA | 833, 1658 | His- | 48 | 0 | 0 |
| hisO i | 1034 | hisA(L169R) | 6139 | His- | 21 | 27 | N/A |
| trpCFB h | 1003 | ΔtrpF | 459, 475 | Trp- | 47 | 0 | 1 |
| gyrA h | 3109 | gyrA(S83F) | 671, 2439 | CipR | 11 | 3 | 0 |
| gyrA i | 638 | gyrA(S83F) | 671 | CipR | 62 | 2 | N/A |
| tufA fusA rpsGL h | 4630 | rpsL(K42N) | 278, 4353 | StrR | 32 | 16 | 0 |
| yheL rpsL i | 523 | rpsL(K42N) | 543 | StrR | 61 | 3 | N/A |
| rplKAJL rpoBC stm4155 h | 11 681 | rpoB(S531L) | 3214, 7146 | RifR | 9 | 15 | 22 |
aFor ΔhisA and ΔtrpF see Figure 2.
bDistance between the cassette and the mutation. When the mutation is contained within the duplication the distances to both junctions are indicated.
cGenotypes were inferred from the phenotypes conferred by the transferred mutations. His-, histidine auxotrophy; Trp-, tryptophan auxotrophy; CipR, Ciprofloxacin resistance; RifR, rifampin resistance; StrR, streptomycin resistance.
dNumber of transductants that displayed the observed segregation pattern as described under e–g.
eAll tested segregants inherited the phenotype of the donor parent (mutant).
fAll tested segregants inherited the phenotype of the recipient parent (wild-type).
gSegregants with both of the parental phenotypes were recovered. Note that the number reported here may be an underrepresentation, as only four segregants from each transductant were tested. N/A, not applicable. The mutation was outside of the duplicated region, so heterozygotes were not expected.
hThe transferred mutation was within the duplicated region.
iThe transferred mutation was outside the duplicated region.
RESULTS
Description of the Dup-In methodology
The method, as outlined in Figure 1, involves three major steps: (i) constructing a Dup-In using λ Red recombineering, (ii) generalized transduction to transfer the Dup-In and the linked mutation to other strains, and (iii) segregating the Dup-In in the transductants to isolate marker-free clones. However, the third step can be done as part of the post-transduction cleanup procedure that in practice translates into five days of low-intensity lab work from transformation to re-constructed mutant.
Figure 1.
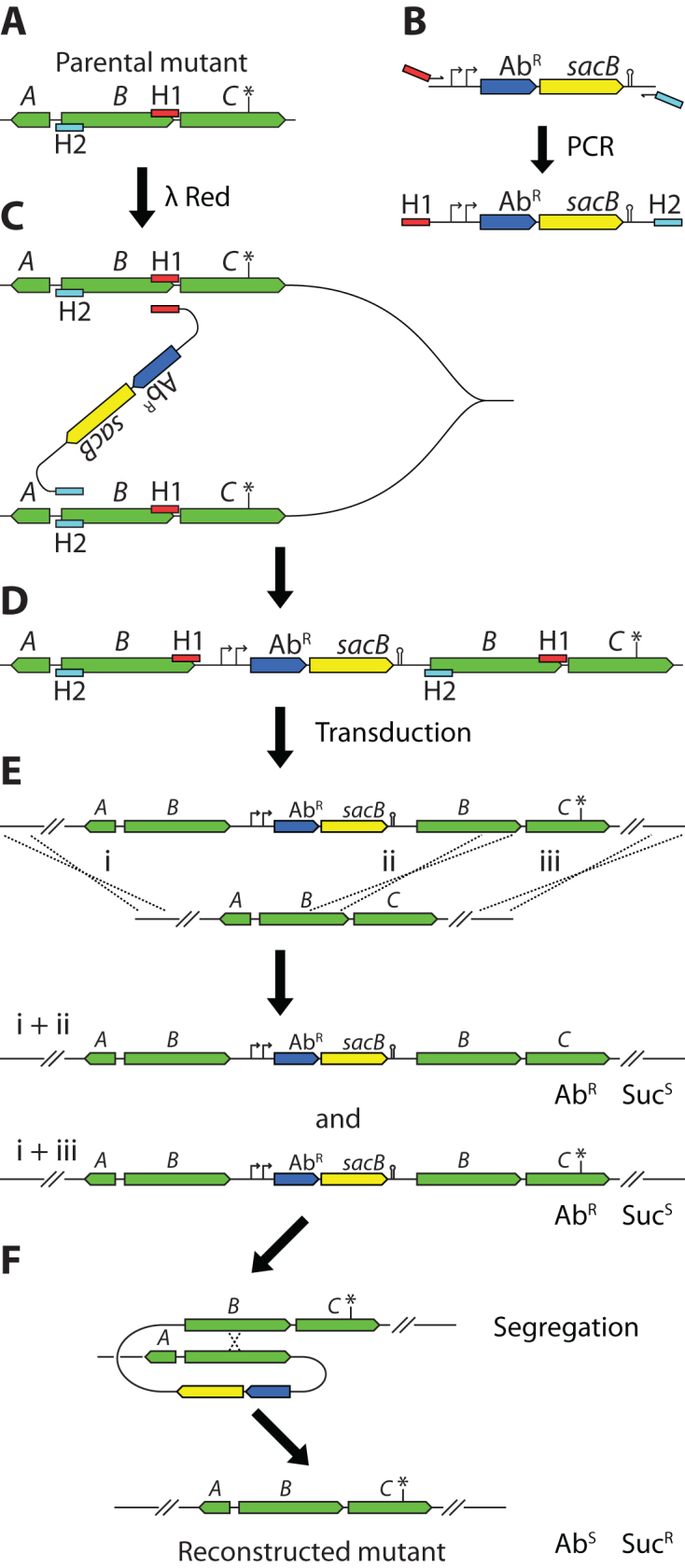
Outline of the method. (A) Hypothetical chromosomal region. The mutation of interest is indicated by an asterisk (*). Recombineering homologies H1 (red rectangles) and H2 (turquoise rectangles) are chosen so that region B will be duplicated. Note that the homology extensions are arranged in an ‘ends-in’ configuration, i.e. the H1 region is on the upper strand and the H2 region is on the lower strand. (B) PCR primers carrying the recombinogenic 5΄-extensions ‘H1’ and ‘H2’ are used to amplify an AbR-sacB cassette. Note that the primer that anneals to the left end of the cassette carries a 5΄-homology extension that directs recombination to the right of the target region (H1, red rectangle), and the primer that anneals to the right end of the cassette carries a 5΄-homology extension that directs recombination to the left of the target region (H2, turquoise rectangle). (C) The homologies H1 and H2 direct λ Red mediated recombination. The exact mechanism of the recombination has not been examined, but is drawn here as if it occurs between two newly replicated sister chromosomes in the same cell. (D) The resulting Dup-In, with one copy of AbR-sacB between two copies of the target region. (E) The Dup-In is transferred to another strain using generalized transduction. For successful transfer, recombination between homologous sequences on each side of the AbR-sacB cassette has to occur. As depicted, one recombination has to occur anywhere to the left of the cassette (i). Recombination to the right of the cassette can occur either between the cassette and the mutation (ii), or somewhere to the right of the mutation (iii), leading to inheritance of the recipient allele (recombinations i and ii) or the donor allele (recombinations i and iii). The relative frequencies of the two types of transductants are dependent on the distance between the cassette and the mutation. (F) Segregants that have lost the Dup-In through homologous recombination are isolated through selection for sucrose-resistance (SucR).
The product of the B. subtilis sacB gene, levansucrase, confers sensitivity to sucrose when present in the periplasm of Gram-negative bacteria, and has been extensively used as counter-selectable marker for allelic replacement (8). To be able to both select and counter-select the Dup-Ins we use cat-sacB or kan-sacB cassettes, conferring sensitivity to sucrose and resistance to chloramphenicol or kanamycin. We also sometimes use variants of these cassettes with additional genes encoding chromoproteins or fluorescent proteins, which provide an extra colorimetric or fluorometric screen for the presence or absence of the cassette (Supplementary Data).
Engineering of a Dup-In
The general strategy for primer design and construction of Dup-Ins is illustrated in Figure 1A–D. Briefly, an antibiotic resistance (AbR)-sacB cassette is amplified with oligos containing in their 3΄-end 20–24 nucleotides that anneal to the template and in the 5΄-end 40 nucleotides that act as recombinogenic homology extensions. The homology extensions are chosen so that the primer that anneals to the right end of the template cassette carries homology to the left border of the duplication and the primer that anneals to the left end of the cassette carries homology to the right border. Furthermore, the homology extensions are arranged in an ‘ends-in’ configuration to direct the duplication of the intervening sequence rather than its replacement (Figure 1A and B). That is, the oligo that anneals to the left end of the cassette serves as the right junction of the duplication and, likewise, the oligo that anneals to the right end of the cassette serves as the left junction of the duplication. Through λ Red recombineering this PCR fragment directs the duplication to the region between the two designed marker-chromosome junctions, presumably by unequal recombination between two sister chromosomes (Figure 1C and D).
Loss of Dup-Ins by counter selection
To test the practical lower limit of useable duplication sizes, we generated a set of cat-sacB Dup-Ins in S. enterica, ranging in size from 100 bp up to 11.6 kb and tested their segregation frequencies in liquid cultures (Figures 2 and 3, Table 1). For duplications between 100 bp and 4.6 kb, a strong positive correlation between duplication size and segregation frequencies was found, whereas above 4.6 kb the segregation frequency plateaued (Figure 3), which is expected for recA-dependent recombination (15). No systematic tests for correlation between duplication size and transformation frequencies were done but we did not notice any differences when constructing strains for the above test.
Figure 2.

Examples of Dup-Ins constructed in S. enterica. Black lines below the gene maps indicate sequences duplicated in the different Dup-In constructs. Numbers next to the lines indicate sizes of the duplicated sequences in base pairs. Dashed lines indicate sequences that are missing in deletion mutants. Vertical lines with text above indicate the position of mutations. (A) Fourteen Dup-Ins in the his-operon. The ΔhisA mutant is missing the entire hisA coding sequence (719 bp). Oligos 1–5 (Supplementary Data) were used with different combinations of oligos 6–11 to generate all variants, except the hisO Dup-In for which oligos 12 and 13 were used. (B) Dup-In in the trp operon. The ΔtrpF mutant is missing the last 569 bp of the fused trpCF gene (oligos 14 and 15, Supplementary Data). (C) Two Dup-Ins in the gyrA region (oligo 18 in combination with oligos 16 or 17, Supplementary Data). (D) Two Dup-Ins in the str operon, containing rpsL (oligos 19 and 20, or 21 and 22, Supplementary Data). (E) Dup-In in the alpha operon, containing the rpoB gene (oligos 23 and 24, Supplementary Data).
Figure 3.
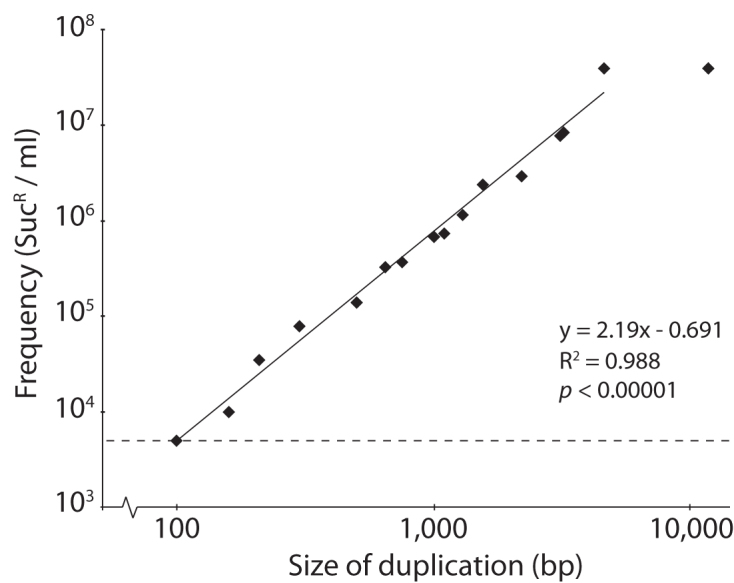
Size-dependence of segregation of Dup-Ins. The dashed line indicates the frequency of spontaneous SucR mutants in a hisA::cat-sacB culture. Inset text: linear equation, R2-value and Pearson correlation p-value for a line fitted to log(frequency) vs. log(duplication size), excluding the largest 11.6 kb Dup-In.
To estimate the background level of loss of SacB function by mechanisms other than segregation, we used a strain with the hisA gene replaced by a cat-sacB cassette. As this cat-sacB cassette was not surrounded by any intentional direct repeats, loss of SacB function was most likely due to point mutations or deletions that inactivated SacB. We found that segregation of the smallest Dup-Ins (100–210 bp) was indistinguishable from the frequency of spontaneous SucR mutants, whereas duplications as small as 300 bp were more than an order of magnitude above the background level (Figure 3). Further tests on plates showed that even for colonies grown in the presence of chloramphenicol to keep selection for the cat-sacB cassette, the segregant frequency was high enough to isolate SucR segregants. Therefore, all further segregant isolations were done by picking colonies containing Dup-Ins directly from the selection plates and streaking for single segregants on sucrose selection plates. This typically resulted in plenty of SucR segregant colonies, and only rarely false positives (Supplementary Data).
Using Dup-Ins as temporary ‘handles’ on mutations
We generated a set of Dup-Ins with cat-sacB cassettes at the junctions, linked to different mutations in both essential and non-essential genes in the S. enterica genome (Figure 2). These Dup-Ins were used to transfer the linked mutations by generalized transduction using phage P22 (Table 1). The mutations used for this test were chosen since they confer easily detected phenotypes, either auxotrophies (mutations in the his and trp operons) or antibiotic resistance (mutations in rpoB, gyrA and rpsL). For different mutations, different construction strategies were tested: the mutation was either within or outside of the duplicated region (Table 1). To move a previously constructed deletion of hisA, we used a Dup-In of the region spanning hisB to hisF, including the hisA deletion (Figure 2A, partially dashed line). A similar strategy was used for moving a trpF deletion (Figure 2B). To move the L169R point mutation in hisA, a Dup-In spanning 1 kb centered at the his attenuator (hisO) was used, placing the nearest marker-chromosome junction approximately 6.1 kb from the mutation (Figure 2A). For the mutations in gyrA and rpsL, both strategies were tested (Figure 2C and D). For the rpoB mutation, only one construct with internal mutation was tested (Figure 2E). In the cases where the mutation was outside of the duplication, the phenotype was tested before segregation and subsequently verified after segregation. In cases when the mutations were within the duplication, the phenotype was tested on four segregants from each transductant to distinguish ‘heterozygous’ transductants that carried the alleles of both parents from ‘homozygous’ transductants. As expected, the frequency of co-inheritance was dependent on the distance between the mutation and the nearest marker-chromosome junction (Table 1). With distances <1 kb, the co-inheritance of homozygous mutants was close to 100% for most mutations, whereas the fraction of transductants that retained the wild-type allele increased with increasing distance.
In addition to the above tests in S. enterica, we have used similar strategies to re-construct S. enterica and E. coli strains with single and multiple mutations found in an experimental evolution study (Table 2). Dup-Ins were constructed as indicated in Supplementary Data. Once constructed, all Dup-Ins could be transferred to other strains by P22 and P1 transductions, for S. enterica and E. coli respectively. In all cases, plating on sucrose selection plates allowed simple isolation of clones that had lost the Dup-Ins. PCR and local sequencing was used to screen the final reconstructed mutants. This was done for between one and nine transductants from each of the 24 different transductions. In only one case did one of the transductants have the wild-type allele and the three other tested clones had the mutation of interest. In all other transductions, all tested clones had the mutations of interest. This allowed us to untangle the effects of individual mutations in multistep evolutionary pathways (Knöppel, Knopp, Albrecht, Lundin, Lustig, Näsvall and Andersson, manuscript in preparation).
Table 2. Multistep strain constructions through step-wise transductions using Dup-Ins.
| Escherichia coli | ||
| Re-constructed strain | Recipient | Donora |
| gltP(-118ΔC) | DA5438 (wt) | dup[gltP(-118ΔC)]*cat-sacB-T0 |
| rpoS(ile128asn) | DA5438 (wt) | dup[rpoS(ile128asn)]*Acatsac1 |
| gltP(-118ΔC) rpoS(ile128asn) | rpoS(ile128asn) | dup[rpoS(ile128asn)]*Acatsac1 |
| nusA(arg258cys) | DA5438 (wt) | dup[nusA(arg258cys)]*cat-sacB-T0 |
| rne(gly172ser) | DA5438 (wt) | dup[rne(gly172ser)]*cat-sacB-T0 |
| glpK(val8phe) | DA5438 (wt) | dup[glpK(val8phe)]*cat-sacB-T0 |
| glpK(val8phe) nusA(arg258cys) | glpK(val8phe) | dup[nusA(arg258cys)]*cat-sacB-T0 |
| glpK(val8phe) rne(gly172ser) | glpK(val8phe) | dup[rne(gly172ser)]*cat-sacB-T0 |
| nusA(arg258cys) rne(gly172ser) | rne(gly172ser) | dup[nusA(arg258cys)]*cat-sacB-T0 |
| glpK(val8phe) nusA(arg258cys) rne(gly172ser) | glpK(val8phe) nusA(arg258cys) | dup[rne(gly172ser)]*cat-sacB-T0 |
| Salmonella enterica | ||
| Re-constructed strain | Recipient | Donora |
| Δ(flhE-flhD) | DA6192 (wt) | dup[Δ(flhE-flhD)]*cat-sacB-T0 |
| barA(gly455cys) | DA6192 (wt) | dup[barA(gly455cys)]*cat-sacB-T0 |
| treB(542+A) | DA6192 (wt) | dup[treB(542+A)]*cat-sacB-T0 |
| barA(gly455cys) treB(542+A) | treB(542+A) | dup[barA(gly455cys)]*cat-sacB-T0 |
| barA(gly455cys) Δ(flhE-flhD) | barA(gly455cys) | dup[Δ(flhE-flhD)]*cat-sacB-T0 |
| Δ(flhE-flhD) treB(542+A) | Δ(flhE-flhD) | dup[treB(542+A)]*cat-sacB-T0 |
| barA(gly455cys) Δ(flhE-flhD) treB(542+A) | Δ(flhE-flhD) treB(542+A) | dup[barA(gly455cys)]*cat-sacB-T0 |
| relA(ile338asn) | DA6192 (wt) | dup[relA(ile338asn)]*cat-sacB-T0 |
| nadR(ala317glu) | DA6192 (wt) | dup[nadR(ala317glu)]*cat-sacB-T0 |
| glpK(arg34his) | DA6192 (wt) | dup[glpK(arg34his)]*cat-sacB-T0 |
| nadR(ala317glu) relA(ile338asn) | nadR(ala317glu) | dup[relA(ile338asn)]*cat-sacB-T0 |
| glpK(arg34his) relA(ile338asn) | glpK(arg34his) | dup[relA(ile338asn)]*cat-sacB-T0 |
| glpK(arg34his) nadR(ala317glu) | glpK(arg34his) | dup[nadR(ala317glu)]*cat-sacB-T0 |
| nadR(ala317glu) relA(ile338asn) glpK(arg34his) | glpK(arg34his) nadR(ala317glu) | dup[relA(ile338asn)]*cat-sacB-T0 |
All Dup-Ins were generated through λ Red in strains isolated from evolving populations in an experimental evolution study (Knöppel, Knopp, Albrecht, Lundin, Lustig, Näsvall and Andersson, manuscript in preparation). The single, double and triple mutants listed in this table were constructed by step-wise transductions of the Dup-Ins, starting with our lab wild-type strains (E. coli DA5438 and S. enterica DA6192) as recipients for the first transduction to re-construct all possible single mutants. After selecting chloramphenicol resistant transductants, the duplication-insertions were allowed to segregate on sucrose selection plates, and the segregants were used as recipients for transductions to introduce the next mutation.
aOnly mutations relevant to these strain constructions are listed.
Efficient recombineering with overlapping PCR fragments
While developing this method, we discovered that recombineering between two overlapping PCR fragments that together make up a functional cassette is equally efficient as recombineering with one large PCR product containing the entire cassette.
When attempting to generate PCR products for constructing a set of very short hisA Dup-Ins (59 and 75 bp duplicated sequence), some of the primer pairs failed to generate any product. As the primers were designed to generate very short duplications, their 5΄-ends carried regions that were complementary within the primer pairs (Supplementary Data). One primer pair (oligos 57 and 58, Supplementary Data) that contained 29 nt of 5΄-complementarity worked, while two primer pairs (oligos 59 and 60, and 61 and 62, Supplementary Data) that contained 41 nt complementarity failed. The reason for the failure of these PCR reactions may be that the 3΄-ends of the new strands after the first few cycles cross-hybridized, causing buildup of multimeric products (Supplementary Data). We therefore first amplified these constructs as two separate PCR products with 277 bp overlap in the cat gene (see example in Supplementary Data), followed by an overlap extension reaction to generate the full-length product. We transformed λ Red induced cells with the DNA from the overlap extension, but included a mix of the two overlapping PCR products (in approximately 1:1 molar ratio) as an intended negative control for the transformation. Surprisingly, both the single long fragment and the ‘negative control’ PCR products resulted in approximately the same number of transformants. Thus, the overlap extension did not significantly affect the success of the transformation (data not shown). To verify that transformation works efficiently with overlapping PCR fragments we repeated these transformations, but without any overlap extension products (Table 3). The difference in transformation frequencies (∼2-fold or less) between the full-length product and two overlapping PCR products in this experiment was well within the variation usually seen between different transformations.
Table 3. Transformation with two overlapping PCR products vs. a single full-length PCR product.
| Construct | Note | 5΄ complementaritya | DNA length | Amount (ng) used in transformation | Total no. of transformants |
|---|---|---|---|---|---|
| hisA(dup 59 bp A)b | Full-length | 29 | 3332 | 250 | ∼1700 |
| hisA(dup 59 bp A)b | Two fragments | 29 | 1647+1962c | 125+147 | ∼1900 |
| hisA(dup 59 bp B)b | Two fragments | 41 | 1653+1968c | 125+147 | ∼1500 |
| hisA(dup 75 bp) | Two fragments | 41 | 1663+1974c | 125+147 | ∼3000 |
aAs the primers direct recombination within the same short sequence (59 or 75 bp) there is considerable complementarity between the 5΄ ends within the primer pair. During PCR cycling, the new 3΄-ends will be complementary, potentially causing multimerization of the products.
bThe primer pairs for generating hisA (dup 59 bp A) and hisA (dup 59 bp B) differed in that the A primer pair (oligos 57 and 58 in Supplementary Data) had shorter 5΄ homologies than the B pair (oligos 59 and 60 in Supplementary Data). The resulting transformants were identical.
cThe two fragments overlapped with each other by 277 bp in the cat gene, requiring recombination between the two fragments to restore a full cat gene and chloramphenicol resistance.
Using Dup-In methodology for precise insertion of foreign genes
We used a slightly modified Dup-In methodology for replacing the native S. enterica and E. coli araBAD and rhaBAD operons with the gene encoding ‘super’ yellow fluorescent protein (SYFP2; Figure 4). This variation of the method was simplified greatly by the observation that transformation frequencies are similar when the construct is transformed as two overlapping PCR fragments as compared to when it is transformed as one PCR fragment. Shortly, to replace the genes, we first generated a template strain containing a Dup-In in a syfp2 gene already present on the chromosome in a S. enterica strain. Transformation with two overlapping PCR fragments that together made up the desired Dup-In successfully generated strains in which YFP expression was controllable with arabinose or rhamnose, respectively (Figure 4 and Supplementary Data). Due to the size of the insertion (4689 bp) and the long internal direct repeats (645 bp of identical syfp2 sequence surrounding the Acatsac1 cassette) it would have been difficult to amplify the entire construct in one piece. Using these Dup-In containing strains as donors in transductions allowed transfer of the foreign gene into other strains without the need to leave any selection marker behind.
Figure 4.

Using a Dup-In for precise replacement of native genes for a foreign gene. (A) From the top: A pre-existing syfp2 gene inserted in the genome of S. enterica. An Acatsac1 cassette is introduced, making an internal Dup-In in syfp2. Oligos 25 and 26 (Supplementary Data) were used for this construction. A strain carrying the resulting Dup-In is used as template for amplification of two PCR products, each carrying 40 bp homology to only one end of the target region (blue and red rectangles) and 277 bp overlap in the cat gene. In order to result in a viable chloramphenicol-resistant transformant, the two fragments need to recombine with each other and the chromosomal target locus. (B) Top line: The structure of the native S. enterica ara operon indicating the 40 bp sequences used as homology for λ Red recombineering (red and blue rectangles). Middle: The resulting recombinant after recombineering with the PCR products from (A). For S. enterica, oligos 27 and 64 were used for one PCR product, and 28 and 63 for the other. For E. coli, oligos 29 and 64, and 30 and 63 were used (Supplementary Data). Bottom line: The segregant after loss of the Dup-In. The entire coding sequences of araB, A and D have been completely replaced by syfp2. The resulting strain is unable to metabolize L-arabinose, but expresses SYFP2 when arabinose is added to the growth medium (Supplementary Data). (C) As in (B) but the rha operon. Oligos 31 and 64, and 32 and 63 (Supplementary Data) were used for the two PCR products for S. enterica. For E. coli oligos 33 and 64, and 34 and 63 were used. The resulting strain is unable to metabolize L-rhamnose, but expresses SYFP2 when rhamnose is added to the growth medium (Supplementary Data). The E. coli ara and rha operons and the corresponding constructs (not shown) are very similar to the S. enterica counterparts, but differ in the surrounding genes.
DISCUSSION
Benefits of using Dup-Ins
Constructing a Dup-In and using it to transfer a mutation between strains requires the same amount of time and effort as using a regular insertion through traditional λ Red (one recombineering plus one transduction). Once a Dup-In is formed, it is easily cured on sucrose selection media, whereas curing an FRT-flanked cassette would require an additional transformation step for introduction of the Flp-expression plasmid, selection for the Flp-expression plasmid, and induction of Flp followed by curing of the plasmid. All in all, the steps involved take at least seven days to perform. If instead using the Dup-In methodology, the process would take about five days. Two days can be saved every time the same mutation is transferred to a new strain. Furthermore, in step-wise strain constructions, two days can be saved for every mutation that is introduced, with the added benefit that the Dup-In leaves no drug resistance marker or scar sequence behind. As there are fewer steps involved, it also requires fewer single-cell bottlenecks and fewer generations of growth, reducing the risk of fixation of unintentional mutations.
Because Dup-Ins can be placed very close to the mutations without causing any additional disruptions in the final (Dup-in free) strains, co-transduction frequencies are high, reducing the screening necessary to find correct clones. In the reconstructions of evolved strains, we screened a very low number of clones (between 1 and 9) but were successful in isolating the correct mutants after all 24 transductions. In only one of the transductions did we find a clone with the wild-type allele. As exemplified by our araBAD::syfp2 and rhaBAD::syfp2 mutations, Dup-In methodology can be used for generating and transferring complex designed mutations in a few simple steps.
A method for reconstruction of evolutionary trajectories
MAGE is a powerful and attractive method for quickly introducing multiple unmarked mutations (16). Iterative cycles of λ Red recombineering using a pool of mutation-containing oligonucleotides can rapidly generate a large set of clones with different combinations of mutations. However, in the presence of functional methyl-directed mismatch repair (MMR), recombineering without selection for transformants typically requires screening in order to find all possible combinations (17). With a disabled MMR on the other hand, unwanted mutations are generated at high frequencies, making analysis of the generated strains problematic. This issue with MAGE can however be efficiently avoided by transiently expressing dominant-negative mutator alleles only when needed (18). With high frequencies of successful recombinants, no need for mutator strains, and selection at every step, our method may overcome these shortcomings, although MAGE may still be the method of choice if many different mutations are to be combined in many different combinations. If screening is thorough during MAGE, all possible mutants can be found within a few transformation cycles. Missing combinations of mutants can either be due to insufficient screening or due to epistasis between different mutations. With the Dup-In methodology, such effects are taken into account by step-wise introduction of one mutation after the other.
General design considerations
Three general guidelines that need to be considered when designing Dup-Ins follow below. In many cases, one may need to decide on a good compromise between conflicting rules. Some hypothetical examples of Dup-In designs are illustrated in Figure 5.
Figure 5.

Strategies for Dup-In design. Thin black lines under the gene maps indicate the duplicated region and double-headed arrows indicate the distances between the cassette and the mutation. (A) Hypothetical genomic region containing a mutation (marked by an asterisk) in the gene C in the BCD operon. (B) Dup-In that leaves one intact and one truncated copy of gene C, with one marker-chromosome junction in gene D and another in C. Note that expression of the downstream gene D may be disrupted by the presence of the cassette. (C) Dup-In that leaves one intact and one truncated copy of the BCD operon. (D) Dup-in with both junctions outside, but on the same side of the BCD operon. (E) Dup-in with both junctions outside, but on different sides of the BCD operon. This leaves two copies of the entire operon. (F) Dup-in that duplicates gene C. (G) Internal Dup-In in gene C. No intact gene copy is present. (H) Large Dup-In with one junction much further away than the other.
Place the marker close to the mutation
When the Dup-Ins are going to be used as linked markers for co-transducing nearby point mutations, it is desirable to keep the marker-chromosome junctions close enough (<1 kb if possible) to the mutation to ensure a high frequency of co-inheritance. This will minimize the need for screening among the transductants.
Keep the duplication small
For genes that are non-essential and in operons that do not have any essential genes, a relatively small duplication (∼0.5–1 kb) either encompassing the mutation or located just next to the mutation would keep the co-transduction frequency high while still allowing a high frequency of segregation (Figure 5B, F and G). As a Dup-In inside a gene or operon is most likely disruptive, this construction may not be possible when the gene itself or downstream genes in an operon are essential. In such cases, it is possible to place the duplication either completely outside the gene or operon, or at the beginning or the end of the gene or operon, in such a way that there is one completely intact copy of the gene or operon (Figure 5B–E).
If the mutation is inside the duplicated area, keep both junctions close to the mutation and screen for the mutant allele after segregation. If the mutation is within the duplicated area (Figure 5E–H), some of the resulting transductants will be heterozygous with one wild-type copy and one mutant copy. Such heterozygous clones will segregate into both of the parental genotypes, with frequencies that depend on the distances between the site of the mutation and the chromosome-marker junctions. It is therefore important to screen for the mutant allele after segregation, as screening before segregation may be misleading. Heterozygotes with a duplication where the mutation is far from one junction but close to the other (Figure 5H) are likely to segregate to the wild-type more often than to the mutant allele. Transductions with such a Dup-In (e.g. Figure 5H) would often form heterozygotes with the wild type allele in the more distant copy (e.g. the left copy of the C gene in Figure 5H), and would segregate to leave the more distant copy more often than the closer copy. Because of this, after segregation, the clones can behave as if the mutation co-transduces only with the more distant of the chromosome-marker junctions.
Using Dup-Ins to temporarily inactivate a gene
A Dup-in with both junctions within a gene is likely to disrupt the function of that gene (Figure 5G). This can be advantageous in cases when an easily revertible knockout is needed. In the presence of antibiotic, the vast majority of the cells in a population would retain the Dup-In and lack a functional copy of the gene. Once the gene is needed again, the Dup-In is easily lost by sucrose selection, and a functional gene copy is restored. We have not specifically tested this idea, but we have demonstrated the principle with our ΔaraBAD::syfp2 and ΔrhaBAD::syfp2 Dup-Ins (Figure 4). As long as the Acatsac1 cassette is present, the syfp2 gene is interrupted, and addition of arabinose or rhamnose does not induce any detectable fluorescence. Allowing the Dup-Ins to segregate restores the syfp2 gene, allowing arabinose- or rhamnose controllable expression of YFP (Supplementary Data).
Mechanism of recombination
We have not attempted to elucidate the mechanism involved in Dup-In formation during recombineering, but it is clear that the mechanism at work is not the same for formation of duplications as for generation of simple insertions or replacements. Single-strand oligo mediated repair (ssOR) and formation of insertions using relatively short dsDNA cassettes (‘ends-out’ recombineering) show a strong lagging-strand bias and evidence for a single-stranded recombination intermediate (19–21). This single-stranded intermediate is the result of complete degradation of one strand of the incoming dsDNA by λ Exo (a 5΄→3΄ exonuclease) while the other strand is left completely intact. With increasing insertion size, transformation frequencies decrease while the strand bias is gradually lost, indicating a gradual transition from an efficient mechanism with lagging strand bias to another less efficient mechanism lacking this bias. Similarly, ‘ends-in’ recombineering (resulting in gap-repair cloning or duplication of the sequence corresponding to the homology arms of the recombineering cassette) does not show any strand bias unless only a single strand is provided (19,22). The reason for the absence of strand bias could indicate a mechanism that uses both strands for the recombination. Our observation that recombineering with two partially overlapping fragments is apparently as efficient as recombineering with the corresponding complete large fragments may indicate that recombineering with large fragments always occurs between several smaller fragments. If this is the case, we imagine that the mechanism involves the 3΄-ends of both DNA strands from the incoming DNA, but not necessarily associated in the same dsDNA molecule prior to recombination. For formation of a duplication, we assume that the recombination event occurs by the ‘bridging’ of two newly replicated sister chromosomes as depicted in Figure 1C. Due to the at least superficial similarity between gap-repair cloning and Dup-In formation, perhaps a Dup-In is formed as an intermediate during gap-repair cloning. With such a mechanism for gap-repair cloning, the integrated copy of the plasmid would be between identical, directly repeated copies of the target sequence. Interference between plasmid replication and chromosome replication could be enough to select segregants that have circularized the plasmid.
Here, we have expanded the λ Red recombineering toolkit with a new utility. We have demonstrated that Dup-In methodology can be used for transferring mutations between strains. Multi-step strain constructions can be made considerably faster, easier, and less problematic by using Dup-Ins compared to other tools. Dup-Ins can be used for replacing native genes or inserting non-native genes without the need to leave any selection cassettes or scar sequences behind in the final strains. In addition, we suggest that Dup-Ins can be used for generating easily revertible gene knockouts. Our observation that recombineering with overlapping PCR fragments is as efficient as single fragment recombineering may give clues to the mechanisms involved in λ Red mediated recombination, and may inspire new ideas for how recombineering can be used.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Jennifer Jagdmann for her help in improving the language in this manuscript, and to Douglas Huseby for construction of the kan-sacB-T0, Ykansac1 and Akansac1 cassettes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Swedish Research Council (Vetenskapsrådet; grant number 2014-4479 to J.N. and 2012-2186 to D.I.A.). Funding for open access charge: The Swedish research council (Vetenskapsrådet).
Conflict of interest statement. None declared.
REFERENCES
- 1. Datsenko K.A., Wanner B.L.. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Datta S., Costantino N., Court D.L.. A set of recombineering plasmids for Gram-negative bacteria. Gene. 2006; 379:109–115. [DOI] [PubMed] [Google Scholar]
- 3. Yu D., Ellis H.M., Lee E.C., Jenkins N.A., Copeland N.G., Court D.L.. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:5978–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Knöppel A., Näsvall J., Andersson D.I.. Compensating the fitness costs of synonymous mutations. Mol. Biol. Evol. 2016; doi:10.1093/molbev/msw028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Herring C.D., Glasner J.D., Blattner F.R.. Gene replacement without selection: regulated suppression of amber mutations in Escherichia coli. Gene. 2003; 311:153–163. [DOI] [PubMed] [Google Scholar]
- 6. Blank K., Hensel M., Gerlach R.G.. Rapid and Highly Efficient Method for Scarless Mutagenesis within the Salmonella enterica Chromosome. PLOS One. 2011; 6:e15763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tischer B.K., von Einem J., Kaufer B., Osterrieder N.. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. BioTechniques. 2006; 40:191–197. [DOI] [PubMed] [Google Scholar]
- 8. Blomfield I.C., Vaughn V., Rest R.F., Eisenstein B.I.. Allelic exchange in Escherichia coli using the Bacillus subtilis sacB gene and a temperature-sensitive pSC101 replicon. Mol. Microbiol. 1991; 5:1447–1457. [DOI] [PubMed] [Google Scholar]
- 9. Skorupski K., Taylor R.K.. Positive selection vectors for allelic exchange. Gene. 1996; 169:47–52. [DOI] [PubMed] [Google Scholar]
- 10. Miller V.L., Mekalanos J.J.. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 1988; 170:2575–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schmieger H. Phage P22 mutants with increased or decreased transduction abilities. Mol. Gen. Genet. MGG. 1972; 119:75–88. [DOI] [PubMed] [Google Scholar]
- 12. Ikeda H., Tomizawa J.. Transducing fragments in generalized transduction by phage P1. J. Mol. Biol. 1965; 14:85–109. [DOI] [PubMed] [Google Scholar]
- 13. Jensen P.R., Hammer K.. The sequence of spacers between the consensus sequences modulates the strength of prokaryotic promoters. Appl. Environ. Microbiol. 1998; 64:82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koskiniemi S., Pränting M., Gullberg E., Näsvall J., Andersson D.I.. Activation of cryptic aminoglycoside resistance in Salmonella enterica. Mol. Microbiol. 2011; 80:1464–1478. [DOI] [PubMed] [Google Scholar]
- 15. Lovett S.T., Hurley R.L., Sutera V.A., Aubuchon R.H., Lebedeva M.A.. Crossing over between regions of limited homology in Escherichia coli: RecA-dependent and RecA-independent pathways. Genetics. 2002; 160:851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang H.H., Isaacs F.J., Carr P.A., Sun Z.Z., Xu G., Forest C.R., Church G.M.. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009; 460:894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Costantino N., Court D.L.. Enhanced levels of λ Red-mediated recombinants in mismatch repair mutants. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:15748–15753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nyerges Á., Csörgő B., Nagy I., Bálint B., Bihari P., Lázár V., Apjok G., Umenhoffer K., Bogos B., Pósfai G. et al. . A highly precise and portable genome engineering method allows comparison of mutational effects across bacterial species. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:2502–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maresca M., Erler A., Fu J., Friedrich A., Zhang Y., Stewart A.F.. Single-stranded heteroduplex intermediates in λ Red homologous recombination. BMC Mol. Biol. 2010; 11:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mosberg J.A., Lajoie M.J., Church G.M.. Lambda red recombineering in Escherichia coli occurs through a fully single-stranded intermediate. Genetics. 2010; 186:791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ellis H.M., Yu D., DiTizio T., Court D.L.. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:6742–6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reddy T.R., Fevat L.M.S., Munson S.E., Stewart A.F., Cowley S.M.. Lambda red mediated gap repair utilizes a novel replicative intermediate in Escherichia coli. PLOS One. 2015; 10:e0120681. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.