


Abstract
Multi-target regulators represent a largely untapped area for metabolic engineering and anti-bacterial development. These regulators are complex to characterize because they often act at multiple levels, affecting proteins, transcripts and metabolites. Therefore, single omics experiments cannot profile their underlying targets and mechanisms. In this work, we used an Integrative FourD omics approach (INFO) that consists of collecting and analyzing systems data throughout multiple time points, using multiple genetic backgrounds, and multiple omics approaches (transcriptomics, proteomics and high throughput sequencing crosslinking immunoprecipitation) to evaluate simultaneous changes in gene expression after imposing an environmental stress that accentuates the regulatory features of a network. Using this approach, we profiled the targets and potential regulatory mechanisms of a global regulatory system, the well-studied carbon storage regulatory (Csr) system of Escherichia coli, which is widespread among bacteria. Using 126 sets of proteomics and transcriptomics data, we identified 136 potential direct CsrA targets, including 50 novel ones, categorized their behaviors into distinct regulatory patterns, and performed in vivo fluorescence-based follow up experiments. The results of this work validate 17 novel mRNAs as authentic direct CsrA targets and demonstrate a generalizable strategy to integrate multiple lines of omics data to identify a core pool of regulator targets.
INTRODUCTION
One of the major challenges of understanding cellular physiology is to decipher the mechanisms and circuitry of regulatory networks (1). Although many high-throughput tools (e.g. proteomics, transcriptomics, metabolomics) have become available to identify and study the effects of global regulators (2,3), on their own these studies often leave unanswered questions regarding the biological importance of any trends observed. For instance, one of the major challenges of analyzing these large overarching regulatory systems is determining the repertoire of targets. Given that a great number of proteins, metabolites, and RNAs vary in response to environmental changes, any single omics technique may produce false positives and any single growth condition may miss gene expression patterns that happen under other conditions (4). As such, two omics studies of the same type performed under different growth conditions can produce different results, making it difficult to draw solid conclusions about the targetome (5,6). To alleviate these problems, multiple omics datasets can be integrated to reduce false positives and elucidate direct targets and the true core responses of the network (7,8). Nevertheless, the use of omics studies for this comprehensive characterization of global response pathways is only beginning to be realized (3,9,10).
Many of the challenges involved in characterizing the targets and responses of a global regulatory network are embodied in the widely conserved bacterial carbon storage regulatory (Csr) system (11–13). The Csr system is one of the few known bacterial sRNA–protein regulators (besides Hfq (14)). It affects a wide array of genes (15–17) that have significant impact on bacterial virulence and metabolism (11). In Escherichia coli, the global regulatory capabilities of the Csr system have attracted attention for their possible use in metabolic engineering applications (18,19) and in engineering complex dynamic control systems (20–22). Likely as a result of these important cellular roles, the Csr system forms a complex cellular network of targets and responses. The Csr system (i) is affected by environmental factors and is sensitive to changes in growth phase, (ii) depends on multiple RNAs and proteins, (iii) affects a large collection of genes and (iv) employs diverse regulatory mechanisms. Importantly, these features suggest that complete behavior of the Csr system cannot be accurately captured by a single omics approach or time point.
In this study, we utilize an Integrative FourD omics approach (INFO) that consists of collecting and analyzing systems data using (i) multiple genetic backgrounds and (ii) multiple omics approaches (transcriptomics, proteomics and coimmunoprecipitations) to evaluate changes in gene expression after (iii) imposing a particular environmental stress to amplify the regulatory features of the network throughout, (iv) multiple time points to monitor dynamic regulation on a global scale (Figure 1A). We use this approach to characterize the Csr system and the cellular responses it elicits starting from the Csr system core components.
Figure 1.

Investigating the Escherichia coli carbon storage regulatory system. (A) The Integrated 4D omics (INFO) approach. This experiment focuses on integrating omics measurements to determine the targets and circuitry of a regulatory network. The four arms of the approach are using multiple mutant strains, multiple time points, multiple omics analyses and a stress to trigger the regulatory system. (B) An illustration of the basic components of the carbon storage regulatory (Csr) system. CsrA binds to a wide variety of cellular mRNAs, which affects target transcript and/or protein levels. CsrA is antagonized by CsrB and CsrC sRNAs, which bind to CsrA and prevent it from interacting with its targets. CsrD promotes degradation of CsrB and CsrC via EIIAGlc activation. The BarA and UvrY two-component system senses environmental stimuli such as formate or acetate levels and activates csrB and csrC transcription.
The Csr system of E. coli has four basic components: the CsrA and CsrD proteins and the CsrB and CsrC small RNAs (23–26) (Figure 1B). CsrA regulates many cellular processes by binding to the 5΄ untranslated region (UTR) of mRNAs and altering their translation, stability, and/or transcription termination (27–32). CsrB and CsrC bind to and sequester CsrA from its RNA targets, as observed in vivo and in vitro (33,34). In association with EIIAGlc, CsrD mediates RNase E cleavage and degradation of CsrB and CsrC (26,35,36). As a highly dynamic network, the Csr system responds to a variety of environmental conditions, such as the availability of nutrients (5), pH (37) and acetate levels (38) through these conditions’ effects on the BarA/UvrY two component system. This system activates csrB/csrC transcription (39,40) in the presence of glucose through the nutrient's effect on CsrD activity (35,36).
Computational predictions (6,41,42), mass spectrometry analyses, and omics studies (5,6,17,43) have suggested a large collection of mRNAs (∼1500) that are potentially regulated through CsrA interaction with an ANGGA consensus motif (42,44). However, these studies only partially overlap and therefore leave the full extent of the network unclear. Differences in hypothesized CsrA targets from these studies have been attributed, in part, to differences in environmental conditions and in growth phases (5,6,17) as it is understood that levels of CsrA, CsrB and CsrC change as the cell transitions from exponential to stationary phase (25,45). As such, we wanted to understand if Csr control of genes was environmentally dependent, specifically if there were sets of condition-specific Csr-controlled genes and/or a core set of genes that displayed Csr system control across varying conditions. Additionally, we aimed to assess sequence features of these genes, particularly ANGGA motifs, to aid in future prediction of CsrA regulated targets.
In this work, we used the INFO approach to identify potential CsrA targets, map their dynamic changes in expression during carbon starvation, and develop mechanistic insights about the roles of CsrA in regulation. We performed proteomics, transcriptomics, and high throughput sequencing crosslinking immunoprecipitation (HITS-CLIP) experiments to track the network dynamically (over the course of 5 h) in response to a carbon starvation-imposed stress where the Csr system is expected to play an important role (5,35,46). This characterization was performed using a csrA transposon mutant strain and a csrB and csrC double deletion strain to determine genes significantly affected by these components. Although a variety of recent multi-omics studies have tracked cellular responses to environmental stress (3,9,47), few studies have examined the contribution of a regulator to that stress response.
An important aspect of our study was to obtain a newly hypothesized targetome based on integrating multiple analyses that generated a total of 126 datasets. Specifically, we demonstrated that the use of integrated dynamic omics experiments expands the value of systems studies to uncover large numbers of CsrA targets that are difficult or impossible to identify with more traditional (lower throughput) strategies and cannot be mechanistically probed with single high-throughput approaches. We identified 136 potential direct CsrA targets, of which 86 were previously proposed based on computational (42) or experimental analysis (5,6) and 50 are novel. Using two different fluorescence-based in vivo assays, we validated 19 of our proposed target genes as true direct CsrA targets in E. coli, adding to the existing pool of direct mRNA targets that includes glgC, cstA, nhaR, sdiA, pnp, pgaA, hfq, flhD, moaA, csrA, relA, ycdT and dgcZ. Collectively, these results substantiate the use of the INFO approach in identifying the targetome of a complex global regulatory system and suggesting potential mechanisms by which individual genes are controlled.
MATERIALS AND METHODS
Stress experiment
Single colonies of E. coli strains CML 377, CML 378 and CML 379 (Supplementary Table S1) were generated using E. coli MG1655 as a base strain (Supplementary Materials and Methods). These strains were inoculated into 5 ml of Luria-Bertani (LB) broth (Becton Dickinson) (48) and cultured at 37°C overnight. 330 μl of overnight culture was added to 33 ml of M9 plus a final concentration of 0.2% glucose and then cultured overnight at 37°C, shaking at 200 rpm (49). Biological triplicates of a single strain were grown on the same day.
The 33 ml culture was added to a Fernbach flask containing 1.65 l of M9 plus 0.2% glucose medium. We chose to perform the experiments in M9 minimal media because it has well defined growth properties and composition. This media choice was different than many previous studies, which have focused on characterizing the Csr system in LB and Kornberg media (23,66). Each culture was grown to an optical density at 600 nm (OD600) of 0.6. We noted that strain CML 379 takes about 10 h longer than CML 377 and CML 378 to reach OD 0.6.
Once the cultures reached OD of 0.6, a carbon starvation stress was imposed by resuspending each culture in glucose free media and incubating these cultures at 37°C (see Materials and Methods, Large scale sample collection). Cell samples were collected ten minutes prior to this stress and at 0, 30, 60, 180 and 300 min after the stress (t-10, t0, t30, t60, t180, t300).
Large scale sample collection
Samples were prepared at the t-10 time point as follows. Cells from 275 ml of culture were collected by centrifugation in 50 ml conical tubes at 4000 rpm (4°C) for 8 min, flash frozen on dry ice, and stored at –80°C for further processing (100 ml for mass spectroscopy, 40 ml for RNA-seq, 135 ml for crosslinking). For the t0 time point, 275 ml of each culture was collected under the same conditions and resuspended in 50 ml of glucose-free M9, before a second centrifugation at 4000 rpm for 8 min and subsequent flash freezing.
The remaining 1.1 l of every culture was divided equally into centrifuge bottles and centrifuged at 5000 rpm (25°C) for 10 min in a super-speed centrifuge (Thermo Sorvall RC 6 Plus). Cell pellets were resuspended in 12.5 ml glucose-free M9. Resuspensions were added separately to four 1-l flasks each containing 262.5 ml glucose-free M9, and were incubated at 37°C. At each remaining time point (t30, t60, t180, t300), a flask containing 275 ml of culture was pelleted, flash frozen, and stored in the same manner as the t-10 sample for cross linking, mass spectrometry and transcriptomic experiments.
Cell samples for mass spectrometry analysis were resuspended in 50 μl of Tris–HCl (pH 8.0), lysed, digested with trypsin and cleaned up according to previously published protocols (10). Prepared samples were sent to the University of Texas ICMB proteomics facility for LC–MS/MS analysis using a Dionex UPLC purification followed by tandem mass spectrometry (Thermo Orbitrap Elite Mass Spec) similar to previously published protocols (10).
To prepare samples for RNA-seq, total RNA was isolated according to a previously published protocol (50) and concentrations were quantified using a NanoDrop (Thermo Scientific). 1 μg of purified RNA was treated with DNase I (RNase-free) (NEB) and repurified with a second total-RNA extraction. Purified samples were submitted to the University of Texas ICMB Genomic Sequencing and Analysis Facility (GSAF) for RNA library preparation and Next-Gen sequencing (Illumina NextSeq 500 platform).
Crosslinking immunoprecipitation was used in combination with high-throughput sequencing (HITS-CLIP) to analyze the RNA bound to CsrA (51). A chromosomally-encoded CsrA-FLAG fusion protein present in strain CML 377 was used to purify CsrA–RNA complexes from the cell. At each time point, 135 ml sample was taken from the larger culture and crosslinked with formaldehyde (0.5% final concentration) and then processed according to a previously published protocol with three minor changes (52). First, samples were temporarily stored at –80°C for later processing after crosslinking and glycine quenching rather than the post-centrifugation wash step as done in the published method. Second, samples were sonicated (Microson XL2000 Ultrasonic Homogenizer) with four five-second pulses as opposed to 40 half-second pulses. Finally, after total-RNA extraction and DNaseI digestion, samples were purified again with ethanol and sent to the UT GSAF facility for RNA library preparation and Next-Gen sequencing (Illumina NextSeq 500 platform).
Northern and western blotting
Northern and western blots were performed according to established techniques to determine if CsrA or CsrB responded to the carbon starvation stress (see Supplementary Materials and Methods) (33,50).
Omics data analysis
Normalization, scaling, and analysis of proteomics, transcriptomics and HITS-CLIP data are described in the Supplementary Materials and Methods.
Classification of Csr system impacted genes
Genes impacted by the Csr system were grouped on the basis of proteomics and transcriptomics data by means of a classification scheme. The basic premise of this classification was that true Csr system targets show different patterns in their gene expression levels between wild type and mutant strains. Given the inherent difficulty of determining what a meaningful difference is in dynamic expression data, we performed four different analyses to search for meaningful differences. First, we started by visually identifying a pool of proteins from our proteomics data whose standard deviations did not overlap in at least four of the time points tested. Second, to evaluate total RNA transcriptomics patterns, we used DESeq2 (53) to determine if a given transcript responded differentially to the stress in the wild type versus the mutant strains. Employing the negative binomial distribution-based Wald test, we considered differences in expression of a gene between a Csr system mutant strain and the wild type strain significant if the adjusted P-value was < 0.02 and the log base 2 fold change > 0.5 or < –0.5 (see Supplementary Materials and Methods). Concurrently, we also used a Fisher exact test (P-value < 0.05 at four time points, see Supplementary Material and Methods) to determine if expression of a given protein was statistically different in the wild type as compared to either mutant strain. Additionally, CsrA HITS-CLIP data were evaluated to identify genes that could have direct interactions with CsrA.
Using the proteomics visual analysis (based on standard deviations) and DESeq2 analysis of total RNA transcriptomics patterns, we separated the genes into five classes as described below. We additionally sub-classified genes as statistically significant at the protein level and ‘associated with CsrA’ based upon the Fisher exact test and analysis of the HITS-CLIP experiment, respectively (see Supplementary Materials and Methods). This multi-layered classification allowed us to develop a pool of genes that would potentially be a mixture of direct and indirect targets of the Csr system. We also wanted to identify genes that are only influenced by Csr components pre-stress. We identified pre-stress impacted genes by examining the mass spectrometry data at time points –10 and 0 min before stress and looking for standard deviation differences between the csrA::kan and the wild type strains that were present only at those two time points. We then binned these genes with a pre-stress designation.
Class I
Classified gene's protein levels are higher in the csrA::kan mutant than in the wild type strain for at least four time points, according to non-overlapping standard deviations. Additionally, the gene's transcript levels are significantly higher in the csrA::kan mutant strain than in the wild type strain by DESeq2 (P-value < 0.02 and log base 2 fold change > 0.5).
Class II
Classified gene's protein levels are higher in the csrA::kan mutant than in the wild type strain for at least four time points, according to non-overlapping standard deviations.
Class III
Classified gene's protein levels are lower in the csrA::kan mutant than in the wild type strain for at least four time points, according to non-overlapping standard deviations. Additionally, the gene's transcript levels are significantly lower in the csrA::kan mutant strain than in the wild type strain by DESeq2 (P-value < 0.02 and log base 2 fold change < –0.5).
Class IV
Classified gene's protein levels are lower in the csrA::kan mutant than in the wild type strain for at least four time points, according to non-overlapping standard deviations.
Class V
Classified gene's protein levels are lower or higher in the ΔcsrBΔcsrC mutant than in the wild type strain for at least four time points, according to non-overlapping standard deviations. Classified gene's transcript levels are significantly different in the ΔcsrBΔcsrC mutant than the wild type strain by DESeq2 with a P-value < 0.02 and the log base 2 fold change > 0.5 or < –0.5 such that the direction of the relationship is the same at the transcript and protein levels. Additionally, the classified gene does not have differences at the protein level, according to standard deviations, for at least three time points when wild type expression is compared to the csrA::kan mutant strain.
Fluorescent reporter assays
We performed fluorescent reporter translational assays to more directly determine if the 5΄ UTRs of potential direct CsrA targets (genes classified I–IV and associated with CsrA in HITS-CLIP experiments) could mediate changes in gene expression in response to CsrA. The reporter consisted of the 5΄ untranslated region (UTR) of the gene of interest and 100 nucleotides of the coding sequence (CDS) of the gene attached in frame to a GFP reporter (46). Given the amount of coding sequence included in this design, this method was likely to have lower success rates in studying membrane and secreted proteins. Design and construction of 5΄ UTRs is described in the Supplementary Materials and Methods.
UTR-GFP fusion plasmids (Supplementary Table S2) were paired with a second plasmid, pHL600, which contained an inducible CsrA gene (Supplementary Table S2). The two plasmids for this system were expressed in the CML 577 strain (Supplementary Table S1) (46). Cells were grown overnight and inoculated into fresh LB media. These cells were grown at 37°C with agitation to an approximate OD600 of 0.3. At this time, half the samples were induced to express CsrA by adding IPTG to a final concentration of 0.1 mM. Fluorescence was measured 3 h later by flow cytometry. Measurements were taken after 3 h to limit visualization of indirect impacts of CsrA induction while allowing enough cell divisions to dilute otherwise stable GFP concentrations.
To determine the relative amount of CsrA repression or activation, a ratio of GFP reporter expression was calculated:
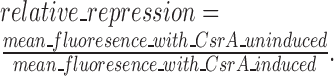 |
Constructs were tested in biological duplicate to verify the observed trends. To evaluate significance, we only counted UTRs as impacted by CsrA if (i) their average fluorescence was above background (> 10 au), and (ii) they appear significant using one tailed heteroscedastic student t-test (P-value < 0.05). Although the experiments were performed in duplicate, our negative control fecA and a variety of other genes do not show significant changes in a t-test indicating that this analysis is capturing real trends.
Tri-fluorescence complementation assays (TriFC)
TriFC experiments were performed according to previously published methods (33) and modified as described in the Supplementary Materials and Methods.
RESULTS
Designing a four dimensional systems profiling approach
Our first goal was to assess the time and stress dimensions of our analysis. To study the Csr system in the transition between two physiologically important states, we imposed a starvation stress on the cells by culturing them in M9 minimal media with 0.2% glucose to mid log-phase (OD600 = 0.6) followed by immediate resuspension in an equal volume of M9 lacking glucose. We then tracked the stress response of CsrA protein and CsrB RNA expression levels over six total time points (Supplementary Figure S1), two before the stress (t = –10 and 0 min) and four post-stress (t = 30, 60, 180 and 300 min). This total time span of 300 min is similar to the timescale of classic proteomics starvation experiments (54,55). We observed that CsrA levels did not change substantially over this timeframe as detected by western blotting analysis (Supplementary Figure S1). This result is consistent with reported observations of CsrA protein stability (46) and with previous reports of only a modest increase in CsrA levels when reaching stationary phase (45). In contrast, CsrB exhibited an observable decrease in cellular levels within 30 min post-stress, as measured by northern blotting analysis (Supplementary Figure S1). We assumed that CsrC would follow a roughly similar trajectory to CsrB (25) given the similarity of the CsrB/C synthesis and turnover pathways. Given the reported low expression levels of CsrD (26), we suspected that this protein might not change drastically in response to stress. These initial assumptions were later confirmed with data from our proteomics and transcriptomics studies, where we profile the Csr components (Supplementary Figure S2).
In addition to the stress and time scale analyses, we integrated two other dimensions in this approach: the use of multiple omics techniques and genetic backgrounds. We conducted proteomics and transcriptomics studies to identify genes that were affected (directly or indirectly by components of the Csr system) at either the transcriptional or translational levels. Furthermore, we used CsrA HITS-CLIP experiments to identify mRNA transcripts that interacted directly or indirectly with the CsrA protein. We used E. coli K-12 MG1655 and two mutants of this strain: one lacking the Csr sRNAs (ΔcsrBΔcsrC) and the second with a transposon inserted at codon 51/61 of the CsrA protein (23). It is referred to hereafter as the csrA::kan strain. The original csrA::kan transposon mutant strain (27) has been studied extensively, because deletion of csrA causes a severe growth defect (56) that significantly affects phenotypes (23). The ΔcsrBΔcsrC and csrA::kan transposon mutants were compared to an E. coli MG1655 strain containing a chromosomally-encoded CsrA-FLAG tagged gene. Note that for the purposes of this study, the CsrA-FLAG tagged MG1655 strain is referred to as wild type. The genetic dimension of this systems profiling approach was designed to distinguish the carbon starvation stress response from the pleiotropic CsrA-dependent cellular response by directly altering CsrA or indirectly affecting its activity by eliminating the CsrB and CsrC sRNAs. With all four design elements considered, we conducted experiments that involved the collection of growth rates for these strains (Supplementary Figure S3) and 126 total samples for proteomics, RNA-seq and CsrA HITS-CLIP analyses.
Resolution of carbon starvation and Csr system dependent changes in gene expression
To differentiate genes that responded to the Csr system from genes that responded generally to the carbon starvation stress, we performed a two-way analysis of variance (ANOVA) on the proteomics data using the strain and the time points, to account for the stress, as the two factors in the analysis. We expected that decoupling these effects at the proteome level would be most relevant due to the post-transcriptional nature of CsrA regulation. The ANOVA identified 1903 proteins that responded significantly (P-value < 0.05) to the genetic differences in the Csr network, the time points, or an interaction of these two factors (Supplementary Table S3; P-values for all proteins given in Supplementary Table S4). This eliminates ∼50% of the total 3800 unique proteins detected with high confidence (5% FDR) in these samples. They were not included in further analysis because their expression changes were determined to be mediated independently from effects of carbon stress or the Csr system. Within the remaining 1903, 88 failed to respond to the strain-imposed differences in the Csr regulatory pathway and were attributed to pure carbon stress responses. Several hundred proteins were impacted by both strain and stress, indicating that the Csr system and the stress were impacting a partially overlapping set of genes. Many of the proteins that responded over time in these analyses have been previously linked to E. coli carbon starvation stress response in literature (57–59). After eliminating the genes that did not show a Csr-dependent response in the ANOVA, we sorted the remaining 1815 genes into different patterns associated with Csr regulation.
Systems level classification of the Csr targetome by integrative FourD profiling
The objective of our pattern classification was to group genes that exhibited similar responses at the protein and transcript levels in the wild type strain compared to the csrA::kan and the ΔcsrBΔcsrC mutant strains. Since CsrA regulates mRNAs by a variety of mechanisms (27,28,30,32), we expected to observe a variety of distinct dynamic patterns. Supported by consistent and complementary proteomics and transcriptomics dynamic signatures, we defined five response categories (Materials and Methods, Classification of Csr system impacted genes) containing 255 genes. We determined that, of the remainder of the 1815 Csr-dependent detected genes from the ANOVA analysis, 1130 genes had no classifiable pattern and 430 were present at levels that were too low in the proteomics data to infer meaningful patterns. The five profiled classes (I–V) are summarized in Supplementary Figure S4. All genes classified from the omics data are referred to hereafter as ‘classified genes’. Proteomics and transcriptomics profiles of all classified genes are presented in Supplementary Figure S5 (see Supplementary Table S5 for list of classified genes and Supplementary Table S6 for specific DESeq2 results).
We also sub-classified genes based on HITS-CLIP data as not all classified genes were expected to directly interact with CsrA. Although not definitive evidence of direct CsrA–mRNA interactions, an association with CsrA in HITS-CLIP analysis suggested genes to serve as prime candidates for follow-up testing. As proof of principle, we confirmed that well characterized CsrA mRNA targets (glgC, cstA, hfq, flhD, sdiA, moaA, nhaR, pnp, pgaA, csrA, relA, ycdT and dgcZ) displayed a consistent signature in the HITS-CLIP experiments. All of these mRNAs were confirmed to interact with the CsrA protein by in vitro binding analyses in previous studies (27–30,60–65). The normalized abundance of 10 of these RNAs ranked in the top 35th percentile in the abundance of all transcripts identified in at least two time points in the HITS-CLIP experiments. Using this signature as a metric, we defined genes as ‘associated with CsrA’ if their transcripts were found in the top 35th percentile of all the transcripts identified in the HITS-CLIP studies in at least two time points (Supplementary Table S5). Although this designation may produce false positives or false negatives when taken by itself, we reasoned that when used in conjunction with the other omics data, this metric would identify genes that are most likely valid CsrA mRNA targets. Henceforth, we refer to genes of classes I-IV that are associated with CsrA in HITS-CLIP experiments as ‘potential direct CsrA targets.’ Altogether, 136 of the 255 classified genes fall into the latter category (Supplementary Table S5, Supplementary Figure S5). Additionally, we sub-classified genes based on Fisher exact test analysis as ‘classed significantly at the protein level’ if they met all the protein-level classification requirements executed with Fisher exact tests (P-value < 0.05) rather than the non-overlapping standard deviation metric (Supplementary Table S7, Materials and Methods, Classification of Csr system impacted genes). We tracked these sub-classified genes throughout the rest of the analyses and follow up assays to assess the impact a stringent protein-level statistical classification measure could impart in integrated analysis of dynamic multi-omics data. In total, 42 class I–IV genes display corresponding class protein-level patterns significantly (Supplementary Table S5). It is important to note that while the five classes were constructed from differences between strains, a variety of the classified genes including, csiD, lsrB, clpB and frdA, also showed a dynamic protein response to the stress, indicating that a subset of Csr influenced genes show dynamic responses to stress as expected. For many of these genes, dynamic responses were also observed on the transcript level. In Supplementary Figure S5, we show all genes that we classified and all other genes within the operon, as recorded in RegulonDB (66) for context when interpreting classified patterns.
Class I
Genes were included in class I if higher protein and transcript expression levels were observed in the csrA::kan mutant relative to the wild type strain (Supplementary Figure S4A). Figure 2A illustrates an example of a potential direct class I CsrA target, the monocistronic gene aidB. In total, we identified 40 class I genes, 23 of which are potential direct CsrA targets and 13 of which show significant class I protein-level patterns by the Fisher exact test. This pool includes novel potential direct CsrA targets, such as aidB and evgA. We also classified several well characterized CsrA targets (e.g. glgC, dgcZ and pgaA) as having a class I pattern. This is consistent with the reported CsrA effect on regulation of these genes, namely that CsrA represses their expression (27,67).
Figure 2.

Genetic responses to Csr components. (A–E) Genes representative of patterns in each class. For each class I–V, we show protein and transcript expression levels of a representative gene. Error bars depict standard deviation of three replicates. Specifically, class I and II class have lower expression of the gene's protein and transcript or just protein respectively in the wild type than the csrA::kan mutant. Class III and IV have the inverse patterns to class I and IV. Class V genes show no recognizable relationship to the csrA::kan mutant; expression effects are observed in the ΔcsrBΔcsrC mutant relative to the wild type.
Class II
Class II genes displayed a strong CsrA dependent response, manifested by higher protein expression levels of the gene in the csrA::kan mutant strain, but no difference in transcript levels in the csrA::kan or the ΔcsrBΔcsrC mutant relative to the wild type strain, Supplementary Figure S4B. One possible mechanism for class II genes is that CsrA inhibits translation of the target gene but does not affect transcript stability as described previously (28). In total, we identified 84 members of this class, including 72 potential direct CsrA targets and 19 genes that show significant protein-level patterns by the Fisher exact test. Notable novel members of this pool of potential direct class II targets are acnA, dcrB and ybeL. As shown in Figure 2B, dacC is an example of a member of this class.
Class III
Class III genes (Supplementary Figure S4C) display lower protein and transcript expression levels in the csrA::kan mutant strain than in the wild type strain. Some possible mechanisms for class III genes include mRNA stabilization by CsrA as shown for flhD (56) and increased RBS availability facilitated by CsrA. In the case of flhD, CsrA binding blocks RNase E mRNA cleavage (32). We were not able to detect the FlhD protein; however, the flhDC transcript displayed increased expression in the presence of CsrA (Supplementary Figure S5). As shown in Figure 2C, pta is an example of a class III potential direct CsrA target which has not been suggested previously in literature. In total, we identified 10 members of this class, all of which are potential direct targets and two of which show significant class III protein-level patterns by the Fisher exact test. It is worth noting that the majority of these potential direct targets were already associated with CsrA (17).
Class IV
Class IV genes displayed lower protein expression, but not lower transcript levels in the csrA::kan strain than in the wild type strain (Supplementary Figure S4D). This pattern has not previously been observed in connection with CsrA regulation, but it is conceivable that CsrA binding could increase protein expression without affecting transcript stability (68). In this class, we identified 54 genes, 31 of which were sub-classified using HITS-CLIP data as potential direct CsrA targets. Eight were sub-classified as showing significant class IV protein-level patterns using the Fisher exact test. An example of a gene in this class is ybiT (Figure 2D). Altogether, 30 members of this potential target pool have not before been hypothesized as CsrA targets. We noted that not as many of these genes were as strongly associated with CsrA in our HITS-CLIP experiments than in the other classes. This could suggest that some of these protein expression patterns were the result of indirect regulation or growth defects caused by the CsrA transposon mutation. To determine if individual genes were indirectly or directly regulated by CsrA, translation fusion and in vivo protein-mRNA interaction assays were performed as described in subsequent sections.
Class V
Class V genes displayed increased or decreased expression levels on both the protein and the transcript levels in ΔcsrBΔcsrC strains, but not in the csrA::kan strain, relative to the wild type strain (Supplementary Figure S4E). The unique dependence on the sRNAs in this class was unexpected given the paradigm that CsrA protein regulates by binding to mRNA transcripts and that the CsrB and CsrC sRNAs sequester the CsrA protein. In total, 67 genes were classified in this category, of which nudK is an example (Figure 2E). Twelve of these genes show significant class V protein-level patterns. Additionally, it should be noted that 55 genes of the 67 genes in this category display a strong association with CsrA in our HITS-CLIP analysis. One potential explanation of the data is that these genes require a higher amount of active CsrA than what is present in the wild type cells grown in our experimental conditions to observe regulatory effects. Under this paradigm, the removal of CsrB and CsrC increases the levels of active CsrA in the cells enough to observe phenotypic differences in this group of genes.
Fluorescent reporter assays to authenticate CsrA regulation of multiple target genes
After generating a list of potential direct CsrA targets, we designed follow up experiments to characterize CsrA-gene interactions in vivo. As a starting point to validate these interactions, we performed translational expression assays to study the impact of CsrA on potential CsrA target genes. This assay was based on the knowledge that CsrA interacts with the 5΄ UTR of its targets to regulate their expression (11). Assuming our potential targets would largely behave the same way, we designed a plasmid that fused the 5΄ UTR of a given gene to a GFP reporter (Figure 3, Supplementary Table S2). This plasmid was transformed into a ΔcsrABCD ΔpgaABCD ΔglgCAP strain (CML 577) along with a second plasmid, pHL600 (Supplementary Table S1, Table S2), which contained an IPTG-inducible CsrA (46). With this experimental setup, we compared the resulting GFP fluorescence of 5΄ UTR-GFP reporters under two conditions: (i) with induction of and (ii) without induction of CsrA. Our hypothesis was that true CsrA targets should display either translational upregulation (consistent with patterns observed for class III and IV) or translational downregulation (consistent with patterns observed for classes I and II) in the presence of CsrA. Importantly, upon quantifying intracellular levels of CsrA relative to the E. coli K-12 MG1655 strain to determine the extent of CsrA overexpression in this experimental system, IPTG-induced levels of CsrA were only ∼2× higher relative to the wild type CsrA (Supplementary Figure S6A). These results indicated that the assay represented only a mild overexpression of CsrA relative to native conditions and was ideal for studying the impact of CsrA under physiological conditions.
Figure 3.

CsrA regulation determined using fluorescent reporter constructs. The fluorescent reporter assay measures fluorescence output of 5΄ UTR-reporter constructs that contain the 5΄ UTR of a candidate CsrA-regulated gene and the first 100 nucleotides of its coding sequence (CDS), expressed as an in-frame GFP fusion, and transcribed from a constitutively active promoter. Flow cytometry is used to determine if CsrA induction has a statistically significant increase or decrease on fluorescence.
We constructed and tested 5΄ UTR-GFP fusions of 132 potential direct CsrA targets (the remaining four were not successfully cloned). We additionally tested 18 constructs of class I–IV genes that showed strong classification patterns but were not associated with CsrA in the HITS-CLIP experiment (Supplementary Table S8). For these experiments, we initially created and tested two 5΄ UTR-GFP fusions as negative controls, phoB and gmk, since CsrA had been shown to not bind these UTRs (5,61). Unfortunately, GFP fluorescence was not observed above background levels in our assay when controlled by these UTRs so we mined our omics data for other UTRs to serve as a control. Among those we considered was the 5΄ UTR of fecA whose protein and transcript levels did not respond to CsrA (Supplementary Figure S5) and was not pulled down in HITS-CLIP experiments (Supplementary Table S5). After cloning this 5΄ UTR into our reporter construct, we observed that the construct did show appreciable GFP expression, but did not respond to CsrA induction so we established this UTR as our negative control (Supplementary Table S8). We also tested well established CsrA targets (sdiA, glgC, hfq, ycdT, pgaA, cstA, and nhaR) from previous literature as positive controls. Although not all of these constructs produced fluorescence above background levels, the fusions that did fluoresce above background showed expected response patterns to CsrA (Supplementary Table S8). With these controls established, we tested samples by growing the cells to OD 0.3, inducing CsrA, and waiting 3 hours prior to fluorescence measurements. We measured the fluorescence using flow cytometry and observed CsrA-dependent upregulation of 10 potential targets, downregulation of 55 potential targets, and no effect on 33 other genes (Figure 4, Supplementary Figure S6B). We also noted 52 additional constructs were tested but they failed to exhibit any fluorescence.
Figure 4.

Differential effects of CsrA on the expression of fluorescent reporter constructs. The number of 5΄ UTR constructs in which CsrA significantly increases, decreases, or has no impact on GFP expression. Colored bars represent genes of class: I, orange; II, blue; III, purple; IV red; V, green (in numerical order, left to right, for each assay result). Flow cytometry data for each individual gene is contained in Supplementary Table S8.
In spite of these limitations, we confirmed that 56% (55/98) of 5΄ UTR constructs that were found to be functional as GFP fusions were affected by CsrA regulation in a manner consistent with their original classification. The disagreement with remaining 44% was largely due to genes which the omics data suggested could interact with CsrA, but did not show a significant shift in our assay. This difference could be a result of indirect effects captured by the omics data or a need for more measurements of some samples to establish statistical significance.
Confirmation of in vivo 5΄ UTR–CsrA interaction using trifluorescence complementation (TriFC)
The ultimate goal of the INFO approach is to identify potential target genes that can be tested for direct regulation by CsrA. The assays and analyses described up to this point (i.e. omics analyses, fluorescent reporter assay) can provide a strong indication that a given gene is functionally affected by CsrA, but do not conclusively show a direct regulatory interaction. To corroborate INFO predictions of potential direct CsrA targets, we conducted additional in vivo RNA–protein interaction experiments. Because few protocols exist for evaluating RNA–protein interactions inside of cells in a high throughput manner, we recently developed an assay for RNA–protein interactions using a three component fluorescence complementation system (TriFC), which was validated specifically in the context of CsrA–CsrB interactions (33). The assay is based on in vivo reconstitution of a split yellow fluorescence protein (YFP) to detect a CsrA–RNA interaction, which has been described previously (33). In brief, the system contains three fusions, a CsrA-nYFP fusion protein, an MS2-cYFP fusion protein, and a transcript containing an MS2-binding domain (MS2BD) conjoined with an RNA of interest. The MS2-cYFP protein fusion strongly interacts with the MS2-binding domain containing transcript and, therefore, if CsrA interacts with the RNA, the two components of the YFP are brought into close proximity, generating a fluorescent signal not present without a CsrA–RNA interaction (Figure 5A).
Figure 5.

TriFC data demonstrate interactions between CsrA and the 5΄ UTR of selected potential direct targets. (A) Schematic of CsrA–mRNA TriFC. The TriFC system consists of two proteins, the CsrA–nYFP fusion and the MS2–cYFP fusion, and one mRNA. The mRNA fusion contains a MS2 coat protein binding site (MS2BD) followed by a 5΄UTR of potential direct CsrA target, the first 100 nucleotides of the gene's coding sequence, and an mstrawberry fluorescence gene in frame with the coding sequence. The MS2–cFYP protein binds to the MS2BD region on the RNA upstream of the 5΄ UTR of interest. If CsrA interacts with the 5΄ UTR sequence, then the two complements of YFP will be in close enough proximity to refold and produce a fluorescence signal. (B) Number of genes in each class whose 5΄ UTR interacts with CsrA in our TriFC assay. We determined that any 5΄ UTR construct with fluorescence higher than the fecA 5΄ UTR negative control and a P-value < 0.1 by t-test was interacting with CsrA (Supplementary Table S9). Blue portion indicates total genes whose 5΄ UTR interacts with CsrA in the TriFC assay AND whose 5΄ UTR shows regulation in the fluorescent reporter assay consistent with its class. Orange portion indicate genes whose 5΄ UTR interacts with CsrA in the TriFC assay but does not have similar fluorescent reporter assay evidence.
The TriFC assay was adapted here to test interactions between 5΄ UTRs and CsrA. Since CsrA is believed to interact primarily with the 5΄ UTR of transcripts, we designed reporter constructs for the TriFC system, which incorporated an MS2 coat protein binding domain followed by the 5΄ UTR of the gene of interest (Figure 5A). In order to confirm that the mRNA was expressed after addition of the 5΄ MS2BD tag, an mstrawberry red fluorescence protein was attached to the end 3΄ end of a control MS2BD-synthetic RBS construct. The control exhibited sufficient red fluorescence, indicating translation of the RBS with the 5΄ MS2BD tag fused. We then focused our attention solely on the yellow fluorescence of the system, as this was indicative of the CsrA-target interaction. To calibrate the assay, we established a no-interaction baseline YFP fluorescence level using the fecA 5΄ UTR tested in our fluorescent reporter assay. Our glgC 5΄ UTR, which is known to have CsrA interactions (46,64), served as a positive control confirming that true direct CsrA targets fluoresce above this baseline. We used this system to test direct interactions between CsrA and 5΄ UTRs.
We identified 31 class I–IV genes as having YFP fluorescence above the established baseline by comparing triplicate fluorescence readings of the test sample to the triplicate readings of the fecA negative control using a one tailed heteroscedastic t-test (P-value < 0.1) (Supplementary Table S9). These results strongly suggest a direct interaction between CsrA and the mRNAs of these 31 genes (Figure 5B, Supplementary Table S9). The TriFC assay is only a positive test for CsrA interaction because multiple factors can affect the refolding of the split YFP fragments, including the unique geometry in which CsrA binds a particular 5΄ UTR. Therefore, it is likely that some mRNAs that do not display a TriFC signal may be direct targets of CsrA binding.
DISCUSSION
In this paper, we demonstrate the genome-wide application of INFO in determining targets of the carbon storage regulatory system. While we present several methods for predicting and characterizing genetic targets of complex regulators, it is important to note that no single test presented would have been adequate to identify potential direct targets of a regulon. The value of this approach is that it weaves together multiple indirect lines of evidence using HITS-CLIP, proteomics and transcriptomics of multiple genetic mutants under the dynamic conditions from an environmentally induced stress to identify potential targets of a global regulatory network. This integration narrowed the CsrA target pool from ∼1600 hypothesized targets in the literature to 136 potential direct CsrA targets that show convincing evidence in our INFO analysis. In the context of previous Csr works, this represents the most information-dense pool of potential targets identified from omics-type datasets (5,17).
To evaluate the novelty of the 136 potential direct CsrA targets identified in this work, we compiled all previous large omics studies and computational predictions on the Csr system that inferred CsrA targets (Supplementary Table S5). We observed that 86 genes (62.3%) had been previously associated with CsrA in the literature and that 50 genes (36.7%) are novel genes that could be associated with CsrA (Figure 6). However, of the 86 genes overlapping with literature, 23 were only previously suggested from computational predictions (6,42) and 31 from only CsrA-pulldown studies (5). The integrated experimental findings of this work add significant evidence for mRNAs of these genes associating with CsrA.
Figure 6.

Genes identified by INFO approach confirm and expand the set of genes impacted by CsrA. Venn diagram comparing genes that are shared between potential direct CsrA targets (yellow circle, left) and hypothesized CsrA literature targets (blue circle, right). Of the 136 candidate CsrA targets, gathered from our omics analysis, 86 confirm potential literature targets (yellow and blue overlap).
After identifying a pool of relevant genes (136) from omics data, our fluorescent reporter assay data provided evidence that CsrA specifically impacted expression from the 5΄ UTR of many of these genes. It also suggested a relative level of CsrA impact on expression, i.e. regulation, of the tested 5΄ UTRs. For instance, our data suggests that CsrA expression has a greater impact on the glgC, glsA and acnA UTRs than it does on evgA and ybaL (Supplementary Table S8). Qualitatively, this assay identifies 5΄ UTRs that are more or less sensitive to CsrA, providing information not only suggesting what is a target, but also a rough hierarchy of which targets are most affected on a genome scale.
Our TriFC experiment allowed us to identify 19 transcripts that we consider bona fide direct CsrA targets, i.e. potential direct CsrA targets that display regulation consistent with their classification in the fluorescent reporter assay and direct interaction with CsrA in the TriFC assay (Table 1). Of these 19, five, glgC, nhaR, dps, proP and ucpA, had previous experimental evidence connecting them with E. coli CsrA. Two of these transcripts, glgC and nhaR, are well studied in their relationship to CsrA and are already known as direct targets (5,6,43,63,67). On the other hand, dps, proP and ucpA were associated with CsrA in a single pulldown study (5). The last of the list, ucpA, has also been associated with CsrA in computational predictions. Six of the 19 transcripts, gstA, ybaL, evgA, gadB, ydhQ and fdoH, were only predicted as CsrA targets in computational studies (6,42), likely for the multiple strong consensus CsrA binding motifs contained in their 5΄ UTRs (42). The current research significantly expands our knowledge of how CsrA impacts these nine targets. The remaining eight, aidB, sdhA, yebE, sucC, glsA, patA, purM and clpB, had not been, to our knowledge, previously suggested in the E. coli CsrA target literature (5,6,16,17,61,69). Our data suggest, in total, 17 new direct CsrA targets (that were fully validated by all our experimental systems), significantly increasing the number of transcripts that have been tested for direct interaction with CsrA. Additionally, patA showed consistent upregulation in both the omics and fluorescent reporter experiments, substantially expanding the number of known transcripts directly upregulated by CsrA (32).
Table 1. Cumulative omics, fluorescent reporter (FR) assay, and TriFC assay evidences for classified genes.
| Omics evidence only | Omics and FR assay disagree | Omics and FR assay agree | Omics and TriFC evidence | Omics and FR assay agree, and TriFC evidence | Total members | |
|---|---|---|---|---|---|---|
| Class I | 14 | 5 | 14 | 0 | 7 | 40 |
| Class II | 38 | 16 | 16 | 4 | 10 | 84 |
| Class III | 2 | 3 | 4 | 1 | 0 | 10 |
| Class IV | 28 | 15 | 2 | 7 | 2 | 54 |
| Total | 82 | 39 | 36 | 12 | 19 | 188 |
Importantly, when these 19 genes are compared to the pool of Csr responsive genes identified just from DESeq2 analysis of transcriptomics data (See Supplementary Materials and Methods, Supplementary Tables S5 and S6), only 8 of 19 direct targets were identified. This observation demonstrates the value of the INFO approach in identifying additional targets not found in traditional single omics approaches. Additionally, considering the sub-classifications based on HITS-CLIP analysis and statistically significant protein-level patterns, only 7 of 19 direct targets have statistically significant protein-level differences in the Fisher exact test. These results suggested that in this study HITS-CLIP experiments were more relevant for identifying true direct CsrA targets.
One of the most interesting novel CsrA targets we identified was the succinate-quinone oxidoreductase gene, sdhA. This gene was consistently impacted by the Csr system in all our experiments, raising the question of how CsrA might be affecting it (Supplementary Figure S5, Tables S5, S8 and S9). We discovered that some of its operon members have been previously associated with CsrA in E. coli (5,17,42,70) and in Salmonella enterica (15). Specifically, a UV crosslinking immunoprecipitation experiment identified a CsrA regulatory element in the 3΄ portion of the sdhD coding sequence in Salmonella. We identified the same regulatory element in the 3΄ portion of the sdhD coding sequence in E. coli. Realizing the proximity (< 100 nucleotides) of the element to the start codon of the following gene on the operon, sdhA, we tested the element's effect on translation of sdhA. Indeed, we observed that the regulatory element inside of the sdhD coding sequence directly interacted with CsrA and affected translation of sdhA in the TriFC and fluorescent reporter assays, respectively. This suggests a mechanism where CsrA, from a single binding site, may directly impact the translation of two proteins simultaneously via different mechanisms. This contrasts with previous examples of CsrA operonic control in which CsrA exhibits regulation by binding in the transcript's 5΄ UTR (27,67) or by binding in the intergenic region between two members of an operon (63). This has significant implications for identification of CsrA regulated transcripts because crosslinking studies may miss regulated transcripts, and suggests a role for CsrA in fine tuning the expression ratios of different proteins from the same operon.
The dual-condition nature of our study played an important role in our understanding of Csr system targets. Imposing a carbon starvation stress revealed a pool of 20 condition-specific CsrA-impacted genes and lead to the identification of two novel CsrA targets, sucC, impacted pre-stress only, and glsA, impacted post-stress only (Supplementary Table S5; Materials and Methods, Classification of Csr system impacted genes). The remaining potential direct targets show Csr system impact in both pre-stress and post-stress conditions; this highlights a core pool of potential CsrA targets relatively insensitive to glucose starvation (Results, Systems level classification of the Csr targetome by integrative FourD profiling). The dynamics also allow us to capture the important role of CsrB and CsrC in altering gene expression profiles when a stress causes growth arrest. For example, the protein levels of class V genes rely on CsrB and CsrC to titrate CsrA when cells cannot dilute CsrA concentration through cell division (46). These results suggest that class V genes may be more sensitive to the levels of CsrB and CsrC than other classified genes because they weakly interact with CsrA. Although the interaction with CsrA is presumably weak, it is surprising that removing CsrB and CsrC results in very strong regulation for genes such as ydcJ, flu and astD (Supplementary Figure S5). These genes represent novel UTR parts for metabolic engineering with the Csr system that are easily titratable because they give strong responses to the CsrB and CsrC regulators.
In addition to identifying new CsrA targets, we were also interested in identifying similar sequence features of these genes that may aid in future prediction of CsrA regulated targets. In addition to analyzing the sequence features of the 5΄ UTRs of all classed genes, we derived an additional approximately 4300 5΄ UTRs sequences (using annotations from regulonDB and the E. coli genome (NCBI accession: CP014225.1)) to compare sequence features of classed genes to the entire genome (66). From this analysis, we observed that class I and II genes have significantly more ANGGA motifs than the ∼4300 non-classified genes; 75% of class I genes have at least one ANGGA motif in their tested sequence, 76% for class II, 50% for class III, 46% for class IV, 61% for class V and 53% for the remaining 4300 genes. This analysis agrees with previous findings that suggest ANGGAs are an effective way to identify CsrA repressed targets (42), with the caveat that an ANGGA motif search would have missed four genes, dps, clpB, patA and purM, which we identified as direct CsrA targets in this study. Considering targets activated by CsrA, most class III/IV genes either did not work (no fluorescence above baseline) or were not impacted by the presence of CsrA when tested in the fluorescent reporter assay. This might suggest that many of these genes were activated indirectly by CsrA in our omics studies, potentially explaining why the amount of ANGGA's present in these groups is more similar to the genome average. However, patA (IV) and purM (IV) were determined to be direct CsrA targets and we did observe CsrA increasing expression of mtlD (III), ytfQ (III), pta (III), ugpB (III), aroD (IV) and uxuB (IV), suggesting that CsrA does have a limited role in direct upregulation of genes, as previously thought (17,30,32) (Supplementary Table S8). Interestingly, of the class III genes, six of its ten members were identified as upregulated by CsrA in a recent work by Esquerre (17), which, in combination with the data in this paper, makes a convincing argument for further characterization of CsrA regulation of class III genes. More generally, these classified genes provide excellent starting points for tailored investigation of specific types of CsrA-transcript control.
Taken together, these findings have broad applicability to the study of gene regulatory networks. While the INFO approach was explicitly aimed at the CsrA post-transcriptional regulator, it can be applied to many transcriptional regulators using ChIP-seq or ChIP-exo (40) in place of the CsrA immunoprecipitation. This study demonstrates the basic design principle that integrating multiple streams of omics data allows for strong conclusions even if a particular data stream is inherently noisy. The strength of combined data facilitated identification of core genes in the Csr regulon, genes that are stress dependent Csr targets, and new avenues of research to further understand the physiological or metabolic impacts of Csr regulon configurations.
Supplementary Material
ACKNOWLEDGEMENTS
We are very grateful to Maria Person and Andre Bui who performed the mass spectrometry analysis described in this paper and to the University of Texas at Austin core transcriptomics facility who performed the next generation sequencing. We also acknowledge Chelsea Clark who helped perform some of the initial experiments in this work and the Contreras lab members who provided helpful conversations and insights. Additionally we gratefully acknowledge the help of Dr. Archana Pannuri for assistance in strain construction for this study.
Author contributions: S.S., G.G. and L.M.C. designed the study. C.A.V. generated strains for omics experiments. S.S., G.G., A.P. and S.L. performed omics experiments. G.G., S.S., A.N.L., A.B., S.L. and A.P. performed large-scale cloning work for follow up assays. G.G., S.S., A.N.L. and A.B. performed follow up assays. S.S., A.N.L. and G.G. analyzed results. S.S., A.N.L., G.G. and L.M.C. wrote the manuscript. T.R. and M.B. reviewed the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [GM059969 to T.R., AI097116 to T.R.]; Welch Foundation [F-1756 to L.M.C.]; The National Science Foundation [MCB 1330862 to L.M.C. and M.B., DGE-1610403 to A.N.L.]; Named and Provost Fellowships from the University of Texas at Austin Graduate School [to S.S. and A.N.L., respectively]; McKetta Department of Engineering Thrust Fellowship [to A.N.L]; Cancer Prevention Research Initiative of Texas [RP110782]. Funding for open access charge: Welch Foundation [F-1756 to L.M.C.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Sowa S.W., Vazquez-Anderson J., Contreras L.M.. Capturing full cellular regulation in silico using ‘Big’ Data: a frontier for systems biology perspectives. Curr. Synth. Syst. Biol. 2013; 1:107. [Google Scholar]
- 2. Tsai C.-H., Liao R., Chou B., Palumbo M., Contreras L.M.. Genome-wide analyses in bacteria show small-RNA enrichment for long and conserved intergenic regions. J. Bacteriol. 2015; 197:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jozefczuk S., Klie S., Catchpole G., Szymanski J., Cuadros-Inostroza A., Steinhauser D., Selbig J., Willmitzer L.. Metabolomic and transcriptomic stress response of Escherichia coli. Mol. Syst. Biol. 2010; 6:364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fondi M., Liò P.. Multi -omics and metabolic modelling pipelines: challenges and tools for systems microbiology. Microbiol. Res. 2015; 171:52–64. [DOI] [PubMed] [Google Scholar]
- 5. Edwards A.N., Patterson-Fortin L.M., Vakulskas C.A., Mercante J.W., Potrykus K., Vinella D., Camacho M.I., Fields J.A., Thompson S.A., Georgellis D. et al. Circuitry linking the Csr and stringent response global regulatory systems. Mol. Microbiol. 2011; 80:1561–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McKee A., Rutherford B., Chivian D., Baidoo E., Juminaga D., Kuo D., Benke P., Dietrich J., Ma S., Arkin A. et al. Manipulation of the carbon storage regulator system for metabolite remodeling and biofuel production in Escherichia coli. Microb. Cell Factories. 2012; 11:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Panda G., Basak T., Tanwer P., Sengupta S., dos Santos V.A.P.M., Bhatnagar R.. Delineating the effect of host environmental signals on a fully virulent strain of Bacillus anthracis using an integrated transcriptomics and proteomics approach. J. Proteomics. 2014; 105:242–265. [DOI] [PubMed] [Google Scholar]
- 8. Zhai Z., Douillard F.P., An H., Wang G., Guo X., Luo Y., Hao Y.. Proteomic characterization of the acid tolerance response in Lactobacillus delbrueckii subsp. bulgaricus CAUH1 and functional identification of a novel acid stress-related transcriptional regulator Ldb0677. Environ. Microbiol. 2014; 16:1524–1537. [DOI] [PubMed] [Google Scholar]
- 9. Buescher J.M., Liebermeister W., Jules M., Uhr M., Muntel J., Botella E., Hessling B., Kleijn R.J., Le Chat L., Lecointe F. et al. Global network reorganization during dynamic adaptations of Bacillus subtilis metabolism. Science. 2012; 335:1099–1103. [DOI] [PubMed] [Google Scholar]
- 10. Houser J.R., Barnhart C., Boutz D.R., Carroll S.M., Dasgupta A., Michener J.K., Needham B.D., Papoulas O., Sridhara V., Sydykova D.K. et al. Controlled measurement and comparative analysis of cellular components in E. coli reveals broad regulatory changes in response to glucose starvation. PLoS Comput. Biol. 2015; 11:e1004400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vakulskas C.A., Potts A.H., Babitzke P., Ahmer B.M.M., Romeo T.. Regulation of bacterial virulence by Csr (Rsm) systems. Microbiol. Mol. Biol. Rev. 2015; 79:193–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tan Y., Liu Z.-Y., Liu Z., Zheng H.-J., Li F.-L.. Comparative transcriptome analysis between csrA-disruption Clostridium acetobutylicum and its parent strain. Mol. BioSyst. 2015; 11:1434–1442. [DOI] [PubMed] [Google Scholar]
- 13. Yakhnin H., Pandit P., Petty T.J., Baker C.S., Romeo T., Babitzke P.. CsrA of Bacillus subtilis regulates translation initiation of the gene encoding the flagellin protein (hag) by blocking ribosome binding. Mol. Microbiol. 2007; 64:1605–1620. [DOI] [PubMed] [Google Scholar]
- 14. De Lay N., Schu D.J., Gottesman S.. Bacterial small RNA-based negative regulation: Hfq and its accomplices. J. Biol. Chem. 2013; 288:7996–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Holmqvist E., Wright P.R., Li L., Bischler T., Barquist L., Reinhardt R., Backofen R., Vogel J.. Global RNA recognition patterns of post-transcriptional regulators Hfq and CsrA revealed by UV crosslinking in vivo. EMBO J. 2016; 991–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morin M., Ropers D., Letisse F., Laguerre S., Portais J.-C., Cocaign-Bousquet M., Enjalbert B.. The post-transcriptional regulatory system CSR controls the balance of metabolic pools in upper glycolysis of Escherichia coli. Mol. Microbiol. 2016; 686–700. [DOI] [PubMed] [Google Scholar]
- 17. Esquerré T., Bouvier M., Turlan C., Carpousis A.J., Girbal L., Cocaign-Bousquet M.. The Csr system regulates genome-wide mRNA stability and transcription and thus gene expression in Escherichia coli. Scientific Rep. 2016; 6:25057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tatarko M., Romeo T.. Disruption of a global regulatory gene to enhance central carbon flux into phenylalanine biosynthesis in Escherichia coli. Curr. Microbiol. 2001; 43:26–32. [DOI] [PubMed] [Google Scholar]
- 19. Yakandawala N., Romeo T., Friesen A.D., Madhyastha S.. Metabolic engineering of Escherichia coli to enhance phenylalanine production. Appl. Microbiol. Biotechnol. 2008; 78:283–291. [DOI] [PubMed] [Google Scholar]
- 20. Sowa S.W., Gelderman G., Contreras L.M.. Advances in synthetic dynamic circuits design: using novel synthetic parts to engineer new generations of gene oscillations. Curr. Opin. Biotechnol. 2015; 36:161–167. [DOI] [PubMed] [Google Scholar]
- 21. Cho S.H., Haning K., Contreras L.M.. Strain engineering via regulatory noncoding RNAs: not a one-blueprint-fits-all. Curr. Opin. Chem. Eng. 2015; 10:25–34. [Google Scholar]
- 22. Vazquez-Anderson J., Contreras L.M.. Regulatory RNAs: charming gene management styles for synthetic biology applications. RNA Biol. 2013; 10:1778–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Romeo T., Gong M., Liu M.Y., Brun-Zinkernagel A.M.. Identification and molecular characterization of csrA, a pleiotropic gene from Escherichia coli that affects glycogen biosynthesis, gluconeogenesis, cell size, and surface properties. J. Bacteriol. 1993; 175:4744–4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu M.Y., Gui G., Wei B., Preston J.F. 3rd, Oakford L., Yuksel U., Giedroc D.P., Romeo T.. The RNA molecule CsrB binds to the global regulatory protein CsrA and antagonizes its activity in Escherichia coli. J. Biol. Chem. 1997; 272:17502–17510. [DOI] [PubMed] [Google Scholar]
- 25. Weilbacher T., Suzuki K., Dubey A.K., Wang X., Gudapaty S., Morozov I., Baker C.S., Georgellis D., Babitzke P., Romeo T.. A novel sRNA component of the carbon storage regulatory system of Escherichia coli. Mol. Microbiol. 2003; 48:657–670. [DOI] [PubMed] [Google Scholar]
- 26. Suzuki K., Babitzke P., Kushner S.R., Romeo T.. Identification of a novel regulatory protein (CsrD) that targets the global regulatory RNAs CsrB and CsrC for degradation by RNase E. Genes Dev. 2006; 20:2605–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang X., Dubey A.K., Suzuki K., Baker C.S., Babitzke P., Romeo T.. CsrA post-transcriptionally represses pgaABCD, responsible for synthesis of a biofilm polysaccharide adhesin of Escherichia coli. Mol. Microbiol. 2005; 56:1648–1663. [DOI] [PubMed] [Google Scholar]
- 28. Dubey A.K., Baker C.S., Suzuki K., Jones A.D., Pandit P., Romeo T., Babitzke P.. CsrA regulates translation of the Escherichia coli carbon starvation gene, cstA, by blocking ribosome access to the cstA transcript. J. Bacteriol. 2003; 185:4450–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yakhnin H., Baker C.S., Berezin I., Evangelista M.A., Rassin A., Romeo T., Babitzke P.. CsrA represses translation of sdiA, which encodes the N-acylhomoserine-L-lactone receptor of Escherichia coli, by binding exclusively within the coding region of sdiA mRNA. J. Bacteriol. 2011; 193:6162–6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Patterson-Fortin L.M., Vakulskas C.A., Yakhnin H., Babitzke P., Romeo T.. Dual posttranscriptional regulation via a cofactor-responsive mRNA leader. J. Mol. Biol. 2013; 425:3662–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Figueroa-Bossi N., Schwartz A., Guillemardet B., D'Heygère F., Bossi L., Boudvillain M.. RNA remodeling by bacterial global regulator CsrA promotes Rho-dependent transcription termination. Genes Dev. 2014; 28:1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yakhnin A.V., Baker C.S., Vakulskas C.A., Yakhnin H., Berezin I., Romeo T., Babitzke P.. CsrA activates flhDC expression by protecting flhDC mRNA from RNase E-mediated cleavage. Mol. Microbiol. 2013; 87:851–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gelderman G., Sivakumar A., Lipp S., Contreras L.. Adaptation of Tri-molecular fluorescence complementation allows assaying of regulatory Csr RNA-protein interactions in bacteria. Biotechnol. Bioeng. 2015; 112:365–375. [DOI] [PubMed] [Google Scholar]
- 34. Dubey A.K., Baker C.S., Romeo T., Babitzke P.. RNA sequence and secondary structure participate in high-affinity CsrA-RNA interaction. RNA. 2005; 11:1579–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leng Y., Vakulskas C.A., Zere T.R., Pickering B.S., Watnick P.I., Babitzke P., Romeo T.. Regulation of CsrB/C sRNA decay by EIIA(Glc) of the phosphoenolpyruvate: carbohydrate phosphotransferase system. Mol. Microbiol. 2016; 99:627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vakulskas C.A., Leng Y., Abe H., Amaki T., Okayama A., Babitzke P., Suzuki K., Romeo T.. Antagonistic control of the turnover pathway for the global regulatory sRNA CsrB by the CsrA and CsrD proteins. Nucleic Acids Res. 2016; 44:7896–7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mondragon V., Franco B., Jonas K., Suzuki K., Romeo T., Melefors O., Georgellis D.. pH-dependent activation of the BarA-UvrY two-component system in Escherichia coli. J. Bacteriol. 2006; 188:8303–8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gonzalez Chavez R., Alvarez A.F., Romeo T., Georgellis D.. The physiological stimulus for the BarA sensor kinase. J. Bacteriol. 2010; 192:2009–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Suzuki K., Wang X., Weilbacher T., Pernestig A.-K., Melefors Ö., Georgellis D., Babitzke P., Romeo T.. Regulatory circuitry of the CsrA/CsrB and BarA/UvrY systems of Escherichia coli. J. Bacteriol. 2002; 184:5130–5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zere T.R., Vakulskas C.A., Leng Y., Pannuri A., Potts A.H., Dias R., Tang D., Kolaczkowski B., Georgellis D., Ahmer B.M. et al. Genomic targets and features of BarA-UvrY (-SirA) signal transduction systems. PLoS One. 2015; 10:e0145035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kulkarni P.R., Cui X., Williams J.W., Stevens A.M., Kulkarni R.V.. Prediction of CsrA-regulating small RNAs in bacteria and their experimental verification in Vibrio fischeri. Nucleic Acids Res. 2006; 34:3361–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kulkarni P.R., Jia T., Kuehne S.A., Kerkering T.M., Morris E.R., Searle M.S., Heeb S., Rao J., Kulkarni R.V.. A sequence-based approach for prediction of CsrA/RsmA targets in bacteria with experimental validation in Pseudomonas aeruginosa. Nucleic Acids Res. 2014; 42:6811–6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Revelles O., Millard P., Nougayrede J.P., Dobrindt U., Oswald E., Letisse F., Portais J.C.. The carbon storage regulator (Csr) system exerts a nutrient-specific control over central metabolism in Escherichia coli strain Nissle 1917. PLoS One. 2013; 8:e66386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Duss O., Michel E., Diarra dit Konte N., Schubert M., Allain F.H.. Molecular basis for the wide range of affinity found in Csr/Rsm protein-RNA recognition. Nucleic Acids Res. 2014; 42:5332–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gudapaty S., Suzuki K., Wang X., Babitzke P., Romeo T.. Regulatory interactions of Csr components: the RNA binding protein CsrA activates csrB transcription in Escherichia coli. J. Bacteriol. 2001; 183:6017–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Adamson D.N., Lim H.N.. Rapid and robust signaling in the CsrA cascade via RNA–protein interactions and feedback regulation. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:13120–13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Borirak O., Rolfe M.D., de Koning L.J., Hoefsloot H.C.J., Bekker M., Dekker H.L., Roseboom W., Green J., de Koster C.G., Hellingwerf K.J.. Time-series analysis of the transcriptome and proteome of Escherichia coli upon glucose repression. Biochim. Biophys. Acta (BBA) - Proteins Proteomics. 2015; 1854:1269–1279. [DOI] [PubMed] [Google Scholar]
- 48. LB (Luria-Bertani) liquid medium. Cold Spring Harbor Protocols. 2006; pdb.rec8141. [Google Scholar]
- 49. M9 minimal medium (standard). Cold Spring Harbor Protocols. 2010; pdb.rec12295. [Google Scholar]
- 50. Cho S.H., Lei R., Henninger T.D., Contreras L.M.. Discovery of ethanol responsive small RNAs in Zymomonas mobilis. Appl. Environ. Microbiol. 2014; 80:4189–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Darnell R.B. HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley Interdiscipl. Rev. RNA. 2010; 1:266–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vakulskas C.A., Pannuri A., Cortes-Selva D., Zere T.R., Ahmer B.M., Babitzke P., Romeo T.. Global effects of the DEAD-box RNA helicase DeaD (CsdA) on gene expression over a broad range of temperatures. Mol. Microbiol. 2014; 92:945–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Groat R.G., Schultz J.E., Zychlinsky E., Bockman A., Matin A.. Starvation proteins in Escherichia coli: kinetics of synthesis and role in starvation survival. J. Bacteriol. 1986; 168:486–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Reeve C.A., Bockman A.T., Matin A.. Role of protein degradation in the survival of carbon-starved Escherichia coli and Salmonella typhimurium. J. Bacteriol. 1984; 157:758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wei B., Shin S., LaPorte D., Wolfe A.J., Romeo T.. Global regulatory mutations in csrA and rpoS cause severe central carbon stress in Escherichia coli in the presence of acetate. J. Bacteriol. 2000; 182:1632–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Raman B., Nandakumar M.P., Muthuvijayan V., Marten M.R.. Proteome analysis to assess physiological changes in Escherichia coli grown under glucose-limited fed-batch conditions. Biotechnol. Bioeng. 2005; 92:384–392. [DOI] [PubMed] [Google Scholar]
- 58. Feng Y., Cronan J.E.. Crosstalk of Escherichia coli FadR with global regulators in expression of fatty acid transport genes. PLoS ONE. 2012; 7:e46275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chang D.E., Smalley D.J., Conway T.. Gene expression profiling of Escherichia coli growth transitions: an expanded stringent response model. Mol. Microbiol. 2002; 45:289–306. [DOI] [PubMed] [Google Scholar]
- 60. Baker C.S., Eory L.A., Yakhnin H., Mercante J., Romeo T., Babitzke P.. CsrA inhibits translation initiation of Escherichia coli hfq by binding to a single site overlapping the Shine-Dalgarno sequence. J. Bacteriol. 2007; 189:5472–5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jonas K., Edwards A.N., Simm R., Romeo T., Römling U., Melefors Ö.. The RNA binding protein CsrA controls cyclic di-GMP metabolism by directly regulating the expression of GGDEF proteins. Mol. Microbiol. 2008; 70:236–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wei B.L., Brun-Zinkernagel A.M., Simecka J.W., Pruss B.M., Babitzke P., Romeo T.. Positive regulation of motility and flhDC expression by the RNA-binding protein CsrA of Escherichia coli. Mol. Microbiol. 2001; 40:245–256. [DOI] [PubMed] [Google Scholar]
- 63. Pannuri A., Yakhnin H., Vakulskas C.A., Edwards A.N., Babitzke P., Romeo T.. Translational repression of NhaR, a novel pathway for multi-tier regulation of biofilm circuitry by CsrA. J. Bacteriol. 2012; 194:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Baker C.S., Morozov I., Suzuki K., Romeo T., Babitzke P.. CsrA regulates glycogen biosynthesis by preventing translation of glgC in Escherichia coli. Mol. Microbiol. 2002; 44:1599–1610. [DOI] [PubMed] [Google Scholar]
- 65. Park H., Yakhnin H., Connolly M., Romeo T., Babitzke P.. CsrA participates in a PNPase autoregulatory mechanism by selectively repressing translation of pnp transcripts that have been previously processed by RNase III and PNPase. J. Bacteriol. 2015; 197:3751–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Salgado H., Peralta-Gil M., Gama-Castro S., Santos-Zavaleta A., Muniz-Rascado L., Garcia-Sotelo J.S., Weiss V., Solano-Lira H., Martinez-Flores I., Medina-Rivera A. et al. RegulonDB v8.0: omics data sets, evolutionary conservation, regulatory phrases, cross-validated gold standards and more. Nucleic Acids Res. 2013; 41:D203–D213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liu M.Y., Yang H., Romeo T.. The product of the pleiotropic Escherichia coli gene csrA modulates glycogen biosynthesis via effects on mRNA stability. J. Bacteriol. 1995; 177:2663–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ren B., Shen H., Lu Z.J., Liu H., Xu Y.. The phzA2-G2 transcript exhibits direct RsmA-mediated activation in Pseudomonas aeruginosa M18. PLoS ONE. 2014; 9:e89653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sabnis N.A., Yang H., Romeo T.. Pleiotropic regulation of central carbohydrate metabolism in Escherichia coli via the gene csrA. J. Biol. Chem. 1995; 270:29096–29104. [DOI] [PubMed] [Google Scholar]
- 70. Cunningham L., Guest J.R.. Transcription and transcript processing in the sdh CDAB-sucABCD operon of Escherichia coli. Microbiology. 1998; 144:2113–2123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.