


Abstract
CRISPR-Cas (clustered regularly interspaced short palindromic repeats and the associated genes) constitute adaptive immune systems in bacteria and archaea and they provide sequence specific immunity against foreign nucleic acids. CRISPR-Cas systems are activated by viral infection. However, little is known about how CRISPR-Cas systems are activated in response to viral infection or how their expression is controlled in the absence of viral infection. Here, we demonstrate that both the transcriptional regulator Csa3b, and the type I-A interference complex Cascade, are required to transcriptionally repress the interference gene cassette in the archaeon Sulfolobus. Csa3b binds to two palindromic repeat sites in the promoter region of the cassette and facilitates binding of the Cascade to the promoter region. Upon viral infection, loading of Cascade complexes onto crRNA-matching protospacers leads to relief of the transcriptional repression. Our data demonstrate a mechanism coupling CRISPR-Cas surveillance of protospacers to transcriptional regulation of the interference gene cassette thereby allowing a fast response to viral infection.
INTRODUCTION
Clustered regularly interspaced short palindromic repeats (CRISPR) and their CRISPR associated genes (cas) constitute the CRISPR-Cas immune system. This system provides immunity against invading genetic elements in most archaeal and many bacterial species (1,2). Short DNA fragments originating from an invading virus or plasmid are incorporated into the repeat-spacer array adjacent to the CRISPR leader, in a process known as adaptation (3–5). The CRISPR leader harboring a promoter initiates transcription of the CRISPR locus and the long pre-crRNA transcript is then processed to generate mature CRISPR RNAs (crRNAs) that are bound by Cas proteins to form ribonucleoprotein complexes for interference (6–9). Interference complexes have been classified into three major types (I, II and III) and two putative minor types (IV and V) (10). Among these, CRISPR-Cas type I is most widespread in nature and the interference complex is termed Cascade (CRISPR-associated complex for antiviral defense) (10,11).
Since the first experimental demonstration of the anti-viral function of the CRISPR-Cas systems (1), major efforts in the field have been dedicated to elucidating the structural and functional characteristics of CRISPR-Cas adaptation, transcription/processing and interference (12). In contrast, much less is known about how CRISPR-Cas systems are regulated. Among the few studies reported, Agari et al. investigated the genome wide transcription profile of Thermus thermophilus HB8 upon infection by the lytic phage ΦYS40. Expression of the majority of cas genes were found to be significantly upregulated at 75 min post phage infection (13). Similarly, Quax et al. also detected upregulation of the majority of CRISPR arrays and interference gene cassettes in Sulfolobus islandicus LAL14/1 upon infection by rudivirus SIRV2. The expression of two type I-A gene cassettes, one type I-D gene cassette and one type III-B gene cassette reached the highest level at 1 h post infection, while expression of another type I-D gene cassette continued increasing (14).
The differential expression of CRISPR-Cas systems upon viral infection strongly indicates the involvement of transcription regulators. However, very few transcription regulators involved in CRISPR-Cas transcriptional control have been studied. In Escherichia coli K12, the heat-stable nucleoid structuring protein (H-NS), which recognizes and silences the expression of foreign DNA with high AT content relative to the resident genome, was also shown to be involved in silencing CRISPR-cas promoters (15). Moreover, H-NS-mediated repression of CRISPR-based immunity could be relieved by a LysR-type transcription factor LeuO through direct binding to two sites flanking the casA promoter and the H-NS nucleation site (16). BaeR activated by envelope stress can also promote release of H-NS mediated cas repression by serving as an antagonist for H-NS binding (17). CRP represses type I-E CRISPR-Cas system in E. coli by competing with the activator LeuO (16,18), whereas in Pectobacterium atrosepticum it activates type I-F CRISPR-Cas transcription. In P. atrosepticum, GalM regulates the cas transcription through CyaA and CRP (19). Apart from transcriptional regulation, the high-temperature protein G (HtpG) chaperone can affect the CRISPR-Cas system in E. coli by stabilizing its client protein Cas3 (20). Even fewer studies have been on archaea where the transcriptional regulator Csa3a (SiRe0764), associated with the CRISPR-Cas adaptation gene cassette (SiRe0763-0760) in the genome of Sulfolobus islandicus Rey15A, was shown to activate the transcription of adaptation genes and trigger de novo CRISPR spacer acquisition (21).
Here, we report on the transcriptional co-regulation of CRISPR-Cas type I-A interference gene cassettes by both Csa3b (SSO1444 and SiRe0765, sharing 87% identity) and the Cascade complex in the archaeon Sulfolobus. The putative transcriptional regulator Csa3b is about 23 kDa carrying a CARF (CRISPR associated Rossmann fold) domain and a HTH (helix-turn-helix) domain (22). It differs significantly from Csa3a and its corresponding gene associates tightly with type I-A interference gene cassettes consisting of csa5, cas7, cas5, cas3΄, cas3″, cas8a and cas6 (10) (Figure 1A). Recently we reported that expression of the type I-A interference gene cassette is regulated (23). We have shown that Csa5 generates conditional toxicity in S. solfataricus. Under the control of its native promoter Pcas (77 bp upstream of the Csa5 start codon) in a shuttle vector pEXA2, Csa5 is toxic in CRISPR-Cas-deficient mutant S. solfataricus Sens1 but not in S. solfataricus InF1 containing functional CRISPR-Cas systems (23). InF1 encodes six CRISPR arrays (A-F), three type I-A gene cassettes and four type III gene cassettes (24). Sens1 contains only arrays E-F, one type III gene cassette and one incomplete type I-A gene cassette (25,26). As the Csa5 toxicity in Sens1 cells was found to be related to its high expression level and protein oligomerization, we inferred that the absence of Csa5 toxicity in InF1 cells is due to, at least partially, the repression of csa5 transcription by CRISPR-Cas related genes.
Figure 1.

The 77 bp Pcas contains sequence elements required for transcriptional repression. (A) Schematic presentation of the CRISPR-Cas type I-A interference gene cassette of S. solfataricus studied in this work, along with the associated CRISPR array (containing 31 spacers) and the following type I-A adaptation gene cassette. Both gene ID (SSO numbers) and cas nomenclature are shown for each gene. The Pcas orientation and start site are indicated (bended arrow). (B) Depiction of csa5-containing inserts cloned into three constructs (pcsa5-39, −62 and −77) based on plasmid vector pEXA2. As reference, the position and orientation of chromosomal Pcas (black bar) in relation to csa3 (gray arrow) and csa5 (black arrow) is indicated on top of the three inserts. (C) Transformation efficiency of different csa5-containing constructs in Sens1 (lacking functional CRISPR-Cas) and InF1 cells.
Here, we demonstrate that both Csa3b and the Cascade complex bind to the promoter region of CRISPR-Cas type I-A interference gene cassettes and function as transcriptional repressors. Upon viral infection, redistribution of the Cascade complex to the crRNA-matching protospacers leads to relief of transcriptional repression thereby allowing a fast transcriptional response, and potentially high interference levels to viral infection.
MATERIALS AND METHODS
Strains, media and growth conditions
Sulfolobus strains used in this study are listed in Supplementary Table S1 with the genotype indicated. S. islandicus LAL549 was isolated by spreading S. islandicus LAL14/1 on plates containing uracil and FOA as described previously (27). It carries a deletion of 549 bp (1 301 868–1 302 416 bp) inside pyrF gene. Sulfolobus strains were grown at 78°C in basic salt medium (28) supplemented with 0.2% Glucose, 0.2% Casamino Acids, 0.005% Yeast extract and a vitamin mixture (GCVY). When needed, arabinose (0.2%) was substituted for glucose to induce gene expression from the arabinose promoter on different plasmid constructs (ACVY). E. coli strain DH5α and Rosetta cells were used for DNA cloning and recombinant protein production. All E. coli strains were cultured at 37°C in Lysogeny Broth supplemented with final concentration of 100 μg/ml ampicillin or 30 μg/ml kanamycin as required.
Plasmid construction
Detailed information about cloning vectors, restriction sites and PCR primers for the construction of the plasmids are indicated in Supplementary Table S2. In case of cloning two or more different fragments into the same vector, fusion PCR was performed before cloning and all the four or six primers are indicated. The genome-editing plasmids were constructed following the protocol described previously (29,30). A DNA fragment carrying two copies of type I-A CRISPR repeat separated by two oppositely oriented SapI recognition sequences was inserted into pEXA2 between NdeI and MluI sites adjacent to the arabinose promoter. To facilitate the insertion of mutant allele, an XhoI restriction enzyme site was added adjacent to SphI to construct the basic genome-editing plasmid pGE1 (Supplementary Figure S1). pGE2 was constructed by replacing type I-A CRISPR repeat with type I-D CRISPR repeat in pGE1. To construct each specific genome-editing plasmid, the spacer DNA fragment was prepared by annealing two complementary oligonucleotides and the resultant dsDNA carrying 3 nt overhangs was inserted into the SapI sites of pGE1 or pGE2. Next, donor DNA fragments containing a mutant allele of target gene were obtained by fusion PCR and the PCR product was inserted between the SphI and XhoI sites of the mini-CRISPR array containing pGE1 or pGE2, giving complete genome editing plasmids named pGE1 (or pGE2)-target site-Strain (Supplementary Table S2).
Construction of gene or CRISPR array knockout mutants
csa3b and cas8a were deleted from E233S based on the marker insertion and unmarked target gene deletion method (MID) (31) using plasmids pMID-csa3b and pUC19-cas8a (Supplementary Table S2), respectively. The CRISPR array deletion mutants were obtained using the CRISPR-based genome editing method (30). To knockout the two arrays in E233S, pGE1-Array12-E233S (Supplementary Table S2) was transformed into E233S and the plasmid was subsequently removed by counterselection (30). The five arrays in LAL549 were deleted sequentially using plasmids pGE1-Array12-LAL, pGE1-Array5-LAL and pGE2-Array34-LAL (Supplementary Table S2), to remove arrays 1+2, array 5 and arrays 3+4, respectively. All knockout mutants were confirmed by PCR with primers listed in Supplementary Table S2 (Supplementary Figure S2).
Pull-down assay using biotinylated DNA
The promoter region of the type I-A interference gene cassette was amplified by PCR using primers NoBiotin-pcsa5-rev and Biotin-pcsa5-fwd (Supplementary Table S3). Sulfolobus Cells were harvested at OD600 around 0.9 and resuspended in DNA-binding buffer (10 mM Tris-HCl, pH 7.6, 150 mM KCl, 2 mM DTT, 10% glycerol) before sonication. The biotinylated DNA (4 μg) and 100 μl M-280 Streptavidin Dynabeads (Invitrogen, 11205D) were added to the cell extract and then mixed gently at 50°C for 20 min. After incubation, the beads were washed three times and dissolved in 100 μl binding buffer. A portion of the beads was boiled and loaded for SDS-PAGE.
Protein purification and antibody preparation
csa3b (sso1444) was cloned downstream of the arabinose promoter and upstream of the histine tag coding sequence in pEXA3 to make pAPCsa3b-His6 for His-tagged Csa3b (Csa3b-His6) protein expression in Sulfolobus. For Csa3b-His6 protein expression in E. coli Rosetta cells, the pET-30a based construct pCsa3b-His6 (Supplementary Table S2) was used. pAPCsa3b-His6 was transformed into S. solfataricus Sens1 for Csa3b-His6 purification. Sulfolobus cells were grown in GCVY medium at 78°C until OD600 reached around 0.6. To induce Csa3b-His6 expression from the arabinose promoter, we transferred the culture from GCVY to ACVY medium (0.2% arabinose was substituted for glucose) and the culture was incubated overnight. Cells were harvested by centrifugation at 8000 g for 10 min, resuspended in buffer (50 mM NaH2PO4 (PH 8.0), 300 mM NaCl, 1 mM EDTA, 1 mM PMSF) and lysed by sonication. The lysate was subjected to His-tag purification using Ni-NTA agarose beads (Qiagen, Germany) according to the manufacturer's instruction. The N-terminal fragment of Csa3b containing 120 aa residues was purified from E. coli transformed with plasmid pCsa3bpartN-His6 (Supplementary Table S2) and the partial Csa3b was used to raise polyclonal antibody in rabbit (Innovagen, Sweden).
DNase I footprinting assay
FAM fluorescence labeling capillary electrophoresis was used for DNase I footprinting (32). First, the wild-type and mutant forms of Pcas were PCR amplified using primers listed in Supplementary Table S3 and the PCR products were cloned into pJET1.2. Using these plasmids as a template, FAM labeled DNAs were obtained by amplifying the insert with the primer Fprintingfwd and the FAM labeled primer FAM-pJET1.2rev (Supplementary Table S3). Next, about 0.03 μM FAM labeled DNA fragments were incubated with 0.17 μM Csa3b-His6 protein (purified from Sulfolobus cells) at 37°C for 30 min in 50 μl binding buffer [20 mM HEPES (pH 7.6), 10 mM (NH4)2SO4, 1 mM DTT, 0.2% Tween-20, 30 mM KCl]. The reaction was subjected to DNase I (0.016 units) (Thermo Scientific, EN0521) digestion for 40 s at 37°C, then stopped by addition of EDTA at a final concentration of 60 mM and incubation at 80°C for 10 min. Samples were subjected to phenol–chloroform extraction, ethanol precipitation and sent to Eurofins Genomics company (Germany) for fragment length analysis by capillary electrophoresis.
β-Galactosidase assay
All constructs including placS-77, placS-39, placS-77 RI L, placS-77 RI R, placS-77 RII L and placS-77 RII R were transformed into suitable Sulfolobus strains individually, and single colonies were isolated. β-Galactosidase activity in an equal amount of total crude proteins was measured for each transformant, as described previously (33).
RNA isolation, quantitative RT-PCR analysis and RNA sequencing
Total RNAs were extracted from Sulfolobus cells (50 ml, OD600 around 0.2) using Trizol reagent (Invitrogen) following the instruction of the manufacturer. Twenty micrograms of RNA was treated by DNase (TURBO DNA-free™ Kit, Life Technologies, AM1907) and 3.5 μg DNase-treated RNA was subjected to first strand cDNA synthesis using Revert Aid First Strand cDNA Synthesis Kit (Thermo Scientific, K1622). Quantitative PCR analysis was conducted using the cDNAs as templates and Maxima SYBR Green/ROX qPCR Master Mix (Thermo Scientific, K0221). Template cDNA, 5 μl Maxima SYBR Green/ROX qPCR Master Mix and forward and reverse primers (each 5 pmol) were mixed in each reaction system (total volume 10 μl). The PCR protocol consisted of 95°C for 5 min, 40 cycles of 95°C for 10 s, 56°C for 10 s and 72°C for 20 s. Relative amounts of RNAs were calculated using the comparative Ct method (34), and amplification efficiencies of csa5 and reference tfbI sequences were validated (Supplementary Figure S3). For RNA sequencing, 20 μg DNase treated RNAs were sent to BGI, Shenzhen, China followed by standardized library construction and Illumina sequencing. The RNA-Seq data were normalized and the ratio of CRISPR-Cas related transcripts between the knockout mutants and the wild-type E233S strain was calculated by BGI according to their standard procedure.
ChIP assay
Sulfolobus cultures (OD600 = 0.4) were fixed in 1% formaldehyde for 10 min at 37°C and then quenched by adding glycine to a final concentration of 125 mM, followed by an additional incubation of 5 min at 37°C. Chromatin immunoprecipitation was performed using methods developed by Jia Guo et al. (35). Following DNA extraction with phenol/chloroform and DNA precipitation, DNA pellet was washed twice with 70% ethanol and resuspended in 50 μl Milli-Q water, and a 0.01 μl DNA solution was subjected to quantitative PCR using the primer sets listed in Supplementary Table S3.
Comparative bioinformatics
Putative Csa3 sequences were identified from genomes present in Refseq, as of 21.03.2016, using the TIGRFAMs hidden Markov model TIGR01884 and hmmsearch from the HMMER 3.0 package and the model's trusted cutoffs (36–38). Muscle version 3.8.31 was used for multiple alignments. The evolutionary relationship was inferred by using the Maximum Likelihood method based on the JTT matrix-based model (39). Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then by selecting the topology with superior log likelihood value. The tree with the highest log likelihood (−26 217.2609) was selected and drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis included 142 aa sequences. There were a total of 336 positions in the final data set. Evolutionary analyses were conducted in MEGA7 (40). Sequences upstream of CRISPR-Cas type I-A interference gene cassettes were extracted manually and motifs were identified using MEME version 4.8.1 (41).
RESULTS
Two tandem palindromic sequences in promoter Pcas are involved in transcriptional regulation of the Sulfolobus type I-A interference gene cassette.
To identify regulatory sequences in the promoter of type I-A interference gene cassette of S. solfataricus, Pcas, we shortened the 77 bp Pcas promoter to 62 or 39 bp upstream of csa5, and the plasmid constructs (Figure 1B) were electroporated individually into InF1 cells. In contrast to the 77 bp promoter sequence, both shortened promoter sequences conferred toxicity to their corresponding construct because no InF1 transformants were obtained (Figure 1C). This suggests that a regulatory sequence in the 77 bp promoter region was absent or incomplete in the shortened promoter sequences. Indeed, two 18-bp tandem imperfect repeats, RI and RII, are present immediately upstream of the basal promoter, each of which contains palindromic ends that could serve as binding sites for transcriptional regulators (Figure 2). Both repeats are missing in the 39 bp promoter whereas the TATA box-proximal repeat (RII) remains intact and most of the distal repeat (RI) is deleted in the 62 bp promoter (Figure 1B).
Figure 2.

Two palindromic repeats, RI and RII, are involved in transcriptional repression of Pcas in InF1 cells. (A) Depiction of inserts cloned into pEXA2-based constructs placS-77 WT, placS-77 RI or RII mutants and placS-39. LacS is fused with Pcas or Pcas derivatives in the constructs. (B) Sequences of Pcas and Pcas derivatives in the six constructs. Repeats RI, RII and the corresponding mutants are underlined with the mutated sequences shown in bold. The TFB recognition element (BRE) and TATA box in the basal promoter of Pcas are indicated. The start codon of lacS gene, ATG, is shown in bold. (C) Relative LacS activity expressed from plasmid-borne Pcas and Pcas derivatives in InF1 cells.
Next, in order to investigate the involvement of the palindromic repeats in the regulation of gene expression from Pcas, we mutated the palindromic sequences individually and the promoter was fused in pEXA2 vector with a reporter gene encoding galactosidase (LacS, SiRe2224) (Figure 2A and B). The plasmid constructs containing the shortened promoter (39 bp) (placS-39), or the 77 bp promoter with or without mutations (placS-77 wild type (WT), RI or RII mutants), were transformed individually into InF1 and the specific β-galactosidase activity was measured for each transformant. The activity was normalized for each strain by arbitrarily setting the activity from InF1/placS-39 as 100. As shown in Figure 2C, activity derived from the 39 bp promoter (placS-39) was more than 10 times higher than that obtained from the 77 bp promoter (placS-77 WT), indicative of a promoter derepression in the former. Since the only difference between the two promoter derivatives is the tandem palindromic repeats carried by the latter, these repeats could function in the repression of the promoter activity. Indeed, mutation of half of each palindromic repeat resulted in an elevated reporter gene activity (Figure 2C), suggesting a reduced or abolished binding of repressor(s) at the mutated sites. RI appears slightly more important than RII as the effect of mutation is more profound for the former.
Csa3b binds specifically to the palindromic repeat sequences in Pcas
A 227 bp sequence upstream of csa5 start codon was amplified by PCR with one of the two primers biotinylated. This biotinylated DNA contains both RI and RII sites and was used for pull-down assay to identify transcriptional regulator(s) of the promoter. Cellular extracts were prepared from Sens1 and InF1 cultures. As transcriptional repression only occurred in InF1, no transcriptional repressor was expected to bind to the sequence in Sens1, which was thus used as a control. One protein was obtained from InF1 cells that was absent from Sens1 cells in the pull-down assay and the size (23 kDa) was similar to that of Csa3b (SSO1444). Western blotting using an antibody against Csa3b (SSO1444) revealed a specific signal at the position of the extra band present in InF1 (Supplementary Figure S4A). Compared to the His-tagged and thus slightly larger Csa3b expressed in and purified from E. coli, the intensity of the Western blot signal was proportional to that observed in Coomassie blue stained PAGE gel (Supplementary Figure S4A). Further, a similar band occurred in the sample obtained from the pull-down assay with Sens1 cells carrying a plasmid-borne csa3b (Supplementary Figure S4A). Therefore, we concluded that the extra band in InF1 sample was Csa3b protein.
DNase I footprinting assays were performed on the promoter region in the presence or absence of Csa3b protein, using a capillary sequencer to analyze the protected regions (32). The template strand of the DNA fragments covering the two repeats, RI and RII, was labeled by FAM and subjected to DNase I footprinting assays. In contrast to the control (black signal), the DNA sample bound by Csa3b (red signal) was protected over most of the repeat region, including both RI and RII sites (Figure 3A). Moreover, four hypersensitive sites occurred upon Csa3b binding, corresponding to the four nucleotides at the ends of the palindromes (G and C in red, Figure 3A). This suggests that binding of Csa3b on the repeat region caused DNA bending making the four nucleotides highly accessible for DNase I digestion. Next, the two regulation sites (RI and RII) were mutated individually and subjected to the same assay. Mutation of the right palindromic part of each regulation site prevented the protection of the corresponding site by Csa3b and the two corresponding hypersensitive sites disappeared (Figure 3B and C). This demonstrates that Csa3b may regulate the expression of CRISPR-Cas type I-A interference gene cassette by binding to the two regulation sites upstream of the promoter Pcas.
Figure 3.

DNase I footprinting of Pcas DNA. DNase I footprinting of Pcas DNA was performed with the template strand (FAM-labeled) in the presence (spectra in red) or absence (spectra in black) of Csa3b. (A) Wild-type or (B and C) mutated Pcas DNA were used. RI and RII sites on the coding strand are underlined and mutated nucleotides are indicated in green. Red colored nucleotides represent DNase I hypersensitive sites on the template strand.
Cascade is involved in the transcriptional repression of the type I-A interference gene cassette
As described above, the presence of the RI and RII sequences in the promoter confers more than 10 times repression in InF1 cells (Figure 2) encoding multiple CRISPR-Cas gene cassettes (42). To establish whether Csa3b was the sole protein responsible for the repression, the reporter gene lacS was fused with the promoter containing both RI and RII (promoter 77) and cloned into pEXA2 with or without the csa3b gene (Supplementary Figure S4B). csa3b was positioned either upstream or downstream of lacS. The three constructs were transformed into Sens1 cells lacking functional CRISPR-Cas systems and the β-galactosidase activity was measured and quantified by arbitrarily setting the activity derived from the plasmid placs-77 as 100. In the presence of csa3b, LacS activity was reduced to about 40% (Supplementary Figure S4C), suggesting that the much stronger repression of the promoter in InF1 cells (>10-fold, Figure 2) resulted from co-operation between Csa3b and other element(s).
To assess the role of Csa3b on transcription regulation, we deleted csa3b from S. islandicus E233S (27) which carries a single type I-A interference gene cassette and an upstream csa3b (Supplementary Figure S5), both of which share high sequence similarities (86–95%) to the corresponding homologs in S. solfataricus InF1. RNAs were isolated from both E233S and the knockout strain Δcsa3b, and the transcript level of csa5, the first gene of the interference gene cassette, was compared by quantitative reverse transcription PCR (RT-PCR). The threshold cycle (Ct) values of samples, with three technical replicates, are shown in Supplementary Table S4A. The analysis showed that csa5 transcription was upregulated 25 times in Δcsa3b, indicating that Csa3b is needed for the repression of the interference gene cassette (Figure 4A). This is further supported by the restored transcriptional repression of csa5 in Δcsa3b cells complemented with a plasmid-borne csa3b (pcsa3b) expressed from its native promoter (Δcsa3b/pcsa3b in Figure 4A).
Figure 4.

Regulation of Pcas in different deletion mutants derived from E233S. (A) Transcriptional level of csa5. (B) Relative LacS activity derived from Pcas. Results from three technical replicates are shown in both A and B, and error bars indicate standard deviation. WT: E233S.
We examined the overall effect of csa3b deletion on transcription by sequencing the transcriptomes of Δcsa3b and the complement strain Δcsa3b/pcsa3b with that of the mother strain E233S as reference. Along with the three samples, we also sequenced the transcriptomes of individual deletion mutants of the type I-A interference gene cassette (Δcsa5, Δcas7, Δcas5, Δcas3΄, Δcas3″, Δcas6) and the transcriptome of two type III-B interference gene cassettes deletion mutants (Δcmrα cmrβ), all of which were generated previously (43). As expected, the expression of all the genes of the type I-A interference gene cassette (csa5–cas6) was upregulated in the Δcsa3b cells by between 14- and 40-fold, and the repression is restored in the complement strain Δcsa3b/pcsa3b (Supplementary Table S5A and B). Surprisingly, deletion of any individual component of the type I-A interference gene cassette also caused upregulated transcription of the gene cassette, with the effect of cas7 deletion and cas6 deletion being most profound (up to 45-fold and 22-fold, respectively). The only exceptions were the last three genes (cas3″, cas8a and cas6) that are moderately downregulated in Δcas5 and Δcas3΄. The upregulation of the type I-A interference gene cassette appears specific, because the adaptation gene cassette was not affected. Importantly, deleting both cmr cassettes did not cause significant transcriptional regulation on the type I-A interference gene cassette. This suggests that the components of the type I-A interference gene cassette are involved in transcriptional repression of the gene cassette.
Next, we performed quantitative RT-PCR on csa5 to verify the upregulation of the type I-A interference gene cassette in the individual deletion mutant. The threshold cycle (Ct) values of samples, with three technical replicates, are shown in Supplementary Table S4A. Values for the tfb I gene were used for normalization. The normalized value for csa5 transcript in WT was set to 1. In accordance with the transcriptomic data, csa5 is upregulated about 45, 9, 13, 18, 19 and 26 times, in Δcas7, Δcas5, Δcas3΄, Δcas3″, Δcas8a and Δcas6, respectively (Figure 4A). As cas6 is responsible for mature crRNA biogenesis in S. islandicus, and deletion of cas6 caused dramatic upregulation of the type I-A interference gene cassette, we set out to check whether crRNAs are also involved in transcriptional repression of the gene cassette. Depletion of crRNA by deleting the CRISPR repeat arrays along with the acquisition gene cassette (E233SΔarrays) (Supplementary Figure S5) caused a 25-fold upregulation of csa5 transcription (Figure 4A). The necessity of crRNA for the transcriptional repression was further supported by the restored transcriptional repression of csa5 in E233SΔarrays/arrayCK cells carrying a plasmid-borne mini-CRISPR composed of two copies of type I-A repeat interspaced with one spacer (arrayCK, Supplementary Table S2). Taken together, the data suggest that all the type I-A interference components, including crRNA, are involved in the transcriptional repression of the gene cassette, possibly through binding of Cascade to the promoter region.
csa5 expression in different deletion mutants could be influenced by other factors in addition to the promoter strength thus causing the observed varied effects (Figure 4A). Therefore, the regulation of Pcas in the different knockout strains was further studied using the construct placS-77 (Figure 2) that carries the fusion of Pcas (77 bp) and the reporter gene lacS. placS-77 was transformed into the deletion mutants and β-Galactosidase activity was quantified by arbitrarily setting the activity from E233S/placS-77 as 1. LacS was upregulated around 40 times in Δcsa3b, Δcsa5, Δcas7, Δcas5, Δcas8a and Δcas6 but only around 10 times in Δarrays, Δcas3΄ and Δcas3″ (Figure 4B). Possibly, two different versions of Cascade are involved in transcriptional repression of Pcas, one comprising all the components and the other missing crRNA, Cas3΄ and Cas3΄'. This possibility is supported by previous studies suggesting the existence of subcomplexes of the type I system (44–47).
Recruitment of Cascade to protospacers leads to Pcas derepression
The requirement of crRNA and all the components of the interference gene cassette for the repression strongly suggest that the Cascade complex binds to the promoter Pcas. However, it is not feasible to test the binding in vitro due to the difficulty of isolating a complete and functional Cascade from Sulfolobus, as experienced in our laboratory and by others (45). Therefore, we employed a virus-host system to investigate the effect of recruiting Cascade to viral protospacers.
Sulfolobus islandicus LAL14/1, the model host of rudivirus SIRV2, encodes five CRISPR arrays as well as type I-A, type I-D and type III-B systems (48), the majority of which were upregulated 1 h post infection by SIRV2 (14). The CRISPR arrays contain 13 spacers matching SIRV2 genome with one to five base pair mismatches. To avoid potential complications caused by these spacers, we knocked out all the five arrays in Sulfolobus islandicus LAL549 (a derivative of LAL14/1 carrying a partial pyrF deletion, Supplementary Table S1) and the resultant strain LALΔarrays carries one type I-A, one type I-D and one type III-B interference gene cassette. Next we constructed two type I-A mini-CRISPR arrays individually in vector pGE1 (Supplementary Table S2) targeting the coding strand of SIRV2 gp38 (Cgp38-CCA) or the template strand of gp38 (Tgp38-CCG). Both spacers are among the 13 natural spacers matching SIRV2 genome, but the mismatches were corrected in the mini-CRISPR arrays so that both spacers match the viral protospacers perfectly with the typical type I-A PAM CCN. To test the effect of the PAM motif, we removed CCA and CCG, respectively, by shifting both spacers 1 bp in mini-CRISPR arrays to construct Cgp38-CAT and Tgp38-CGT (Supplementary Figure S6). The four constructs were transformed individually into LALΔarrays with arrayCK being a control which does not match either the genome of LALΔarrays or that of SIRV2. The transformants were grown in arabinose-containing ACVY liquid medium until OD600 around 0.2, infected with SIRV2 (multiplicity of infection around 5) and the level of csa5 transcript at 1 h post infection was compared by quantitative RT-PCR. The threshold cycle (Ct) values of samples, with three technical replicates, are shown in Supplementary Table S4B. In comparison with the transcript level in the corresponding uninfected cultures (Figure 5), csa5 was upregulated around 6 and 4-fold post infection in cultures containing Cgp38-CCA and Tgp38-CCG, respectively, demonstrating a transcriptional derepression of the type I-A interference gene cassette. No upregulation of csa5 was detected in infected cells containing the control spacer carried on arrayCK. Compared to the CCN PAM for type I-A interference complex, the use of CGT did not lead to a release of the repression while the use of CAT resulted in a mild release (∼1.8-fold) of the repression. The slight release observed with Cgp38-CAT correlates with an observed low interference activity accompanied by CAN PAM (49). Moreover, in accordance with the transcriptomic data from SIRV2 infected wild-type LAL14/1 cells (14), quantitative RT-PCR revealed about 13-fold upregulation of csa5 in SIRV2 infected cells (Figure 5). Further, 6 to 8-fold transcriptional upregulation of csa5 was observed in SIRV2 infected cells carrying plasmid-borne spacers that target either the coding or the template strand of randomly selected SIRV2 genes (gp01, gp29, gp49) (Supplementary Figure S7 and Supplementary Table S4C). The plasmid-borne spacers do not match to the chromosome of LALΔarrays including the interference gene cassette, and thus, the observed transcriptional regulation is unlikely due to direct binding of the corresponding crRNAs to the gene cassette. Taken together, this result suggests that recruitment of Cascade to protospacers with preferred CCN PAM reduces the amount of Cascade binding at Pcas thereby releasing transcriptional repression.
Figure 5.
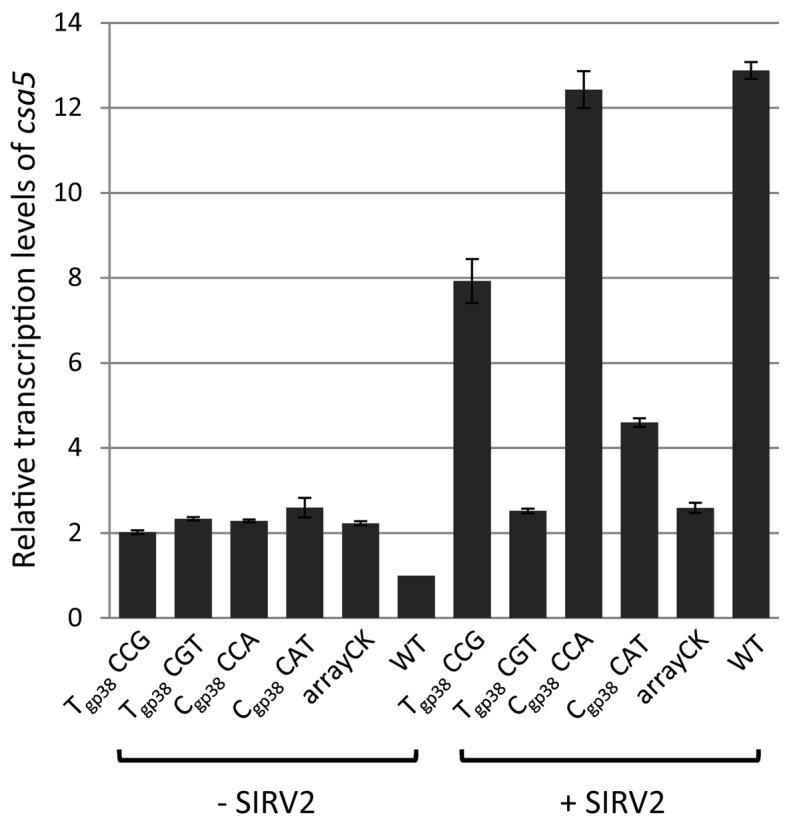
Transcriptional regulation of csa5 in LALΔarrays carrying different plasmid-borne spacers upon SIRV2 infection. LALΔarrays transformed with construct Tgp38-CCG, Tgp38-CGT, Cgp38-CCA, Cgp38-CAT or arrayCK, as well as the wild-type (WT) strain LAL549, were incubated with (+ SIRV2) or without SIRV2 (- SIRV2) before RNA extraction and RT-qPCR. Sample normalization was done as indicated for Figure 4A. Results from three technical replicates are shown, and error bars indicate standard deviation.
Csa3b-dependent binding of Cascade complex to Pcas
To further confirm the binding of Cascade to the promoter Pcas, we performed ChIP assay with an anti-Csa5 antibody. S. islandicus E233S and its corresponding Δcas7, Δcas8, Δarrays and Δcsa3b cultures were treated with 1% formaldehyde to crosslink interacting macromolecules, and DNA bound to Cascade was co-immunoprecipitated using the anti-Csa5 antibody following DNA fragmentation. The size distribution of the fragmented DNA, a loading control (SDS PAGE of 5 μl cell lysate) and the specificity of Csa5-Ab were shown in Supplementary Figure S8. The amount of Pcas DNA was then quantified by qPCR using tfbI gene (704 kbp from the Pcas) as reference. The threshold cycle (Ct) values of samples, with three technical replicates, are shown in Supplementary Table S4D. As shown in Figure 6, ∼7-fold more DNA from the promoter region was detected in sample E233S precipitated by Csa5 antibody relative to the control where Csa5 antibody was not added. This supports the inference that Cascade binds to Pcas and this is further supported by the failure to detect significant amounts of Pcas DNA in samples Δcas7, Δcas8 or Δarrays and by the detection of ∼5-fold more DNA in sample Δarrays complemented by a plasmid-borne mini-CRISPR (Figure 6). Interestingly, the Csa5-antibody failed to precipitate Pcas DNA from the csa3b deletion mutant Δcsa3b, whereas complementation of csa3b deficiency by a plasmid-borne csa3b (Δcsa3b/pcsa3b) allowed precipitation of 11-fold more Pcas DNA by Csa5 antibody than the control (Figure 6). This suggests that Csa3b is needed for Cascade binding to Pcas.
Figure 6.

In vivo binding of Cascade to Pcas. ChIP-qPCR values for the tfbI gene were used for normalization. The normalized value for Pcas derived from E233S (WT) without antibody addition was set to 1. Results from three technical replicates are shown, and error bars indicate standard deviation. +, with Csa5-Ab; −, without Cas5-Ab.
Conservation of csa3 and Pcas in archaea
We searched for Csa3 in all genomes present in the Refseq database and retrieved 142 Csa3 sequences using the TIGRFAMs hidden Markov model specific for Csa3. More than half (53%) of the csa3 genes are immediately adjacent to CRISPR-Cas type I-A gene cassettes, and the remainder are associated with CRISPR-Cas type I-D, type I-B, type I-C, type I-E and/or type III (in total ∼26%) as well as with non-CRISPR related genes (∼21%) (Supplementary Table S6). Phylogenetic analysis demonstrates that most of the Csa3 sequences associated with type I-A or type I-D gene cassettes are clustered together (Supplementary Figure S9).
Further analysis was performed for a major cluster comprising Csa3 sequences that are closely related to SSO1444 and SiRe0765, and are therefore designated Csa3b (Figure 7). These csa3b genes are all derived from genomes of Sulfolobaceae and all except two are proximal to a type I-A interference gene cassette (Figure 7, Supplementary Table S6). Moreover, the promoter regions of the immediately adjacent type I-A interference gene cassettes are conserved and mostly carry two highly similar palindromic repeats (Figure 7). On the other hand, more than 90% of CRISPR-Cas type I-A gene cassettes have an adjacent csa3 gene, previously designated casR (50). Taken together, this suggests that Csa3b/Cascade mediated transcriptional repression is likely applicable to most CRISPR-Cas type I-A systems.
Figure 7.

Phylogenetic tree of (A) Csa3b and (B) the associated palindromic sequences in Pcas of 25 members of the Sulfolobaceae. Strain names and Csa3b gene IDs are given in the tree. Twenty strains labeled in red carry two copies of the palindromic repeat and the other five (in black) contain only one copy. The palindromic sequences within each subbranch are almost identical and only those from the strains shown in bold in the tree were used to build the sequence logo.
DISCUSSION
We have shown that the Sulfolobus transcriptional repressor Csa3b binds to two imperfect palindromic repeats (TTTTCN8GAAAA/T) upstream of the promoter of the type I-A interference gene cassette (Figure 3). However, Csa3b alone can only confer a moderate level of repression on the expression of the type I-A interference gene cassette in Sulfolobus. All components of the type I-A Cascade complex, including crRNAs, are needed for complete repression (Figure 4A and B, Supplementary Table S5A and B). Importantly, recruitment of the type I-A Cascade complex to crRNA-matching protospacers leads to transcriptional derepression that requires the type I-A PAM, CCN (Figure 5). Further, ChIP assay strongly suggested that the Cascade complex binds to the promoter (Figure 6). Taken together, the data allow us to propose the following model for transcriptional regulation of CRISPR-Cas type I-A interference gene cassette in Sulfolobus (Figure 8). In the absence of viral infection, Cascade binds at the promoter region and maintains transcriptional repression in a Csa3b-dependent manner. Upon viral infection Cascade is redistributed to protospacer sites on the viral genome, and its binding to the promoter is thus reduced allowing transcriptional activation of the interference gene cassette.
Figure 8.

Model of transcriptional regulation of CRISPR-Cas type I-A interference gene cassette. Stage 1, Pcas is repressed by Csa3b and Cascade or Cascade subcomplex in the absence of viral infection; stage 2, a protospacer is introduced into the host cell upon viral infection; stage 3, binding of Cascade to the protospacer reduces the amount of Cascade on Pcas, leading to transcriptional derepression. The subcomplex lacks crRNA, Cas3΄ and Cas3″, and is thus not expected to direct viral infection surveillance (stage 2΄).
Our data also suggest that a subcomplex of Cascade containing Csa5, Cas7, Cas5, Cas8a and Cas6 is involved in transcriptional repression of Pcas (Figures 4B and 8). This subcomplex is not expected to direct viral infection surveillance due to the lack of crRNA, Cas3΄ and Cas3″ (Figure 8). It remains an interesting question as to how this subcomplex-mediated transcriptional repression is regulated. Further, future studies are needed to elucidate whether the complete Cascade and the subcomplex interact in binding and repressing Pcas.
Bacterial and archaeal species often carry large arrays of CRISPR repeat-spacer units and encode multiple cas gene cassettes (51,52), and their expression is expected to be maintained at a low level in the absence of invading viruses in order to minimize energy consumption. On the other hand, the defence system should be able to respond quickly to viral infections. In support of this scenario, we have shown that both Csa3b and the Cascade complex cooperate in transcriptional repression of the type I-A interference gene cassette in Sulfolobus in the absence of viral infection. Upon rudiviral SIRV2 infection, Cascade redistributes to crRNA-matching protospacers on the viral DNA thereby allowing derepression of the interference gene cassette. This strategy ensures a direct coupling between CRISPR-Cas surveillance of an infecting virus and transcriptional activation of the interference gene cassette and thereby, enables rapid defence against the infecting virus.
To date, <10 global transcription regulators have been identified from several bacterial organisms including E. coli, Salmonella Typhi, Myxococcus xanthus and Thermus thermophilus, which repress or activate CRISPR-Cas transcription (53). However, all except one were investigated in the absence of viral infection (13). Therefore, it remains an open question whether, and how, these regulators respond to viral infection. The tight association of csa3b with type I-A interference gene cassettes and the prevalence of palindromic sequences directly upstream of the gene cassettes in some archaea suggests that transcriptional regulation of type I-A interference gene cassettes by Csa3b and the Cascade complex may be a highly conserved and important regulation mechanism.
S. islandicus LAL549 carries 13 spacers matching SIRV2 genome with one to five base pair mismatches. Despite being susceptible to SIRV2 infection, LAL549 wild-type cells exhibit a degree of CRISPR-Cas interference against the virus, as evidenced by much lower plaquing efficiency in the wild-type cells than in the CRISPR array deletion mutant LALΔarrays (data not shown). The partial defence against SIRV2 could be due to the absence of spacers that could confer strong interference activities or unknown viral counteractive mechanisms (54,55). Nevertheless, activation of Cascade expression is expected to confer higher interference activity.
Virus induced transcriptional activation of CRISPR-Cas systems has also been described for bacteria (13). Although CRP-dependent upregulation of two cas operons was observed in Thermus thermophiles HB8 upon infection by the lytic phage PhiYS40, the regulatory mechanism remains unknown. Moreover, upregulation of several cas genes and multiple CRISPR loci in the infected cells was independent of CRP. Further, CRP activates type I-F CRISPR-Cas system in P. atrosepticum (19), whereas it represses type I-E CRISPR-Cas system in E. coli (16,18). This suggests that different regulatory pathways operate in different prokaryotes.
To date, a complete and functional type I-A Cascade complex has been isolated only from one organism, the crenarchaeon Thermoproteus tenax, which contains all the six proteins encoded in the interference gene cassette, Csa5, Cas7, Cas5, Cas8a, Cas3΄ and Cas3″ (44). In Sulfolobus solfataricus, only a single subcomplex of the type I-A Cascade has been isolated containing Cas7 and Cas5 (45). Therefore, since we find that all the six protein components, and the crRNA, are involved in the complete repression of its own promoter, Sulfolobus type I-A Cascade probably has a similar composition to that of T. tenax.
Regardless of the classification of the CRISPR-Cas interference (types I-V), they are all involved in protospacer recognition and subsequent degradation of DNA/RNA targets (10,56). In this study, we demonstrate, for the first time, that the type I-A Cascade complex is directly involved in transcriptional repression of its own gene cassette. This mechanism enables the coupling of viral DNA surveillance to transcriptional activation, and thereby facilitates a rapid response to viral infections.
Supplementary Material
ACKNOWLEDGEMENTS
Ling Deng is thanked for construction of pGE1 and S. islandicus LAL549.
Footnotes
Present address: Wenfang Peng, Hubei Collaborative Innovation Center for Green Transformation of Bio-resources, Hubei Key Laboratory of Industrial Biotechnology, College of Life Sciences, Hubei University, Wuhan 430062, PR China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
EU FP7 project HotZyme [265933 to X.P.]; The Danish Council for Independent Research/Natural Sciences [DFF-4181-00274B to Q.S and X.P.]; Humboldt Research Fellowship for postdoctoral researchers [to G. V.]. Funding for open access charge: Copenhagen University.
Conflict of interest statement. None declared.
REFERENCES
- 1.Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P.. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007; 315:1709–1712. [DOI] [PubMed] [Google Scholar]
- 2.Makarova K.S., Haft D.H., Barrangou R., Brouns S.J., Charpentier E., Horvath P., Moineau S., Mojica F.J., Wolf Y.I., Yakunin A.F. et al. . Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011; 9:467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yosef I., Goren M.G., Qimron U.. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012; 40:5569–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nunez J.K., Kranzusch P.J., Noeske J., Wright A.V., Davies C.W., Doudna J.A.. Cas1-Cas2 complex formation mediates spacer acquisition during CRISPR-Cas adaptive immunity. Nat. Struct. Mol. Biol. 2014; 21:528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nunez J.K., Lee A.S., Engelman A., Doudna J.A.. Integrase-mediated spacer acquisition during CRISPR-Cas adaptive immunity. Nature. 2015; 519:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garrett R.A., Vestergaard G., Shah S.A.. Archaeal CRISPR-based immune systems: exchangeable functional modules. Trends Microbiol. 2011; 19:549–556. [DOI] [PubMed] [Google Scholar]
- 7.Horvath P., Barrangou R.. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010; 327:167–170. [DOI] [PubMed] [Google Scholar]
- 8.Terns M.P., Terns R.M.. CRISPR-based adaptive immune systems. Curr. Opin. Microbiol. 2011; 14:321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiedenheft B., Sternberg S.H., Doudna J.A.. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012; 482:331–338. [DOI] [PubMed] [Google Scholar]
- 10.Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J., Charpentier E., Haft D.H. et al. . An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015; 13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brouns S.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J., Snijders A.P., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J.. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008; 321:960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van der Oost J, Westra E.R., Jackson R.N., Wiedenheft B.. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat. Rev. Microbiol. 2014; 12:479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agari Y., Sakamoto K., Tamakoshi M., Oshima T., Kuramitsu S., Shinkai A.. Transcription profile of Thermus thermophilus CRISPR systems after phage infection. J. Mol. Biol. 2010; 395:270–281. [DOI] [PubMed] [Google Scholar]
- 14.Quax T.E., Voet M., Sismeiro O., Dillies M.A., Jagla B., Coppee J.Y., Sezonov G., Forterre P., van der Oost J., Lavigne R. et al. . Massive activation of archaeal defense genes during viral infection. J. Virol. 2013; 87:8419–8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pul U., Wurm R., Arslan Z., Geissen R., Hofmann N., Wagner R.. Identification and characterization of E. coli CRISPR-cas promoters and their silencing by H-NS. Mol. Microbiol. 2010; 75:1495–1512. [DOI] [PubMed] [Google Scholar]
- 16.Westra E.R., Pul U., Heidrich N., Jore M.M., Lundgren M., Stratmann T., Wurm R., Raine A., Mescher M., Van Heereveld L. et al. . H-NS-mediated repression of CRISPR-based immunity in Escherichia coli K12 can be relieved by the transcription activator LeuO. Mol. Microbiol. 2010; 77:1380–1393. [DOI] [PubMed] [Google Scholar]
- 17.Perez-Rodriguez R., Haitjema C., Huang Q.Q., Nam K.H., Bernardis S., Ke A.L., DeLisa M.P.. Envelope stress is a trigger of CRISPR RNA-mediated DNA silencing in Escherichia coli. Mol. Microbiol. 2011; 79:584–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang C.D., Chen Y.H., Huang H.Y., Huang H.D., Tseng C.P.. CRP represses the CRISPR/Cas system in Escherichia coli: evidence that endogenous CRISPR spacers impede phage P1 replication. Mol. Microbiol. 2014; 92:1072–1091. [DOI] [PubMed] [Google Scholar]
- 19.Patterson A.G., Chang J.T., Taylor C., Fineran P.C.. Regulation of the Type I-F CRISPR-Cas system by CRP-cAMP and GalM controls spacer acquisition and interference. Nucleic Acids Res. 2015; 43:6038–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yosef I., Goren M.G., Kiro R., Edgar R., Qimron U.. High-temperature protein G is essential for activity of the Escherichia coli clustered regularly interspaced short palindromic repeats (CRISPR)/Cas system. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:20136–20141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu T., Li Y., Wang X., Ye Q., Li H., Liang Y., She Q., Peng N.. Transcriptional regulator-mediated activation of adaptation genes triggers CRISPR de novo spacer acquisition. Nucleic Acids Res. 2015; 43:1044–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makarova K.S., Anantharaman V., Grishin N.V., Koonin E.V., Aravind L.. CARF and WYL domains: ligand-binding regulators of prokaryotic defense systems. Front. Genet. 2014; 5:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He F., Chen L., Peng X.. First experimental evidence for the presence of a CRISPR toxin in sulfolobus. J. Mol. Biol. 2014; 426:3683–3688. [DOI] [PubMed] [Google Scholar]
- 24.Gudbergsdottir S., Deng L., Chen Z., Jensen J.V., Jensen L.R., She Q., Garrett R.A.. Dynamic properties of the Sulfolobus CRISPR/Cas and CRISPR/Cmr systems when challenged with vector-borne viral and plasmid genes and protospacers. Mol. Microbiol. 2011; 79:35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okutan E., Deng L., Mirlashari S., Uldahl K., Halim M., Liu C., Garrett R.A., She Q., Peng X.. Novel insights into gene regulation of the rudivirus SIRV2 infecting Sulfolobus cells. RNA Biol. 2013; 10:875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deng L., He F., Bhoobalan-Chitty Y., Martinez-Alvarez L., Guo Y., Peng X.. Unveiling cell surface and type IV secretion proteins responsible for archaeal rudivirus entry. J. Virol. 2014; 88:10264–10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng L., Zhu H., Chen Z., Liang Y.X., She Q.. Unmarked gene deletion and host-vector system for the hyperthermophilic crenarchaeon Sulfolobus islandicus. Extremophiles. 2009; 13:735–746. [DOI] [PubMed] [Google Scholar]
- 28.Zillig W., Kletzin A., Schleper C., Holz I., Janekovic D., Hain J., Lanzendorfer M., Kristjansson J.K.. Screening for sulfolobales, their plasmids and their viruses in icelandic solfataras. Syst. Appl. Microbiol. 1994; 16:609–628. [Google Scholar]
- 29.Peng W., Feng M., Feng X., Liang Y.X., She Q.. An archaeal CRISPR type III-B system exhibiting distinctive RNA targeting features and mediating dual RNA and DNA interference. Nucleic Acids Res. 2015; 43:406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y., Pan S., Zhang Y., Ren M., Feng M., Peng N., Chen L., Liang Y.X., She Q.. Harnessing Type I and Type III CRISPR-Cas systems for genome editing. Nucleic Acids Res. 2016; 44:e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang C., Guo L., Deng L., Wu Y., Liang Y., Huang L., She Q.. Revealing the essentiality of multiple archaeal pcna genes using a mutant propagation assay based on an improved knockout method. Microbiology. 2010; 156:3386–3397. [DOI] [PubMed] [Google Scholar]
- 32.Zianni M., Tessanne K., Merighi M., Laguna R., Tabita F.R.. Identification of the DNA bases of a DNase I footprint by the use of dye primer sequencing on an automated capillary DNA analysis instrument. J. Biomol. Tech. 2006; 17:103–113. [PMC free article] [PubMed] [Google Scholar]
- 33.Peng N., Deng L., Mei Y., Jiang D., Hu Y., Awayez M., Liang Y., She Q.. A synthetic arabinose-inducible promoter confers high levels of recombinant protein expression in hyperthermophilic archaeon Sulfolobus islandicus. Appl. Environ. Microbiol. 2012; 78:5630–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng N., Xia Q., Chen Z., Liang Y.X., She Q.. An upstream activation element exerting differential transcriptional activation on an archaeal promoter. Mol. Microbiol. 2009; 74:928–939. [DOI] [PubMed] [Google Scholar]
- 35.Guo J., Zhao J., Li L., Chen Z., Wen Y., Li J.. The pathway-specific regulator AveR from Streptomyces avermitilis positively regulates avermectin production while it negatively affects oligomycin biosynthesis. Mol. Genet. Genomics. 2010; 283:123–133. [DOI] [PubMed] [Google Scholar]
- 36.Haft D.H., Selengut J.D., White O.. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003; 31:371–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eddy S.R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009; 23:205–211. [PubMed] [Google Scholar]
- 38.Pruitt K.D., Brown G.R., Hiatt S.M., Thibaud-Nissen F., Astashyn A., Ermolaeva O., Farrell C.M., Hart J., Landrum M.J., McGarvey K.M. et al. . RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014; 42:D756–D763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones D.T., Taylor W.R., Thornton J.M.. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992; 8:275–282. [DOI] [PubMed] [Google Scholar]
- 40.Kumar S., Stecher G., Tamura K.. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016; 33:1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J., White M.F.. Hot and crispy: CRISPR-Cas systems in the hyperthermophile Sulfolobus solfataricus. Biochem. Soc. Trans. 2013; 41:1422–1426. [DOI] [PubMed] [Google Scholar]
- 43.Peng W., Li H., Hallstrom S., Peng N., Liang Y.X., She Q.. Genetic determinants of PAM-dependent DNA targeting and pre-crRNA processing in Sulfolobus islandicus. RNA Biol. 2013; 10:738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Plagens A., Tripp V., Daume M., Sharma K., Klingl A., Hrle A., Conti E., Urlaub H., Randau L.. In vitro assembly and activity of an archaeal CRISPR-Cas type I-A Cascade interference complex. Nucleic Acids Res. 2014; 42:5125–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lintner N.G., Kerou M., Brumfield S.K., Graham S., Liu H., Naismith J.H., Sdano M., Peng N., She Q., Copie V. et al. . Structural and functional characterization of an archaeal clustered regularly interspaced short palindromic repeat (CRISPR)-associated complex for antiviral defense (CASCADE). J. Biol. Chem. 2011; 286:21643–21656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sinkunas T., Gasiunas G., Waghmare S.P., Dickman M.J., Barrangou R., Horvath P., Siksnys V.. In vitro reconstitution of Cascade-mediated CRISPR immunity in Streptococcus thermophilus. EMBO J. 2013; 32:385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Westra E.R., van Erp P.B., Kunne T., Wong S.P., Staals R.H., Seegers C.L., Bollen S., Jore M.M., Semenova E., Severinov K. et al. . CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol. Cell. 2012; 46:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jaubert C., Danioux C., Oberto J., Cortez D., Bize A., Krupovic M., She Q., Forterre P., Prangishvili D., Sezonov G.. Genomics and genetics of Sulfolobus islandicus LAL14/1, a model hyperthermophilic archaeon. Open Biol. 2013; 3:130010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mousaei M., Deng L., She Q., Garrett R.A.. Major and minor crRNA annealing sites facilitate low stringency DNA protospacer binding prior to Type I-A CRISPR-Cas interference in Sulfolobus. RNA Biol. 2016; 13:1166–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vestergaard G., Garrett R.A., Shah S.A.. CRISPR adaptive immune systems of Archaea. RNA Biol. 2014; 11:156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garrett R.A., Shah S.A., Erdmann S., Liu G., Mousaei M., Leon-Sobrino C., Peng W., Gudbergsdottir S., Deng L., Vestergaard G. et al. . CRISPR-Cas adaptive immune systems of the sulfolobales: Unravelling their complexity and diversity. Life (Basel). 2015; 5:783–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rousseau C., Gonnet M., Le Romancer M., Nicolas J.. CRISPI: a CRISPR interactive database. Bioinformatics. 2009; 25:3317–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Richter C., Chang J.T., Fineran P.C.. Function and regulation of clustered regularly interspaced short palindromic repeats (CRISPR) / CRISPR associated (Cas) systems. Viruses. 2012; 4:2291–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maniv I., Jiang W., Bikard D., Marraffini L.A.. Impact of different target sequences on type III CRISPR-Cas immunity. J. Bacteriol. 2016; 198:941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bondy-Denomy J., Pawluk A., Maxwell K.L., Davidson A.R.. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013; 493:429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Plagens A., Richter H., Charpentier E., Randau L.. DNA and RNA interference mechanisms by CRISPR-Cas surveillance complexes. FEMS Microbiol. Rev. 2015; 39:442–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.