


Abstract
The genomic loci bound by the glucocorticoid receptor (GR), a hormone-activated transcription factor, show little overlap between cell types. To study the role of chromatin and sequence in specifying where GR binds, we used Bayesian modeling within the universe of accessible chromatin. Taken together, our results uncovered that although GR preferentially binds accessible chromatin, its binding is biased against accessible chromatin located at promoter regions. This bias can only be explained partially by the presence of fewer GR recognition sequences, arguing for the existence of additional mechanisms that interfere with GR binding at promoters. Therefore, we tested the role of H3K9ac, the chromatin feature with the strongest negative association with GR binding, but found that this correlation does not reflect a causative link. Finally, we find a higher percentage of promoter–proximal GR binding for genes regulated by GR across cell types than for cell type-specific target genes. Given that GR almost exclusively binds accessible chromatin, we propose that cell type-specific regulation by GR preferentially occurs via distal enhancers, whose chromatin accessibility is typically cell type-specific, whereas ubiquitous target gene regulation is more likely to result from binding to promoter regions, which are often accessible regardless of cell type examined.
INTRODUCTION
Transcription factors (TFs) play a pivotal role in regulating the expression of genes by binding to genomic response elements. The recognition sequences for eukaryotic TFs encoded within these response elements are generally short and degenerate (1). Consequently, only a fraction of all potential binding sites of a TF present in the genome is occupied. Moreover, for several TFs, the genomic binding pattern shows little overlap between cell types (2), further emphasizing that the presence of a recognition sequence alone provides insufficient information to specify where in the genome TFs bind. Additional information is provided by the combinatorial architecture of response elements that typically harbor recognition sequences for multiple TFs. These TFs can, for example, act cooperatively, when the binding of one TF is stabilized by the presence of another, thereby influencing its genome-wide binding pattern (3,4). In addition to sequence, the local chromatin environment plays an important role in defining where in the genome TFs bind. For example, many TFs preferentially bind to regions of accessible chromatin as assayed by DNase I sensitivity assays (5). Accordingly, TF-bound regions are enriched for histone modifications that are associated with active promoters (e.g. H3K4me3 and H3K9ac) and enhancer elements (H3K4me1 and H3K27ac) (6). Consistent with an important role for the chromatin environment in specifying where TFs bind, genomic TF binding site prediction benefits from incorporating both sequence and chromatin accessibility data (7,8). Similarly, histone modification data is highly informative when predicting genomic TF binding (8).
The dynamic nature of the chromatin environment provides a mechanism to facilitate cell type-specific TF binding. For example, regions of accessible chromatin show little overlap between cell types, especially regions that do not locate to the transcriptional start site (TSS) of genes (7,9). Similarly, binding of the glucocorticoid receptor (GR), a hormone-activated TF that predominantly binds to regions of open chromatin, shows little overlap between cell types (10,11). The cell type-specific binding of GR likely also explains the limited overlap in genes regulated by GR when comparing different cell types ((12), this study) and thus could facilitate the diverse range of biological responses to glucocorticoid signaling ranging from gluconeogenesis in the liver, to the suppression of inflammation in chronic inflammatory diseases like asthma.
Here, we set out to study the role of sequence and the chromatin environment in specifying the genomic loci occupied by GR. Specifically, we explored available data from the ENCODE (13) and the Epigenomics roadmap (14) consortia regarding several histone modifications, the histone variant H2A.Z and DNase I sensitivity data in three cell types for which we have genome-wide GR occupancy data. Similar to previous studies (10), we find that GR predominantly (94%) binds to accessible (‘open’) chromatin decorated with histone modifications found at transcriptionally active regions. Since GR almost exclusively binds to open chromatin, we decided to focus only on the universe of DNase I Hypersensitive Sites (DHSs) to study the role of chromatin environment and sequence in specifying which of the open sites (GR binds ∼15% of all DHSs) are bound by GR. We used a hierarchical Bayesian model to quantitatively describe GR binding in several cell lines based on sequence and chromatin features. The model is interpreted as an exploratory, hypothesis-generating step towards further investigation of how genomic sequence combines with cell type-specific chromatin state to produce a diversity of cellular responses to hormone. This approach uncovered that not all open chromatin is equal and that GR preferentially binds DHSs decorated with chromatin features found at enhancers, whereas binding at DHSs decorated with chromatin features found at promoters was disfavored. Further, we found that the depletion of promoter–proximal binding could be explained in part by changes in sequence composition, with fewer GR recognition sequences mapping to promoter–proximal DHSs. Together, our studies uncovered a preferential bias against promoter proximal GR binding, and we provide evidence that this may play a role in facilitating the cell type-specific transcriptional consequences of GR signaling.
MATERIALS AND METHODS
Cell lines
Immortalized wild type mouse embryonic fibroblasts (MEFs) and GCN5/PCAF double knockout (dko) MEFs (15) were a kind gift of Kai Ge. Cells were grown in DMEM supplemented with 10% FBS. A549 cells were grown in DMEM supplemented with 5% FBS, whereas Nalm6 cells were grown in RPMI 1640 supplemented with 10% FBS.
Chromatin immunoprecipiptation (ChIP) and ChIP sequencing (ChIP-seq)
ChIP assays were done essentially as described using either the N499 GR-antibody ((16), 2 μl/ChIP) an H3K9ac antibody (C5B11, Cell Signaling, 6 μl/ChIP) or as control IgG (cat#: C15410206, Diagenode, 6 μl/ChIP). For each ChIP assay, ∼5 million cells were treated with 0.1% ethanol vehicle or 1 μM dexamethasone for 1.5 h. For ChIP-seq experiments, the material of multiple ChIP experiments was pooled to obtain enough material (∼10 ng) for library preparation. Primers used for qPCR analysis of ChIP experiments are listed in Table 1.
Table 1. qPCR primers used in this study (all target mouse).
| Gene/Locus: | Primer fw: | Primer rev: |
|---|---|---|
| FKBP5 | TCTGGTCACTGCAGACATCAC | GTCAGCACATCGAGTTCATGTG |
| RAC1 | TGACCTTGGCATTGGTTCTG | TTTGAGTCTCCAGTGGCTCTTG |
| RBM16 | CCAGTTAAGTTCTCAGGCACAC | AACAGCTCACTATGCCAGACTG |
| PHACTR1 | AGAGAGATCCCCTGCCAAAG | GCCTTGTGAAGCTCATGTTTG |
| SCN9a | GGCAGAAAAGAAACCAACATCC | TCTTAGACACAGTGCCTGCTG |
| SUMF1 | TGCACGGTAGCTTTCATATTCAG | CCAGCCTGTGTGATGTTCTTTG |
| GIG18 | TAAGCACCAGTTCACAAGCC | CTGCCTGTGACTAAAGGATGAC |
| MAGI2 | AACCACGTCTAAGTGACTCACC | AGAACCACTGCTAGACACTGC |
| TXK | AATTCCTGGACTCCTGAATCATG | GACGTGTCATAACTCACTGCAG |
| CDCa7 | TGTCACACGGCTAAGCTCTG | ACAGAGAGTGTTAGGCCAGAC |
| FAM76b | AACAGCTCCTGTAGTCAGTCAG | TTGCCAGAGTCCAGAGGAAATG |
| EEF2 | GCATCTATTGTGTGCAGAAGGC | TTTCAAGAGAGCCCTCGATGC |
| GAPDH | GGGCGCCCTTCCAATTTTATC | TCACTTGTTGGACAGCACTG |
Immunoblotting
Total protein isolated from equal amounts of cells were separated with SDS-PAGE gels, transferred to membranes and incubated with either anti-GR (N499, dilution 1:5000, (16)), anti H3K9ac antibody (C5B11, Cell Signaling, dilution 1:1000) or anti-actin (Sc-1616R, Santa Cruz Biotechnology, dilution 1:1000) antibodies followed by incubation with secondary antibodies conjugated with horseradish peroxidase. Proteins were visualized using an ECL detection system (Amersham Biosciences).
DNase I assays
DNase I assays were done essentially as described (17) by growing MEFs or dko MEFs in six-well plates (500.000 cells/well), washing cells with PBS and scraping them into 1 ml DNase I buffer (20 mM HEPES pH 7.4, 0.5 mM CaCl2, 5 % glycerol, 3 mM MgCl2, 0.2 mM spermine, 0.2 mM spermidine) plus 0.2% NP40 alternative. Nuclei from three wells were pooled, isolated by centrifugation and resuspended in 300 μl DNase I buffer containing 2 μg/ml RNase A. 55 μl aliquots were DNase I treated (or mock treated to normalize for the amount of chromatin in input) by the addition of 2.05 μl DNase I (Qiagen, 2.7 u/μl) and then incubated at 37°C for 10 min. The reaction was stopped by addition 1 ml stop buffer (50 mM Tris pH 8.0; 200 mM NaCl; 12.5 mM EDTA; 1% SDS, 200 μg/ml proteinase K) and samples were incubated at 65°C for 4 h to remove proteins. Finally, DNA was purified using a PCR purification kit (Qiagen) and regions of interest were analyzed by qPCR (primers listed in Table 1).
Computational analysis
Sequencing data
ChIP-seq data GR
GR ChIP-seq data for GR (U2OS, IMR90, K562) were previously generated by us and deposited at EBI ArrayExpress under accession number E-MTAB-2955 (18). ChIP-seq data for A549, Nalm-6 and MEFs were deposited at EBI ArrayExpress with this study under accession number E-MTAB-5113.
ChIP-seq processing
GR ChIP-seq data for A549 and Nalm-6 was processed as described previously (16). For wild type and dko MEFs, the paired-end sequence reads derived from GR ChIP-seq experiments were mapped with Bowtie 2 (19) onto the Mus musculus assembly mm9. Number of allowed mismatches was set to zero; length of seed substrings to align was set to 20; default setting were used for all other parameters. Peak calling was done using MACS2 (20) using the input DNA from each respective cell line as control. Genome size was set to –mm; the keep-dup parameter to 1 and default was used for all remaining parameters. Peaks were called using a FDR cutoff of 0.01.
Overlap GR peaks between wild type and dko MEFs
The differential GR binding analysis between wild type and dko MEFS was done using the R package DiffBind (21, http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf.). Differentially bound peaks where called using the Mann–Whitney-U test with a significance cut off of 0.05. For qPCR validation experiments, loci representing the three classes of GR binding sites: common, dko-specific and wt-specific where picked based on peak height (for common peaks we chose ones with high signal) and peak ratio (for dko- and wt-specific loci, we chose peaks with relatively large differences in peak height between dko and wt MEFs).
ChIP-seq data histone variants, histone modifications and TFs
ChIP-seq mapped sequencing data (BAM files) and annotated peaks from the ENCODE project (13) (A549, K562) were downloaded from the UCSC genome browser website. ChIP-seq data for IMR90 was from the Roadmap Epigenomics Mapping project (14) and downloaded from the NCBI GEO website under the series number GSE16256. H3K9ac ChIP data from primary MEFs was accessed via GEO database series numbers GSM863802 and GSM863804.
The following files were downloaded from the ENCODE project's Data Coordination Center FTP site and used for comparative analysis:
(http://hgdownload.cse.ucsc.edu/goldenpath/hg19/encodeDCC/wgEncodeOpenChromChip/)
Pol2: wgEncodeOpenChromChipA549Pol2PkRep1.narrowPeak ETS1: wgEncodeHaibTfbsA549Ets1V0422111Etoh02PkIntersect.broadPeak
CEBPB: wgEncodeHaibTfbsA549Cebpbsc150V0422111PkIntersect.broadPeak
FOXA1: wgEncodeHaibTfbsA549Foxa2V0416102Etoh02PkIntersect.broadPeak FOSL2: wgEncodeHaibTfbsA549Fosl2V0422111Etoh02PkIntersect.broadPeak
TCF12: wgEncodeHaibTfbsA549Tcf12V0422111Etoh02PkIntersect.broadPeak
DHS data
DNase I-seq mapped sequencing data (BAM files) from the ENCODE project (13) (A549, K562) were downloaded from the UCSC genome browser website. DNase I data for IMR90 was from the Roadmap Epigenomics Mapping project (14) and downloaded from the NCBI GEO website under the series number GSE16256. The DHSs as annotated by the ENCODE project (13) were used to compare GR binding patterns at distinct genomic regions (promoters, exons, introns, distal) and are available from the UCSC Genome Browser as ‘narrowPeak’ files. DHS regions mapping to repeats (RepeatMasker (22) score >1000, track downloaded from repeatmasker.org, February 2012) were removed and resulted in 105 121 remaining DHSs for A549, 127 803 for IMR90 and 97 304 for K562 cells.
Motif score calculations
Motif scores were calculated using the GR motif MA0113.1 from the JASPAR database (23) and scanning both strands in a 160 bp range centered on the DHS peak. The maximum log odds score was assigned to each DHS location. Scanning was performed using the PWMscoreStartingAt function of the Biostrings R/Bioconductor package (24), with a zero-order background model for DNA with 42% GC content.
% Deviance explained by promoter proximity
The effect of promoter proximity on GR binding was tested, controlling for lack of sequence motif, using an analysis of deviance (the change in −2 × log likelihood for a nested statistical model). A binary variable was defined for each DHS, indicating if the DHS overlapped a GR peak. The GR peak presence variable was then modeled using a logistic regression. Two models were fit: (0) motif score as the sole independent variable to explain GR peak presence, and (1) motif score plus a binary indicator of promoter proximity of the DHS as independent variables to explain GR peak presence. The deviance explained by introducing the indicator variable of promoter proximity (model 1) was expressed as a percent of the deviance explained by motif score alone (model 0) (Figure 4B). The significance test for all cell lines rejected the null hypothesis that promoter proximity did not help to explain depletion of GR binding beyond the motif score (P < 2 × 10−16).
Figure 4.

Role of sequence composition in explaining the promoter–proximal depletion of GR binding observed. (A) Distribution of GR motif score (GR motif MA0113.1 from JASPAR, (23)) for the following groups: All promoter–proximal DHSs (salmon), all distal DHSs (green), for GR-bound proximal DHSs (turquoise) and GR-bound distal DHSs (purple) for cell lines as indicated. (B) Percent deviance of GR peak presence at DHSs explained by promoter proximity while controlling for motif score, as a percent of the deviance explained by the motif score alone indicates that motif score distribution explains some, but not all promoter–proximal depletion of GR binding.
Calculation of the overlap between GR peaks and DHSs
The overlap of GR peaks with DHSs (Figure 1B) and the overlap of DHSs with GR peaks (Figure 2A) was calculated using the GR peak regions as defined by the MACS2 software (20) for the IMR90 ChIP-seq data, and the DNase I regions reads from the following file for the IMR90 cell line as defined by the ENCODE project: DNase I: wgEncodeOpenChromDnaseImr90Pk.narrowPeak.
Figure 1.

Interaction of GR with the chromatin landscape. (A) Normalized tag density from ChIP-seq for GR after hormone treatment and from DNase-seq and H3K27ac, H3K4me1 and H3K36me3 ChIP-seq prior to hormone treatment in IMR90 cells is shown for a region of the X-chromosome around the GR target gene TSC22D3. (B) Distribution of GR ChIP-seq peaks between DNase I hypersensitive sites (DHSs) as annotated by the ENCODE consortium (accessible chromatin) and the rest of the genome (inaccessible chromatin, non-DHS) in IMR90 cells. (C) Median read count difference of chromatin features as indicated between GR ChIP-seq peaks and genomic control regions 5kb away (randomly up- or down-stream of the GR peaks) is shown for three cell lines: A549, IMR90 and K562.
Figure 2.

GR binding in the universe of open chromatin. (A) Pie chart showing the percentage of all DHSs in IMR90 cells occupied by GR (15%) in black. (B) Left: The hierarchical Bayesian model for a single cell type to model the read density of GR binding (tall colored columns), based on various chromatin features (colored rectangles). Right: The full hierarchical model. The grey node at the bottom indicates the data. As output, the model generates coefficients (β) for each chromatin feature and for each experiment. Experiments on the same cell type share a common effect (ν), and all cell types share a common effect λ. (C) Estimated coefficients for all levels of the model, with cell type, lab and replicate number indicated e.g. A549 TRG. The cell type parameters (ν) are indicated with only the cell type e.g. A549. The parameter λ is labeled ‘all’. Dots mark the posterior mean; whiskers indicate the 95% quantile-based interval. P (positive) from 0 to 1 indicates the posterior probability of the coefficient having a positive impact on GR binding, according to the posterior distributions obtained from the hierarchical Bayesian model. (D) % variance explained by the model for each of the cell lines.
Calculation of the overlap with different functionally annotated genomic regions for GR bound regions, GR-bound DHSs and all DHSs
The overlap of DHSs and GR-bound DHSs with promoters, exons, introns and distal regions (Figure 3A) was calculated using the genomic annotations defined by the Bioconductor package ‘TxDb.Hsapiens.UCSC.hg19.knownGene’ (24). Promoter regions were defined as ±2.5 kb from the TSS of a known gene. Because the genomic features can overlap, the following hierarchy was defined: (i) promoters, (2) exons, (3) intronic, (4) distal. In case a GR peak overlapped multiple features, it was assigned exclusively to one category according to this hierarchy.
Figure 3.

Genomic distribution of DHSs and of GR-bound DHSs. (A) Distribution of all DHSs and of GR-bound DHSs in cell lines as indicated relative to annotated genomic features (promoters: ±2.5 kb from TSS; exons, introns and distal (rest)). (B) Same as for a) except that ChIP-seq peaks for TFs as indicated (A549 cells) were analyzed. (C) Comparison of GR-bound and unbound promoter DHSs in IMR90 cells uncovered that GR-bound regions have higher levels of enhancer-associated marks (H3K4me1, H3K12ac and H3K20ac) whereas levels for H3K4me3, a typical promoter mark, are lower when compared to their unbound DHS counterparts.
The overlap of TF peaks with promoters, exons, introns and distal regions (Figure 3B) was calculated similarly, but using the ENCODE defined peaks from files downloaded from the Data Coordination Center FTP site.
Overlap MEF common, wt-specific, dko-specific mapping to promoters (Figure 5E): The overlap of MEF common, wt-specific, dko-specific GR peaks is based on the same definition of promoter regions described above. A GR peak was considered to map to promoters when at least one promoter region overlapped the middle of the peak region.
Figure 5.

Functional analysis of the correlation between H3K9ac levels and GR binding: correlation ≠ causation. (A) Schematic diagram depicting the design of the study to test if the correlation between H3K9ac levels and GR binding reflects a causative link. (B) Comparison of % input precipitated by ChIP with an H3K9ac-specific antibody versus IgG control at GR-bound genomic loci as indicated was assayed by qPCR (average enrichment + error from 7 pooled biological replicates) in either wild type MEFs (blue) or in GCN5/PCAF double knock-out (dko) MEFs (salmon). (C) Normalized tag density from ChIP-seq for GR after hormone treatment for wild type MEFs (blue) and dko MEFs (salmon) for genomic region near RAC1 gene: GR-bound in both wt MEFs and dko MEFS; SUMF1: dko-specific GR binding and TXK: wt-specific GR binding. (D) Venn diagram showing the number and overlap of GR-bound loci in wt and dko MEFs. (E) Comparison of the % of GR ChIP-seq peaks mapping to promoter regions for wt-specific, common and dko-specific peaks.
Calculation of read count difference for histone marks
To compare the number of ChIP-seq reads within GR peak regions to random regions of the genome (Figure 1C), we took the 1000 GR peak regions with the lowest p-value as calculated by MACS (20) and counted the reads within them for each of the histone modifications, DNase I and Input, then took the median across all 1000 regions. We then defined a background set of non-peak regions by shifting each of the 1000 GR peak regions 5kb away in a random direction (upstream or down), counted the reads there and again calculated the median across all 1000 background regions. The ratio of these two medians (GR / background) was then calculated, converted to log2 scale and plotted.
Average read count profiles for histone marks ±2 kb from the center of DHS (Supplementary Data) were calculated using the R/Bioconductor GenomicRanges package (25), taking the average of read counts at each position relative to the DHS center over the set of DHSs which overlapped GR peaks, or the set of DHSs which did not overlap a GR peak.
To compare GR-bound promoter DHSs with unbound promoter DHSs in IMR90 cells (Figure 3c), reads from ENCODE histone marks ChIP-seq experiments in IMR90 cells were counted using Bioconductor core packages in 1600 bp windows centered on promoter–proximal DHSs (DHSs within 200 bp of a transcriptional start site defined by RefSeq gene annotation). Shown are boxplots of log10 counts of reads across all DHSs, split by whether or not the DHS overlapped a GR peak as identified by MACS software (20).
H3K9ac abundance for different classes of GR ChIP-seq peaks in MEFs (common, dko-specific and wt-specific) was computed from datasets GSM863802 and GSM863804 retrieved from the GEO database. Both datasets were pooled for the analysis. To avoid biases from different widths of GR peaks, we set the width for each peak to 500 bp for each class of GR ChIP-seq peak (middle of peak region -249 bp, +250 bp). A single-end read from H3K9ac ChIP-seq was considered to fall into the 500 bp GR peak region when at least 1bp overlapped.
Gene regulation and promoter–proximal binding
Microarray datasets for U2OS, A549 and Nalm6 cells comparing dexamethasone treatment (1 μM, 3h) with vehicle control were downloaded from E-GEOD-38971 (EBI ArrayExpress) and processed as described previously (16). To call genes regulated upon treatment with dexamethasone, we used an adjusted p-value cut-off of 0.05 and 1.5 for the fold change (either up- or down-regulated). The overlap of the regulated gene sets between cell lines is visualized as Venn diagram (Figure 6A). The percentage of genes with GR binding in the promoter was computed for cell type-specific genes and for genes regulated in all three cell lines examined (common target genes). Promoters were defined as regions ±2.5 kb around all transcript start sites annotated in the Ensembl database (grch37) of Homo sapiens genes, accessed via Bioconductor package BioMart (26). In case a gene had several transcripts, all of them were considered. A gene was marked as having a GR peak in the promoter, if at least one of its promoter regions overlapped a GR peak position.
Figure 6.

Link between promoter–proximal binding and cell type-specific gene regulation by GR. (A) Venn diagram showing the overlap of genes regulated in response to dexamethasone between three cell lines: U2OS stably expressing GR, A549 and Nalm6 cells. (B) % of promoter regions with a GR-peak is higher for genes regulated across cell types than for cell type-specific target genes in all three cell types examined.
Hierarchical bayesian model
A hierarchical Bayesian model (27) was constructed to correlate binding of GR (as measured by ChIP-Seq read counts) with chromatin features and motif score in DHSs across experiments and across cell types. The log of read counts for various chromatin features plus a pseudocount of 1 and the motif score over the annotated DHSs of a cell type were arranged as columns of a matrix X (Figure 2B). The chromatin feature matrix X was identical for experiments of the same cell type, except the Input feature, which was paired with the GR ChIP experiment: each of the IMR90 ChIP-Seq experiments was paired with its own Input experiment, and the A549 ENCODE GR ChIP experiments were paired with the A549 ENCODE Input experiment. This matrix X was then centered and scaled to have columns with zero mean and unit standard deviation. For an experiment k and a genomic range i centered on a DHS, the count of GR ChIP-Seq reads was modeled as following a Poisson distribution with a varying mean:
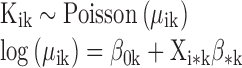 |
The ‘*’ indicates iteration over all j explanatory features. The coefficient β0k provides a sample-specific intercept, or base level of GR ChIP-seq read counts. The coefficient βjk is interpreted as the multiplicative effect of chromatin feature j on the GR ChIP-Seq read counts for experiment k. A positive coefficient implies an inductive contribution to GR binding and a negative coefficient implies an inhibitory contribution to GR binding, while fixing the contribution of all other chromatin features.
We specify that βjk for experiments k of the same cell type are related, with ct(k) denoting the cell type of experiment k. These coefficients are given a shared prior mean νj,ct(k), and experiment-specific variance σ2β:
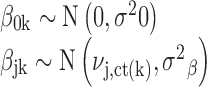 |
The log of the mean for a given experiment and genomic range, log(μik), is therefore distributed as a normal random variable (the sum of a number of normal random variables). Each experiment k has its own set of coefficients, allowing for variability in the mean across samples, similar to the over-dispersion allowed by the Gamma-Poisson (Negative Binomial) distribution.
The parameters of the distributions for the main model coefficients βjk are themselves distributed as follows:
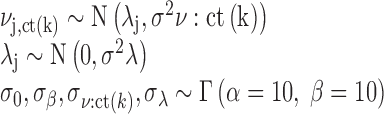 |
The term λj gives the overall contribution of chromatin feature j on GR binding – across all cell types included in the hierarchical model. The zero-mean normal prior for λj in practice produces a shrinkage of terms towards zero (no contribution to GR binding) unless the data provides evidence to the contrary.
The posterior of the model parameters conditioning on the observed data was sampled using the Stan C++ MCMC package and the rstan R package (28). The model was run for four chains for 4000 iterations (the first 2000 iterations discarded as burn-in) using the ‘no U-turn’ setting. R-hat values near 1 were used as a convergence diagnostic. Example code for the Bayesian model is provided as a separate Supplementary Data (bayes_model_Loveteal_2016.txt).
Variance explained by the hierarchical Bayesian model
An estimate of the percent variance of log-scale GR ChIP-seq read counts explained by the model was calculated by squaring the Pearson correlation coefficient between the log observed signal and the log of the predicted GR read count for each DHS, using the coefficients estimated by the hierarchical Bayesian model. The estimated coefficients used for prediction were the posterior means from the Bayesian model.
RESULTS
GR preferentially binds accessible chromatin decorated with chromatin features associated with transcriptional activity
Sequences matching the GR recognition sequence are ubiquitously found in the genome, yet only a cell type-specific minority are actually bound. To understand the mechanisms that specify which of these potential binding sites are in fact occupied by GR, we studied the role of the chromatin landscape in which these binding sites are embedded. Specifically, we used the wealth of information regarding chromatin features available from the ENCODE and the Epigenomics roadmap in three cell types (A549, K562 and IMR90) for which we have genome-wide GR ChIP-seq data ((18) and this study). Notably, the data regarding the chromatin landscape was generated in the absence of GR ligand and thus represents the chromatin landscape GR encounters upon ligand binding. Previous studies have shown that chromatin accessibility pre-determines GR binding patterns, with the majority of binding occurring at preexisting loci of accessible chromatin (10,29). Accordingly, we found that the majority of GR binding in IMR90 cells (94%) overlaps with DHSs as annotated by the ENCODE consortium (13) (Figure 1A and B). Next, we compared the levels of chromatin features at GR-bound regions with those found at random genomic regions. As expected, this analysis showed that GR-bound regions have increased levels of DNase I sensitivity for each of the cell lines examined (Figure 1C). Furthermore, the DHS signal displays a distinctive pair of peaks flanking the GR-bound regions for all enriched histone modifications (H3K27ac, H3K4me1/2/3, H3K9ac) with the exception of H3K4me3, which showed a single peak overlapping the GR ChIP-seq peak (Supplementary Data). The pair of peaks is indicative of GR binding in between nucleosomes, whereas the single peak for the H3K4me3 modification suggests that GR might bind on top of the nucleosome when this modification is present (Supplementary Data). No striking enrichment at GR-bound regions was observed for histone modifications associated with either transcriptionally inactive regions (H3K9me3 and H3K27me3) or with transcriptional elongation (H3K36me3, H3K79me2). Interestingly, we found that H4K20me1, a histone mark linked to transcription-linked histone turn-over (30) and Polycomb repression (31), showed a marked enrichment at GR-bound regions in IMR90 and K562 cells, whereas no enrichment was observed in A549 cells (Figure 1C), indicative of a possible role of H4K20me1 in generating cell type-specific GR binding patterns.
Hierarchical Bayesian modeling of GR binding within universe of accessible chromatin
Together, our findings corroborate previous studies showing that GR predominantly binds to regions of accessible chromatin decorated with chromatin features found at transcriptionally active regions. However, GR only binds a minority (15% in IMR90, Figure 2A) of all DHSs found in a cell raising the question: what distinguishes DHSs bound by GR from those that are not bound? Notably, the occurrence of a GR consensus motif does not explain which sites GR binds with great accuracy because many bound sites do not contain a consensus motif and conversely, because many unbound sites contain GR consensus motifs ((18,32) and Figure 4A). Therefore, we constructed a hierarchical Bayesian model which incorporates both sequence and chromatin features to quantitatively describe the binding of GR (as measured by ChIP-seq read counts) based on the level of cell type-specific chromatin features and the score of the GR motif in DHSs across experiments and across cell types (Figure 2B). As output, the model generates estimates for coefficients (β) for each feature and each cell type, which can be compared between features to determine their relative contribution to explaining GR binding levels. Furthermore, these coefficients can be compared between cell lines to find potential differences between cell types. Positive estimated coefficients indicate a positive association with GR binding whereas negative estimated coefficients indicate a negative association with GR binding. To assess how accurately our model describes GR binding, we calculated the percentage of variance of GR binding explained by using the fitted hierarchical model coefficients to predict log counts of ChIP-seq reads. The percent variance explained was relatively high for IMR90 and K562 (48–57%), while for A549, the percent variance explained was 22%. This difference may have be accounted for by generally lower read counts in the A549 ChIP-seq experiment, leading to more technical, sampling variability at low counts values.
Next, we analyzed the coefficients for individual chromatin features and found that they are mostly consistent across cell types (Figure 2C). In general, chromatin features that show the highest positive correlations with GR binding are marks of distal regulatory elements (H3K27ac, H3K4me1) and the GR motif score. Further, within the universe of DHSs, GR preferentially binds to regions with high DNase I and input read counts. Within the model, the features are scaled to facilitate direct comparisons. Therefore, the similar positive values for the DNase I coefficient and the motif score coefficient indicate that they have a similar positive association with GR binding, whereas the highest positive coefficient is observed for H3K27ac levels indicating that this feature is more important in quantitatively explaining the level of GR binding than any other feature including motif score (Figure 2C).
Four consistently negatively associated chromatin features are H3K36me3, H3K79me2, H2A.Z and H3K9ac. H3K36me3 is typically found along the gene body of transcribed genes and has been linked to suppression of cryptic transcription initiation (33). The negative association with GR binding may thus reflect a link between H3K36me3 levels and prevention of TF binding in transcribed regions. H3K79me2 is also enriched in the gene body of transcribed genes and linked to various biological processes including telomeric silencing, DNA repair and cell cycle checkpoints (34), if and how this could be linked to GR binding however is unclear. The consistent negative association of H2A.Z with GR binding might at first appear incompatible with the findings reported by John et al. (29) that H2A.Z is highly enriched at GR binding sites. However, this study compared H2A.Z levels at GR binding sites with nearby regions that are not DHSs. Similarly, our genome-wide analysis (Figure 1C) shows a strong positive correlation between H2A.Z levels and GR binding. In contrast, the hierarchical Bayesian model presented here focuses only on the universe of DHSs and suggests that of all accessible sites, GR preferentially binds DHSs with low H2A.Z levels. Similar to H2A.Z, H3K9ac is enriched at the transcription start site of genes (35) and we find that within the universe of DHSs, GR binding is associated with low levels of H3K9ac (Figure 2C). Interestingly, the coefficients of two chromatin features that show a strong association with GR binding, H3K4me2 and H3K4me3, have a different sign depending on the cell line examined. H3K4me2 is positively associated with binding in A549 and IMR90 cell lines, whereas its negatively associated in K562 cells. In contrast, H3K4me3 levels correlate negatively with GR binding in A549 and IMR90 cells whereas they are a positive predictor of GR binding in K562 cells.
Together, our hierarchical Bayesian modeling of GR binding within the universe of accessible chromatin uncovered that GR preferentially binds regions with a recognition motif and chromatin marks associated with (active) enhancers. Further, some of the chromatin features showed a cell type-specific link to GR binding indicative of context-specific effects of the chromatin environment on GR binding. Unexpectedly, chromatin marks associated with promoter regions (H3K9ac, H2A.Z and to some degree H3K4me3) showed a negative association with GR binding, suggesting that mechanisms exist that interfere with promoter–proximal GR binding.
GR binding is depleted at promoter–proximal accessible chromatin
The negative link between GR binding and chromatin marks found at promoter regions suggests that GR preferentially binds to open regions that do not map to the promoters of genes. To test this hypothesis, we first determined for each cell line, the fraction of genome-wide DHSs that map to promoters (±2.5 kb around TSS of genes), to exons, to introns and to the rest of the genome (distal). This analysis showed that the percentage of all DHSs mapping to promoter regions ranged from 24% for IMR90 cells to 27% for A549 and K562 cells (Figure 3A). In comparison, the percentage of GR-bound DHSs mapping to promoters is strikingly lower, especially for A549 and IMR90 cells (A549: 13%; IMR90 12%; K562 17%). Similarly, the percentage of GR-bound DHSs mapping to exons is lower than the percentage observed for all DHSs whereas GR binding is relatively enriched at intronic and distal DHSs. Together, our analysis shows that GR preferentially binds to intronic and distal DHSs rather than DHSs mapping to promoter and exonic regions.
To test if the relative depletion of promoter–proximal DHS binding is GR-specific, we examined the distribution of other TFs across different genomic locations for ChIP-seq peaks mapping to DHSs. Similar to what we observed for GR, we observed promoter–proximal depletion of binding for FOSL2 and TCF12 TFs (Figure 3B). In contrast, ETS1, a member of the ETS family of transcription factors and RNA polymerase II preferentially bind promoter–proximal DHS regions with little binding to either exons, introns or distal DHSs (Figure 3B; similar patterns observed for ATF3, BHLHE40, c-MYC, E2F6, ELF1, PBX3, USF1, YY1c and ZBTB33, data not shown). A third group of TFs showed no marked bias for DHSs at specific genomic regions as their binding distribution followed the distribution observed for all DHSs (Figure 3B, examples shown for FOXA2 and CEBPB; similar pattern seen for JUND and TEAD4, data not shown). Together, our analysis indicates that, depending on the TF examined, binding can either be preferentially promoter–proximal, relatively unbiased toward a specific genomic location or, as observed for GR, depleted at promoter regions when compared to the distribution of all DHSs found in a cell.
Notably, GR does bind promoter–proximal in a number of cases (337). To determine if the chromatin features found at GR-bound promoter DHS-regions differ from those present at unbound promoter DHSs, we compared their levels. This analysis showed that the level of certain histone modifications differs between GR-bound and unbound promoter DHSs. First, GR-bound promoter DHSs have higher average levels of H3K4me1, whereas H3K4me3 levels were lower than those observed for unbound promoter DHSs (Figure 3C). Moreover, H2BK12 and H2BK20 acetylation levels, which are markers of active enhancers (36), are higher at GR bound promoter DHSs.
Together, we found that GR preferentially binds non-promoter DHSs and that the subset of GR-bound promoters shows chromatin characteristics typical for enhancer regions.
GR motif score explains part of the observed promoter proximal depletion of GR binding
A simple explanation for the depletion of promoter–proximal GR binding is that many promoters lack a proper sequence motif to support GR binding. To investigate the role of sequence composition, we calculated the distribution of GR motif scores for DHSs grouped by GR peak presence and promoter proximity. As expected, for each of the cell lines, GR-bound DHSs have a slightly higher average motif score than unbound DHSs (Figure 4A). Further, the average motif score for distal DHSs is higher than for their promoter–proximal counterparts arguing that sequence composition contributes to the promoter–proximal depletion of GR binding observed. However, overall the promoter proximal and distal groups of DHSs have largely overlapping motif score distributions and a more formal approach was needed to determine the contribution of motif score in explaining the promoter proximal depletion observed (Figure 4A). Therefore, the contribution of motif score and promoter proximity to GR binding was quantified and statistically tested using an analysis of deviance. When controlling for motif score, promoter proximity could still explain a significant portion of the depletion of GR peaks (Figure 4B) arguing for the existence of additional mechanisms responsible for the observed promoter proximal depletion of GR binding. Together, our analysis indicates that the relatively low frequency of GR consensus motif matches explains part, but not all, of the depletion of GR binding at promoter regions.
Increased genome-wide GR binding in the absence of GCN5 and PCAF
One unexpected finding from our Bayesian modeling was the striking negative association between H3K9ac levels and GR binding. This finding was unexpected because acetylation of H3K9 is linked to transcriptionally active genomic regions and several studies have shown a positive correlation between H3K9ac levels and TF binding (37,38). H3K9ac is preferentially found at the TSS of genes and accordingly, H3K9ac levels can be used to accurately classify enhancers and promoters (39). The majority of H3K9 acetylation is carried out by GCN5 and PCAF, two enzymes that acetylate histones and other proteins including transcription factors (15,40). Of note, consistent with a role of these enzymes in modulating GR binding, overexpression of GCN5 results in a reduced activation of endogenous GR target genes and reduced DNA binding of GR (40). To explore the possible causative and mechanistic connection between H3K9ac levels and GR binding, we examined how the absence of GCN5 and PCAF influences the genome-wide binding pattern of GR (Figure 5A). For these assays, we used mouse embryonic fibroblast (MEF) GCN5/PCAF double knock-out cells (dko MEFs) which show an almost complete loss of global H3K9ac levels whereas acetylation and methylation levels of other lysines in the tails of histones, including H3K27ac, are largely unaffected ((15), Supplementary Data). Accordingly, we found that chromatin-associated H3K9ac levels, as assayed by ChIP, were markedly reduced in dko MEFs when compared to wild type (Figure 5B). Next, we assayed genome-wide GR binding in wild type and in dko MEFS by ChIP-seq. These experiments showed that the majority of GR peaks in wild type were also bound in dko MEFs (Figure 5C and D). In addition, we find that some peaks are wild type-specific (Figure 5C). Most strikingly however and consistent with a role of these enzymes in suppressing GR binding, we find a 3.7 fold increase in the number of genomic GR binding sites in the dko MEFs (Figure 5D). Subsequent analysis of GR binding at the three classes of bound loci (common; dko MEF-specific; wild type MEF-specific) by qPCR verified what we observed in the ChIP-seq experiments for each of the loci tested (Supplementary Data), arguing that the observed increase in GR binding in dko MEFs is not simply a consequence of differences in the quality of the ChIP-seq experiments. Further, analysis of two genes each near wild type and dko-specific GR peaks showed differences in GR-dependent regulation consistent with the observed cell type-specific GR binding (Supplementary Data). The increase in binding cannot be explained by changes in GR protein levels which are comparable for wild type and dko MEFs (Supplementary Data). Previous studies have shown that deletion of GCN5 results in a redistribution of DHSs in yeast (40). Therefore, we tested if changes in chromatin accessibility could explain the changes in GR binding upon GCN5/PCAF deletion by comparing DNase I sensitivity for each of the three classes of binding sites. The analysis of control ‘closed’ and ‘open’ regions showed that the assay can be used to distinguish regions with different chromatin accessibility and that these control regions yielded comparable results for wild type and dko MEFs (Supplementary Data). Further, we found that changes in GR binding levels correlated with changes in DNase I sensitivity prior to GR binding for some of the loci examined (Supplementary Data). For example, the dko-specific binding sites showed higher levels of accessibility in dko MEFs than their wild type counterpart for each of the loci examined, arguing that chromatin accessibility might contribute to the changes in GR binding we observed upon GCN5/PCAF deletion.
Next, we set out to determine if the increased genome-wide GR binding in the absence of GCN5 and PCAF might explain the promoter–proximal depletion of GR binding we observe in wild type cells. Therefore, we calculated the percentage of binding sites mapping to promoter regions for each of the three classes of GR binding sites: dko-specific, wild type-specific and common. If GCN5 and PCAF indeed play a key role in preventing promoter–proximal GR binding, we would predict that a higher percentage of dko-specific peaks map to promoter regions than for common or wild type-specific peaks. What we observed however is that a smaller percentage of dko-specific peaks maps to promoter regions than for common and wild type-specific peaks (Figure 5E). Further, the gained dko sites do not have higher H3K9ac levels prior to GR activation than those observed for common and wild type-specific peaks (Supplementary Data, Figure 5B) indicating that gained binding sites in the dko MEFS are not preferentially regions with high H3K9ac levels in wild type MEFs.
Together, our findings indicate that the negative association between H3K9ac levels and GR binding does not reflect a causative link explaining the depletion of promoter–proximal GR binding observed. Nonetheless, PCAF and GCN5 appear to modulate genome-wide GR binding, possibly by inducing locus-specific changes in chromatin accessibility or by acetylating GR, which in turn can influence GR's ability to interact with DNA (41).
Cell type-specific GR target genes have fewer promoter proximal GR peaks
The observed bias against GR binding at promoter–proximal DHSs matches findings by others (10,42) and prompts the question what the underlying biological significance might be. One possibility could be that the depletion is linked to the cell type-specific functions of the glucocorticoid receptor, which regulates vastly different sets of genes in different cell types (Figure 6A, (12)). Given the strong link between GR binding and chromatin accessibility, we hypothesized that cell type-specific regulation might preferentially occur via distal enhancers, whose chromatin accessibility is typically cell type-specific, whereas the chromatin of promoter regions is often accessible regardless of tissue examined (7,9). To test this hypothesis, we first compared the transcriptional responses to GR activation between three cell types (A549, U2OS and Nalm6) for which we also generated genome-wide GR binding data. Consistent with other studies (12), we find that GR target genes show little overlap between cell types (30 out of a total of 2974 regulated genes in three cell lines combined are regulated in all three cell types). If cell type-specific gene regulation is preferentially driven by distal binding, the expectation would be that cell type-specific target genes have fewer promoter–proximal GR binding events than common target genes. Accordingly, we find that the percentage of genes with a promoter–proximal GR peak is smaller for cell type-specific target genes than for common target genes for each of the three cell lines examined (Figure 6B) and the same trend is seen when cell-type-specific target genes are compared to genes that are regulated in two out of three cell lines, indicating that depletion of promoter proximal binding might play a role in safe-guarding GR's ability to regulate target genes in a cell type-specific manner.
DISCUSSION
Prior to the advent of methods to map TF occupancy genome-wide, studies primarily focused on promoter–proximal regions to identify candidate TF binding sites responsible for the regulation of genes. Subsequent genome-wide analysis of TF binding have shown that the majority of binding for certain TFs, including GR, actually occurs at promoter-distal sites suggesting that certain TFs might preferentially regulate transcription from remote locations. Notably, the majority of open chromatin maps to promoter-distal locations and findings presented here and by others (10) show that most GR binding occurs at chromatin that is accessible prior to hormone treatment. Thus, one explanation for the genomic distribution of GR binding is that it simply follows the genomic distribution of DHSs. Interestingly however, our approach, which focused on the universe of DHSs, uncovered that GR binding is biased against promoter proximal regions (Figure 3A). In agreement with our findings, another study also found that GR binding is biased against promoter proximal DHSs with 39% of all DHSs mapping to promoter regions whereas the percentage of GR binding sites mapping to promoter regions was only 7% (10). Our analysis of other TFs showed different biases towards binding at DHSs located at distinct genomic regions (Figure 2B). For some TFs (for example ETS1, c-MYC and E2F6) the majority of binding (>75%) occurs at promoter–proximal DHSs whereas other TFs follow the distribution of DHSs or, like GR, show a bias against promoter–proximal binding. This prompts the question what the biological significance of the promoter–proximal depletion of TF binding might be. We propose that gene regulation by distal regulatory elements might play a role in allowing GR to regulate different target genes in different tissues. Our hypothesis is motivated by the fact that promoter–proximal binding sites are typically accessible across cell types, whereas promoter-distal sites are predominantly accessible in a cell type-specific manner (7,9). Accordingly, ChIP-seq peaks in promoters frequently show a larger percentage overlap between cell types than peaks promoter-distal sites (2,43). We propose that GR might bind and regulate transcription from promoter–proximal regions regardless of cell type, whereas binding and regulation from promoter-distal regions is more likely to direct cell type-specific transcriptional programs (Figure 7). Consistent with our hypothesis, a larger percentage of genes regulated by GR across cell types harbor a promoter–proximal GR peak than genes regulated in a cell type-specific manner (Figure 6B). In contrast, the preferential promoter–proximal binding of other TFs involved in fundamental processes like DNA repair and cell-cycle progression (E2F6, YY1 and c-MYC) might facilitate the regulation of largely overlapping sets of genes across cell types. One way to further test our hypothesis would be to use genome-editing techniques to introduce GR response elements in the promoter–proximal regions of genes and to test if these genes are more prone to be regulated by GR regardless of cell type than genes whose expression is regulated by GR via distal regulatory elements.
Figure 7.

Cartoon depiction of our model how shared and cell type-specific chromatin accessibility direct gene regulation by GR. (top) Shared regulation of GR targets across cell types preferentially occurs via promoter–proximal DHSs which are accessible regardless of cell type. (bottom) In contrast, the expression of cell type-specific GR target genes preferentially occurs via cell type-specific GR binding to distal DHSs, which are accessible in a cell type-specific manner.
Many bioinformatic approaches have integrated sequence and chromatin features to predict the genome-wide binding patterns of TFs (2,8,44). What is unique about the method presented here is that we modeled GR binding in the universe of accessible chromatin to uncover what distinguishes GR-bound from GR-unbound open chromatin. This approach helped us uncover that not all open chromatin is equal and that GR preferentially binds DHS regions with chromatin marks found at active enhancers rather than DHSs with promoter marks. Further, we found a negative association of H2A.Z with GR binding which at first appears incompatible with a previous study (29) showing that H2A.Z is highly enriched at GR binding sites. However, this study compared H2A.Z levels at GR binding sites with two nearby regions which where not DHSs. Similarly, when we compared GR bound sites (which are predominantly DHSs) with other genomic regions (which are mostly not DHSs) we found a strong enrichment of H2A.Z levels at GR bound loci. This probably reflects the strong enrichment at DHSs of H2A.Z and of other chromatin marks associated with active chromatin (45) when compared to the rest of the genome, which is mostly not DHSs. Consequently, chromatin accessibility and histone modification state might provide redundant information when TF binding is modeled genome-wide. Accordingly, a previous study has shown that addition of information regarding post-translational histone modification state did not provide additional predictive power when DNase I data was already included in the model to predict genome-wide TF binding (8). In contrast, our approach showed that certain chromatin features are specifically enriched or depleted at GR-bound accessible regions and the same approach can be applied to any TF to study the role of chromatin features in specifying which DHSs are preferentially bound by a TF of interest. The hierarchical Bayesian model allows comparisons to be made directly across experiments and across cell types. This showed, for example, that H3K27ac levels show a stronger association with the levels of GR binding than motif score. The parameters are mostly consistent across cell types with the exception of H3K4me2, H3K4me3 and to some degree H4K20me1 although the ß coefficients for this modification are quite small (Figure 2C). H3K4me3 marks actively transcribed promoters and shows a negative association with GR binding in IMR90 and A549 cells. In contrast, for K562 cells, H3K4me3 positively correlates with GR binding suggesting that some of the mechanisms that specify where GR binds might be cell type-specific. Notably, depletion of promoter proximal GR binding in K562 cells is less pronounced than in A549 and IMR90 cells (Figure 3A). Furthermore, GR-bound regions in K562 cells harbor fewer GR consensus motifs ((18), Figure 4A). Together, this argues for the existence of K562-specific mechanisms of genomic GR binding, perhaps involving indirect (tethered) promoter–proximal binding mediated by interactions with other TFs. Our current model only included the GR consensus motif and therefore does not pick-up potential contributions of cell type-specific sequence motifs bound by TFs that tether GR to the genome or act cooperatively. Similarly, sequences that prevent GR binding to nearby response elements (46) were not included in the model. Therefore, future iterations of our model, to include additional sequence motifs, might better explain the observed GR binding variance.
The results of our modeling are useful to generate hypotheses, which guide the design of subsequent follow-up experiments. As detailed above, this approach uncovered a depletion of promoter proximal GR binding and yielded indications that this might play a role in facilitating cell type-specific cellular responses to glucocorticoid signaling. In addition, the modeling uncovered several striking correlations between chromatin features and GR binding. However, whether observed correlations between histone modifications and TF binding reflect a causal relationship is often unclear. Arguing for the existence of additional mechanisms that interfere with promoter–proximal GR binding, we found that motif score distribution only partially explains the depletion. Here, we followed up on the negative correlation between H3K9ac and GR binding and tested the effect of deletion of the two enzymes responsible for H3K9 acetylation, GCN5 and PCAF. Although we found that GR binds to many more genomic loci when PCAF and GCN5 are missing, the gained GR-bound genomic loci did not preferentially map to regions with high H3K9ac levels or promoter regions arguing that the correlation between GR binding and H3K9ac levels does not reflect a causative link. A likely alternative explanation for the observed anti-correlation between H3K9ac levels and GR binding is that high H3K9ac levels are found at promoter regions which have few GR binding sites which is the actual cause for the low levels of GR binding observed. This stresses that observed correlations between binding and histone marks do not necessarily imply causation. Nonetheless, the approach to disrupt the enzymes responsible for depositing specific histone marks provides a way to test for causality, which can also be applied to analyze other observed correlations between TF binding and histone modifications.
Taken together, our results uncovered that although GR preferentially binds accessible chromatin, its binding is biased against accessible chromatin located at promoter regions. This depletion can be explained in part by the sequence composition at promoters which harbor fewer binding sites and we hypothesize that depletion of promoter–proximal GR binding might play a role in safe-guarding cell type-specific transcriptional consequences of glucocorticoid signaling (Figure 7). Given that many TFs preferentially bind accessible chromatin, modeling their binding in the universe of open chromatin, as we did here for GR, might provide additional insights into the mechanisms that direct TFs to specific genomic loci.
ACCESSION NUMBERS
ChIP-seq data for A549, Nalm-6 and MEFs were deposited at EBI ArrayExpress with this study under accession number E-MTAB-5113.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Edda Einfeldt for excellent technical support and Kai Ge for sharing immortalized wild type and dko MEFs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft [ME4154/1-1 to M.J.]; National Institutes of Health [R01 CA20535 to KRY]. Funding for open access charge: Core Max Planck Institute.
Conflict of interest statement. None declared.
REFERENCES
- 1. Mathelier A., Fornes O., Arenillas D.J., Chen C.Y., Denay G., Lee J., Shi W., Shyr C., Tan G., Worsley-Hunt R. et al. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2016; 44:D110–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Handstad T., Rye M., Mocnik R., Drablos F., Saetrom P.. Cell-type specificity of ChIP-predicted transcription factor binding sites. BMC Genomics. 2012; 13:372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Siersbaek R., Rabiee A., Nielsen R., Sidoli S., Traynor S., Loft A., La Cour Poulsen L., Rogowska-Wrzesinska A., Jensen O.N., Mandrup S.. Transcription factor cooperativity in early adipogenic hotspots and super-enhancers. Cell Rep. 2014; 7:1443–1455. [DOI] [PubMed] [Google Scholar]
- 4. Jolma A., Yin Y., Nitta K.R., Dave K., Popov A., Taipale M., Enge M., Kivioja T., Morgunova E., Taipale J.. DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature. 2015; 527:384–388. [DOI] [PubMed] [Google Scholar]
- 5. Li X.Y., Thomas S., Sabo P.J., Eisen M.B., Stamatoyannopoulos J.A., Biggin M.D.. The role of chromatin accessibility in directing the widespread, overlapping patterns of Drosophila transcription factor binding. Genome Biol. 2011; 12:R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D., Barrera L.O., Van Calcar S., Qu C., Ching K.A. et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genetics. 2007; 39:311–318. [DOI] [PubMed] [Google Scholar]
- 7. Natarajan A., Yardimci G.G., Sheffield N.C., Crawford G.E., Ohler U.. Predicting cell-type-specific gene expression from regions of open chromatin. Genome Res. 2012; 22:1711–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pique-Regi R., Degner J.F., Pai A.A., Gaffney D.J., Gilad Y., Pritchard J.K.. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 2011; 21:447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Song L., Zhang Z., Grasfeder L.L., Boyle A.P., Giresi P.G., Lee B.K., Sheffield N.C., Graf S., Huss M., Keefe D. et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011; 21:1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. John S., Sabo P.J., Thurman R.E., Sung M.H., Biddie S.C., Johnson T.A., Hager G.L., Stamatoyannopoulos J.A.. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 2011; 43:264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gertz J., Savic D., Varley K.E., Partridge E.C., Safi A., Jain P., Cooper G.M., Reddy T.E., Crawford G.E., Myers R.M.. Distinct properties of cell-type-specific and shared transcription factor binding sites. Mol. Cell. 2013; 52:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. John S., Johnson T.A., Sung M.H., Biddie S.C., Trump S., Koch-Paiz C.A., Davis S.R., Walker R., Meltzer P.S., Hager G.L.. Kinetic complexity of the global response to glucocorticoid receptor action. Endocrinology. 2009; 150:1766–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bernstein B.E., Stamatoyannopoulos J.A., Costello J.F., Ren B., Milosavljevic A., Meissner A., Kellis M., Marra M.A., Beaudet A.L., Ecker J.R. et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 2010; 28:1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jin Q., Yu L.R., Wang L., Zhang Z., Kasper L.H., Lee J.E., Wang C., Brindle P.K., Dent S.Y., Ge K.. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011; 30:249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thomas-Chollier M., Watson L.C., Cooper S.B., Pufall M.A., Liu J.S., Borzym K., Vingron M., Yamamoto K.R., Meijsing S.H.. A naturally occurring insertion of a single amino acid rewires transcriptional regulation by glucocorticoid receptor isoforms. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:17826–17831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shipp L.E., Lee J.V., Yu C.Y., Pufall M., Zhang P., Scott D.K., Wang J.C.. Transcriptional regulation of human dual specificity protein phosphatase 1 (DUSP1) gene by glucocorticoids. PLoS One. 2010; 5:e13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Starick S.R., Ibn-Salem J., Jurk M., Hernandez C., Love M.I., Chung H.R., Vingron M., Thomas-Chollier M., Meijsing S.H.. ChIP-exo signal associated with DNA-binding motifs provides insight into the genomic binding of the glucocorticoid receptor and cooperating transcription factors. Genome Res. 2015; 25:825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stark R., Brown G.. DiffBind: differential binding analysis of ChIP-Seq peak data. 2011; http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf.
- 22. Tarailo-Graovac M., Chen N.. Baxevanis AD. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics. 2009; doi:10.1002/0471250953.bi0410s25. [DOI] [PubMed] [Google Scholar]
- 23. Sandelin A., Alkema W., Engstrom P., Wasserman W.W., Lenhard B.. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004; 32:D91–D94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huber W., Carey V.J., Gentleman R., Anders S., Carlson M., Carvalho B.S., Bravo H.C., Davis S., Gatto L., Girke T. et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods. 2015; 12:115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lawrence M., Huber W., Pages H., Aboyoun P., Carlson M., Gentleman R., Morgan M.T., Carey V.J.. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013; 9:e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Durinck S., Moreau Y., Kasprzyk A., Davis S., De Moor B., Brazma A., Huber W.. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005; 21:3439–3440. [DOI] [PubMed] [Google Scholar]
- 27. Gelman A. Bayesian Data Analysis. 2004; 2nd ednBoca Raton: Chapman & Hall/CRC. [Google Scholar]
- 28. Carpenter B., Gelman A., Hoffman M., Lee D., Goodrich B., Betancourt M., Brubaker M.A., Guo J., Li P., Riddell A.. Stan: A probabilistic programming language. J. Stat. Softw. 2016; 76, doi:10.18637/jss.v076.i01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. John S., Sabo P.J., Johnson T.A., Sung M.H., Biddie S.C., Lightman S.L., Voss T.C., Davis S.R., Meltzer P.S., Stamatoyannopoulos J.A. et al. Interaction of the glucocorticoid receptor with the chromatin landscape. Mol. Cell. 2008; 29:611–624. [DOI] [PubMed] [Google Scholar]
- 30. Yang H., Kwon C.S., Choi Y., Lee D.. Both H4K20 mono-methylation and H3K56 acetylation mark transcription-dependent histone turnover in fission yeast. Biochem. Biophys. Res. Commun. 2016; 476:515–521. [DOI] [PubMed] [Google Scholar]
- 31. Perner J., Lasserre J., Kinkley S., Vingron M., Chung H.R.. Inference of interactions between chromatin modifiers and histone modifications: from ChIP-Seq data to chromatin-signaling. Nucleic Acids Res. 2014; 42:13689–13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vockley C.M., D'Ippolito A.M., McDowell I.C., Majoros W.H., Safi A., Song L., Crawford G.E., Reddy T.E.. Direct GR Binding Sites Potentiate Clusters of TF Binding across the Human Genome. Cell. 2016; 166:1269–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wagner E.J., Carpenter P.B.. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012; 13:115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nguyen A.T., Zhang Y.. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011; 25:1345–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Z., Zang C., Rosenfeld J.A., Schones D.E., Barski A., Cuddapah S., Cui K., Roh T.Y., Peng W., Zhang M.Q. et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008; 40:897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kumar V., Rayan N.A., Muratani M., Lim S., Elanggovan B., Xin L., Lu T., Makhija H., Poschmann J., Lufkin T. et al. Comprehensive benchmarking reveals H2BK20 acetylation as a distinctive signature of cell-state-specific enhancers and promoters. Genome Res. 2016; 26:612–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu L., Jin G., Zhou X.. Modeling the relationship of epigenetic modifications to transcription factor binding. Nucleic Acids Res. 2015; 43:3873–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Calo E., Wysocka J.. Modification of enhancer chromatin: what, how, and why?. Mol. Cell. 2013; 49:825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rajagopal N., Ernst J., Ray P., Wu J., Zhang M., Kellis M., Ren B.. Distinct and predictive histone lysine acetylation patterns at promoters, enhancers, and gene bodies. G3. 2014; 4:2051–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zelin E., Zhang Y., Toogun O.A., Zhong S., Freeman B.C.. The p23 molecular chaperone and GCN5 acetylase jointly modulate protein-DNA dynamics and open chromatin status. Mol. Cell. 2012; 48:459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nader N., Chrousos G.P., Kino T.. Circadian rhythm transcription factor CLOCK regulates the transcriptional activity of the glucocorticoid receptor by acetylating its hinge region lysine cluster: potential physiological implications. FASEB J. 2009; 23:1572–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Grontved L., John S., Baek S., Liu Y., Buckley J.R., Vinson C., Aguilera G., Hager G.L.. C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J. 2013; 32:1568–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oldfield A.J., Yang P., Conway A.E., Cinghu S., Freudenberg J.M., Yellaboina S., Jothi R.. Histone-fold domain protein NF-Y promotes chromatin accessibility for cell type-specific master transcription factors. Mol. Cell. 2014; 55:708–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Arvey A., Agius P., Noble W.S., Leslie C.. Sequence and chromatin determinants of cell-type-specific transcription factor binding. Genome Res. 2012; 22:1723–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shu W., Chen H., Bo X., Wang S.. Genome-wide analysis of the relationships between DNaseI HS, histone modifications and gene expression reveals distinct modes of chromatin domains. Nucleic Acids Res. 2011; 39:7428–7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Telorac J., Prykhozhij S.V., Schone S., Meierhofer D., Sauer S., Thomas-Chollier M., Meijsing S.H.. Identification and characterization of DNA sequences that prevent glucocorticoid receptor binding to nearby response elements. Nucleic Acids Res. 2016; 44:6142–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.